Novel Schiff Bases Based on the Quinolinone Skeleton: Syntheses, X-ray Structures and Fluorescent Properties


Abstract
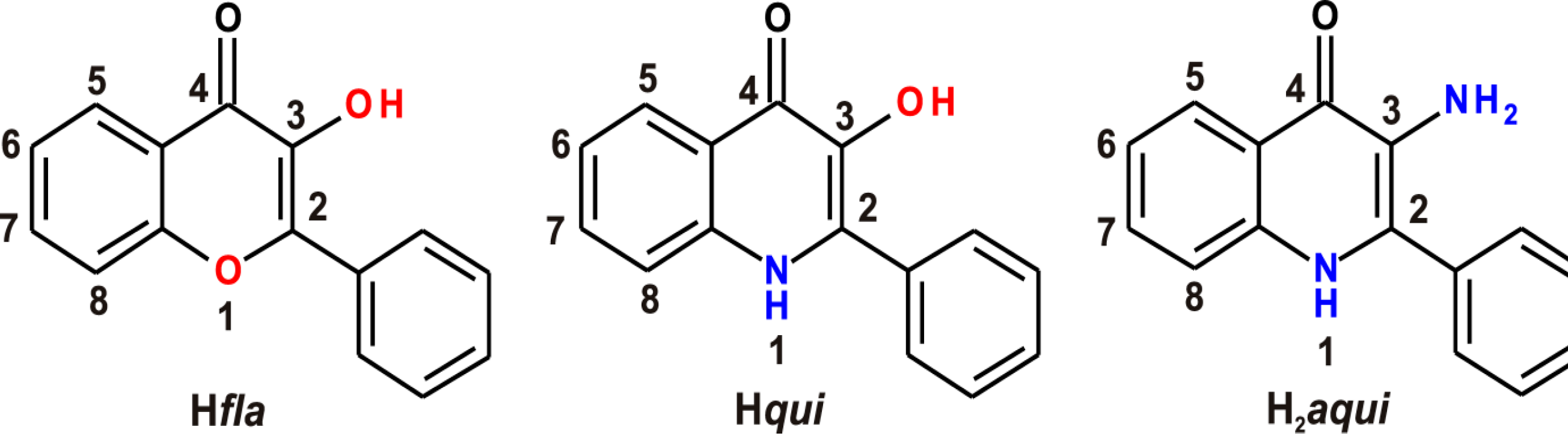
:1. Introduction

2. Results and Discussion
2.1. Synthesis
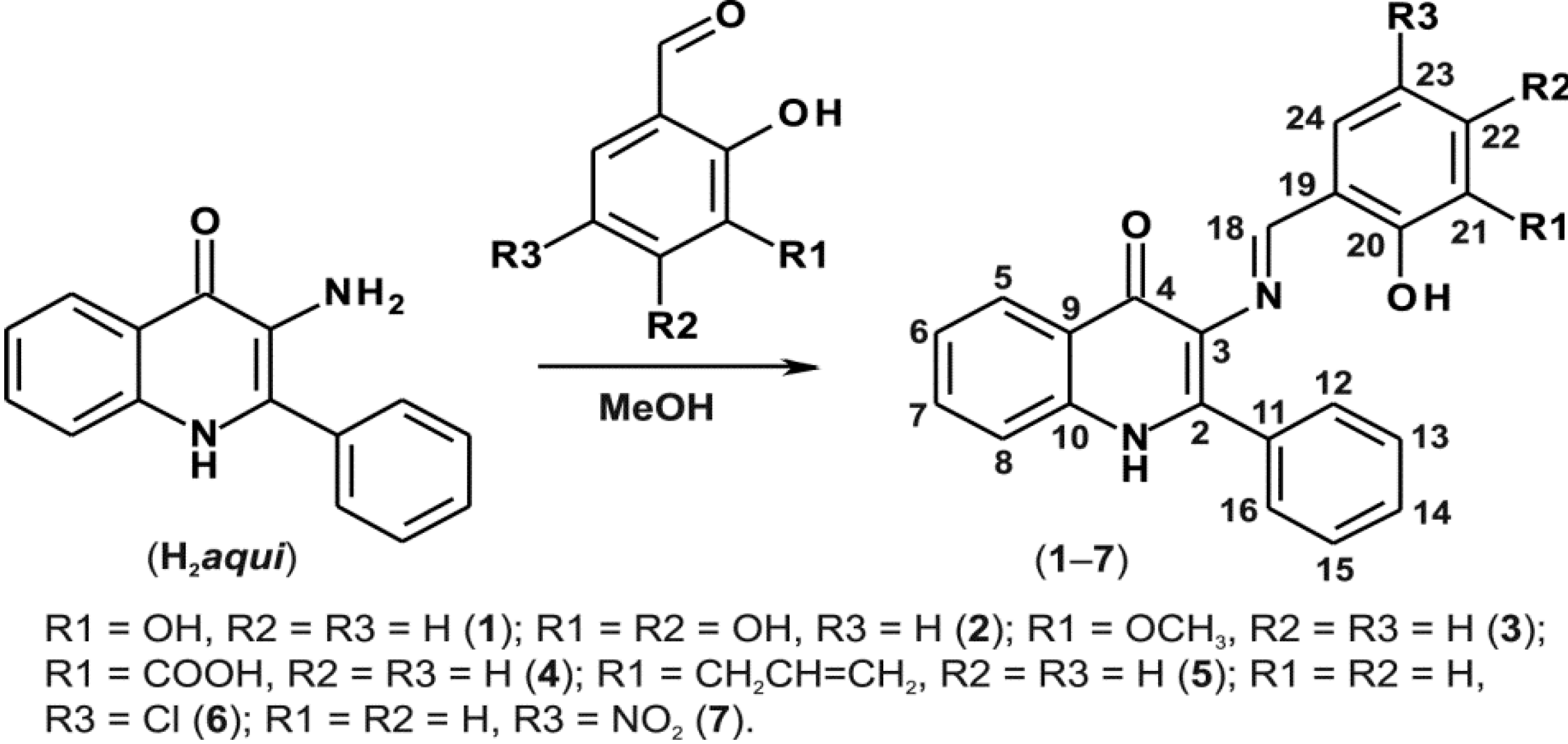
2.2. General Characterizations
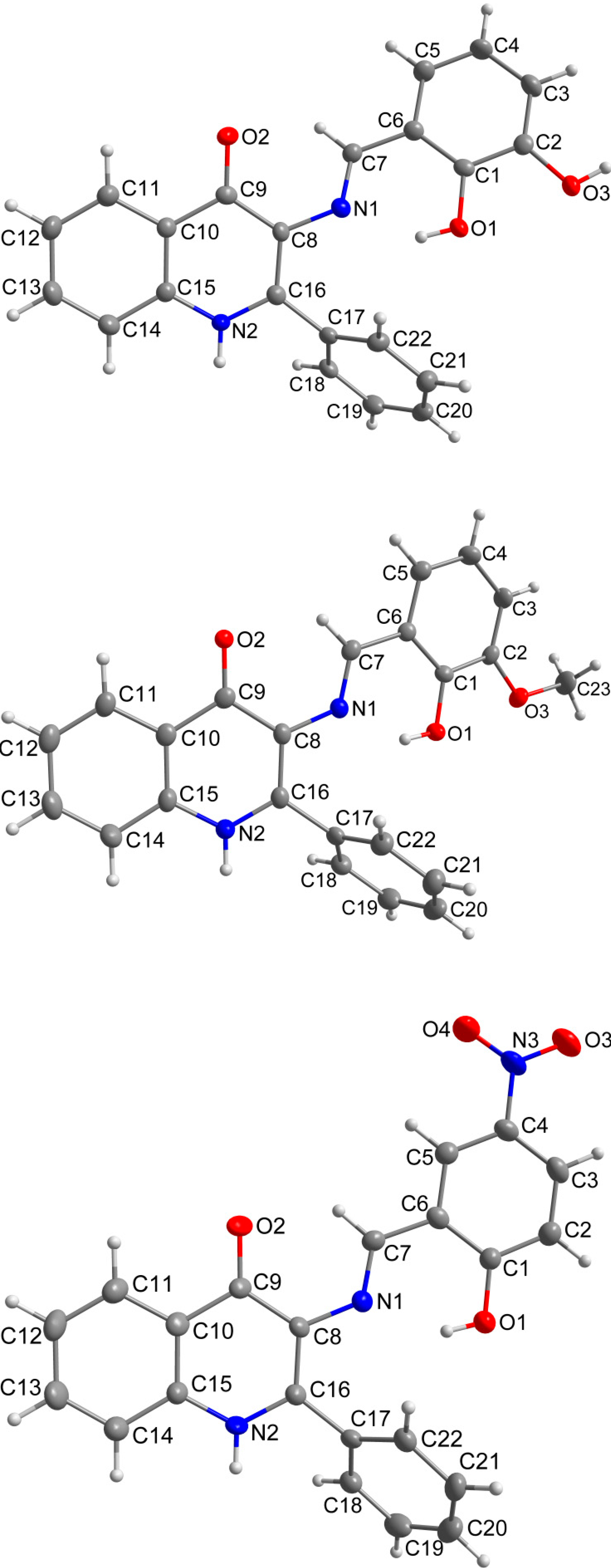
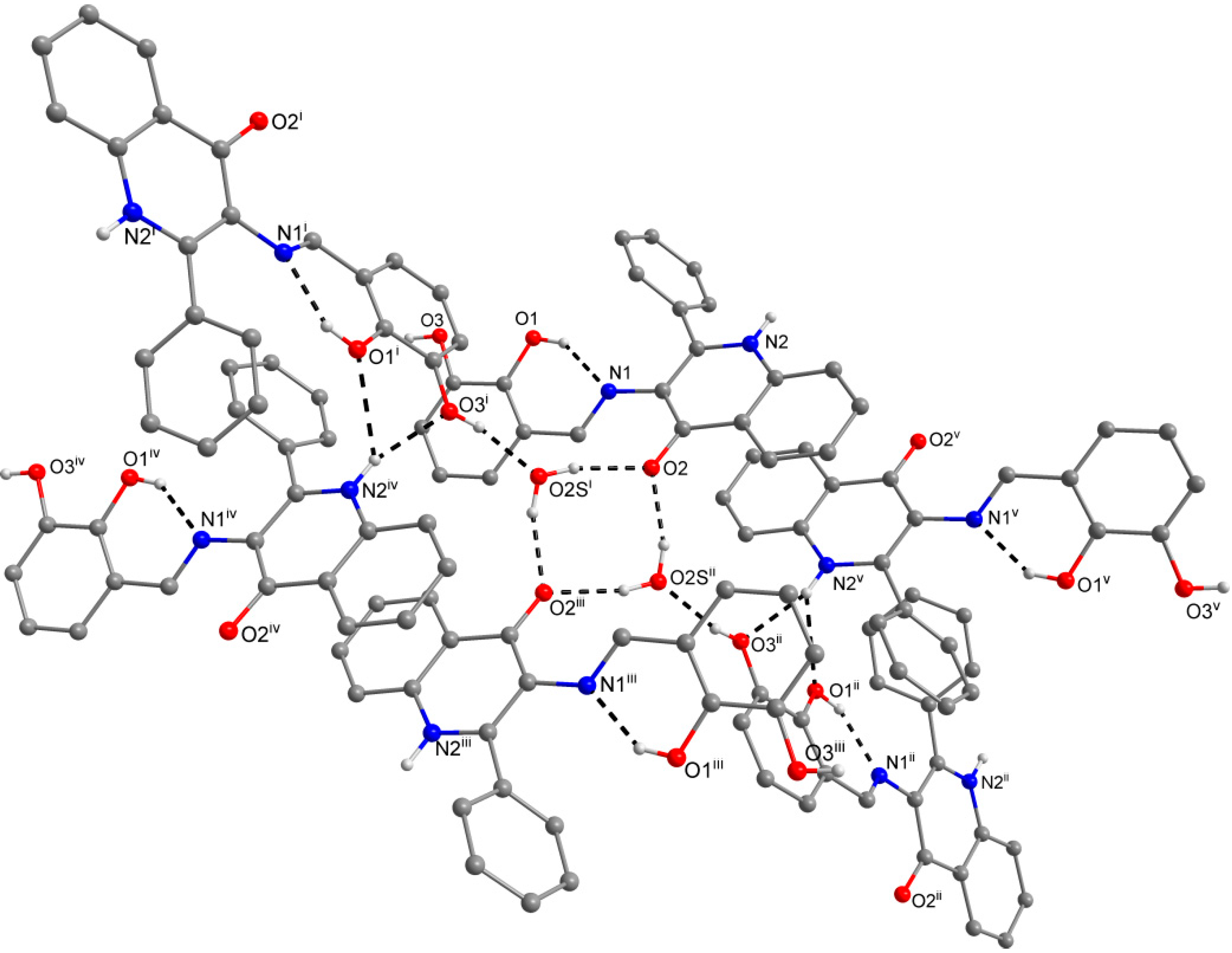
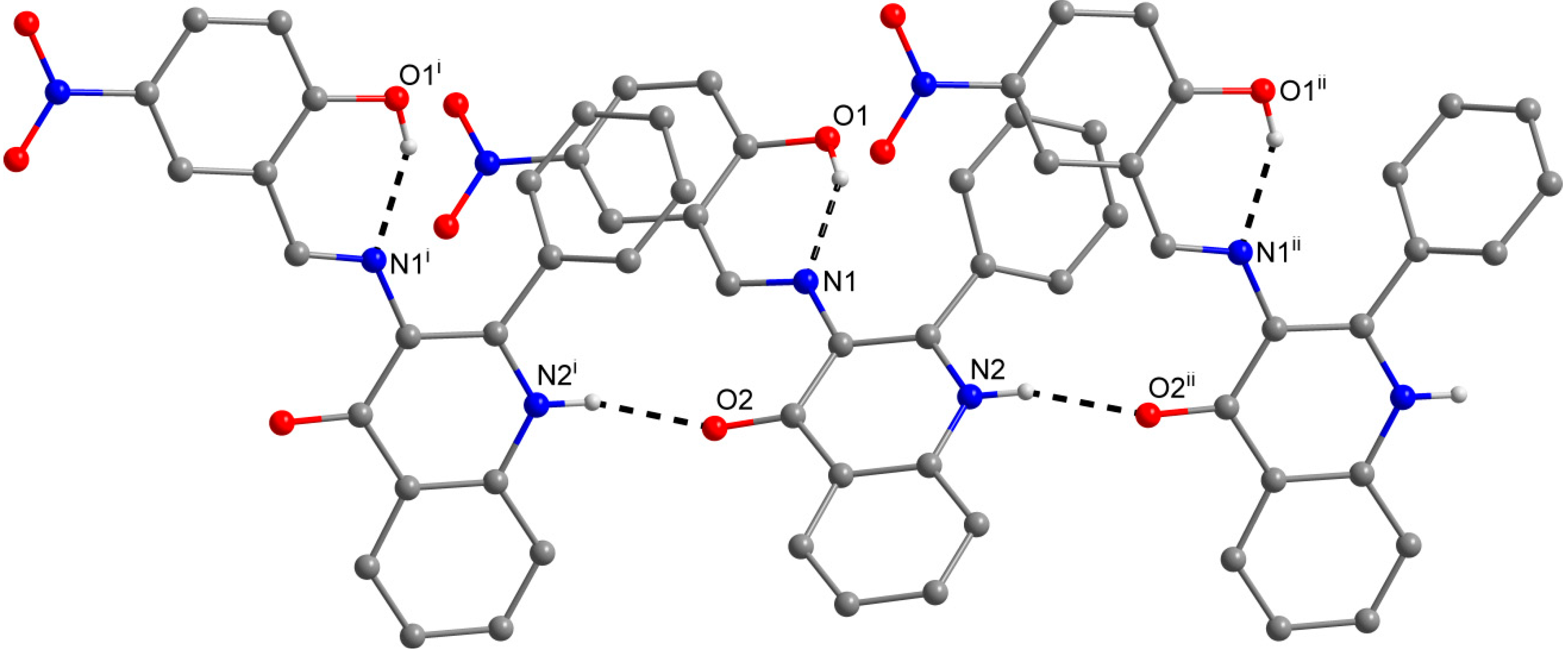
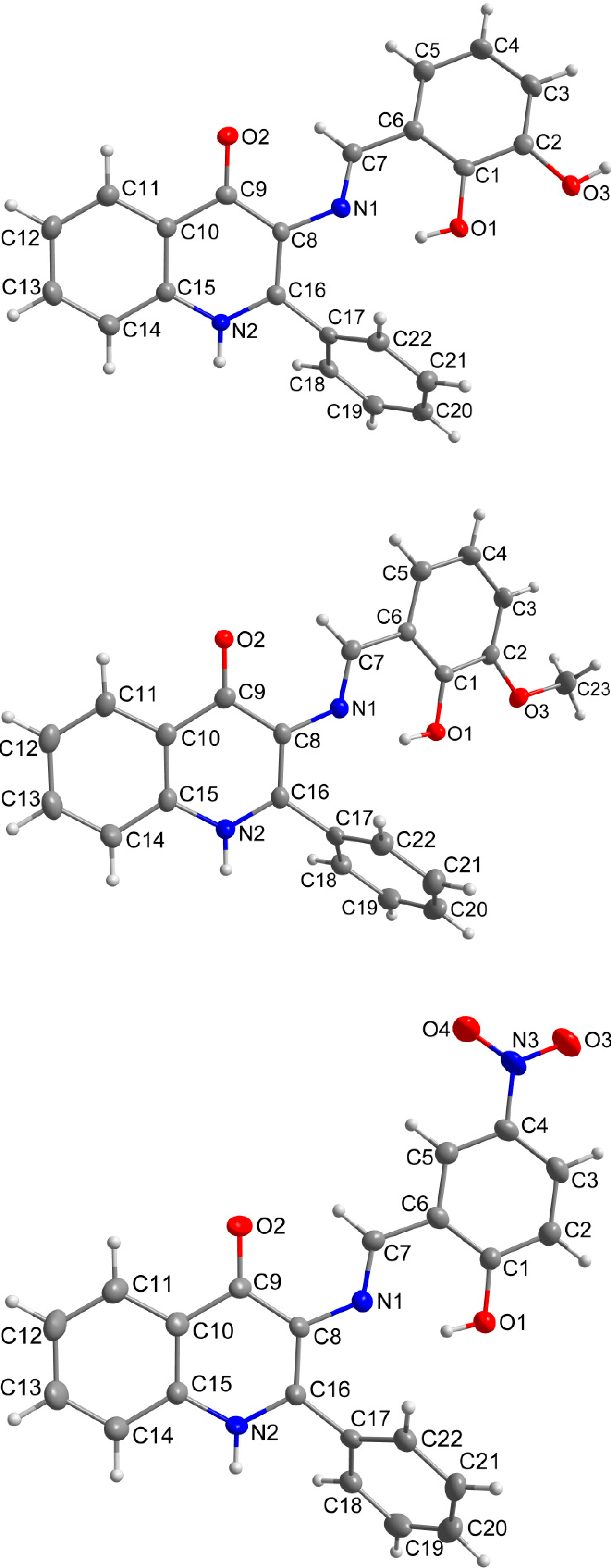
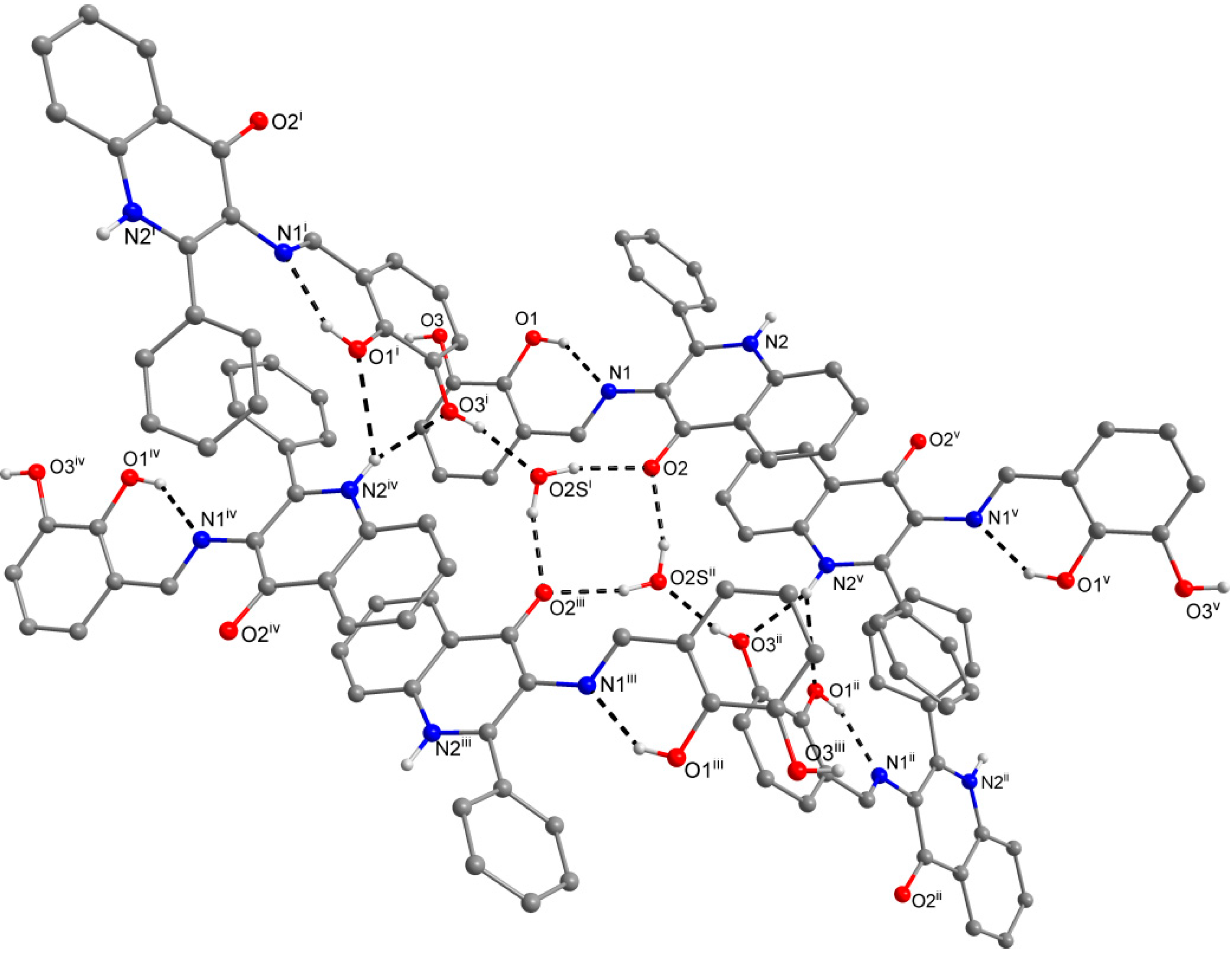
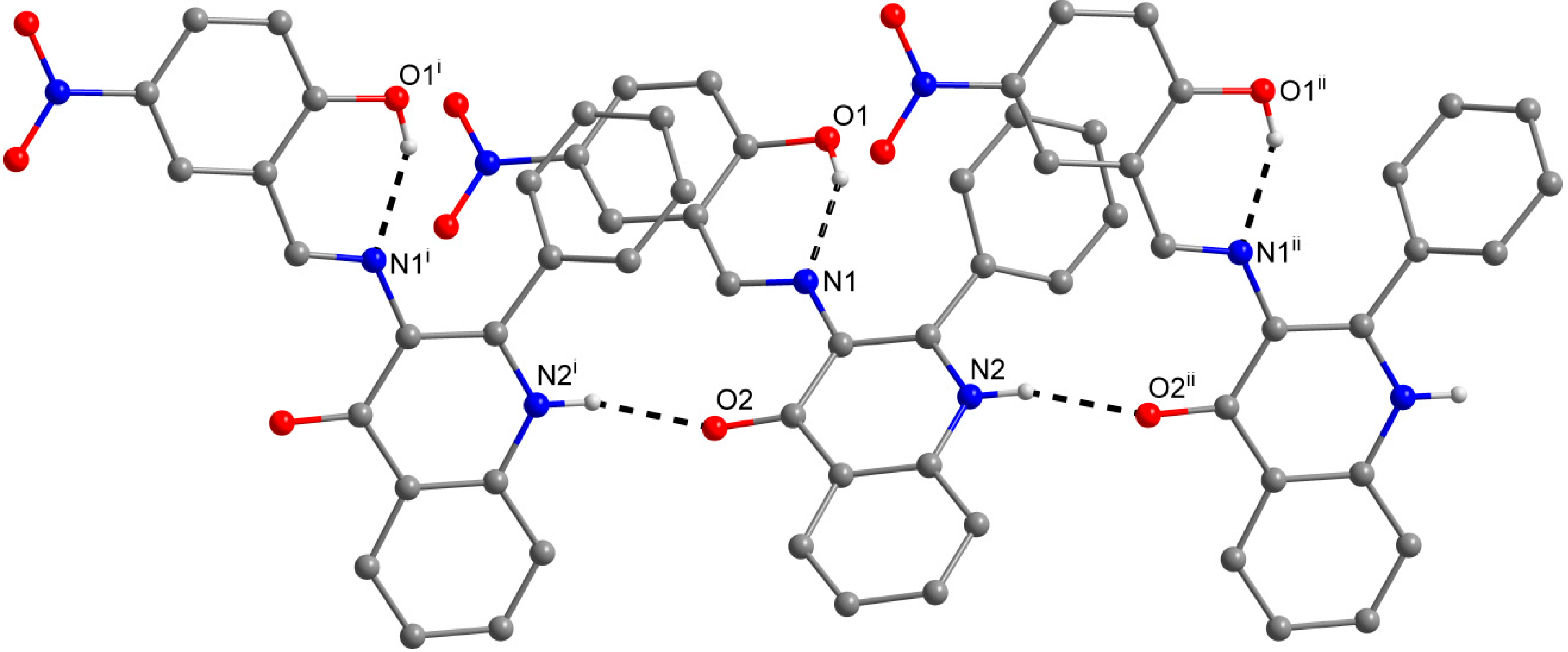
2.3. X-ray Structure Characterizations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (1) | (3) | (7) | |
|---|---|---|---|
| Formula | C22 H18 N2 O4 | C23 H18 N2 O3 | C22 H15 N3 O4 |
| M (g mol−1) | 374.38 | 370.39 | 385.38 |
| T (K) | 100(2) | 100(2) | 120(2) |
| Crystal system, space group | Monoclinic, P21/c | Orthorhombic, P212121 | Orthorhombic, Fdd2 |
| Unit cell dimensions | |||
| a (Å) | 10.5110(4) | 9.4567(4) | 12.6476(10) |
| b (Å) | 8.4997(3) | 11.5154(6) | 23.833(4) |
