Cyclin-Dependent Kinase Inhibitors as Marketed Anticancer Drugs: Where Are We Now? A Short Survey

Abstract
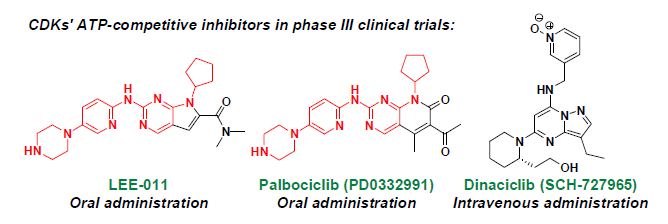
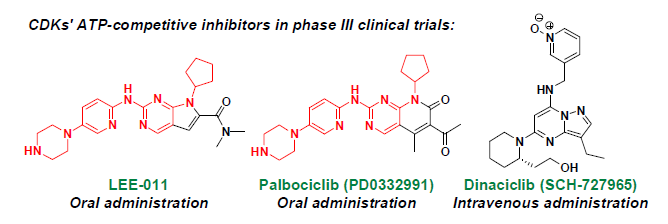
:
1. Introduction
2. ATP-Competitive Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Candidate | Company | Administration Mode | CDK Inhibition Profile: IC50 | Clinical Trial Stage |
|---|---|---|---|---|
| Flavopiridol | Sanofi-Aventis | intravenous | CDK1: 30 nM, CDK2: 100 nM CDK4: 20 nM, CDK6: 60 nM CDK7: 10 nM, CDK9: 10 nM | II |
| Roscovitine | Cyclacel | oral | CDK1: 2.7 μM, CDK2: 0.1 μM CDK7: 0.5 μM, CDK9: 0.8 μM | II |
| Dinaciclib | Merck | intravenous | CDK1: 3 nM, CDK2: 1 nM CDK5: 1 nM, CDK9: 4 nM | III |
| SNS032 | Sunesis | intravenous | CDK2: 38 nM, CDK7: 62 nM CDK9: 4 nM | I |
| AT7519 | Astex/Novartis | intravenous | CDK1: 190 nM, CDK2: 44 nM CDK4: 67 nM, CDK5: 18 nM CDK9: <10 nM | I/II |
| PD0332991 | Pfizer | oral | CDK4: 11 nM, CDK6: 16 nM | III |
| EM-1421 | Erimos | intravenous | CDK1: N/A | I/II |
| RGB-286638 | Agennix | intravenous | CDK1: 2 nM, CDK2: 3 nM CDK3: 5 nM, CDK4: 4 nM CDK9: 1 nM | I |
| P276-00 | Nicholas Piramal | intravenous | CDK9: 20 nM, CDK1: 79 nM CDK2: 224 nM, CDK4: 63 nM | II |
| BAY-1000394 | Bayer | oral | CDK1-4, 7, 9: 5-25 nM; | I |
| TG02/SG1317 | S*BIO/Tragara | oral | CDK9: 3nM, CDK5: 4 nM, CDK2: 5 nM, CDK3: 8 nM, CDK1: 9nM | I |
| PHA-848125 AC | Nerviano | oral | CDK1: 2 nM, CDK2: 3 nM CDK4: 5 nM, CDK5: 4 nM | II |
| LEE-011 | Novartis/Astex | oral | CDK4, 6: N/A | III |
| LY2835219 | Eli Lilly | oral | CDK4, 6: N/A | I/(III) |
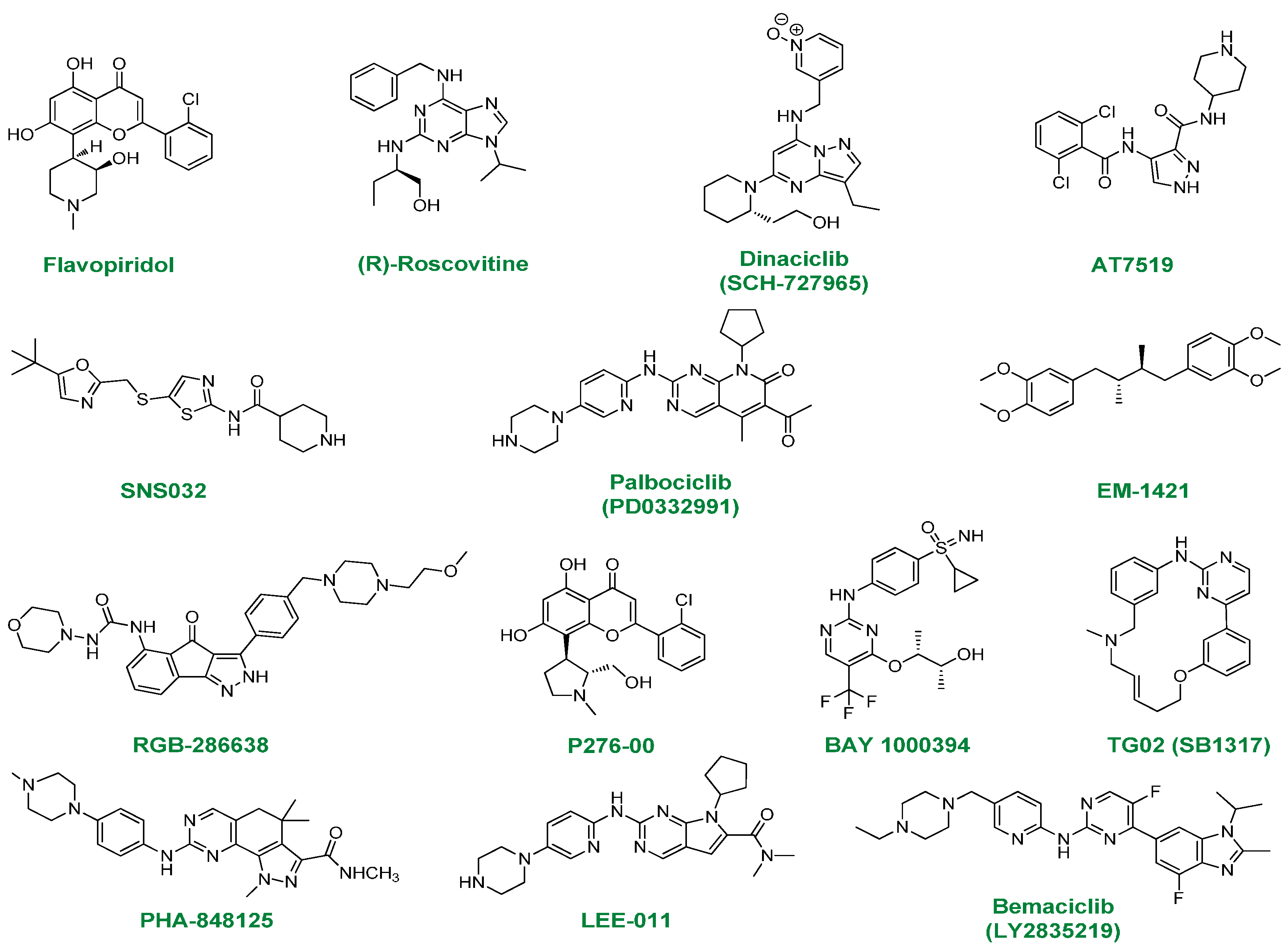
