The Intramolecular Radical Cation Induced Diels-Alder Reaction in the Diene - Diene Format

1
Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, USA
2
Department of Chemistry, University of Wisconsin - River Falls 410 S. Third St., River Falls, WI 54022, USA
*
Author to whom correspondence should be addressed.
Molecules 1997, 2(5), 80-86; https://doi.org/10.3390/20500080
Submission received: 8 April 1997
/
Accepted: 18 April 1997
/
Published: 15 May 1997
{kind=link}
{kind=link}
{kind=link}
Abstract
:Two 1,3,8,10-undecatetraenes were synthesized to test the viability of the intramolecular radical cation induced Diels-Alder reaction in the diene - diene format. Cyclization was successful only for the substituted tetraene with the lower oxidation potential. The major hexahydroindene product possessed a trans ring juncture demonstrating a selectivity for the endo stereochemistry.
Introduction
Knowledge about the radical cation induced Diels-Alder reaction has increased significantly in the past fifteen years. [3,4,5]. A chain mechanism was originally proposed [6], however, more recent evidence suggests a conventional catalytic mechanism [7,8,9].
Although a [4+1] cycloaddition pathway is symmetry allowed [10], the reaction appears virtually insensitive to symmetry considerations and the products of both [4+1] and [3+2] pathways have been observed [11]. Calculation of a minimum energy reaction pathway for the [3+2] cycloaddition using MINDO/3 revealed the likelihood that the reaction proceeds by a non-synchronous "bis-allylic" pathway [12]. The reaction shows a strong preference for formation of products having an endo stereochemical disposition. Most importantly, the radical cation induced reaction adds a new dimension to the synthetic utility of the Diels-Alder reaction since it is effective with substrates that are not amenable to reaction under neutral thermal conditions.
Bauld has categorized the starting materials that are known to participate in the radical cation Diels-Alder reaction into three groups: diene - styrene, diene - electron rich alkene, and diene - diene [13]. To date, Bauld has reported examples of intramolecular radical cation induced Diels-Alder reactions using the diene - styrene and diene - electron rich alkene formats [14,15]. Hertel et al. [16] have described a copper(I) catalyzed photo-induced cycloaddition of several bis-diene ethers which gave rise to some Diels-Alder [4+2] products as well as mixtures of [2+2], [4+4] and rearranged products. Herein we report our success with a radical cation initiated intramolecular Diels-Alder reaction in the diene - diene format.
Methods of initiation of radical cation reactions include anodic oxidation [17], photoinduced electron transfer (PET) [18], and introduction of a stable radical cation salt. This last method is particularly convenient, and the substance tris(p-bromophenyl)aminium hexachloroantimonate, (1), has become widely used for this purpose [19]. One limitation in its use is its low potential for oxidation of substrates (for the neutral amine, Eox= 1.05 V vs SCE)[20] which restricts its effectiveness as an initiator to substrates that have oxidation potentials close to or less than this value.

In selecting substrates with which to study the radical cation induced Diels-Alder reaction, the intramolecular arrangement of diene and dienophile reactants is attractive because of the facility with which bicyclic systems can be constructed. Gassman and coworkers [21,22] have previously observed protic acid catalyzed formation of Diels-Alder adducts from tetraenes such as (2). Treatment of (2) with the radical cation initiator (1) also induced the formation of Diels-Alder adducts, but this outcome was completely prevented by the inclusion of the hindered base, 2,6-di-t- butyl pyridine, in the reaction mixture. This result indicated that product formation in this example was due to the formation of catalytic amounts of hydrogen ion, and not the result of a radical cation mediated process. Lack of initiation by (1) in the presence of base has subsequently become one of several mechanistic criteria used to ensure that a reaction is truly proceeding by a radical cation pathway [13].
For our investigation of the radical cation induced Diels-Alder reaction in the diene - diene format, (3) was selected because of its optimal placement of a substituted electron rich diene together with an unhindered diene. In addition, Gorman and Gassman [22] have shown that this compound does not give Diels-Alder products under protic conditions. Compound (4), which lacks stabilizing methyl substituents, was included for comparison.

