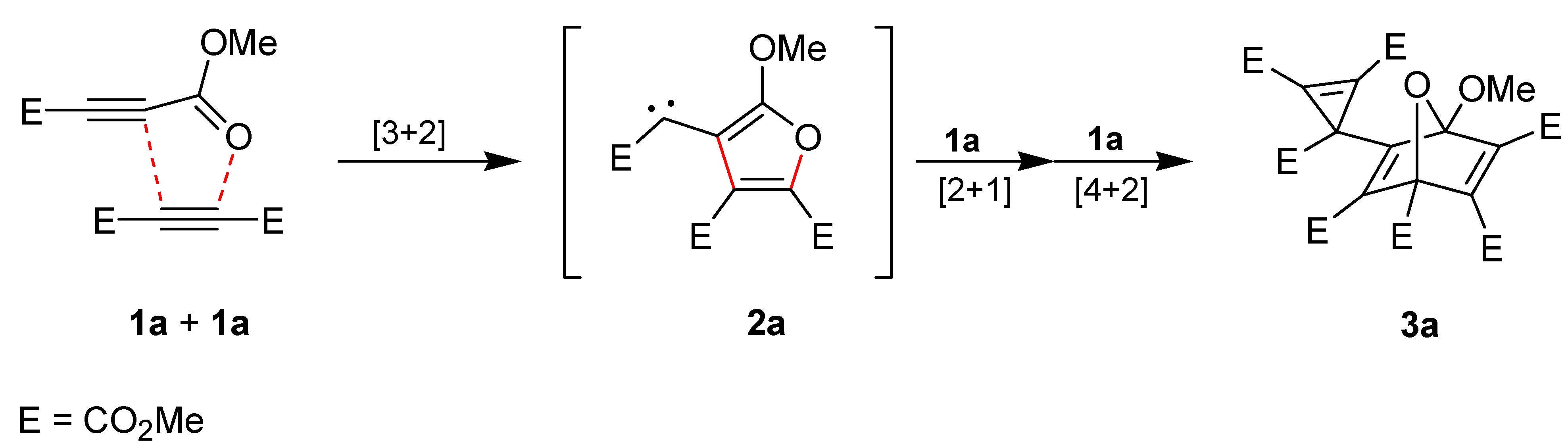
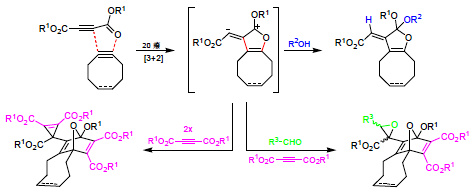
2. Results and Discussion
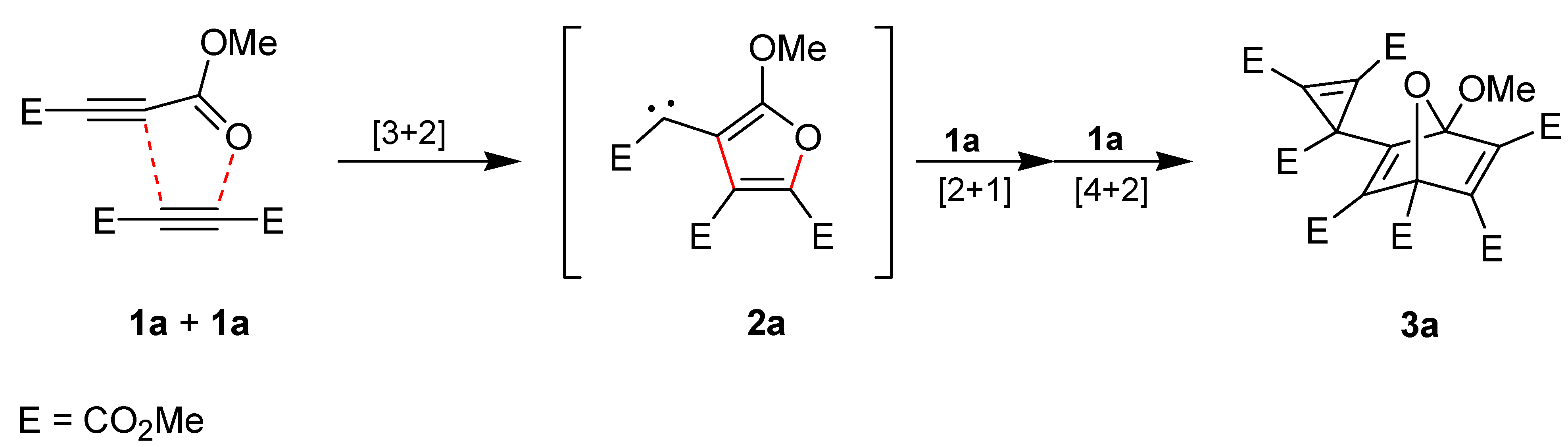
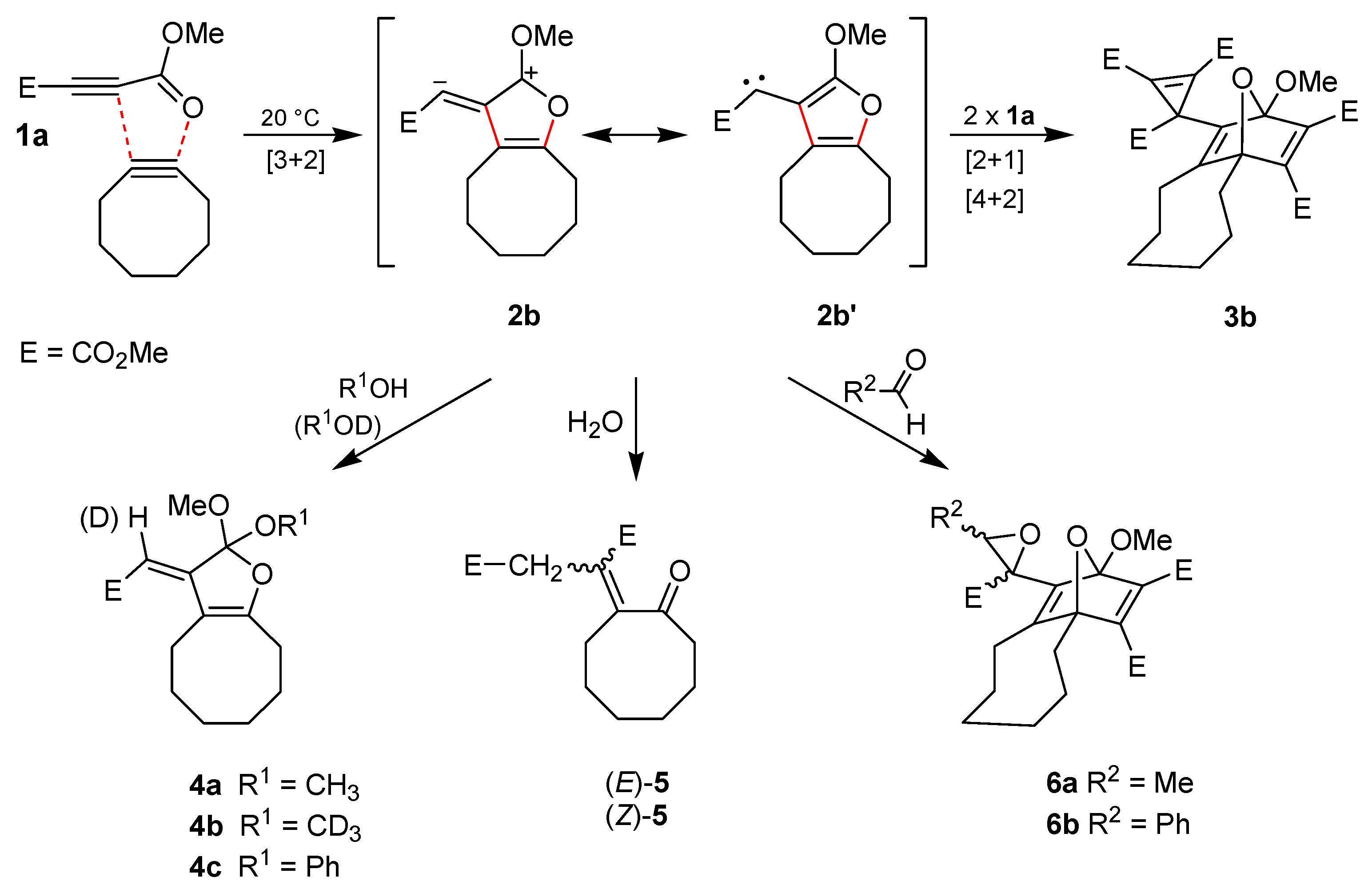
We accidentally found that the alkynes
1a and cyclooctyne [
19] undergo an exothermic reaction at room temperature [
20,
21,
22,
23]. Thus, the transformation was conveniently performed in dichloromethane (20 h/20 °C) and led to the crystalline product
3b with 79% yield (
Scheme 2). The structure of
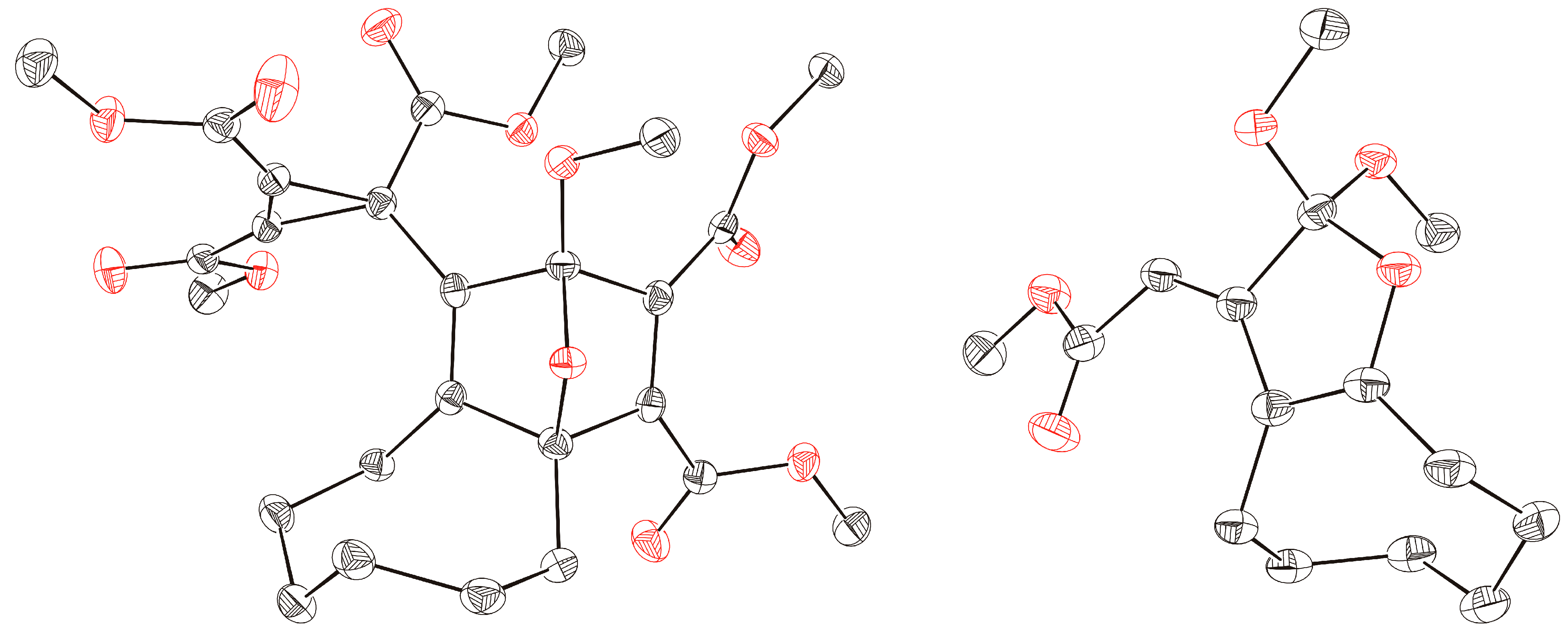