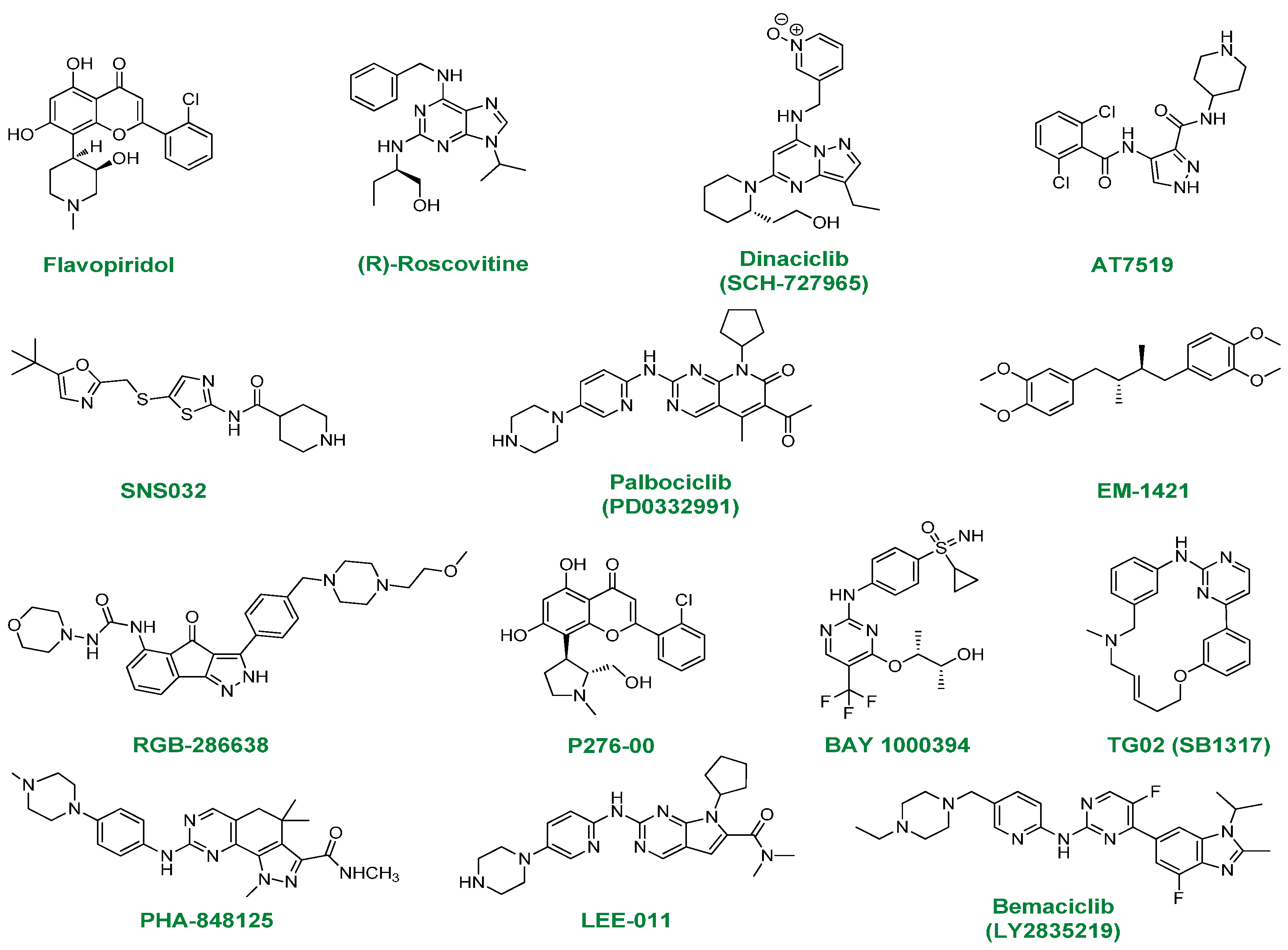
2.1. Flavopiridol
2.2. (R)-Roscovitine
2.3. Dinaciclib
2.4. AT7519
2.5. SNS032
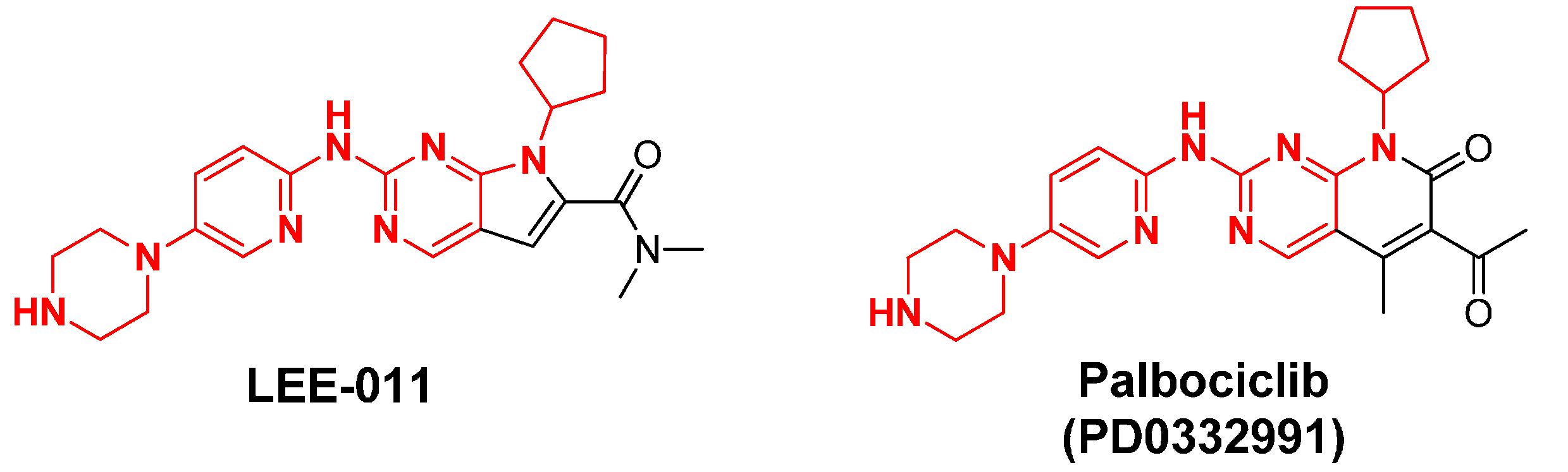
2.6. PD0332991
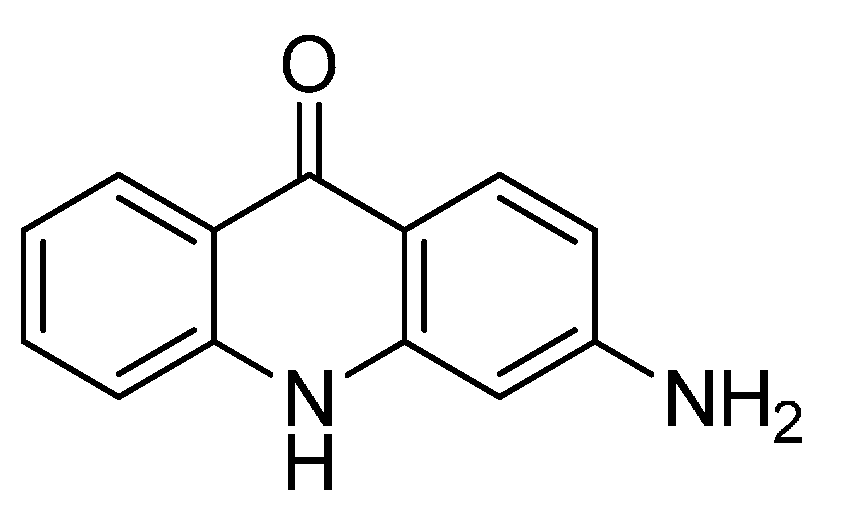
2.7. EM-1421
2.8. RGB-286638
2.9. P276-00
2.10. BAY-1000394
2.11. TG02
2.12. PHA-848125
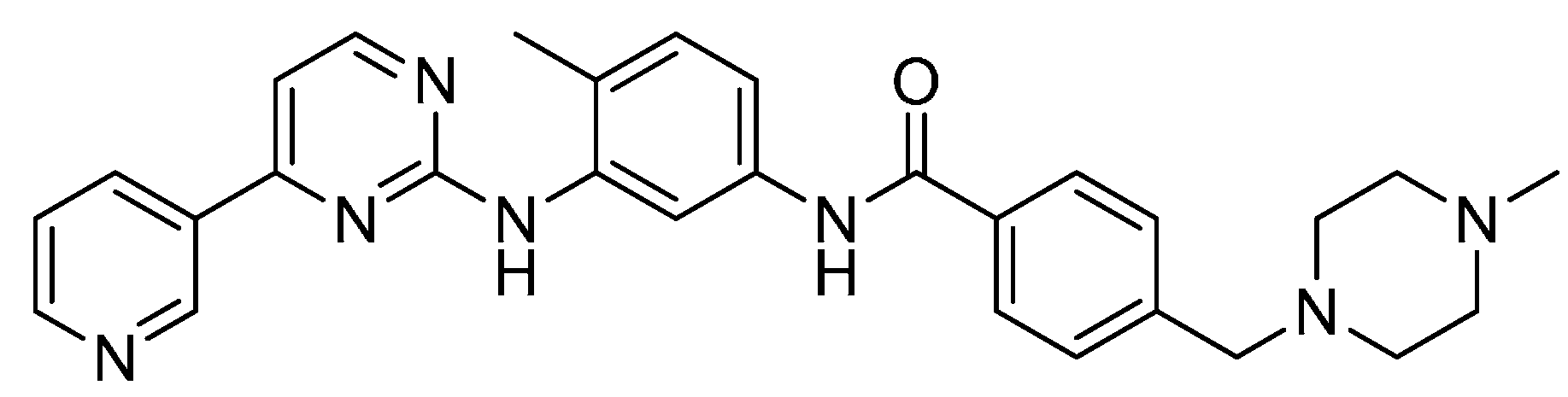
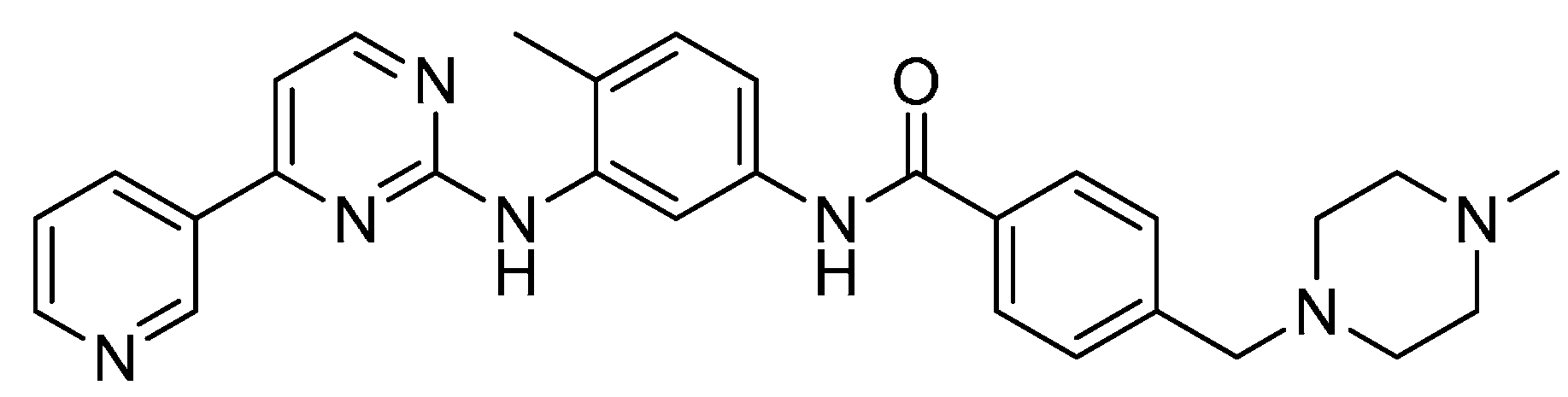
2.13. LEE-011
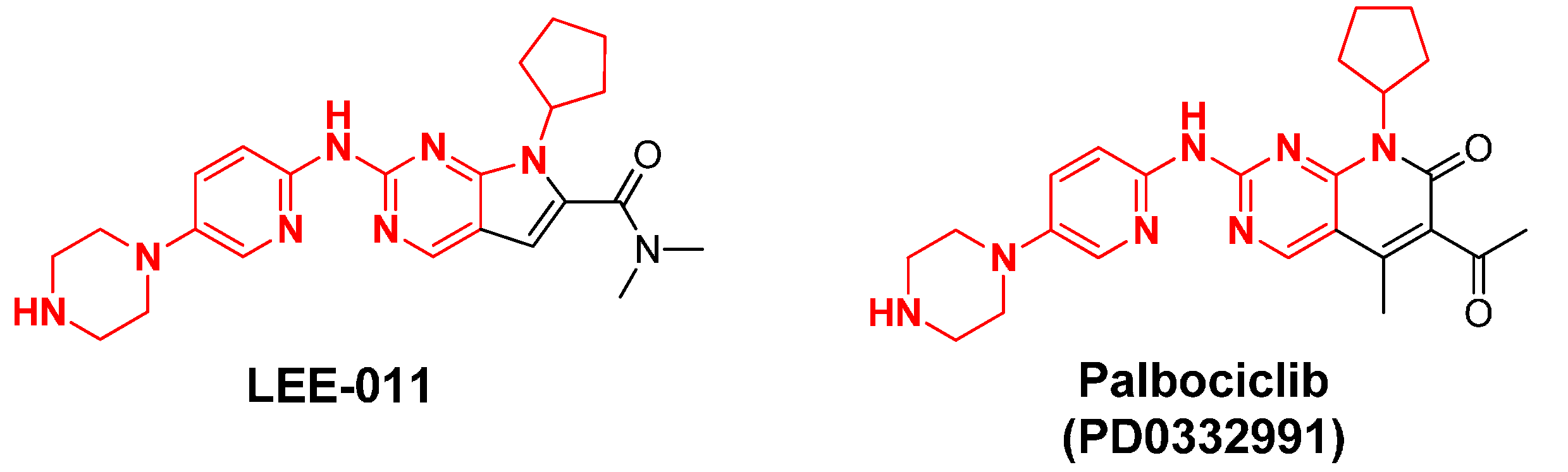
2.14. LY2835219
2.15. An Overview
3. ATP Non-Competitive Inhibitors
3.1. Small Molecules Inhibitors
3.2. Small Peptide Inhibitors
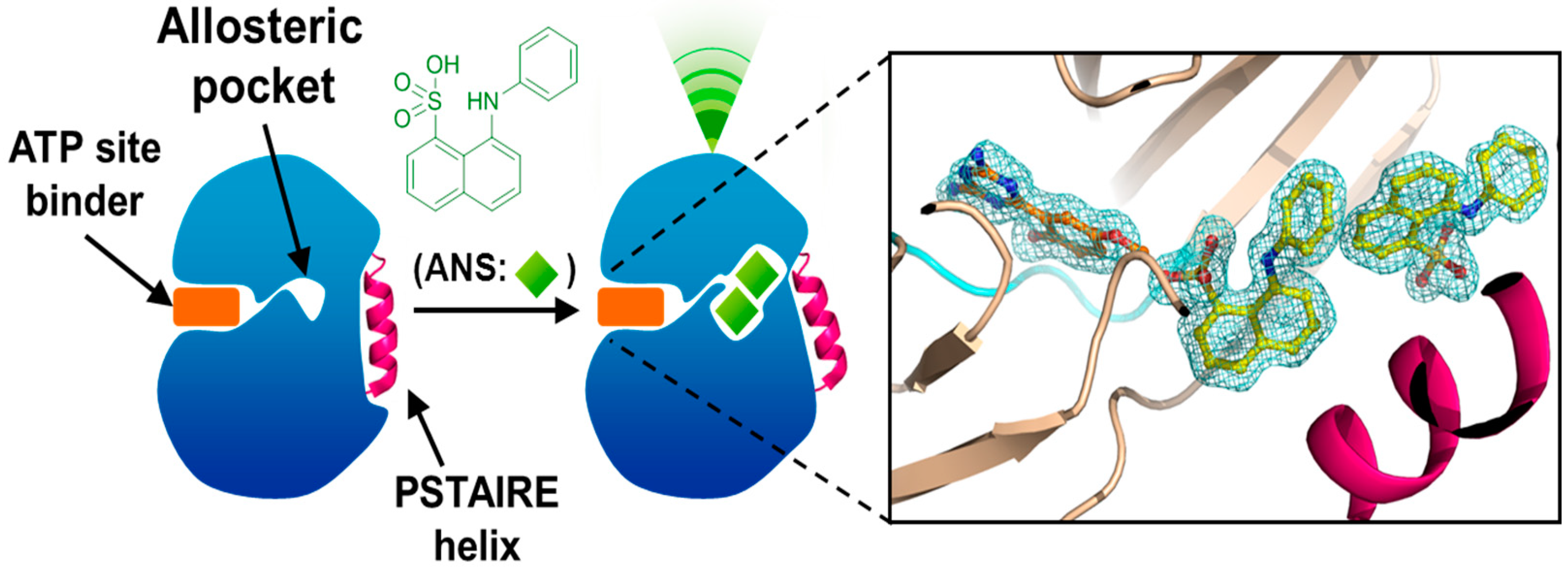
4. Allosteric Inhibitors
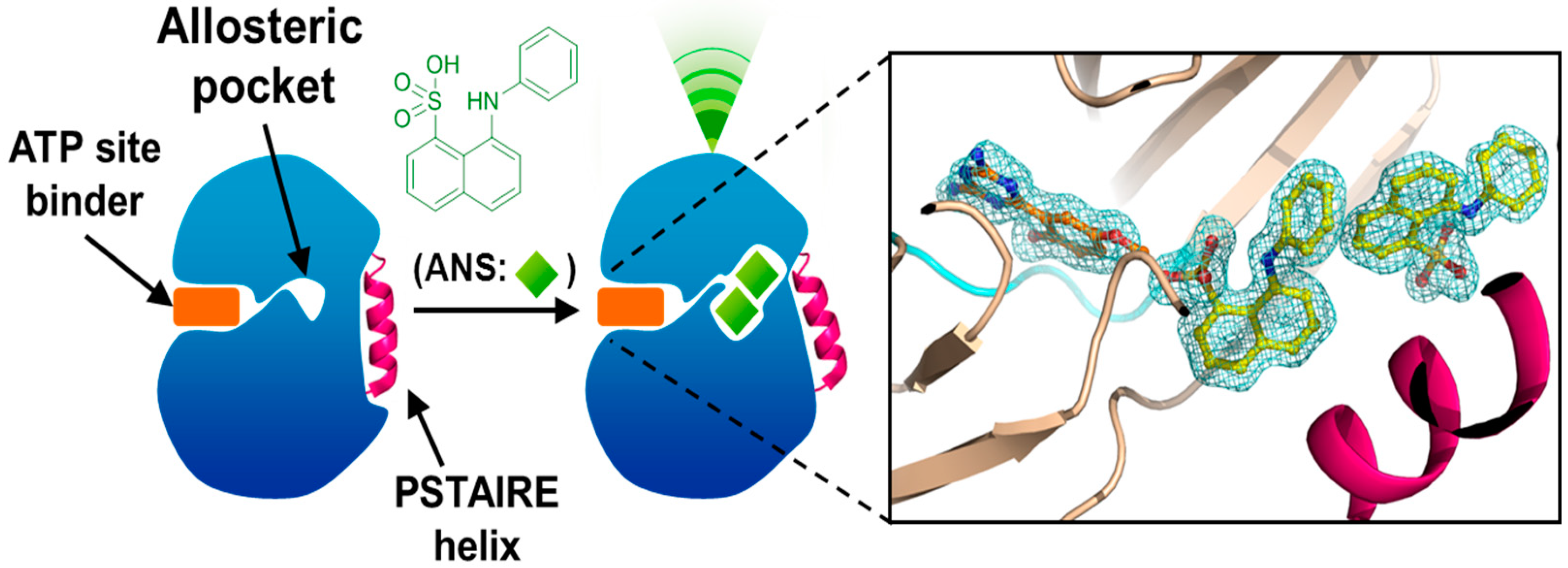
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Matthews, D.J.; Gerritsen, M.E. Targeting Protein Kinases for Cancer Therapy; John Wiley & sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Moen, M.D.; McKeage, K.; Plosker, G.L.; Siddiqui, M.A. Imatinib: A review of its use in chronic myeloid leukaemia. Drugs 2007, 67, 299–320. [Google Scholar] [CrossRef]
- Meijer, L. Le cycle de division cellulaire et sa régulation. Oncologie 2003, 5, 311–326. [Google Scholar]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Schwob, E. Nobel Prize in Medicine 2001: The universal key to cell division. Bull. Cancer 2001, 88, 937. [Google Scholar]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several CDKs, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef]
- Kohoutek, J.; Blazek, D. Cyclin K goes with CDK12 and CDK13. Cell Div. 2012. [Google Scholar] [CrossRef]