| c (Å) | 22.1971(8) | 16.8507(7) | 23.837(3) |
| α (°) | 90 | 90 | 90 |
| β (°) | 102.487(4) | 90 | 90 |
| γ (°) | 90 | 90 | 90 |
| V (A3) | 1934.62(12) | 1835.00(14) | 7185.2(16) |
| Z, Dc (g cm−3) | 4, 1.285 | 4, 1.341 | 16, 1.425 |
| F (000) | 784 | 776 | 3199 |
| θ range for data collection (°) | 3.02 ≤ θ ≤ 25.00 | 3.00 ≤ θ ≤ 24.99 | 3.15 ≤ θ ≤ 25.00 |
| Reflections collected/unique | 17,842/3400(0.0324) | 12,396/3225(0.0425) | 15,283/2946(0.0952) |
| Data/restraints/parameters | 3400/3/261 | 3225/0/255 | 2946/1/263 |
| Goodness-of-fit on F2 | 1.065 | 0.975 | 0.948 |
| Final R indices [I > 2σ(I)] | 0.0393, 0.1104 | 0.0322, 0.0702 | 0.0431, 0.0975 |
| R indices (all data) | 0.0574, 0.1152 | 0.0418, 0.0723 | 0.0593, 0.1020 |
| Largest peak and hole (e Å−3) | 0.306, −0.406 | 0.152, −0.194 | 0.189, −0.227 |
| CCDC Number | 1008032 | 1008033 | 1008034 |

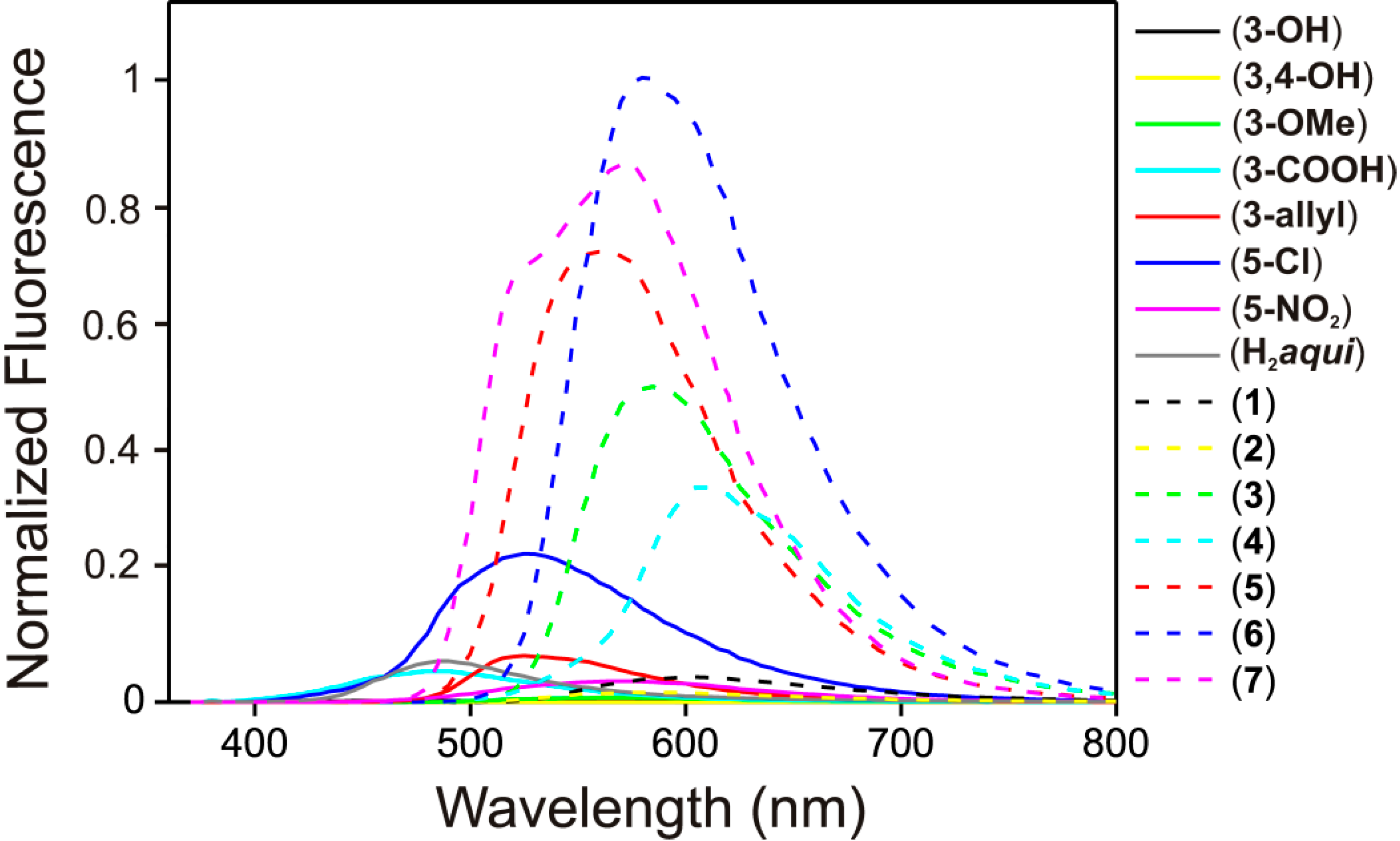
2.4. Fluorescence Spectroscopy Studies

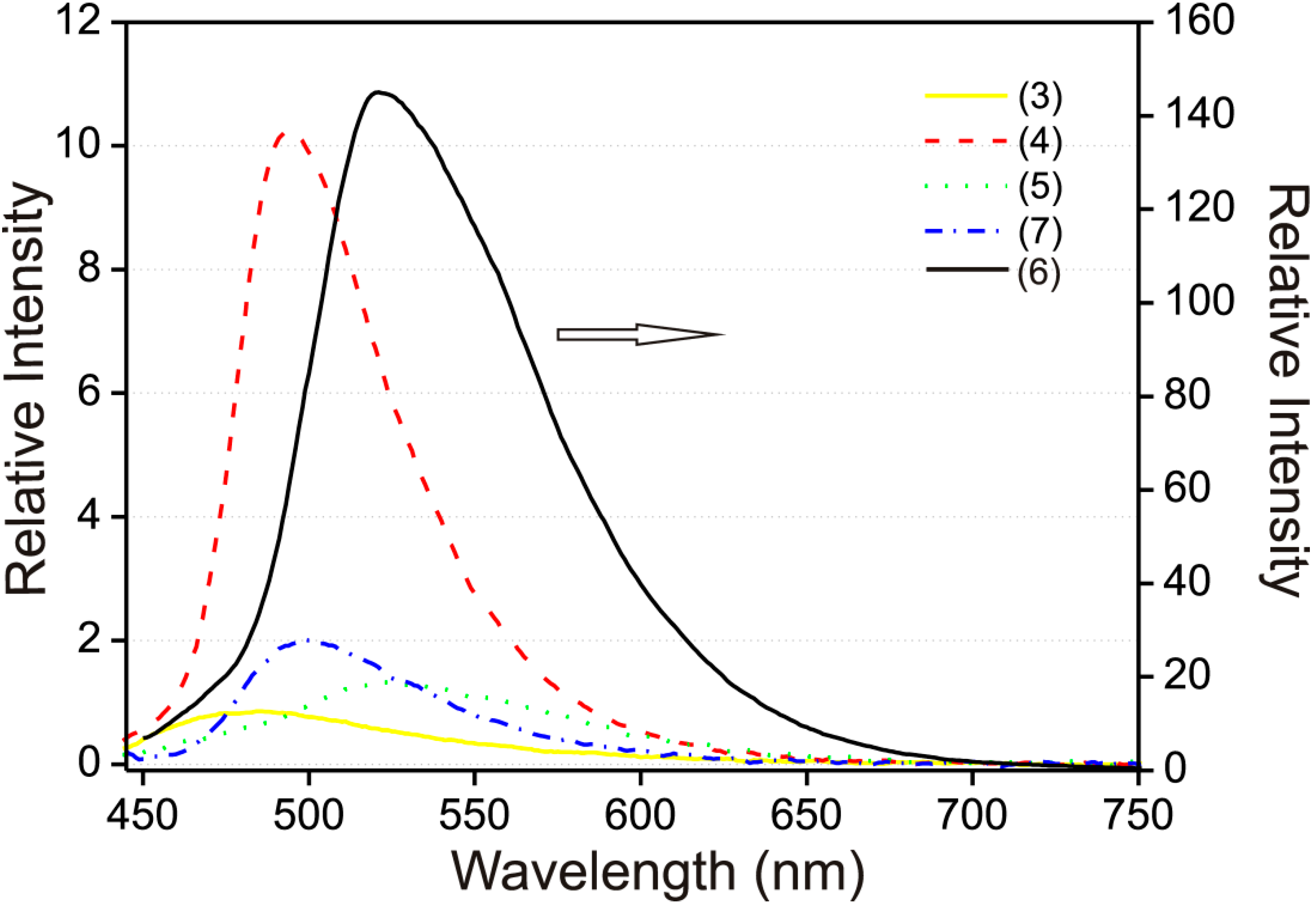