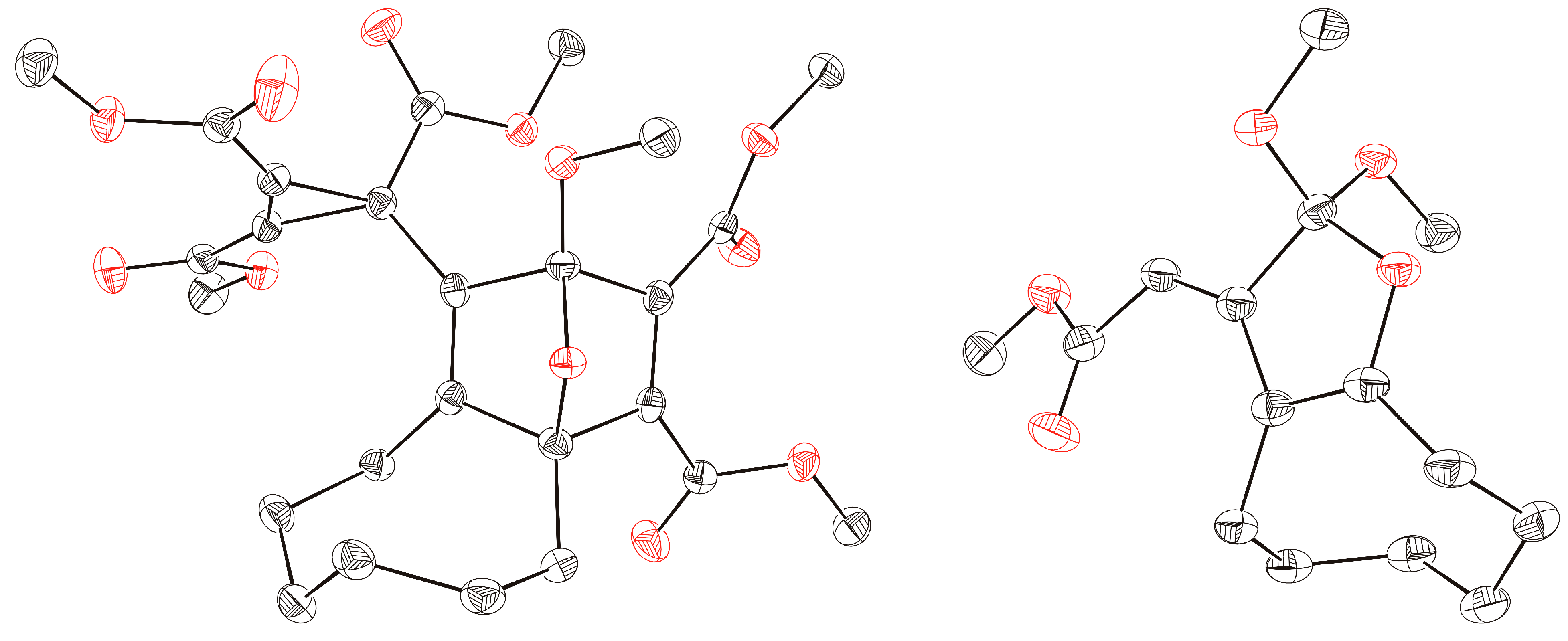
3b was confirmed not only by NMR spectroscopic data but also by single crystal X-ray diffraction analysis (
Figure 1). Obviously, the 1,3-dipolar cycloaddition of
1a at the ring-strained dipolarophile cyclooctyne to generate the intermediate
2b is much more rapid than the dimerization of
1a to produce
2a. This allows the synthesis of the interception product
3b without heating or long reaction times and also formation of several other trapping products of
2b.