- De Bondt, H.L.; Rosenblatt, J.; Jancarik, J.; Jones, H.D.; Morgan, D.O.; Kim, S.H. Crystal structure of cyclin-dependent kinase 2. Nature 1993, 363, 596–602. [Google Scholar]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef]
- Solomon, M.J. Activation of the various cyclin/CDC2 protein kinases. Curr. Opin. Cell Biol. 1993, 5, 180–186. [Google Scholar] [CrossRef]
- Karlsson-Rosenthal, C.; Millar, J.B. CDC25: Mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006, 16, 285–292. [Google Scholar]
- Elledge, S.J.; Winston, J.; Harper, J.W. A question of balance: The role of cyclin-kinase inhibitors in development and tumorigenesis. Trends Cell Biol. 1996, 6, 388–392. [Google Scholar] [CrossRef]
- Kaldis, P. The CDK-activating kinase (CAK): From yeast to mammals. Cell. Mol. Life Sci. 1999, 55, 284–296. [Google Scholar] [CrossRef]
- Galons, H.; Oumata, N.; Meijer, L. Cyclin-dependent kinase inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2010, 20, 377–404. [Google Scholar] [CrossRef]
- Sharma, P.S.; Sharma, R.; Tyagi, R. Inhibitors of cyclin dependent kinases: Useful targets for cancer treatment. Curr. Cancer Drug Targets 2008, 8, 53–75. [Google Scholar] [CrossRef]
- Cicenas, J.; Valius, M. The CDK inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2011, 137, 1409–1418. [Google Scholar] [CrossRef]
- Fischer, P.M.; Gianella-Borradori, A. CDK inhibitors in clinical development for the treatment of cancer. Expert Opin. Investig. Drugs 2003, 12, 955–970. [Google Scholar] [CrossRef]
- Canavese, M.; Santo, L.; Raje, N. Cyclin dependent kinases in cancer: Potential for therapeutic intervention. Cancer Biol. Ther. 2012, 13, 451–457. [Google Scholar] [CrossRef]