| Compound | Solid λmax (nm) | EtOH–H2O (95:5 v/v) λmax (nm) | Quantum Yield (%) * |
|---|---|---|---|
| 3 | 585 | 485 | 0.6 |
| 4 | 605 | 495 | 3.4 |
| 5 | 560 | 520 | 2.5 |
| 6 | 580 | 500 | 3.6 |
| 7 | 575 | 520 | 1.3 |
2.5. In Vitro Cytotoxicity
3. Experimental Section
3.1. General Methods
3.2. Materials
3.3. Synthesis: General preparation of Compounds 1–7
3.4. Single Crystal X‑ray Crystallography
3.5. Fluorescence Measurements

3.6. In Vitro Cytotoxicity
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Staskun, B. 2-Phenyl-3-(1-methyl-2-indolyl)-4(1H)-quinolone. J. Org. Chem. 1966, 31, 2674–2676. [Google Scholar] [CrossRef]
- Hradil, P.; Grepl, M.; Hlaváč, J.; Soural, M.; Maloň, M.; Bertolasi, V. some new routes for the preparation of 3-amino-2-phenyl-4(1h)-quinolinones from anthranilamides. J. Org. Chem. 2006, 71, 819–822. [Google Scholar] [CrossRef]
- Agati, G.; Azzarello, E.; Pollastri, S.; Tattini, M. Flavonoids as antioxidants in plants: Location and functional significance. Plant Sci. 2012, 196, 67–76. [Google Scholar] [CrossRef]
