Chemo-Enzymatic Synthesis of Silybin and 2,3-Dehydrosilybin Dimers

 ,
,  ,
,
Abstract
:
1. Introduction
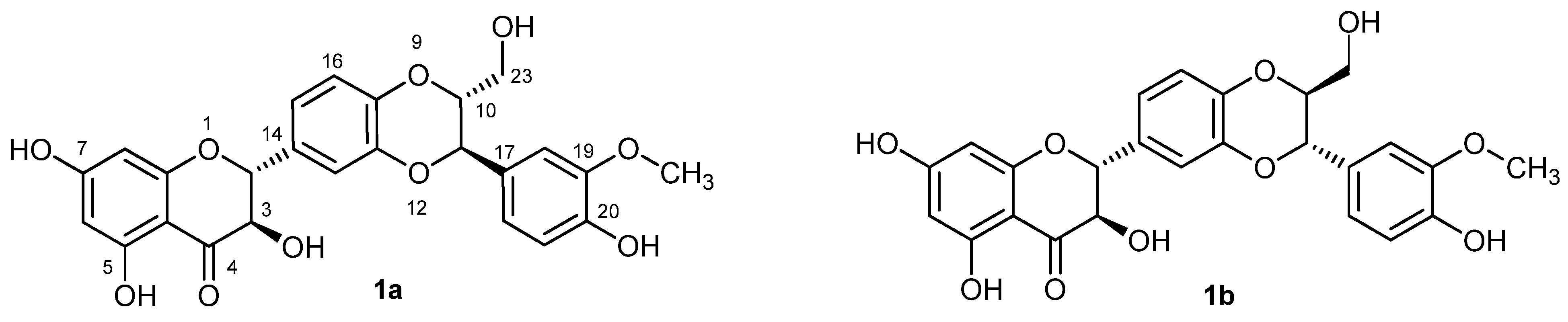
2. Results and Discussion
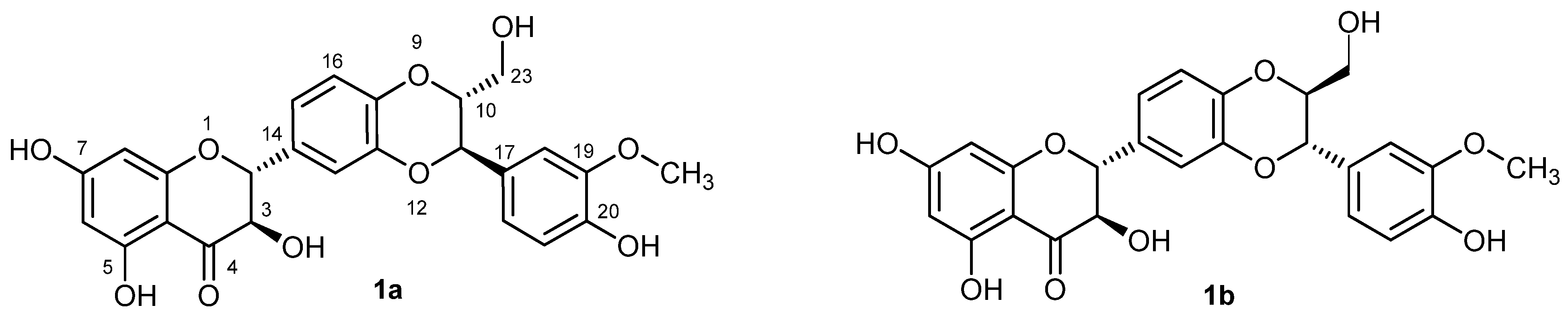