- Diaz-Padilla, I.; Siu, L.L.; Duran, I. Cyclin-dependent kinase inhibitors as potential targeted anticancer agents. Investig. New Drugs 2009, 27, 586–594. [Google Scholar]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef]
- Galons, H.; Oumata, N.; Gloulou, O.; Meijer, L. Cyclin-dependent kinase inhibitors closer to market launch? Expert Opin. Ther. Pat. 2013, 23, 945–963. [Google Scholar] [CrossRef]
- Stone, A.; Sutherland, R.L.; Musgrove, E.A. Inhibitors of cell cycle kinases: Recent advances and future prospects as cancer therapeutics. Crit. Rev. Oncog. 2012, 17, 175–198. [Google Scholar] [CrossRef]
- De Azevedo, W.F.; Mueller-Dieckmann, H.J.; Schulze-Gahmen, U.; Worland, P.J.; Sausville, E.; Kim, S.H. Structural basis for specificity and potency of a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 2735–2740. [Google Scholar]
- Christian, B.A.; Grever, M.R.; Byrd, J.C.; Lin, T.S. Flavopiridol in the treatment of chronic lymphocytic leukemia. Curr. Opin. Oncol. 2007, 19, 573–578. [Google Scholar] [CrossRef]
- De Azevedo, W.F.; Leclerc, S.; Meijer, L.; Havlicek, L.; Strnad, M.; Kim, S.H. Inhibition of cyclin-dependent kinases by purine analogues: Crystal structure of human CDK2 complexed with roscovitine. FEBS J. 1997, 243, 518–526. [Google Scholar]
- Meijer, L.; Raymond, E. Roscovitine and other purines as kinase inhibitors. From starfish oocytes to clinical trials. Acc. Chem. Res. 2003, 36, 417–425. [Google Scholar]
- Whittaker, S.R.; Walton, M.I.; Garrett, M.D.; Workman, P. The Cyclin-dependent kinase inhibitor CYC202 (R-roscovitine) inhibits retinoblastoma protein phosphorylation, causes loss of Cyclin D1, and activates the mitogen-activated protein kinase pathway. Cancer Res. 2004, 64, 262–272. [Google Scholar] [CrossRef]