- Lafuente, A.G.; Guillamón, E.; Villares, A.; Rostagno, M.A.; Martínez, J.A. Flavonoids as anti-infammatory agents: Implications in cancer and cardiovascular disease. Inflamm. Res. 2009, 58, 537–552. [Google Scholar] [CrossRef]
- Sui, Z.; Jason, A.; Nguyen, V.N.; Fernandez, J.; Bernstein, J.I.; Hilliard, J.J.; Barrett, J.F.; Podlogar, B.L.; Ohemeng, K.A. Synthesis and inhibitory activity of novel Tri- and Tetracyclic quinolines against topoisomerases. Bioorg. Med. Chem. 1998, 6, 735–742. [Google Scholar] [CrossRef]
- Sui, Z.H.; Nguyen, V.N.; Altom, J.; Fernandez, J.; Hilliard, J.J.; Bernstein, J.I.; Barrett, J.F.; Ohemeng, K.A. Synthesis and topoisomerase inhibitory activities of novel aza-analogues of flavones. Eur. J. Med. Chem. 1999, 34, 381–387. [Google Scholar] [CrossRef]
- Iwanowicz, J.E.; Watterson, S.H.; Murali, T.G.; Pitts, W.J.; Gu, H.H. Heterocycles that are inhibitors of IMPDH enzyme. WO 2001081340, 1 November 2001. [Google Scholar]