Results and Discussion
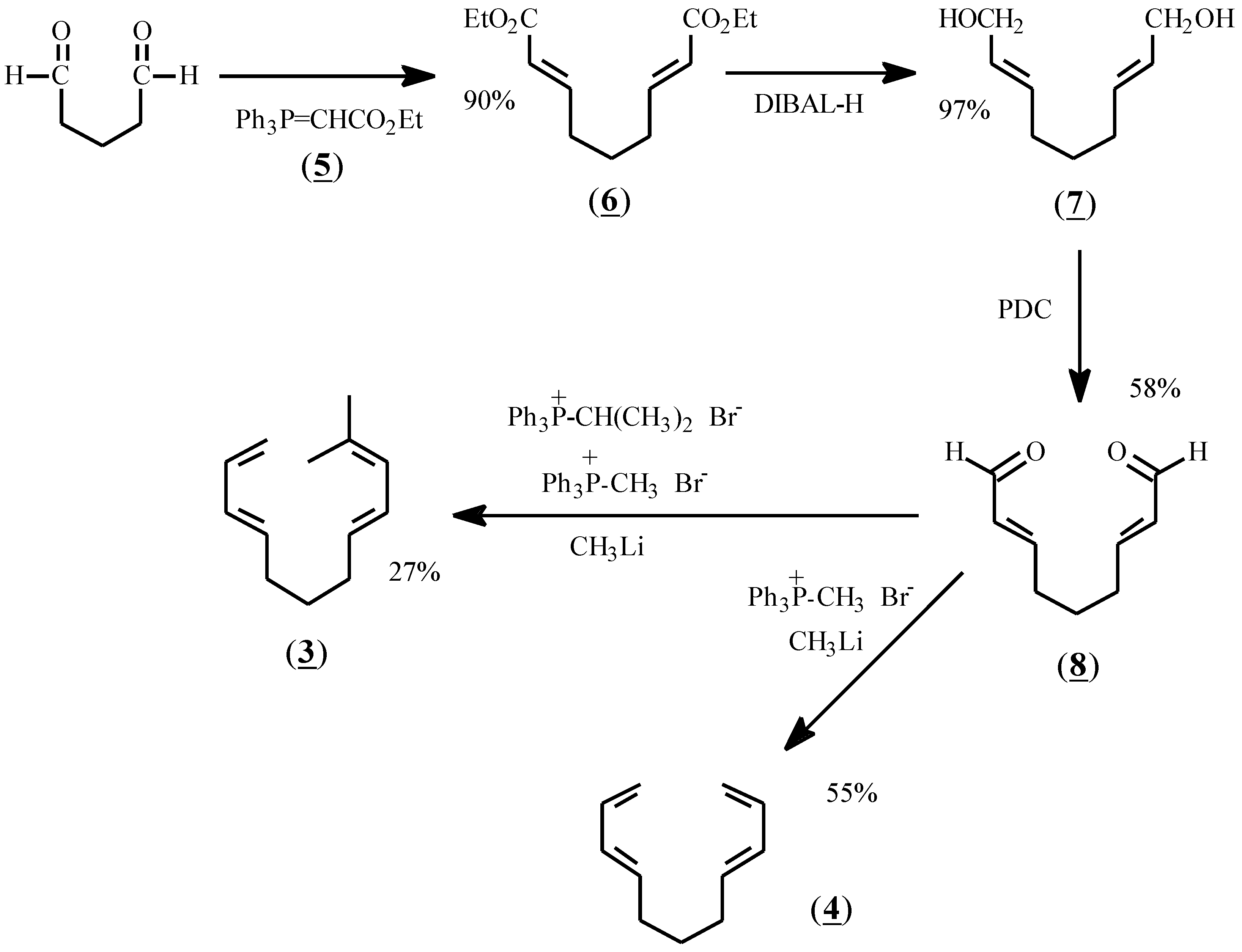
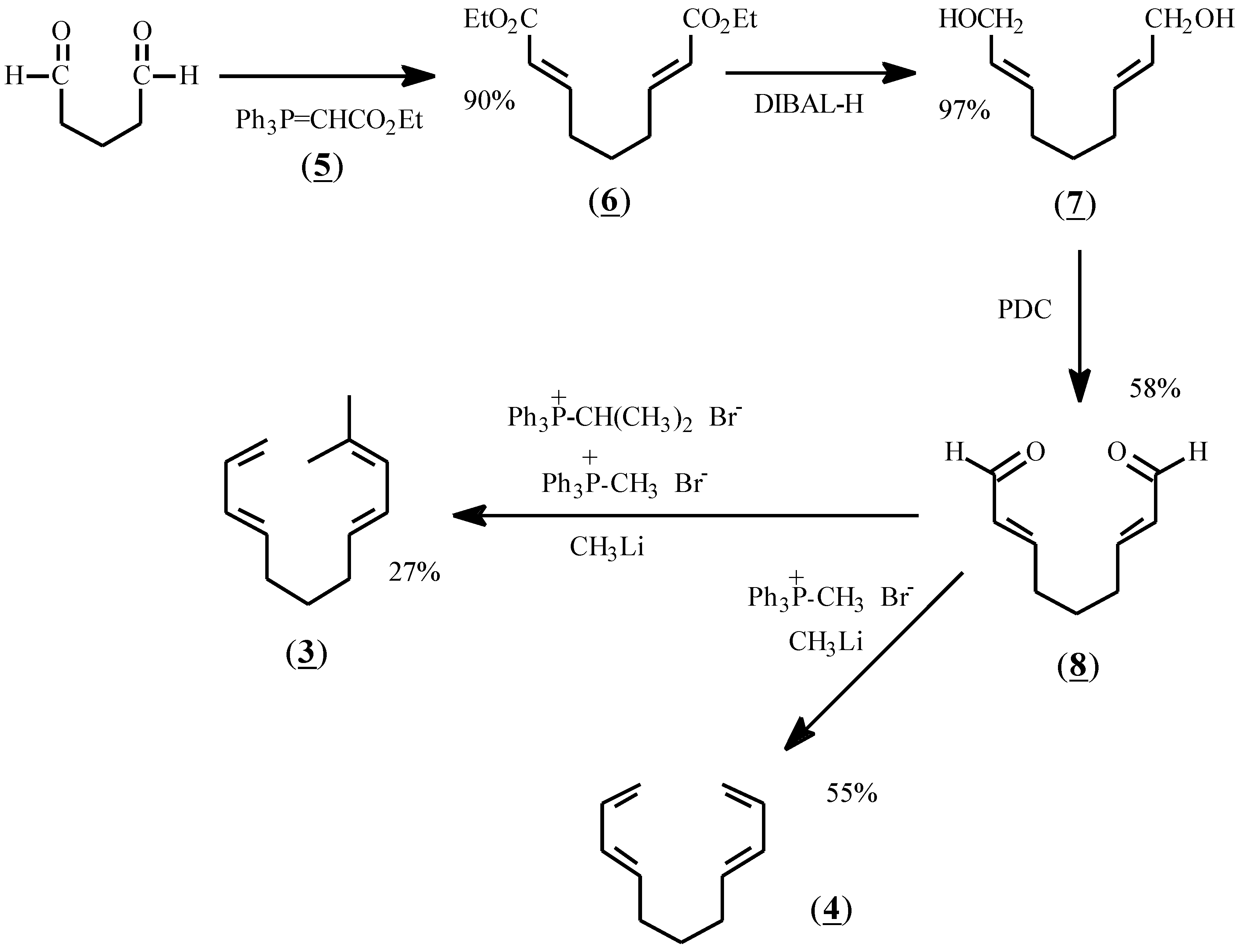
The reactants chosen for the present study were prepared according to Scheme I. Thus, glutaraldehyde was treated with stabilized ylide (5) to give diester (6) in 90% yield. Reduction with diisobutylaluminum hydride (DIBAL-H) afforded dialcohol (7), and oxidation with pyridinium dichromate (PDC) gave the dialdehyde, (8). Treatment of (8) with methylene phosphonium ylide in dry THF at -78 °C gave (4) as a colorless oil after flash chromatography. Treatment of (8) with a mixture of methylene and isopropylidene ylides afforded (3) in 27% yield after purification by preparative gas liquid chromatography.
The oxidation potentials of compounds (3) (Eox=1.20 V) and (4) (Eox=1.61 V) were determined in acetonitrile by cyclic voltammetry versus a standard calomel electrode using ferrocene as internal standard. Attempts to induce radical cation initiated reactions were conducted in dry CH2Cl2 at 0 °C. A solution of (1) in the same solvent was added at once to a solution of the tetraene and samples were withdrawn periodically for glc analysis.
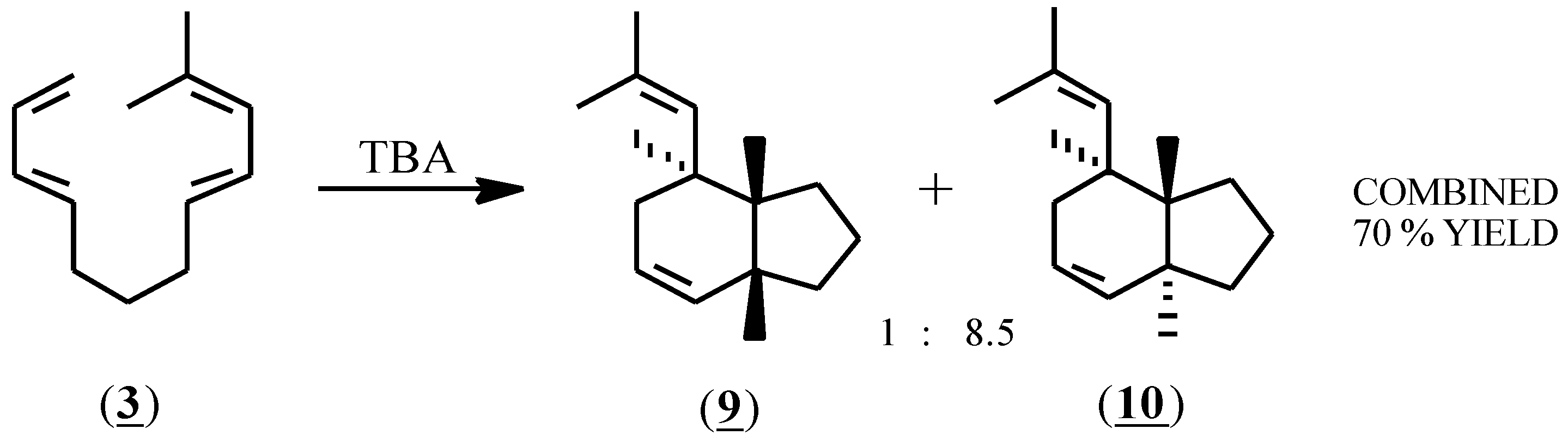