Scheme 2.
Reaction of 1a with cyclooctyne in the absence or in the presence of other reagents.
Scheme 2.
Reaction of 1a with cyclooctyne in the absence or in the presence of other reagents.
Figure 1.
ORTEP representation (50% probability level) of the molecular structures of products 3b (left) and 4a (right); hydrogen atoms are omitted for clarity.
Figure 1.
ORTEP representation (50% probability level) of the molecular structures of products 3b (left) and 4a (right); hydrogen atoms are omitted for clarity.
After treatment of cyclooctyne with
1a in methanol instead of dichloromethane, the orthoester
4a was isolated in 60% yield. The surprising structure and especially the stereochemistry of this product were proved by spectroscopic data and X-ray diffraction analysis (
Figure 1). When
1a and cyclooctyne were analogously reacted in an excess of deuterated methanol (CD
3OD), the product
4b, which indicated the selective incorporation of exactly one equivalent of the deuterated reagent, was obtained with 62% yield. In the presence of phenol instead of methanol, the conversion of
1a and cyclooctyne only led to a low yield (10%) of the corresponding orthoester
4c. We did not get any orthoester trapping product with a structure similar to that of
4 after heating
1a alone in pure methanol (up to 80 °C) because addition of the solvent at the C≡C bond to form dimethyl 2-methoxybut-2-enedioates dominated.
In the case of 4a–c, we exclusively isolated the depicted (E)-stereoisomer, whereas a mixture of (E)-5 and (Z)-5 resulted after exposure of cyclooctyne to 1a in aqueous tetrahydrofuran. The latter products were separated by chromatography to yield (E)-5 (21%) and (Z)-5 (20%), which were assigned with the help of NOE-NMR experiments. The genesis of 5 can be explained through interception of dipolar intermediate 2b by water. The product of this step is similar to orthoesters 4 but includes the substructure of a cyclic hemiacetal, which is transformed into 5 by ring opening followed by tautomerism.
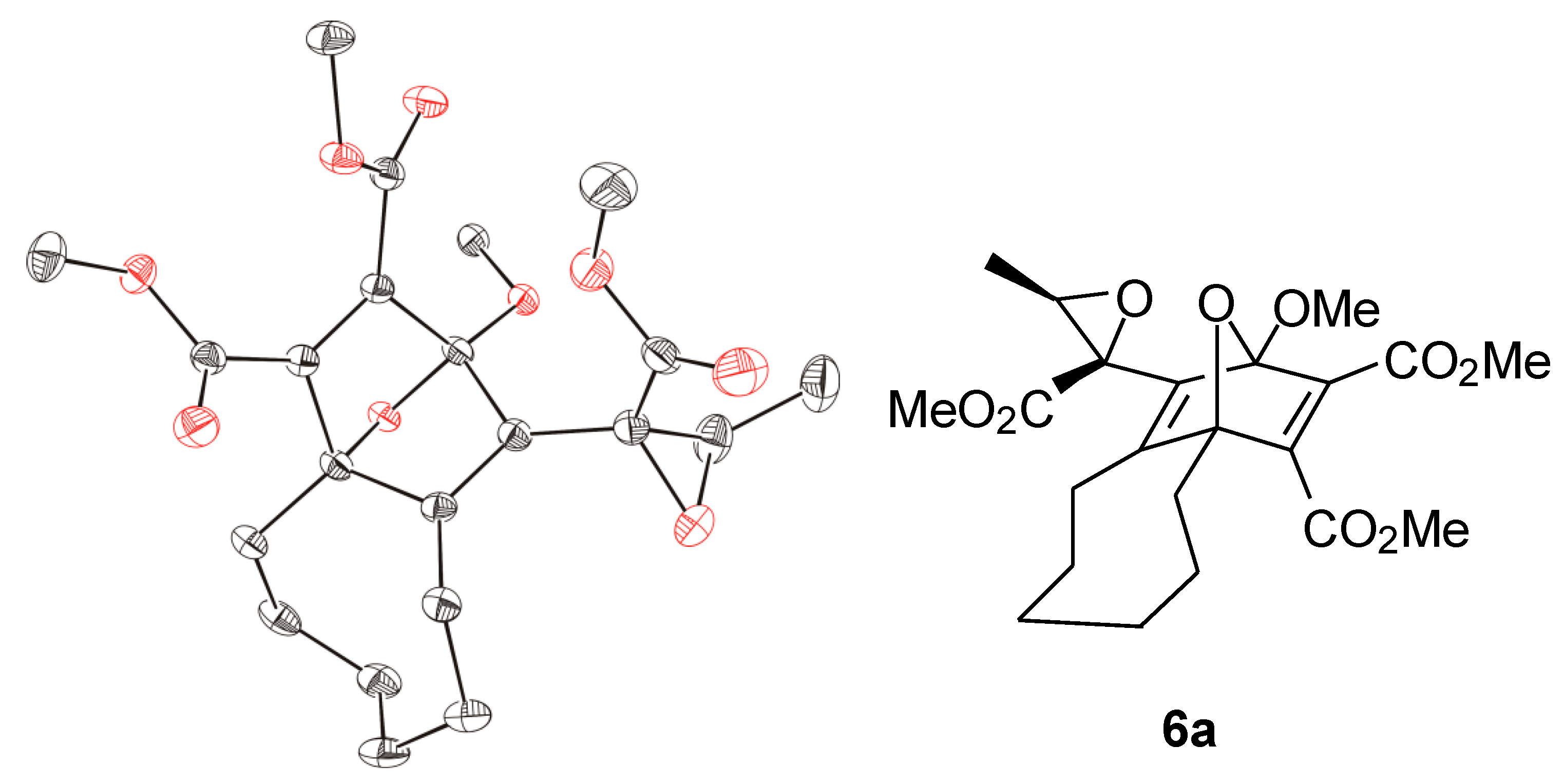
When aldehydes such as acetaldehyde or benzaldehyde were used as solvents for the reaction of cyclooctyne with
1a, epoxides
6a and
6b, respectively, were formed (
Scheme 2). In the case of
6a, the
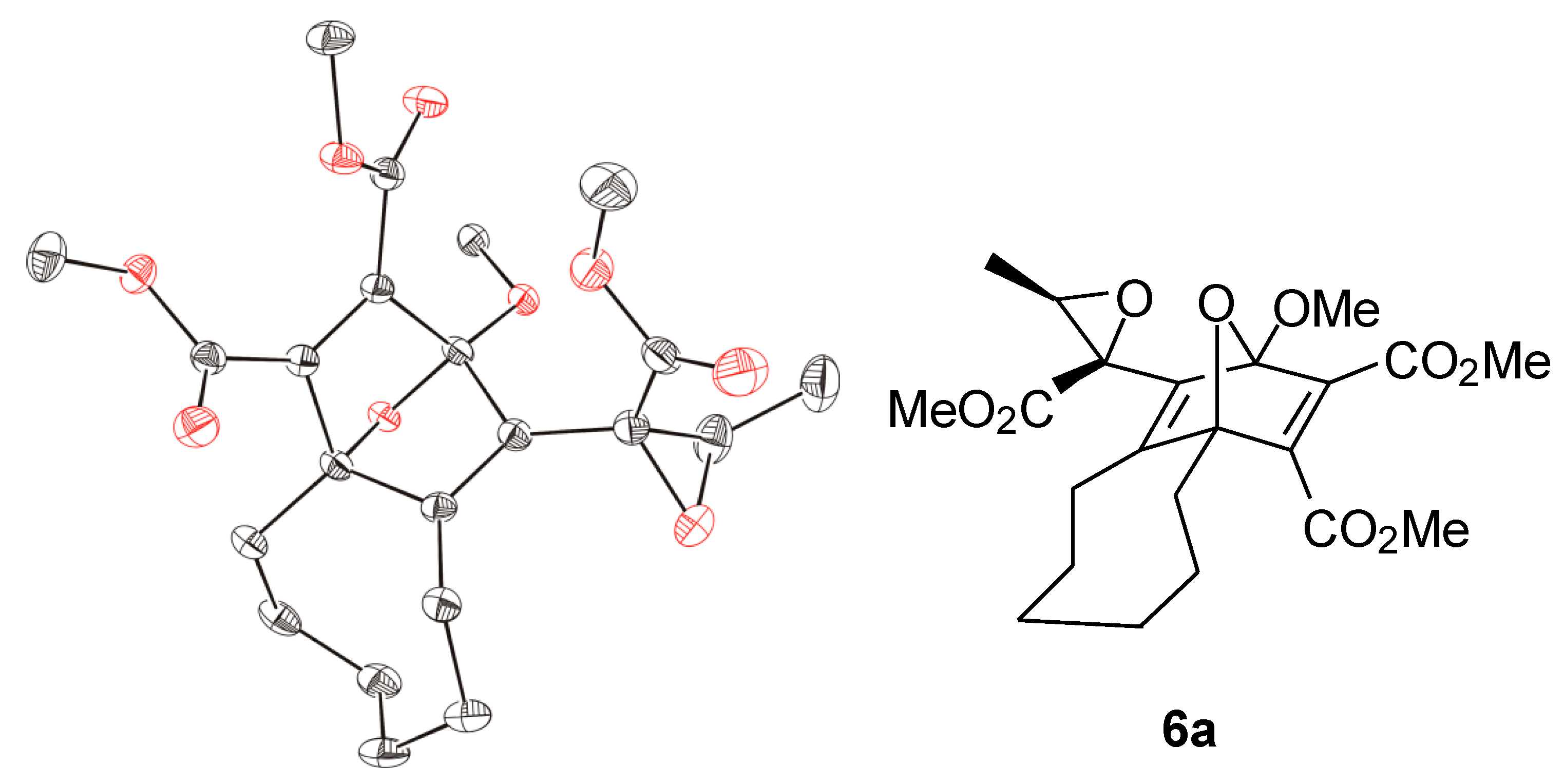
1H-NMR spectrum of the crude reaction mixture indicated the generation of two diastereomers in a ratio of about 3.5:1 although four diastereomers are possible. Only the main product, however, could be isolated by chromatography (22% yield), and the relative configurations of its stereocenters were determined by single crystal X-ray diffraction analysis (
Figure 2). The epoxide
6b was obtained in 21% yield as a mixture of two diastereomers, which could be separated by chromatography.
Figure 2.
ORTEP representation (30% probability level) of the molecular structure of 6a; hydrogen atoms are omitted for clarity; only one of the two enantiomers of the asymmetric unit is shown.
Figure 2.