- Soural, M.; Hlaváč, J.; Funk, P.; Džubák, P.; Hajdúch, M. 2-Phenylsubstituted-3-Hydroxyquinolin-4(1H)-one-Carboxamides: Structure-cytotoxic activity relationship study. ACS Comb. Sci. 2011, 52, 39–44. [Google Scholar]
- Buchtík, R.; Trávníček, Z.; Vančo, J.; Herchel, R.; Dvořák, Z. Synthesis, characterization, DNA interaction and cleavage, and in vitro cytotoxicity of copper(II) mixed-ligand complexes with 2-phenyl-3-hydroxy-4(1H)-quinolinone. Dalton Trans. 2011, 40, 9404–9412. [Google Scholar] [CrossRef]
- Buchtík, R.; Trávníček, Z.; Vančo, J. In vitro cytotoxicity, DNA cleavage and SOD-mimic activity of copper(II) mixed-ligand quinolinonato complexes. J. Inorg. Biochem. 2012, 116, 163–171. [Google Scholar] [CrossRef]
- Trávníček, Z.; Vančo, J.; Hošek, J.; Buchtík, R.; Dvořák, Z. Cellular responses induced by Cu(II) quinolinonato complexes in human tumor and hepatic cells. Chem. Centr. J. 2012, 6, 160. [Google Scholar] [CrossRef]