Compound (3) reacted rapidly with (1) producing one major and one minor product in an 8.5 to 1 ratio in a combined 70% yield (glc) (Scheme 2). Purification by preparative HPLC (reversed phase, 90% acetonitrile - water) afforded the purified components in 57% combined isolated yield. A variety of reaction conditions was investigated. At -78 °C no reaction occurred. At higher temperatures, (-23 °C, 0 °C and 22 °C), the reaction was typically complete within 3 minutes using 3 to 50 mol % of (1). Rapid addition of the reactant (3) to 20 mol % TBA afforded the highest combined yield (77%) by glc. The relative proportions of the products were the same in all cases. In acetonitrile solvent, lower yields (40-60%) were obtained and a larger proportion of (1) (35-50%) was required for complete consumption of the starting material. Support for a radical cation mediated mechanism was provided by the observation that the reaction was unimpaired by inclusion of 2,6-di-t-butylpyridine.
Scheme 1.
Synthetic sequence for the preparation of (3) and (4).
Scheme 2.

The cis ring juncture of the minor product (9) was verified using 1H-NMR spectroscopy and a series of decoupling experiments; a coupling constant of J = 7.25 Hz was found for the cis ring juncture hydrogens. For the major isomer (10), nuclear magnetic resonance spectra (1H-NMR, 13C-NMR, COSY, HETCOR, NOESY) and decoupling experiments were consistent with the proposed structure (10) but were unable to verify the trans ring