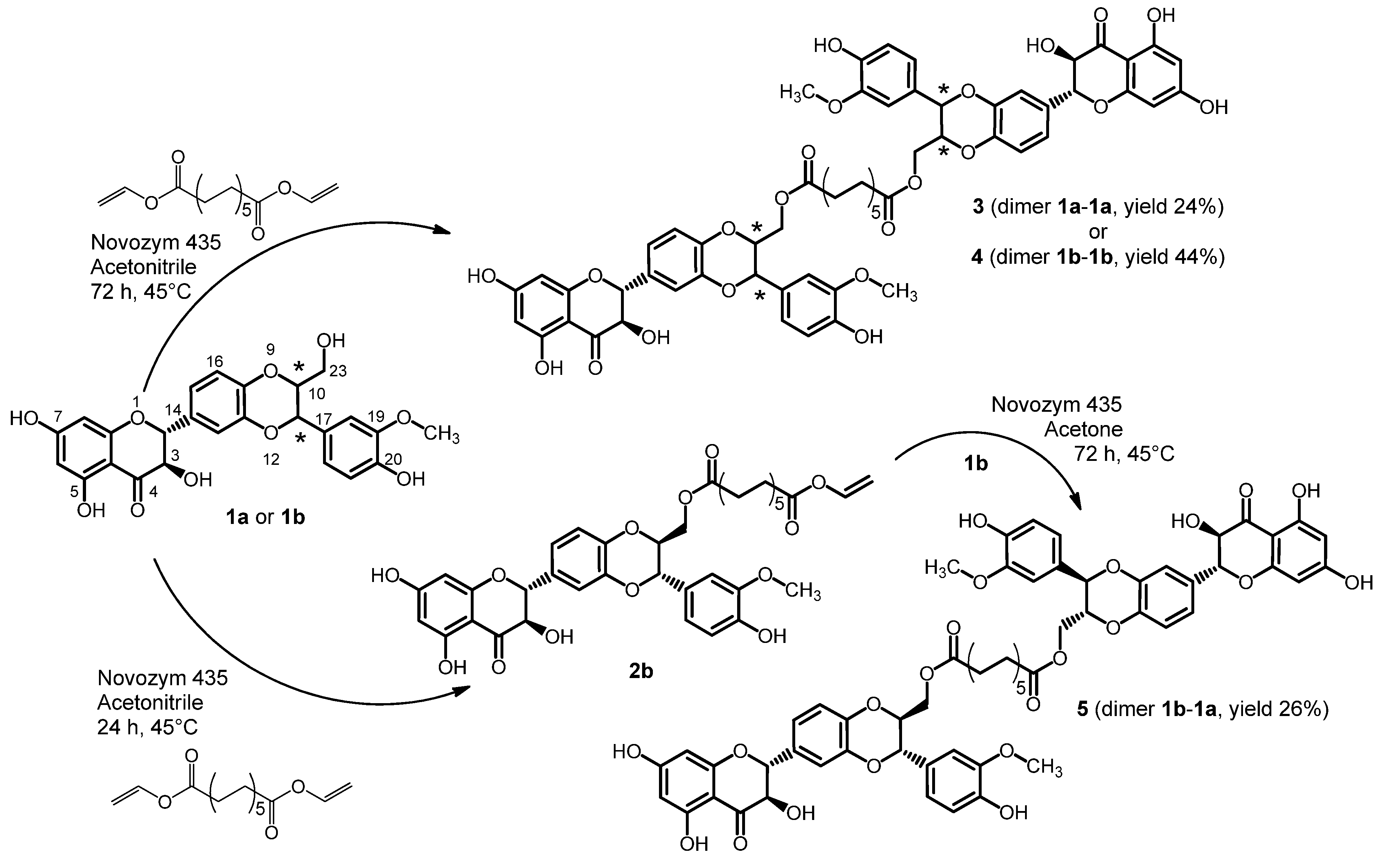
2.1. Synthesis of Dimers
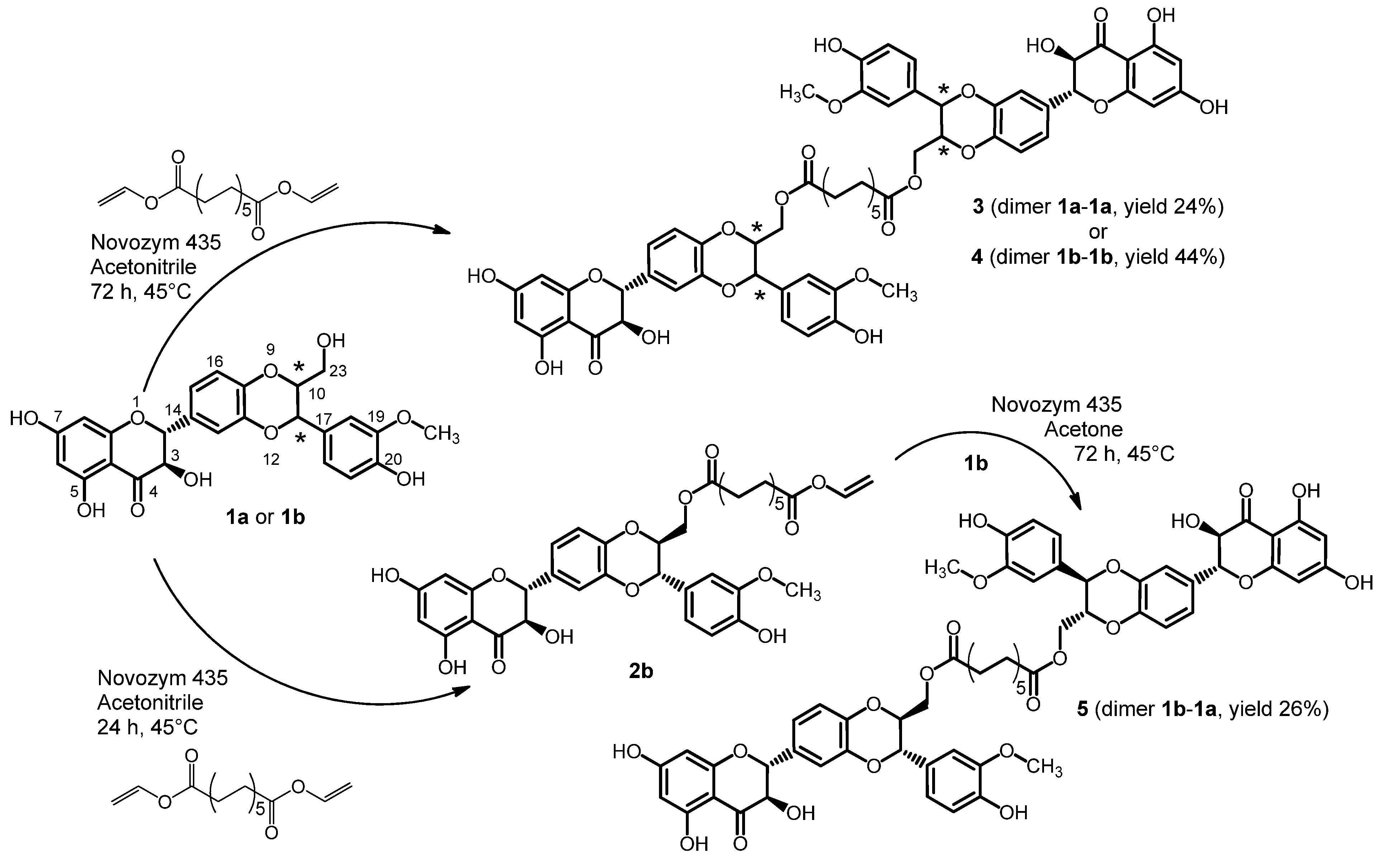
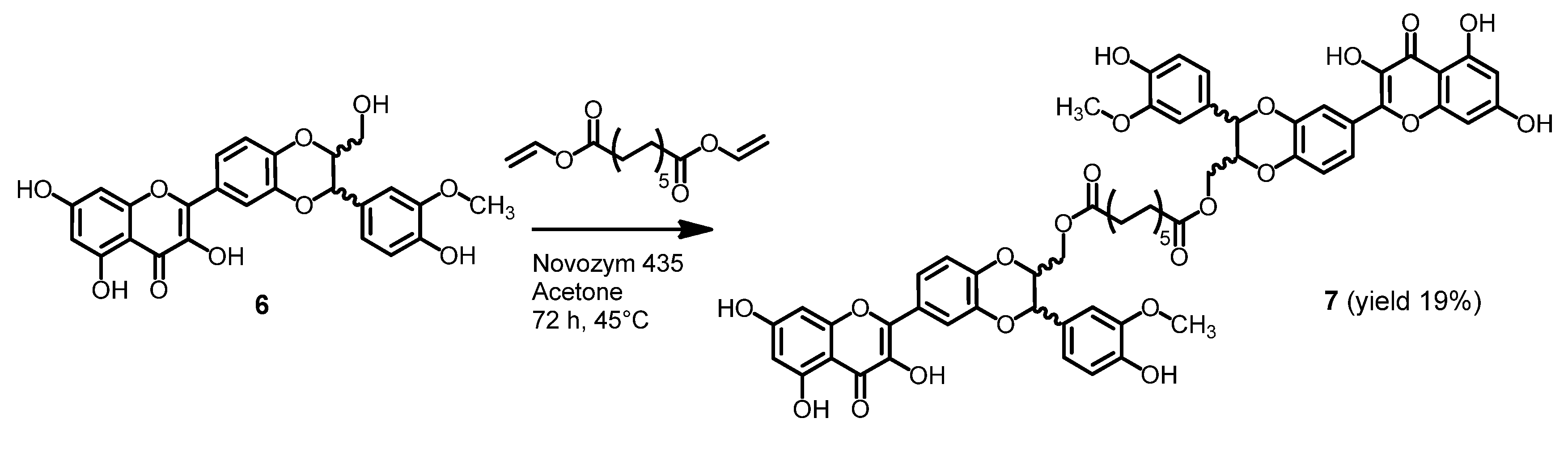
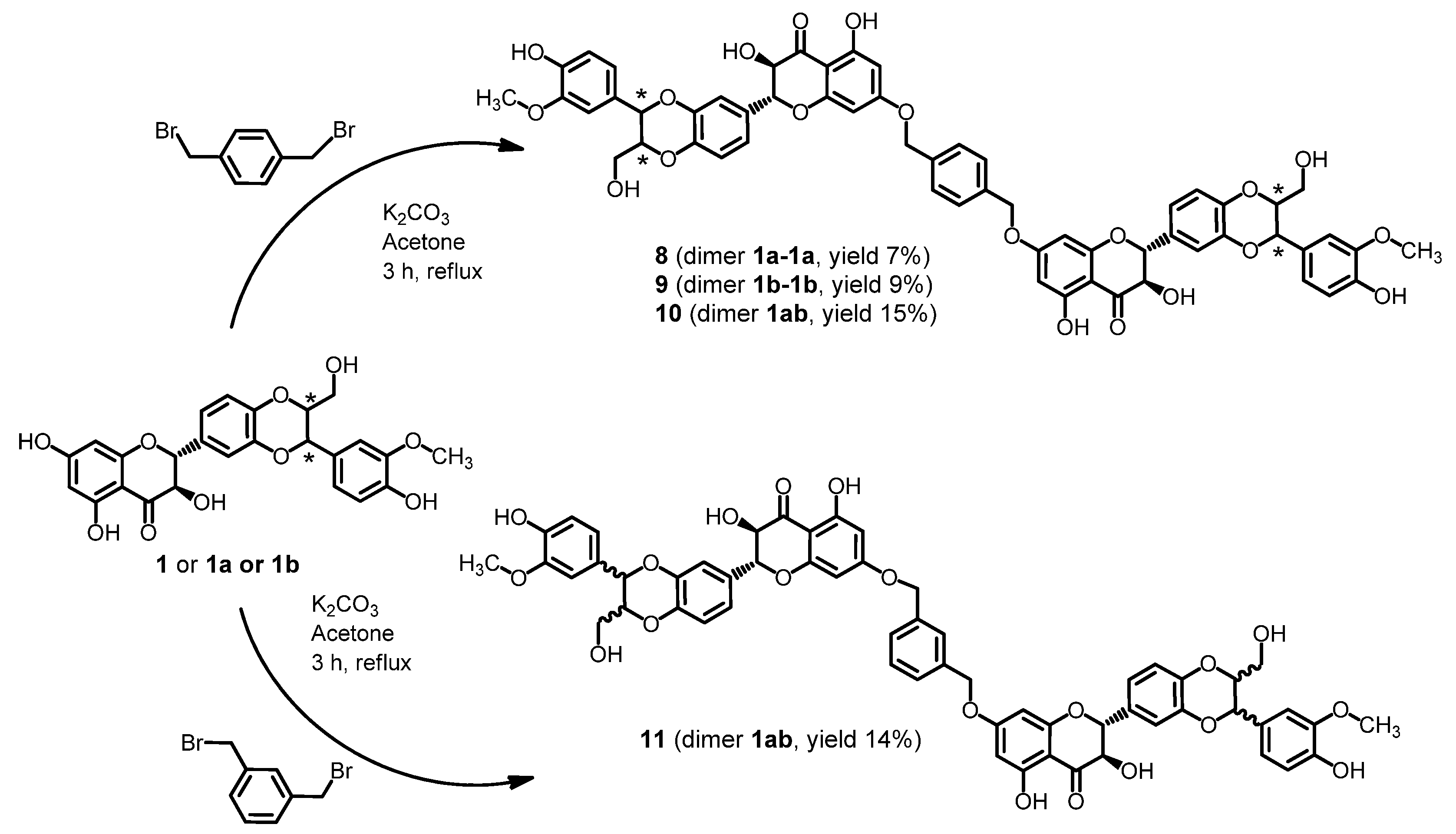
2.2. Stability of Dimers