- Kaur, G.; Stetler-Stevenson, M.; Sebers, S.; Worland, P.; Sedlacek, H.; Myers, C.; Czech, J.; Naik, R.; Sausville, E. Growth inhibition with reversible cell cycle arrest of carcinoma cells by flavone L86–8275. J. Natl. Cancer Inst. 1992, 84, 1736–1740. [Google Scholar] [CrossRef]
- Abate, A.A.; Pentimalli, F.; Esposito, L.; Giordano, A. ATP-noncompetitive CDK inhibitors for cancer therapy: An overview. Expert Opin. Investig. Drugs 2013, 22, 895–906. [Google Scholar] [CrossRef]
- Guha, M. Cyclin-dependent kinase inhibitors move into Phase III. Nat. Rev. Drug Discov. 2012, 11, 892–894. [Google Scholar] [CrossRef]
- Zeidner, J.F.; Foster, M.C.; Blackford, A.; Litzow, M.R.; Morris, L.; Strickland, S.A.; Lancet, J.E.; Bose, P.; Levy, M.Y.; Tibes, R.; et al. Randomized multicenter phase II trial of timed-sequential therapy with flavopiridol (alvocidib), cytarabine, and mitoxantrone (FLAM) versus “7+3” for adults with newly diagnosed acute myeloid leukemia (AML). In Proceedings of the American Society of Clinical Oncology Conference, Chicago, IL, USA, 3 June 2014.
- McClue, S.J.; Blake, D.; Clarke, R.; Cowan, A.; Cummings, L.; Fischer, P.M.; MacKenzie, M.; Melville, J.; Stewart, K.; Wang, S.; et al. In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine). Int. J. Cancer 2002, 102, 463–468. [Google Scholar] [CrossRef]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.G.; Moulinoux, J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases CDC2, CDK2 and CDK5. FEBS J. 1997, 243, 527–536. [Google Scholar] [CrossRef]