juncture. Conversion of (10) to a bis(dichlorocarbene) adduct using chloroform, KOH and a phase transfer catalyst, followed by slow crystallization from acetonitrile produced crystals of the tetrachloro adduct (11), which by X-ray crystallography confirmed the trans ring juncture.

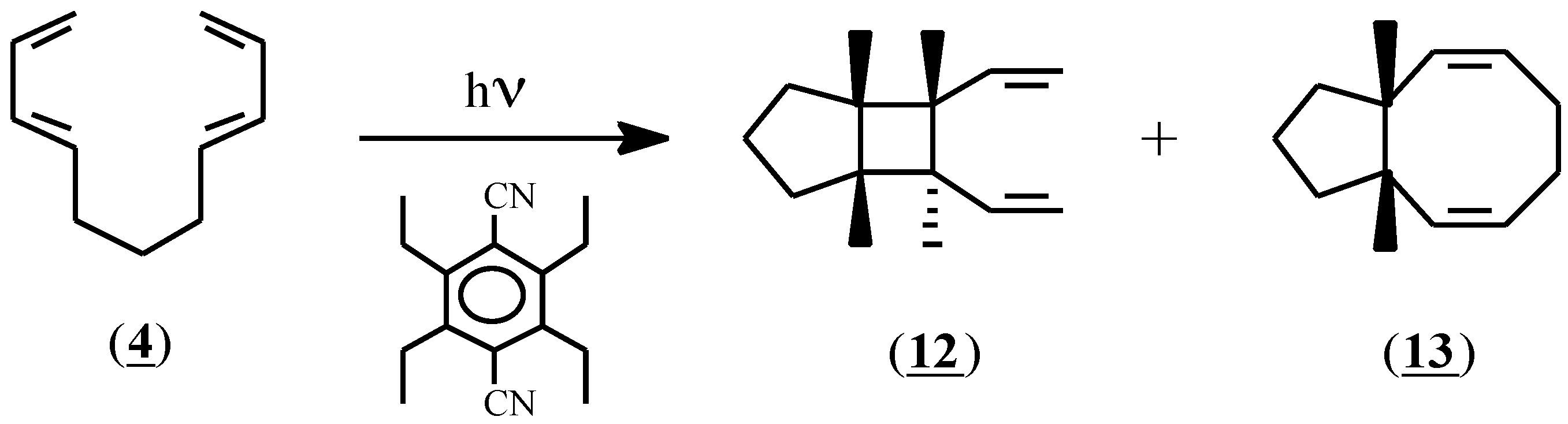
As expected, based on the differences in oxidation potential, the initiator (1) failed to induce Diels-Alder adduct formation with compound (4); no reaction had occurred after stirring for 24 hours at room temperature. In an attempt to expose (4) to more vigorous conditions, this compound was irradiated at 300nm in acetonitrile solution in the presence of the sensitizer 1,4-dicyano-2,3,5,6-tetraethylbenzene (Eox=1.96 V) [23]. In addition to conspicuous amounts of polymer, two soluble products were isolated by preparative gas liquid chromatography and were identified as (12) and (13) in 8% and 13% yield respectively (Scheme 3). Since these compounds are exactly those that would be predicted to result from an excited state reaction, the irradiation experiment was repeated with the sensitizer omitted. The same products were obtained, thereby indicating that a radical cation process was not involved in their formation, and that compound (4) does not convert to a Diels-Alder adduct under radical cation conditions.