ORTEP representation (30% probability level) of the molecular structure of 6a; hydrogen atoms are omitted for clarity; only one of the two enantiomers of the asymmetric unit is shown.
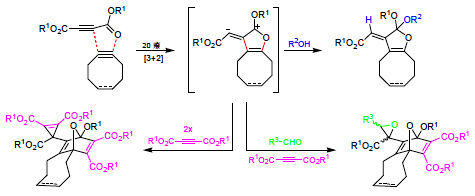
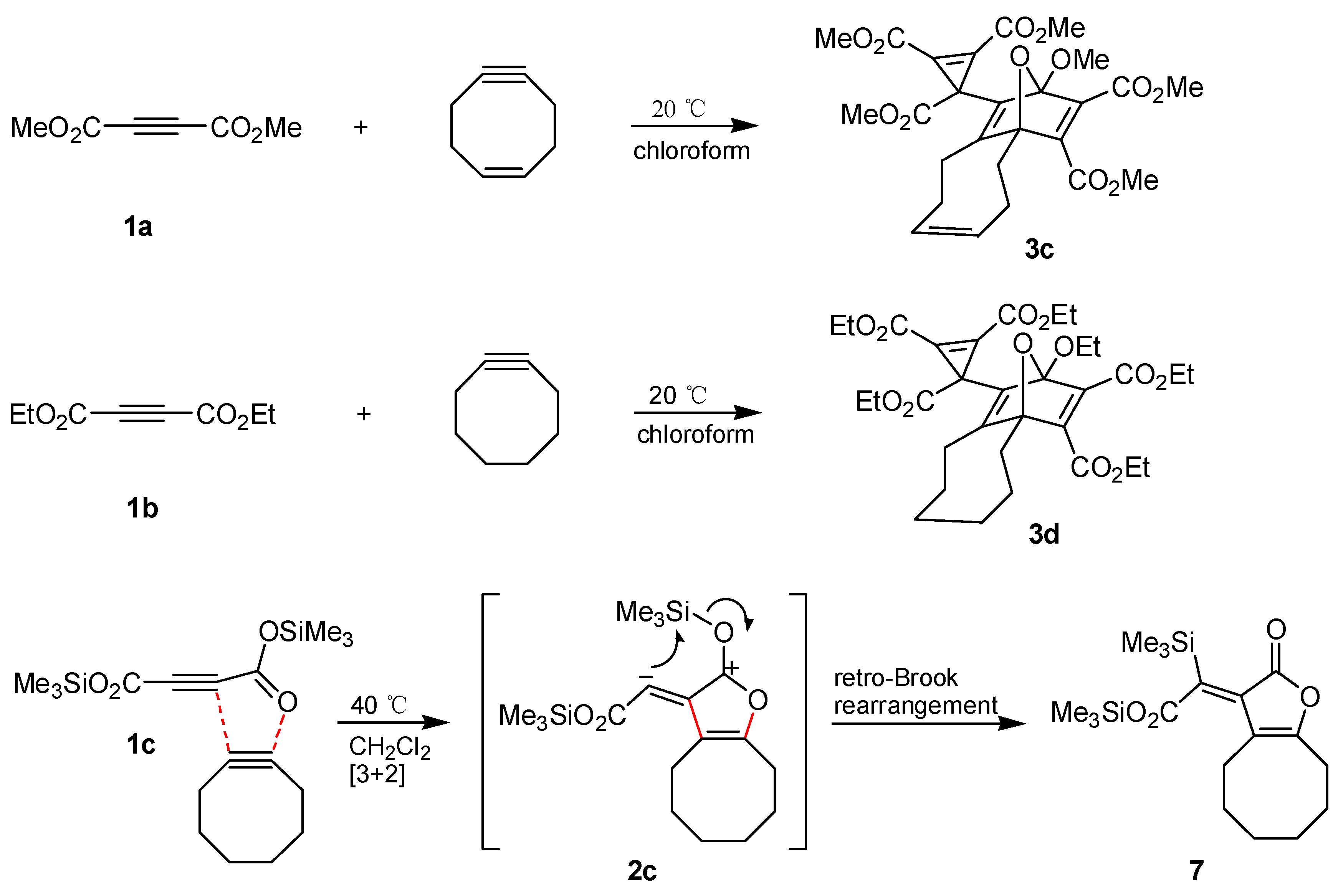
The synthesis of
3b from
1a and cyclooctyne can be transferred to other acetylenedicarboxylates and cycloalkynes. Thus,
1a underwent a similar cascade of cycloaddition reactions in the presence of cycloocten
-5
-yne [
24,
25,
26] to afford the product
3c in 37% yield (
Scheme 3). On the other hand, diethyl acetylenedicarboxylate (
1b) reacted only slightly slower than
1a with cyclooctyne to give the polycycle
3d with 61% yield. We attempted to perform the analogous transformation with di-
tert-butyl acetylenedicarboxylate, however, not any characterizable product was obtained, even on heating the reaction mixture to 40 °C or on utilizing methanol as solvent. We assume that the sterically hindered diester is not able to enter into the rate-determining first step, which prevents not only the generation of the intermediate
2 but also the formation of products of type
3 or
4. However, treatment of bis(trimethylsilyl) acetylenedicarboxylate (
1c) with cyclooctyne in anhydrous dichloromethane at 40 °C led to the oily 1:1 adduct
7 (85% yield). The
13C,
1H long-range correlation 2D-NMR spectra and especially the
29Si
-NMR data indicated that the structure of
7 includes an oxygen-bound trimethylsilyl group and also such a group directly connected with carbon (
Scheme 3). The genesis of
7 is easily explained by a 1,3
-dipolar cycloaddition of the substrates and subsequent retro-Brook rearrangement of the intermediate
2c. This means that the corresponding vinylic carbon atom of
2c should possess nucleophilic properties to allow an intramolecular attack at silicon. Thus, the question arises whether intermediates of type
2 have to be generally described by dipolar resonance structures such as
2b and
2c or by carbene structures like
2a and
2b'. The formation of the three-membered ring in the products
3a–
d and
6a,
b, respectively, can be interpreted via nucleophilic attack of the negatively charged carbon atom of
2 at the π-system of
1 or the aldehydes followed by ring closure of the resulting dipolar species or alternatively by a cheletropic reaction of the carbene version of
2 [
27]. Furthermore, generation of orthoesters
4a–
c is possible through trapping of dipole
2b and is less easily explained via carbene
2b'. Finally, the retro-Brook rearrangement
2c→
7 and also the fact that interception of
2 to prepare products with three-membered rings is successful only in case of electron-poor π-systems but not with simple alkenes or alkynes, are strong arguments to prefer the dipolar resonance structure of intermediates
2 [
28,
29,
30,
31,
32].
Scheme 3.
Furan derivatives from acetylenedicarboxylates and cyclooctynes.
Scheme 3.
Furan derivatives from acetylenedicarboxylates and cyclooctynes.
3. Experimental
3.2. Synthesis of Trimethyl 3-[2-Methoxy-3,4-bis(methoxycarbonyl)-5,6,7,8,9,10-hexahydro-2H-2,4a-epoxybenzo[8]annulen-1-yl]cycloprop-1-ene-1,2,3-tricarboxylate (3b)