- Cyclacel.com. Available online: http://www.cyclacel.com/research_programs_oncology_cyc202.shtml (accessed on 19 July 2014).
- A Phase 3 Study Comparing Dinaciclib versus Ofatumumab in Patients with Refractory Chronic Lymphocytic Leukemia (P07714 AM2). Available online: http://clinicaltrials.gov/ct2/show/NCT01580228?term=dinaciclib&rank=14 (accessed on 20 July 2014).
- i>Mahadevan, D.; Plummer, R.; Squires, M.S.; Rensvold, D.; Kurtin, S.; Pretzinger, C.; Dragovich, T.; Adams, J.; Lock, V.; Smith, D.M.; et al. A phase I pharmacokinetic and pharmacodynamic study of AT7519, a cyclin-dependent kinase inhibitor in patients with refractory solid tumors. ESMO meeting. Ann. Oncol. 2011, 22, 2137–2143. [Google Scholar]
- AT7519. Available online: http://clinicaltrials.gov/ct2/results?term=AT7519 (accessed on 20 July 2014).
- Tong, W.G.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.D.; Badros, A.Z.; Popplewell, L.; Coutre, S.; Fox, J.A.; et al. Phase I and pharmacologic study of SNS-032, a potent and selective CDK2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. ASCO Annual Meeting. J. Clin. Oncol. 2010, 28, 3015–3022. [Google Scholar] [CrossRef]
- Guha, M. Blockbuster dreams for Pfizer’s CDK inhibitor. Nat. Biotechnol. 2013, 31, 187. [Google Scholar] [CrossRef]
- Smolewski, P. Terameprocol, a novel site-specific transcription inhibitor with anticancer activity. IDrugs 2008, 11, 204–214. [Google Scholar]
- Phase 1 Study of Terameprocol (EM-1421) a Survivin and Cyclin-Dependent Kinase-1 (Cdc2) Inhibitor, in Patients with Leukemia. Available online: http://www.clinicaltrials.gov/ct2/results?term=EM-1421&Search=Search (accessed on 20 July 2014).