Scheme 3.

Conclusion
To summarize, tetraenes (3) and (4) have been synthesized and have been exposed to reaction conditions expected to generate radical cation intermediates. Successful cyclization of (3) demonstrates the viability of the diene - diene format for the intramolecular radical cation initiated Diels-Alder reaction. In keeping with previous observations [14], a high endo to exo stereoselectivity (8.5:1) was observed. The contrast in reactivity of (3) versus (4) clearly demonstrates the importance of having at least one easily oxidized diene in the reactant.
Experimental
General
All nuclear magnetic resonance (NMR) spectra (1H-NMR, 13C-NMR) were recorded on an IBM NR/200 FT, IBM NR/300 FT or a Varian Unity 500 Nuclear Magnetic Resonance Spectrometer. Infrared spectra were recorded using a Mattson Polaris FT-IR spectrometer. Analytical GC was performed on a Hewlett Packard Model 5890A capillary gas chromatograph equipped with a 25 m x 0.2 mm HP-5 (0.33 micrometer cross-linked 5% Ph-Me silicone) column and a Hewlett Packard 3390A Reporting Integrator. Mass spectral analyses were performed on a Hewlett Packard 5995 Gas Chromatograph-Mass Spectrometer. Preparative gas chromatography was done using a Varian Aerograph Model 900 with a Fisher Recordall Series 5000 chart recorder. Preparative HPLC was accomplished using a Waters Model 6000A Solvent Delivery System with a 5 micron Lichrosorb RP-18 reversed phase 250 x 10 mm column, a Waters Model R401 differential refractometer detector and an Omniscribe D5000 strip chart recorder.
Diethyl (2E,7E)-nonadien-1,9-dioate (6)
Glutaraldehyde was purified by extraction (CH2Cl2) from aqueous solution (Aldrich), concentration and distillation in vacuo. A suspension of 90.1 g (0.21 mol) of (carbethoxymethyl)triphenylphosphonium bromide in 0.33 L of water was treated with 80 mL of 2.75 M aq. NaOH and extracted with 4 x 125 mL dichloromethane. The combined extracts were dried (MgSO4), filtered, and roto-evaporated to give 65.5 g (0.188 mol, 89.5% yield) of (carbethoxymethylene) triphenylphosphorane (5). This material was dissolved in 200 mL CH2Cl2 and added rapidly dropwise to a stirred solution of freshly distilled glutaraldehyde 8.96 g (0.090 mol) in 100 mL CH2Cl2 at 0 °C. The resulting solution was stirred at 0 °C for 0.5 h and at room temperature for 96 h, whereupon the solution was concentrated in vacuo. The solids (Ph3PO) were triturated in 300 mL Et2O, filtered, and the filtrate concentrated in vacuo. The resulting solids were treated again in a like manner with first 100 mL Et2O and then 100 mL of 10% EtOAc in hexane. The combined organic extracts were filtered and evaporated. The crude product (23 g) was flash chromatographed through a 6″ x 1″ column of silica eluted with 20% EtOAc in hexane. Evaporation of the first two 75 mL fractions and drying at 4 torr afforded 19.4 g (90.3%) of 6 as a clear colorless oil: 1H NMR (CDCl3) delta 6.85 (2H, d of t, J = 15.6, 7 Hz), 5.75 (2H, d, J = 15.6 Hz), 4.10 (4H, q, J = 7 Hz), 2.16 (4H, m), 1.56 (2H, quintet, J = 7 Hz), 1.21 (3H, t, J = 7 Hz); 13C NMR (CDCl3) delta 166.41 (s), 147.92 (d), 122.00 (d), 60.15 (t), 31.38 (t), 26.34 (t), 14.23 (q); IR (neat) 2983, 2937, 1722 (C=O), 1657 (C=C), 853. Anal. Calcd for C13H20O4: C, 64.96; H, 8.39. Found: C, 65.03; H, 8.30.