To a solution of 1a (1.00 g, 7.04 mmol, freshly distilled) in anhydrous CH2Cl2 (2.5 mL), cyclooctyne (189 mg, 1.75 mmol) was added with stirring at 0 °C. After 20 h at ambient temperature, the solvent and the excess of 1a were removed at reduced pressure (finally 1 h at 40 °C and 0.1 mbar), and the residue was purified by flash chromatography (SiO2, CHCl3/ethyl acetate 15:1) to give 3b (0.74 g, 79%) as a pale yellow solid, which was repeatedly crystallized from cyclohexane to yield colorless crystals with m.p. 90 °C that are appropriate for X-ray diffraction analysis. 1H-NMR (CDCl3): δ = 1.15–1.55 (m, 4H), 1.58–1.82 (m, 4H), 1.97–2.08 (m, 2H, 10'-H or 5'-H), 2.42‒2.62 (m, 2H, 10'-H or 5'-H), 3.52 (s, 3H, O-C-OCH3), 3.64 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 3.85 (s, 3H, OCH3). 13C-NMR (CDCl3): δ = 23.12 (t), 23.26 (t, 2C), 25.33 (t), 25.61 (t), 29.03 (t), 32.27 (s, C-3), 52.15 (q, 2C, OCH3), 52.75 (q, OCH3), 53.14 (q, OCH3), 53.17 (q, OCH3), 54.87 (q, O-C-OCH3), 89.42 (s, C-4a'), 114.19 (s), 115.65 (s, O-C-OCH3), 116.49 (s), 140.36 (s), 150.82 (s), 157.01 (s), 157.12 (s), 157.36 (s), 160.62 (s), 163.23 (s), 164.18 (s), 170.19 (s). IR (CDCl3): = 2954 (m), 1732 (s), 1268 (s) cm−1. Elemental Analysis calcd. for C26H30O12: C: 58.42%, H: 5.66%. Found: C: 58.45%, H: 5.63%.
Crystal Data for 3b: C26H30O12, M = 534.50 g·mol−1, monoclinic, C2/c, λ = 0.71073 Å, a = 31.6876 (13) Å, b = 10.1200 (3) Å, c = 16.1580 (6) Å, β = 101.909 (4) °, V = 5070.0 (3) (4) Å3, Z = 8, ρcalcd = 1.400 Mg∙m−3, μ = 0.112 mm−1, T = 100 (2) K, θ range 3.23°–25.00°, 9688 reflections collected, 4448 independent reflections (Rint = 0.0204), R1 = 0.0462, wR2 = 0.0865 (I > 2σ(I)).
3.3. Synthesis of Trimethyl 3-[2-Methoxy-3,4-bis(methoxycarbonyl)-5,6,9,10-tetrahydro-2H-2,4a-epoxybenzo[8]annulen-1-yl]cycloprop-1-ene-1,2,3-tricarboxylate (3c)
To cycloocten-5-yne (74 mg, 0.70 mmol) in CDCl3 (0.7 mL), 1a (400 mg, 2.79 mmol) was added. The mixture was allowed to stand at room temperature for 20 h. After removal of the solvent at reduced pressure, the residue was purified by chromatography (silica gel, dichloromethane/ethyl acetate 15:1) to give 3c (138 mg, 37%) as a slightly yellow oil. 1H-NMR (CDCl3): δ = 1.99–2.32 (m, 4H, CH2), 2.64–2.94 (m, 4H, CH2), 3.45 (s, 3H, O-C-OCH3), 3.61 (s, 3H, OCH3), 3.73 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 5.47–5.54 (m, 2H, CH=CH). 13C-NMR (CDCl3): δ = 24.03 (t), 24.07 (t), 27.55 (t), 27.64 (t), 31.53 (s), 52.03 (q, OCH3), 52.16 (q, OCH3), 52.63 (q, OCH3), 52.96 (q, OCH3), 53.07 (q, OCH3), 54.62 (q, O-C-OCH3), 91.06 (s, C-4a'), 113.51 (s), 115.23 (s, O-C-OCH3), 115.47 (s), 128.18 (d, CH=CH), 129.35 (d, CH=CH), 142.64 (s), 150.62 (s), 156.81 (s), 156.90 (s), 157.25 (s), 159.43 (s), 162.99 (s), 164.16 (s), 170.07 (s). IR (CDCl3): = 2954 (m), 1732 (s), 1270 (s) cm−1. HRMS: m/z calcd. for C26H28O12 ([M+H]+): 533.1659, found: 533.1592; calcd. for ([M+Na]+): 555.1478, found: 555.1470; calcd. for ([M+K]+): 571.1218, found: 571.1169.