- Cirstea, D.; Hideshima, T.; Santo, L.; Eda, H.; Mishima, Y.; Nemani, N.; Hu, Y.; Mimura, N.; Cottini, F.; Gorgun, G.; et al. Small-molecule multi-targeted kinase inhibitor RGB-286638 triggers P53-dependent and -independent anti-multiple myeloma activity through inhibition of transcriptional CDKs. Leukemia 2013, 27, 2366–2375. [Google Scholar] [CrossRef]
- Phase 1 Open-label, Dose-escalation Clinical Study of the Safety and Tolerability of RGB-286638, a Novel, Multi-targeted Kinase Inhibitor, Administered to Patients with Selected, Relapsed or Refractory Hematological Malignancies. Available online: http://clinicaltrials.gov/ct2/show/NCT01168882?term=RGB-286638&rank=1 (accessed on 20 July 2014).
- Agennix.com. Available online: http://www.agennix.com (accessed on 20 July 2014).
- Kim, K.S.; Sack, J.S.; Tokarski, J.S.; Qian, L.; Chao, S.T.; Leith, L.; Kelly, Y.F.; Misra, R.N.; Hunt, J.T.; Kimball, S.D.; et al. Thio- and Oxoflavopiridols, Cyclin-Dependent Kinase 1-Selective Inhibitors: Synthesis and Biological Effects. J. Med. Chem. 2000, 43, 4126–4134. [Google Scholar] [CrossRef]
- Murthi, K.K.; Dubay, M.; McClure, C.; Brizuela, L.; Boisclair, M.D.; Worland, P.J.; Mansuri, M.M.; Pal, K. Structure-activity relationship studies of flavopiridol analogues. Bioorg. Med. Chem. Lett. 2000, 10, 1037–1041. [Google Scholar] [CrossRef]
- Joshi, K.S.; Rathos, M.J.; Mahajan, P.; Wagh, V.; Shenoy, S.; Bhatia, D.; Chile, S.; Sivakumar, M.; Maier, A.; Fiebig, H.H.; et al. P276–00, a novel cyclin-dependent inhibitor induces G1-G2 arrest, shows antitumor activity on cisplatin-resistant cells and significant in vivo efficacy in tumor models. Mol. Cancer Ther. 2007, 6, 926–934. [Google Scholar] [CrossRef]
- Siemeister, G.; Lucking, U.; Wengner, A.M.; Lienau, P.; Steinke, W.; Schatz, C.; Mumberg, D.; Ziegelbauer, K. BAY 1000394, a novel cyclin-dependent kinase inhibitor, with potent antitumor activity in mono- and in combination treatment upon oral application. Mol. Cancer Ther. 2012, 11, 2265–2273. [Google Scholar] [CrossRef]
- Lücking, U.; Jautelat, R.; Krüger, M.; Brumby, T.; Lienau, P.; Schäfer, M.; Briem, H.; Schulze, J.; Hillisch, A.; Reichel, A.; et al. The Lab Oddity Prevails: Discovery of Pan-CDK Inhibitor (R)-S-Cyclopropyl-S-(4-{[4-{[(1R,2R)-2-hydroxy-1-methylpropyl]oxy}-5-(trifluoromethyl)pyrimidin-2-yl]amino}phenyl)sulfoximide (BAY 1000394) for the Treatment of Cancer. ChemMedChem 2013, 8, 1067–1085. [Google Scholar]
- William, A.D.; Lee, A.C.H.; Blanchard, S.; Poulsen, A.; Teo, E.L.; Nagaraj, H.; Tan, E.; Chen, D.; Williams, M.; Sun, E.T.; et al. Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triazatetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa1(25),2(26),3,5,8,10,12(27),16,21, 23- decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma. J. Med. Chem. 2011, 54, 4638–4658. [Google Scholar]
- Goh, K.C.; Novotny-Diermayr, V.; Hart, S.; Ong, L.C.; Loh, Y.K.; Cheong, A.; Tan, Y.C.; Hu, C.; Jayaraman, R.; William, A.D.; et al. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia 2012, 26, 236–243. [Google Scholar] [CrossRef]
- Poulsen, A.; William, A.; Blanchard, S.; Nagaraj, H.; Williams, M.; Wang, H.; Lee, A.; Sun, E.; Teo, E.-L.; Tan, E.; et al. Structure-based design of nitrogen-linked macrocyclic kinase inhibitors leading to the clinical candidate SB1317/TG02, a potent inhibitor of cyclin dependant kinases (CDKs), Janus kinase 2 (JAK2), and Fms-like tyrosine kinase-3 (FLT3). J. Mol. Model. 2013, 19, 119–130. [Google Scholar] [CrossRef]
- Phase 1 Dose-Escalation and Pharmacokinetic Study of TG02 Citrate in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma. Available online: http://www.clinicaltrials.gov/ct2/show/NCT01699152?term=TG02&rank=2 (accessed on 24 July 2014).
- Phase 1 Dose-Escalation and Pharmacokinetic Study of TG02 Citrate in Patients with Advanced Hematological Malignancies. Available online: http://www.clinicaltrials.gov/ct2/show/NCT01204164?term=TG02&rank=1 (accessed on 24 July 2014).
- Phase II Study of Oral PHA-848125AC in Patients with Thymic Carcinoma Previously Treated with Chemotherapy. Available online: http://clinicaltrials.gov/show/NCT01011439 (accessed on 22 July 2014).
- Besse, B.; Garassino, M.C.; Rajan, A.; Novello, S.; Mazieres, J.; Weiss, G.J.; Ciomei, M.; Martignoni, M.; Petroccione, A.; Davite, C.; et al. A phase II study of milciclib (PHA-848125AC) in patients with thymic carcinoma. In Proceedings of the American Society of Clinical Oncology Conference, Chicago, IL, USA, 30 May 2014.