(2E,7E)-Nonadien-1,9-diol (7)
A solution of 9.71 g (0.04 mol) of 6 in 435 mL anhydrous ether was cooled to -78 °C and stirred magnetically under nitrogen while 162 mL (0.24 mol) of 1.5 M diisobutylaluminum hydride (DIBAL-H) in toluene (Aldrich) was added dropwise. The mixture was stirred for 1.25 h at -78 °C, 30 mL of MeOH was cautiously added and the mixture allowed to warm slowly to room temperature. The mixture was shaken vigorously with 435 mL of 25% aqueous sodium potassium tartrate. The aqueous layer was washed with 3 x 100 mL Et2O and the combined organic layers were dried (MgSO4), filtered, and roto-evaporated to give, after drying at 4 torr, 6.19 g (98%) of 7 as a clear colorless oil: 1H NMR (CDCl3) delta 5.53 (4H, m), 3.95 (4H, m), 3.46 (2H, t, J = 7.2 Hz), 1.98 (4H, m), 1.39 (2H, pentet, J = 7.7 Hz); 13C NMR (CDCl3) delta 132.37 (d), 129.37 (d), 63.20 (t), 31.67 (t), 28.51 (t); IR (neat) 3327 (O-H), 3008, 2926, 1670 (C=C), 1003, 969. Anal. Calcd for C9H16O2: C, 69.19; H, 10.32. Found: C, 69.11; H, 10.37.
(2E,7E)-Nonadien-1,9-dial (8)
To a magnetically-stirred suspension of 16.47 g (0.044 mol) pyridinium dichromate (PDC) in 70 mL CH2Cl2 under nitrogen at room temperature was added dropwise a solution of 2.28 g (0.015 mol) of 7 in 75 mL CH2Cl2. The mixture was stirred for 22 h, diluted with 225 mL ether, and filtered. The ppt was washed with 100 mL ether and the combined filtrates were concentrated in vacuo to give 1.63 g (73%) of crude product. The material was flash chromatographed through a 6″ x 1″ column of silica eluted with 50% EtOAc in hexane. Roto-evaporation of the combined eluates gave, after drying at 5 torr, 1.30 g (58.6%) of 8 as a faintly yellow clear oil: 1H NMR (CDCl3) delta 9.42 (2H, d, J = 7.8 Hz), 6.76 (2H, d of t, J= 15.6, 6.8 Hz), 6.04 (2H, d of d, J = 15.6, 7.8 Hz), 2.32 (4H, m), 1.67 (2H, pentet, J = 7.5 Hz); 13C NMR (CDCl3) delta 193.71 (d), 156.99 (d), 133.40 (d), 31.85 (t), 25.88 (t); IR (neat) 3358, 3028, 2935, 2740, 1688 (C=O), 1638 (C=C), 975. Anal. Calcd for C9H12O2: C, 71.02; H, 7.95. Found: C, 70.72; H, 8.03.
(3E,8E)-1,3,8,10-Undecatetraene (4)
To an ice-cold magnetically-stirred suspension of 5.37 g (0.015 mol) of oven-dried methyltriphenylphosphonium bromide (Aldrich) in 80 mL of dry THF under nitrogen was added in portions 11.4 mL (0.016 mol) of MeLi as a 1.36 M solution in ether (Aldrich). The resulting deep amber solution was stirred at room temperature for 0.5 h and then chilled to -78 °C. A solution of 1.09 g (0.00716 mol) of 8 in 35 mL dry THF was added dropwise to the cold ylide solution, stirred for 1.5 h, and allowed to warm to room temperature. The mixture was poured into 120 mL sat’d aqueous NH4Cl, shaken and the layers separated. The aqueous layer was washed with 3 x 60 mL ether and the combined organic layers were dried (MgSO4), filtered, and roto-evaporated. The residue was triturated in 45 mL of 2% EtOAc in hexane, filtered, concentrated and the crude product flash chromatographed through a 6″ x 1″ column of silica eluted with 2% EtOAc in hexane. Evaporation of the eluates and drying at 5 torr gave 0.58 g (54.7%) of 4 as a clear colorless oil: 1H NMR (CDCl3) delta 6.32 (2H, d of t, J = 16.8, 10.2 Hz), 6.05 (2H, m), 5.70 (2H, m), 5.05 (4H, m), 2.10 (4H, m), 1.53 (2H, pentet, J = 8.5 Hz); 13C NMR (CDCl3) delta 137.31 (d), 134.89 (d), 131.36 (d), 114.96 (t), 32.04 (t), 28.71 (t); IR (neat) 3087, 2928, 1799, 1653, 1602, 1001, 898. Anal. Calcd for C11H16: C, 89.12; H, 10.88. Found: C, 89.12; H, 11.00.