- Hirano, J.; Hamase, K.; Fukuda, H.; Tomita, T.; Zaitsu, K. Novel stable fluorophore, 6-methoxy-4-quinolone, with strong fluorescence in wide pH range of aqueous media, and its application as a fluorescent labeling reagent. J. Chromatogr. A 2004, 1059, 225–231. [Google Scholar] [CrossRef]
- Yushchenko, D.A.; Bilokin, M.D.; Pyvovarenko, O.V.; Duportail, G.; Mély, Y.; Pivovarenko, G.V. Synthesis and fluorescence properties of 2-aryl-3-hydroxyquinolones, a new class of dyes displaying dual fluorescence. Tetrahedron Lett. 2006, 47, 905–908. [Google Scholar] [CrossRef]
- Bilokin, M.D.; Shvadchak, V.V.; Yushchenko, D.A.; Klymchenko, A.S.; Duportail, G.; Mely, Y.; Pivovarenko, G.V. 3-Hydroxybenzo[g]quinolones: Dyes with red-shifted absorption and highly resolved dual emission. Tetrahedron Lett. 2009, 50, 4714–4719. [Google Scholar] [CrossRef]
- Issa, R.M.; Khedr, A.M.; Rizk, H. 1H-NMR, IR and UV/VIS spectroscopic studies of some Schiff bases derived from 2-aminobenzothiazole and 2-amino-3-hydroxypyridine. J. Chin. Chem. Soc. 2008, 55, 875–884. [Google Scholar]