3.4. Synthesis of Triethoxy 3-[2-Ethoxy-3,4-bis(ethoxycarbonyl)-5,6,7,8,9,10-hexahydro-2H-2,4a-epoxybenzo[8]annulen-1-yl]cycloprop-1-ene-1,2,3-tricarboxylate (3d)
To cyclooctyne (20 mg, 0.19 mmol) in CDCl3 (0.7 mL), 1b (0.13 g, 0.74 mmol) was added. The mixture was allowed to stand at room temperature for 20 h. After removal of the solvent at reduced pressure, the residue was purified by chromatography (silica gel, dichloromethane/ethyl acetate 15:1) to give 3d (70 mg, 61%) as a slightly yellow oil. 1H-NMR (CDCl3): δ = 1.13–1.20 (m, 6H, CH3), 1.25–1.35 (m, 12H, CH3), 1.39–1.83 (m, 8H, CH2), 2.01–2.12 (m, 2H, CH2), 2.48–2.67 (m, 2H, CH2), 3.70–3.78 (m, 1H, OCH2), 3.84–3.91 (m, 1H, OCH2), 3.99–4.08 (m, 1H, OCH2), 4.11–4.25 (m, 5H, OCH2), 4.27–4.36 (m, 4H, OCH2). 13C-NMR (CDCl3): δ = 13.99 (q, 2 C, CH3), 13.94 (q, CH3), 13.98 (q, CH3), 14.07 (q, CH3), 15.02 (q, CH3), 23.27 (t, CH2), 23.31 (t, CH2), 23.33 (t, CH2), 25.46 (t, CH2), 25.68 (t, CH2), 29.02 (t, CH2), 32.36 (s, C-3), 61.01 (t, OCH2), 61.08 (t, OCH2), 61.53 (t, OCH2), 62.22 (t, OCH2), 62.37 (t, OCH2), 63.59 (t, OCH2), 89.29 (s, C-4a'), 114.65 (s), 115.40 (s), 116.20 (s), 140.68 (s), 151.17 (s), 156.87 (s), 156.91 (s), 157.17 (s), 160.07 (s), 162.96 (s), 163.91 (s), 169.68 (s). IR (CDCl3): = 2984 (m), 1727 (s), 1260 (s) cm−1. HRMS: m/z calcd. for C32H42O12 ([M+H]+): 619.2755, found: 619.2697; calcd. for ([M+Na]+): 641.2574, found: 641.2608; calcd. for ([M+K]+): 657.2313, found: 657.2263.
3.5. Synthesis of Methyl (E)-2-(2,2-Dimethoxy-4,5,6,7,8,9-hexahydrocycloocta[b]furan-3(2H)-ylidene)acetate (4a)
To cyclooctyne (271 mg, 2.51 mmol) in anhydrous MeOH (2.5 mL), 1a (1.40 g, 9.85 mmol) was added. The mixture was stirred at room temperature for 20 h. After removal of the solvent at reduced pressure, the residue was purified by chromatography (silica gel, cyclohexane/tert-butyl methyl ether 14:1) to give 4a (426 mg, 60%) as a colorless solid with m.p. 60–61 °C (from cyclohexane/tert-butyl methyl ether). 1H-NMR (CDCl3): δ = 1.39–1.46 (m, 2H, 6'-CH2), 1.49–1.57 (m, 2H, 7'-CH2), 1.61–1.69 (m, 2H, 5'-CH2), 1.68–1.75 (m, 2H, 8'-CH2), 2.46 (m, 2H, 9'-CH2), 2.73 (t, J = 6.3, 2H, 4'-CH2), 3.33 (s, 6H, 2'-(OCH3)2), 3.69 (s, 3H, CO2CH3), 5.50 (s, 1H, 2-CH). 13C-NMR (CDCl3): δ = 22.2 (t, C-4'), 25.6 (t, C-6'), 26.5 (t, C-7'), 27.1 (t, C-9'), 28.7 (t, C-8'), 29.3 (t, C-5'), 51.0 (q, 1-OCH3), 51.3 (q, 2'-(OCH3)2), 106.3 (d, C-2), 112.5 (s, C-3a'), 121.7 (s, C-2'), 152.1 (s, C-3'), 166.3 (s, C-1), 170.3 (s, C-9a'). IR (film): = 2928 (s), 2853 (m), 1718 (s), 1642 (m), 1595 (s), 1458 (m), 1441 (m), 1353 (m), 1324 (m), 1277 (s), 1226 (s), 1167 (s), 1126 (s), 1086 (m), 1050 (s), 1022 (m), 1009 (m), 969 (m), 931 (m), 911 (m), 886 (w), 843 (m), 778 (w) cm−1. HRMS: m/z calcd. for C15H22O5 ([M+H]+): 283.1521, found: 283.1540; calcd. for ([M+Na]+): 305.1360, found: 305.1359. Elemental Analysis calcd. for C15H22O5: C: 63.81%, H: 7.85%, found: C: 63.82%, H: 7.90%.
Crystal Data for 4a: C15H22O5, M = 282.32 g·mol−1, monoclinic, I2/a, λ = 1.54184 Å, a = 18.1382 (3) Å, b = 11.3690 (2) Å, c = 15.9193 (4) Å, β = 118.261 (2) °, V = 2891.44(12) Å3, Z = 8, ρcalcd = 1.297 Mg∙m−3, μ = 0.798 mm−1, T = 110 (2) K, θ range 3.10°–61.986°, 4314 reflections collected, 2243 independent reflections (Rint = 0.0233), R1 = 0.0452, wR2 = 0.1172 (I > 2σ(I)).
3.6. Synthesis of Methyl d4-(E)-2-(2,2-Dimethoxy-4,5,6,7,8,9-hexahydrocycloocta[b]furan-3(2H)-ylidene)acetate (4b)
As described for 4a, cyclooctyne was analogously treated with CD3OD and 1a, and workup led to 4b (447 mg, 62%) as a colorless solid with m.p. 63–64 °C (from cyclohexane/tert-butyl methyl ether). 1H-NMR (CDCl3): δ = 1.35–1.42 (m, 2H, 6'-CH2), 1.46–1.53 (m, 2H, 7'-CH2), 1.57–1.65 (m, 2H, 5'-CH2), 1.64–1.71 (m , 2H, 8'-CH2), 2.42 (m, 2H, 9'-CH2), 2.69 (t, J = 6.3, 2H, 4'-CH2), 3.27 (s, 3H, 2'-OCH3), 3.63 (s, 3H, CO2CH3). 2H-NMR (CHCl3:CDCl3 = 9:1): δ = 3.30 (s, 3D, 2'-OCD3), 5.53 (s, 1D, 2-CD). 13C-NMR (CDCl3): δ = 22.0 (t, C-4'), 25.5 (t, C-6'), 26.4 (t, C-7'), 26.9 (t, C-9'), 28.6 (t, C-8'), 29.2 (t, C-5'), 50.4 (s (DEPT), sept with 1JCD = 22.0, 2'-OCD3), 50.9 (q, OCH3), 51.1 (q, OCH3), 105.8 (s (DEPT), t with 1JCD = 24.5, C-2), 112.4 (s, C-3a'), 121.6 (s, C-2'), 152.0 (s, C-3'), 166.1 (s, C-1), 170.2 (s, C-9a'). HRMS: m/z calcd. for C15H18D4O5 ([M+H]+): 287.1791, found: 287.1772; calcd. for ([M+Na]+): 309.1611, found: 309.1633; calcd. for ([M+K]+): 325.1350, found: 325.1367.