(3E,8E)-11-Methyl-1,3,8,10-dodecatetraene (3)
To a stirred suspension of 8.13 g (0.0228 mol) of methyltriphenyl phosphonium bromide (Aldrich) and 8.77 g (0.0228 mol) of isopropyl triphenylphosphonium bromide (Alfa) in 190 mL dry THF at room temperature under nitrogen was added 34.4 mL (0.0468 mol) of MeLi as a 1.36 M solution in ether. The deep red solution was cooled to -78 °C and a solution of 3.36 g (0.0217 mol) of 8 in 100 mL dry THF was added dropwise. The mixture was stirred at -78 °C for 25 min, allowed to warm to room temperature, and poured into 290 mL of sat’d aqueous NH4Cl. After shaking vigorously, the layers were separated and the aqueous portion extracted with 3 x 100 mL ether. The combined organic layers were dried (MgSO4), filtered, and roto-evaporated. The residue was repeatedly triturated in hexane, filtered and roto-evaporated. The crude product mixture was flash chromatographed through a 6″ x 1″ column of silica eluted with hexane. Evaporation of the solvent afforded 3.0 g (78.5%) of a mixture of 3, 4, and (4E,9E)-1,12-dimethyl-2,4,9,11-tridecatetraene (14). These were separated by repeatedly injecting 400 microliter aliquots onto a 13′ x .25″ column packed with 14% SE-30 on Chromosorb-W NAW. The carrier gas was helium with a flow rate of approximately 60 mL per min. The oven temperature was 210 °C. The components,3, 4 and 14 were recovered in the ratio of 3.2 to 1.0 to 3.0 respectively. The spectral characteristics of 4 have been described above. For 3 which appeared as a clear colorless oil: 1H-NMR (CDCl3) delta 6.4-6.0 (3H, m), 5.8-5.4 (3H, m), 5.09 (1H, d, J = 16.7), 4.97 (1H, d, J = 9.1), 2.10 (4H, m), 1.76 (6H, two singlets), 1.50 (2H, pentet, J = 7.7); 13C-NMR (CDCl3) delta 137.4 (d), 135.1 (d), 132.9 (s), 131.4 (d), 131.3 (d), 127.2 (d), 125.1 (d), 114.7 (t), 32.4 (t), 32.1 (t), 29.1 (t), 25.9 (q), 18.2 (q); IR (neat) 3086, 2930, 1653, 1603, 1440, 1002, 958, 897. Anal. Calcd for C13H20: C, 88.56; H, 11.44. Found: C, 88.53; H, 11.39.
[3aS,7R,7aR]- and [3aR,7S,7aS]-7-(2-Methylpropenyl)- 2,3,3a,6,7,7a-hexahydro-(1H)-indene (9) and [3aR,7R,7aR]- and [3aS,7S,7aS]-7-(2-Methylpropenyl)-2,3,3a,6,7,7a-hexahydro-(1H)-indene (10)
To a solution of 83.7 mg (0.475 mmol) of 3 in 50 mL of dry CH2Cl2 under nitrogen at room temperature was added dropwise with stirring 77.5 mg (0.095 mmol, 20 mol%) of tris(p-bromophenyl)aminium hexachloroanti-monate (1) in 10 mL CH2Cl2. Samples (0.5 mL) for glc analysis were withdrawn by pipette and filtered through a 10 mm column of silica gel in a disposable pipette. Complete consumption of the starting material was observed when a blue or bluish-green color was persistent in the reaction mixture. In some cases excess initiator was quenched by addition of a solution of sodium methoxide in methanol. Finally, the reaction mixture was roto-evaporated, the residue triturated in pentane, and the organic extracts filtered and concentrated to give the product as an oil contaminated with tris(p-bromophenyl) amine. Passage of the crude through a 13′ x 0.25″ column packed with 14% SE-30 on Chromosorb-W NAW at 210 °C eluted with He at 60 mL/min gave 47.4 mg (56.6%) of product (approx. 85% compound 10 by glc). The product mixture was dissolved in acetonitrile and passed through a 5 micron Lichrosorb RP-18 reversed phase 250 x 10 mm column eluted with 90% acetonitrile - 10% water at 2 kpsi. The minor fraction eluted first at 12.8 min, the major fraction at 16.6 min. The collected fractions were diluted with an equal volume of water and extracted with an equal volume of pentane which was back-washed with water, dried, and evaporated to give 4.7 mg (5.6%) of (9) and 40.4 mg (48.3%) of (10). For compound (9), 1H-NMR (CDCl3) delta 5.73 (1H, d of t, J= 9.5, 3.0 Hz), 5.64 (1H, m), 4.91 (1H, d, J=9 Hz), 2.35 (1H m), 2.13 (1H, q of d, J= 9.8, 4.9 Hz), 1.93 (1H, d of t, J=17.6, 4.9 Hz), 1.80 (2H, m), 1.75 (3H, s), 1.70 (3H, m), 1.66 (3H, s), 1.44 (2H, m) 1.26 (1H, m); 13C NMR (CDCl3) delta 131.0 (s), 130.8 (d), 129.8 (d), 125.4 (d), 42.4 (d), 40.0 (d), 35.1 (d), 32.6 (t), 31.1 (t), 30.0 (t), 25.9 (q), 23.9 (t), 18.1 (q); IR (neat) 3018 2970 2890 1650 1448 1394; Anal. Calcd for C13H20: C, 88.56; H, 11.44. Found: C, 88.41; H, 11.45. For compound (10), 1H-NMR (CDCl3) delta 5.80 (1H, d of d, J= 2.0, 10.0 Hz), 5.58 (1H, m), 4.95 (1H, d, J=10.0), 2.39 (1H, q of d, J= 10.3, 6.0 Hz), 1.82 (3H, m), 1.69 (3H, s), 1.66 (3H, m), 1.62 (3H, s), 1.18 (2H, m), 1.05 (1H, m); 13C NMR (CDCl3) delta 130.4 (s), 130.0 (d), 129.6 (d), 127.1 (d), 49.5 (d), 44.7 (d), 39.0 (d), 34.2 (t), 29.7 (t), 28.5 (t), 25.8 (q), 21.9 (t), 18.2 (q); IR (neat) 3017 2970 2876 1637 1453 1398; Anal. Calcd for C13H20: C, 88.56; H, 11.44. Found: C, 88.57; H, 11.35.