- Ciomei, M.; Scaburri, A. CDK Inhibitor for the Treatment of Mesothelioma. WO2010058006 A1, 27 May 2010. [Google Scholar]
- Kurt, S. LEE011 CDK Inhibitor Showing Early Promise in Drug-Resistant Cancers. Oncol. Times 2014, 36, 39–40. [Google Scholar]
- Macmillan Publishers Limited. CDK inhibitors speed ahead. Nat. Rev. Drug Discov. 2014, 13, 323. [Google Scholar] [CrossRef]
- A Phase 1b Study of LY2835219 in Combination with Endocrine Therapies for Patients with Hormone Receptor Positive, HER2 Negative Metastatic Breast Cancer. Available online: http://clinicaltrials.gov/ct2/show/NCT02057133?term=LY2835219&rank=5 (accessed on 25 July 2014).
- A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Study of Fulvestrant with or without LY2835219, a CDK4/6 Inhibitor, for Women with Hormone Receptor Positive, HER2 Negative Locally Advanced or Metastatic Breast Cancer. Available online: http://clinicaltrials.gov/ct2/show/NCT02107703?term=LY2835219&rank=9 (accessed on 25 July 2014).
- Kubo, A.; Nakagawa, K.; Varma, R.K.; Conrad, N.K.; Cheng, J.Q.; Lee, W.C.; Testa, J.R.; Johnson, B.E.; Kaye, F.J.; Kelley, M.J. The p16 status of tumor cell lines identifies small molecule inhibitors specific for cyclin-dependent kinase 4. AACR meeting. Clin. Cancer Res. 1999, 5, 4279–4286. [Google Scholar]
- Liggett, W.H., Jr.; Sidransky, D. Role of the p16 tumor suppressor gene in cancer. ASCO Annual Meeting. J. Clin. Oncol. 1998, 16, 1197–1206. [Google Scholar]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Adams, P.D.; Sellers, W.R.; Sharma, S.K.; Wu, A.D.; Nalin, C.M.; Kaelin, W.G., Jr. Identification of a cyclin-CDK2 recognition motif present in substrates and p21-like cyclin-dependent kinase inhibitors. Mol. Cell. Biol. 1996, 16, 6623–6633. [Google Scholar]
- Harper, J.W.; Adams, P.D. Cyclin-Dependent Kinases. Chem. Rev. 2001, 101, 2511–2526. [Google Scholar] [CrossRef]
- Gondeau, C.; Gerbal-Chaloin, S.; Bello, P.; Aldrian-Herrada, G.; Morris, M.C.; Divita, G. Design of a novel class of peptide inhibitors of cyclin-dependent kinase/cyclin activation. J. Biol. Chem. 2005, 280, 13793–13800. [Google Scholar] [CrossRef]
- Andrews, M.J.; McInnes, C.; Kontopidis, G.; Innes, L.; Cowan, A.; Plater, A.; Fischer, P.M. Design, synthesis, biological activity and structural analysis of cyclic peptide inhibitors targeting the substrate recruitment site of cyclin-dependent kinase complexes. Org. Biomol. Chem. 2004, 2, 2735–2741. [Google Scholar] [CrossRef]
- McInnes, C.; Andrews, M.J.; Zheleva, D.I.; Lane, D.P.; Fischer, P.M. Peptidomimetic design of CDK inhibitors targeting the recruitment site of the cyclin subunit. Curr. Med. Chem. Anti-Cancer Agents 2003, 3, 57–69. [Google Scholar]
- Liu, S.; Premnath, P.N.; Bolger, J.K.; Perkins, T.L.; Kirkland, L.O.; Kontopidis, G.; McInnes, C. Optimization of Non-ATP Competitive CDK/Cyclin Groove Inhibitors through REPLACE-Mediated Fragment Assembly. J. Med. Chem. 2013, 56, 1573–1582. [Google Scholar] [CrossRef]
- Pargellis, C.; Tong, L.; Churchill, L.; Cirillo, P.F.; Gilmore, T.; Graham, A.G.; Grob, P.M.; Hickey, E.R.; Moss, N.; Pav, S.; et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 2002, 9, 268–272. [Google Scholar] [CrossRef]
- Ohren, J.F.; Chen, H.; Pavlovsky, A.; Whitehead, C.; Zhang, E.; Kuffa, P.; Yan, C.; McConnell, P.; Spessard, C.; Banotai, C.; et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat. Struct. Mol. Biol. 2004, 11, 1192–1197. [Google Scholar] [CrossRef]
- Betzi, S.; Alam, R.; Martin, M.; Lubbers, D.J.; Han, H.; Jakkaraj, S.R.; Georg, G.I.; Schonbrunn, E. Discovery of a potential allosteric ligand binding site in CDK2. ACS Chem. Biol. 2011, 6, 492–501. [Google Scholar] [CrossRef]
- Ember, S.W.J.; Zhu, J.-Y.; Olesen, S.H.; Martin, M.P.; Becker, A.; Berndt, N.; Georg, G.I.; Schonbrunn, E. Acetyl-lysine Binding Site of Bromodomain-Containing Protein 4 (BRD4) Interacts with Diverse Kinase Inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. [Google Scholar] [CrossRef]
- Martin, M.P.; Olesen, S.H.; Georg, G.I.; Schonbrunn, E. Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains. ACS Chem. Biol. 2013, 8, 2360–2365. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mariaule, G.; Belmont, P. Cyclin-Dependent Kinase Inhibitors as Marketed Anticancer Drugs: Where Are We Now? A Short Survey. Molecules 2014, 19, 14366-14382. https://doi.org/10.3390/molecules190914366
Mariaule G, Belmont P. Cyclin-Dependent Kinase Inhibitors as Marketed Anticancer Drugs: Where Are We Now? A Short Survey. Molecules. 2014; 19(9):14366-14382. https://doi.org/10.3390/molecules190914366
Chicago/Turabian StyleMariaule, Gaëlle, and Philippe Belmont. 2014. "Cyclin-Dependent Kinase Inhibitors as Marketed Anticancer Drugs: Where Are We Now? A Short Survey" Molecules 19, no. 9: 14366-14382. https://doi.org/10.3390/molecules190914366