3.7. Synthesis of Methyl (E)-2-(2-methoxy-2-phenoxy-4,5,6,7,8,9-hexahydrocycloocta[b]furan-3(2H)-ylidene)acetate (4c)
To a solution of phenol (3.00 g, 31.9 mmol) and cyclooctyne (0.40 g, 3.7 mmol) in anhydrous THF (4 mL), 1a (1.05 g, 7.4 mmol) was added with the help of a syringe within 2 h. Thereafter, the mixture, which changed its color to deep red, was stirred at ambient temperature for 16 h. After removal of the solvent at reduced pressure, the residue was dissolved in Et2O (40 mL) and washed with aqueous sodium hydroxide (10%, 2 L). After drying of the organic phase (MgSO4) and removal of the solvent, the residue was purified by chromatography (first SiO2, CH2Cl2, then basic Al2O3, CH2Cl2) to furnish a green oil that was treated with hexane. This led to the precipitation of a colorless solid, which was removed by filtration. After removal of the solvent, 4c (247 mg, 10%) was obtained as a green oil that can be further purified by HPLC (LiChrospher Si 60 (5μ), 20 × 2 cm, CH2Cl2, 20 mL/min). 1H-NMR (CDCl3, relaxation delay d1 = 15 s): δ = 1.00–1.72 (m, 8H), 2.38–2.41 (m, 2H), 2.53–2.60 (m, 1H), 2.77–2.83 (m, 1H), 3.44 (s, 3H, 2'-OCH3), 3.70 (s, 3H, CO2CH3), 5.74 (s, 1H, 2-H), 7.06–7.25 (m, 5H, O-Ph). 13C-NMR (CDCl3): δ = 22.0 (t, C-4'), 25.1 (t, C-6'), 26.2 (t, C-7'), 26.9 (t, C-9'), 28.3 (t, C-8'), 29.1 (t, C-5'), 51.1 (q, 1-OCH3), 51.5 (q, 2'-OCH3), 107.5 (d, C-2), 112.7 (s, C-3a'), 121.1 (d, Ph), 122.3 (s, C-2'), 124.4 (d, Ph), 128.8 (d, Ph), 152.3 (s, C-3'), 152.4 (s, Ph), 166.2 (s, C-1), 169.7 (s, C-9a'). IR (CDCl3): = 2931 (s), 1705 (s, C=O) 1506 (s), 1490 (s) cm−1.
3.8. Synthesis of Dimethyl (E)-2-(2-oxocyclooctylidene)succinate [(E)-5] and Dimethyl (Z)-2-(2-oxocyclooctylidene)succinate [(Z)-5]
To THF (2 mL) and water (1 mL), cyclooctyne (0.41 g, 3.84 mmol) and 1a (0.56 g, 3.94 mmol) were added with stirring at 0 °C. Thereafter, the mixture was stirred at ambient temperature for 21 h. This led to the formation of an inhomogeneous mixture, which was diluted with diethyl ether (50 mL) and dried (MgSO4). After removal of the solvent under reduced pressure, the resulting yellow liquid was analyzed by 1H-NMR, which indicated a 5:1 ratio of (E)-5 and (Z)-5. By using flash chromatography (SiO2, Et2O/hexane 1:2), (E)-5 (0.22 g, 21%) and (Z)-5 (0.20 g, 20%) were isolated as colorless liquids.
(E)-5: 1H-NMR (CDCl3): δ = 1.42‒1.48 (m, 2H), 1.52‒1.58 (m, 2H), 1.61‒1.68 (m, 2H), 1.78‒1.86 (m, 2H), 2.46‒2.51 (m, 2H, 8'-H), 2.72‒2.76 (m, 2H, 3'-H), 3.25 (s, 2H, 3-H), 3.64 (s, 3H, OCH3), 3.74 (s, 3H, OCH3). 13C-NMR (CDCl3): δ = 23.64 (t, CH2), 25.51 (t, CH2), 25.77 (t, CH2), 27.56 (t, CH2), 32.44 (t, C-8'), 36.04 (t, C-3), 43.46 (t, C-3'), 52.03 (q), 52.14 (q), 122.34 (s), 154.13 (s), 167.33 (s), 170.79 (s), 212.43 (s, C-2'). IR (CCl4): = 1745 (s) cm−1. Elemental Analysis calcd. for C14H20O5: C: 62.67%, H: 7.51%. found: C: 62.37%, H: 7.46%.
(Z)-5: 1H-NMR (CDCl3): δ = 1.44–1.63 (m, 6H), 1.75‒1.82 (m, 2H), 2.40‒2.44 (m, 2H, 8'-H), 2.62‒2.66 (m, 2H, 3'-H) 3.38 (s, 2H, 3-H), 3.68 (s, 6H, 2 × OCH3). 13C-NMR (CDCl3): δ = 23.62 (t, CH2), 25.49 (t, CH2), 25.74 (t, CH2), 27.53 (t, CH2), 32.40 (t, C-8'), 36.00 (t, C-3), 43.43 (t, C-3'), 51.99 (q, OCH3), 52.09 (q, OCH3), 122.32 (s), 154.08 (s), 167.28 (s), 170.74 (s), 212.36 (s, C-2'). IR (CCl4): = 1745 (s) cm‒1. Elemental Analysis calcd. for C14H20O5: C: 62.67%, H: 7.51%, found: C: 62.43%, H: 7.36%.
3.9. Synthesis of Dimethyl 2-Methoxy-1-[(2-methoxycarbonyl)-3-methyloxiran-2-yl]-5,6,7,8,9,10-hexahydro-2H-2,4a-epoxybenzo[8]annulene-3,4-dicarboxylate (6a)
To a solution of cyclooctyne (410 mg, 3.79 mmol) in acetaldehyde (3 mL, freshly distilled), a solution of 1a (540 mg, 3.79 mmol) in anhydrous THF (2 mL) was added with the help of a syringe within 2 h. The yellow mixture was stirred at room temperature overnight. Thereafter, the solvent and the excess of acetaldehyde were removed at reduced pressure, and the resulting yellow oil was purified by flash chromatography (SiO2, Et2O/hexane 1:1). The oily product was recrystallized twice from Et2O/hexane (1:3) to give 6a (180 mg, 22%) as colorless crystals with m.p. 66–67 °C. Single crystals, which were appropriate for X-ray diffraction analysis, were obtained by slow evaporation of a solution of 6a in cyclohexane. 1H-NMR (CDCl3): δ = 1.38 (d, 3J = 5.6, 3H, C–CH3), 1.25–1.91 (m, 8H), 2.11 (m, 2H, 5-H), 2.51 (ddd, 3J10-H(a),9-H(a) = 15, 2J10-H(a),10-H(b) = 13.2, 3J10-H(a),9-H(b) = 4.8, 1H, 10-H(a)), 2.66 (dt, 2J10-H(a),10-H(b) = 13.2, 3J10-H(b),9-H(a) = 4, 3J10-H(b),9-H(b) = 4, 1H, 10-H(b)), 3.61 (s, 3H, OCH3), 3.68 (q, 3J3'-H,CH3 = 5.6, 1 H, 3'-H), 3.73 (s, 3H, OCH3), 3.779 (s, 3H, OCH3), 3.783 (s, 3H, OCH3). 13C-NMR (CDCl3): δ = 13.74 (q, CH3), 23.01 (t, CH2), 23.21 (t, CH2), 23.37 (t, CH2), 25.13 (t, CH2), 25.58 (t, CH2), 29.60 (t, CH2), 52.24 (q, 2 OCH3), 52.54 (q, OCH3), 55.01 (q, OCH3), 57.60 (s, C-2'), 57.86 (d, C-3'), 89.53 (s, C-4a), 115.48 (s, C-2), 139.33 (s), 150.29 (s), 156.52 (s), 163.22 (s), 163.91 (s), 164.18 (s), 168.35 (s). IR (CCl4): = 1709 (s) cm−1. Elemental Analysis calcd. for C22H28O9: C: 60.54%, H: 6.47%. found: C: 60.55%, H: 6.45%.