Acknowledgment
The authors thank the National Science Foundation (CHE-8921744) for support of this research.
References
- Current Address: Dow Chemical USA, Midland, MI 48674, USA.
- Deceased April 21, 1993. Formerly of The Department of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA.
- Bauld, N. L. Tetrahedron 1989, 45, 5307.
- Yueh, W.; Bauld, N.L. J. Chem. Soc., Perkin Trans. 2 1996, 1761.
- Schepp, N. P.; Johnston, L. J. J. Am. Chem. Soc. 1996, 118, 2872.
- Ledwith, A. Accts. Chem. Res. 1972, 5, 133.
- Eberson, L.; Olofsson, B. Acta Chem. Scand. 1991, 45, 316. [Google Scholar]
- Schmittel, M.; von Seggern, H. J. Am. Chem. Soc. 1993, 115, 2165.
- Yueh, W.; Bauld, N. L. J. Phys. Org. Chem. 1996, 9, 529.
- Bellville, D. J.; Bauld, N. L. J. Am. Chem. Soc. 1982, 104, 2665.
- Chockalingam, K.; Pinto, M.; Bauld, N. L. J. Am. Chem. Soc. 1990, 112, 447.
- Bauld, N. L.; Bellville, D. J.; Pabon, R. A. J. Am. Chem. Soc. 1983, 105, 2378.
- Reynolds, D. W.; Lorenz, K. T.; Chiou, H-S.; Bellville, D. J.; Pabon, R. A.; Bauld, N. L. J. Am. Chem. Soc. 1987, 109, 4960.
- Harirchian, B.; Bauld, N.L. Tetrahedron Lett. 1987, 28, 927.
- Mirafzal, G. A.; Kim, T.; Liu, J.; Bauld, N. L. J. Am. Chem. Soc. 1992, 114, 10968.
- Hertel, R.; Mattay, J.; Runsink, J. J. Am. Chem. Soc. 1991, 113, 657.
- Mlcoch, J.; Steckhan, E. Tetrahedron Lett. 1987, 28, 1081.
- Muller, F.; Mattay, J. Chem. Rev. 1993, 93, 99.
- Bell, F. A.; Ledwith, A.; Sherrington, D.C. J. Chem. Soc. (C) 1969, 2719.
- Seo, E. T.; Nelson, R. F.; Fritsch, J. M.; Marcoux, L. S.; Leedy, D. W.; Adams, R. N. J. Am. Chem. Soc. 1966, 88, 3498.
- Gassman, P. G.; Singleton, D. A. J. Am. Chem. Soc. 1984, 106, 6085.
- Gorman, D. B.; Gassman, P. G. J. Org. Chem. 1995, 60, 977.
- deSilva, S. personal communication.
- Sample Availability: Not available.
© 1997 MDPI. All rights reserved
Share and Cite
MDPI and ACS Style
Rusterholz, D.B.; Gorman, D.B.; Gassman, P.G. The Intramolecular Radical Cation Induced Diels-Alder Reaction in the Diene - Diene Format. Molecules 1997, 2, 80-86. https://doi.org/10.3390/20500080
AMA Style
Rusterholz DB, Gorman DB, Gassman PG. The Intramolecular Radical Cation Induced Diels-Alder Reaction in the Diene - Diene Format. Molecules. 1997; 2(5):80-86. https://doi.org/10.3390/20500080
Chicago/Turabian StyleRusterholz, David B., David B. Gorman, and Paul G. Gassman. 1997. "The Intramolecular Radical Cation Induced Diels-Alder Reaction in the Diene - Diene Format" Molecules 2, no. 5: 80-86. https://doi.org/10.3390/20500080