- Smith, B. Infrared Spectral Interpretation; CRC Press: Boca Ranton, FL, USA, 1999. [Google Scholar]
- Biczok, L.; Berces, T.; Linschitz, H. Quenching processes in hydrogen-bonded pairs: Interactions of excited fluorenone with alcohols and phenols. J. Am. Chem. Soc. 1997, 119, 11071–11077. [Google Scholar] [CrossRef]
- CrysAlis CCD and CrysAlis RED software; Version 1.171.33.52; Oxford Diffraction Ltd.: Abingdon, Oxfordshire, UK, 2009.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. Crystal Impact GbR. Available online: http://www.crystalimpact.com/diamond/references.htm (accessed on 26 February 2008).
- Van der Sluis, P.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. 1990, A46, 194–201. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Melhuish, W.H.J. Quantum efficiencies of fluorescence of organic substances: Effect of solvent and concentration of the fluorescent solute. J. Phys. Chem. 1961, 65, 229–235. [Google Scholar] [CrossRef]
- Vrzal, R.; Štarha, P.; Dvořák, Z.; Trávníček, Z. Evaluation of in vitro cytotoxicity and hepatotoxicity of platinum(II) and palladium(II) oxalato complexes with adenine derivatives as carrier ligands. J. Inorg. Biochem. 2010, 104, 1130–1132. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–7 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Trávníček, Z.; Buchtík, R.; Nemec, I. Novel Schiff Bases Based on the Quinolinone Skeleton: Syntheses, X-ray Structures and Fluorescent Properties. Molecules 2014, 19, 13509-13525. https://doi.org/10.3390/molecules190913509
Trávníček Z, Buchtík R, Nemec I. Novel Schiff Bases Based on the Quinolinone Skeleton: Syntheses, X-ray Structures and Fluorescent Properties. Molecules. 2014; 19(9):13509-13525. https://doi.org/10.3390/molecules190913509
Chicago/Turabian StyleTrávníček, Zdeněk, Roman Buchtík, and Ivan Nemec. 2014. "Novel Schiff Bases Based on the Quinolinone Skeleton: Syntheses, X-ray Structures and Fluorescent Properties" Molecules 19, no. 9: 13509-13525. https://doi.org/10.3390/molecules190913509