Crystal Data for 6a: C22H28O9, M = 436.44 g∙mol−1, monoclinic, P21/c, λ = 1.54184 Å, a = 8.8577 (4) Å, b = 28.5456 (16) Å, c = 17.3589 (9) Å, β = 90.063 (4) °, V = 4389.2 (4) Å3, Z = 8, ρcalcd = 1.321 Mg∙m−3, μ = 0.863 mm−1, T = 100 (2) K, θ range 3.10°−61.77°, 28823 reflections collected, 6811 independent reflections (Rint = 0.0557), R1 = 0.0548, wR2 = 0.1478 (I > 2σ (I)).
3.10. Synthesis of Dimethyl 2-Methoxy-1-[(2-methoxycarbonyl)-3-phenyloxiran-2-yl]-5,6,7,8,9,10-hexahydro-2H-2,4a-epoxybenzo[8]annulene-3,4-dicarboxylate (6b)
To a solution of cyclooctyne (270 mg, 2.53 mmol) in benzaldehyde (4 mL, freshly distilled), a solution of 1a (1.10 g, 7.58 mmol) in anhydrous THF (2 mL) was added with the help of a syringe within 2 h. The orange mixture was stirred overnight at room temperature. Thereafter, the solvent was evaporated and the excess of benzaldehyde was removed at 50 °C and 9 × 10−2 mbar. The resulting yellow oil was purified by flash chromatography (SiO2, Et2O/hexane 1:1). The oily product was crystallized from Et2O/hexane (ratio 1:3) to give 6b (260 mg, 21%) as colorless crystals. 1H NMR data indicated that the substance consisted of two diastereomers in a 4:1 ratio. IR (mixture of isomers): = 1711 (s) cm−1. Elemental Analysis (mixture of isomers) calcd. for C27H30O9: C: 65.05%, H: 6.07%, found: C: 64.77%, H: 6.02%. The diastereomers of 6b were (partly) separated by flash chromatography (SiO2, CH2Cl2); the minor isomer was eluted before the main isomer.
Minor isomer: 1H-NMR (CDCl3): δ = 1.25–2.10 (m, 8H), 2.18 (m, 2H, 5-H), 2.54 (ddd, 3J10-H(a),9-H(a) = 15.2, 2J10-H(a),10-H(b) = 13, 3J10-H(a),9-H(b) = 4.8, 1H, 10-H(a)), 2.72 (dt, 2J10-H(a),10-H(b) = 13, 3J10-H(b), 9-H(a) = 4, 3J10-H(b),9-H(b) = 4, 1H, 10-H(b)), 3.39 (s, 3H, OCH3), 3.75 (s, 3H), 3.779 (s, 3H), 3.783 (s, 3H), 4.32 (s, 1H, 3'-H), 7.28–7.41 (m, 5 H, Ph). 13C-NMR (CDCl3): δ = 23.05 (t, CH2), 23.44 (t, CH2), 23.69 (t, CH2), 25.37 (t, CH2), 25.77 (t, CH2), 30.11 (t, CH2), 52.08 (q, CH3), 52.24 (q, 2C), 55.40 (q, CH3), 60.66 (s, C-2'), 63.11 (d, C-3'), 89.18 (s, C-4a), 115.82 (s, C-2), 126.51 (d, 2C), 128.00 (d, 2C), 128.29 (d, CH), 133.07 (s), 139.82 (s), 150.77 (s), 157.34 (s), 162.24 (s), 163.01 (s), 164.35 (s), 166.51 (s).
Major isomer: M.p. 116–118 °C. 1H-NMR (CDCl3): δ = 1.31–2.22 (m, 10 H), 2.55 (ddd, 3J10-H(a),9-H(a) = 15.2, 2J10-H(a),10-H(b) = 13, 3J10-H(a),9-H(b) = 4.8, 1 H, 10-H(a)), 2.85 (dt, 2J10-H(a),10-H(b) = 13, 3J10-H(b),9-H(a) = 4, 3J10-H(b),9-H(b) = 4, 1H, 10-H(b)), 3.42 (s, 3H, OCH3), 3.70 (s, 3H), 3.79 (s, 6H), 4.71 (s, 1H, 3'-H), 7.28–7.41 (m, 5H, Ph). 13C-NMR (CDCl3): δ = 22.94 (t, CH2), 23.20 (t, CH2), 23.45 (t, CH2), 25.08 (t, CH2), 25.72 (t, CH2), 29.82 (t, CH2), 52.16 (q, CH3), 52.28 (q, 2C), 55.10 (q, CH3), 60.54 (s, C-2'), 61.89 (d, C-3'), 89.67 (s, C-4a), 115.54 (s, C-2), 126.28 (d, 2C), 128.06 (d, 2C), 128.34 (d, CH), 133.25 (s), 138.99 (s), 150.43 (s), 156.48 (s), 163.21 (s), 163.80 (s), 165.26 (s), 166.65 (s).
3.11. Synthesis of (Z)-Trimethylsilyl 2-(2-Oxo-4,5,6,7,8,9-hexahydrocycloocta[b]furan-3(2H)-ylidene)-2-(trimethylsilyl)acetate (7)
To a solution of bis(trimethylsilyl) but-2-ynedioate (0.10 g, 0.37 mmol) in anhydrous methylene chloride (2 mL), cyclooctyne (0.06 g, 0.55 mmol) was added under nitrogen atmosphere. The mixture was stirred for 48 h at 40 °C. The solvent and the excess of cyclooctyne were evaporated under reduced pressure to give 0.17 g (85%) of an unstable yellow oil. 1H-NMR (CDCl3): δ = 0.23 (s, 9H, CSi(CH3)3), 0.30 (s, 9H, CO2Si(CH3)3), 1.44–1.60 (m, 6H, CH2), 1.69–1.75 (m, 2 H, CH2), 2.31 (t, J = 6.4, 2 H, CH2), 2.48 (t, J = 7.1, 2 H, CH2). 13C-NMR(CDCl3): δ = −1.48 (q, CSi(CH3)3), −0.23 (q, OSi(CH3)3), 21.11 (t, CH2), 25.30 (t, CH2), 25.85 (t, CH2), 26.00 (t, CH2), 27.02 (t, CH2), 28.15 (t, CH2), 114.48 (s, C-3a'), 133.46 (s, C=C-COOC), 146.84 (s, C=CCOOC), 158.85 (s, C-9a'), 168.35 (s, CO), 171.09 (s, CO). Signal assignment and determination of the stereochemistry were supported by HMBC and NOESY experiments, respectively. 29Si-NMR (CDCl3): δ = −3.86 (CSi(CH3)3), 25.59 (OSi(CH3)3). IR (CCl4): = 3010 (w), 2933 (s), 1785 (s), 1688 (s), 1249 (s) cm−1. HRMS: m/z calcd. for: C18H30O4Si2 ([M+Na]+): 389.1580, found: 389.1643.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}