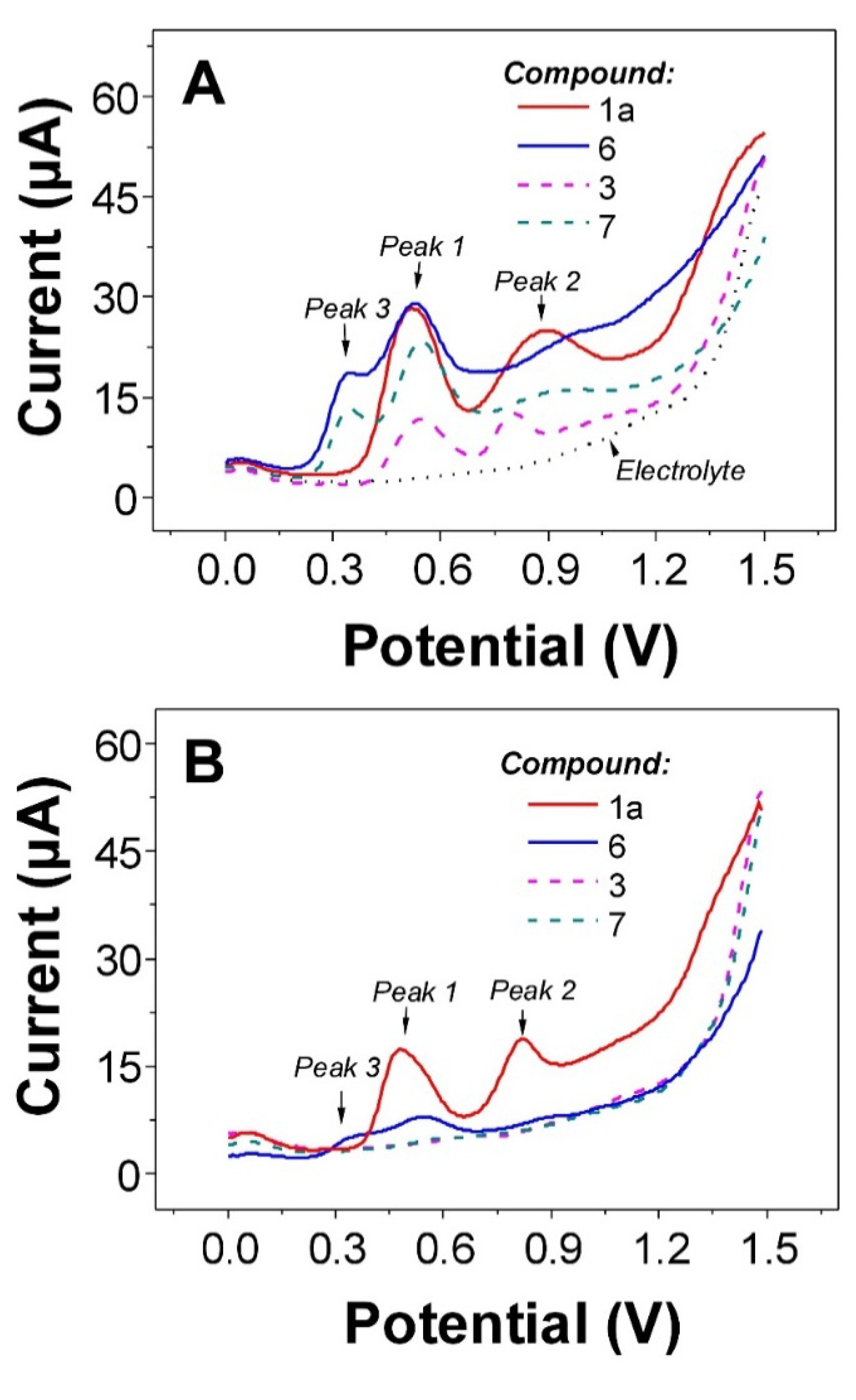
2.3. Electrochemistry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Oxidation potential [V] * vs. Ag/AgCl/3 M KCl n = 3 | DPPH [% of inhibition] ** n = 4 | ||
|---|---|---|---|---|
| Ep3 | Ep1 | Ep2 | 33 µM | |
| 1a | 0.55 | 0.90 | 6.6 ± 1.7 a | |
| 3 | 0.56 | 0.83 | 7.1 ± 2.8 a | |
| 4 | 0.57 | 0.83 | 9.0 ± 3.0 a | |
| 5 | 0.55 | 0.83 | 7.0 ± 2.9 a | |
| 6 | 0.37 | 0.54 | 0.92 | 82.9 ± 0.2 b |
| 7 | 0.36 | 0.57 | 0.99 | 33.4 ± 3.1 c |
| 8 | 0.56 | 0.81 | 8.5 ± 3.7 a | |
| 9 | 0.57 | 0.83 | 8.9 ± 4.0 a | |
| 10 | 0.56 | 0.80 | 8.1 ± 3.4 a | |
| 11 | 0.52 | 0.87 | 10.7 ± 3.1 a | |
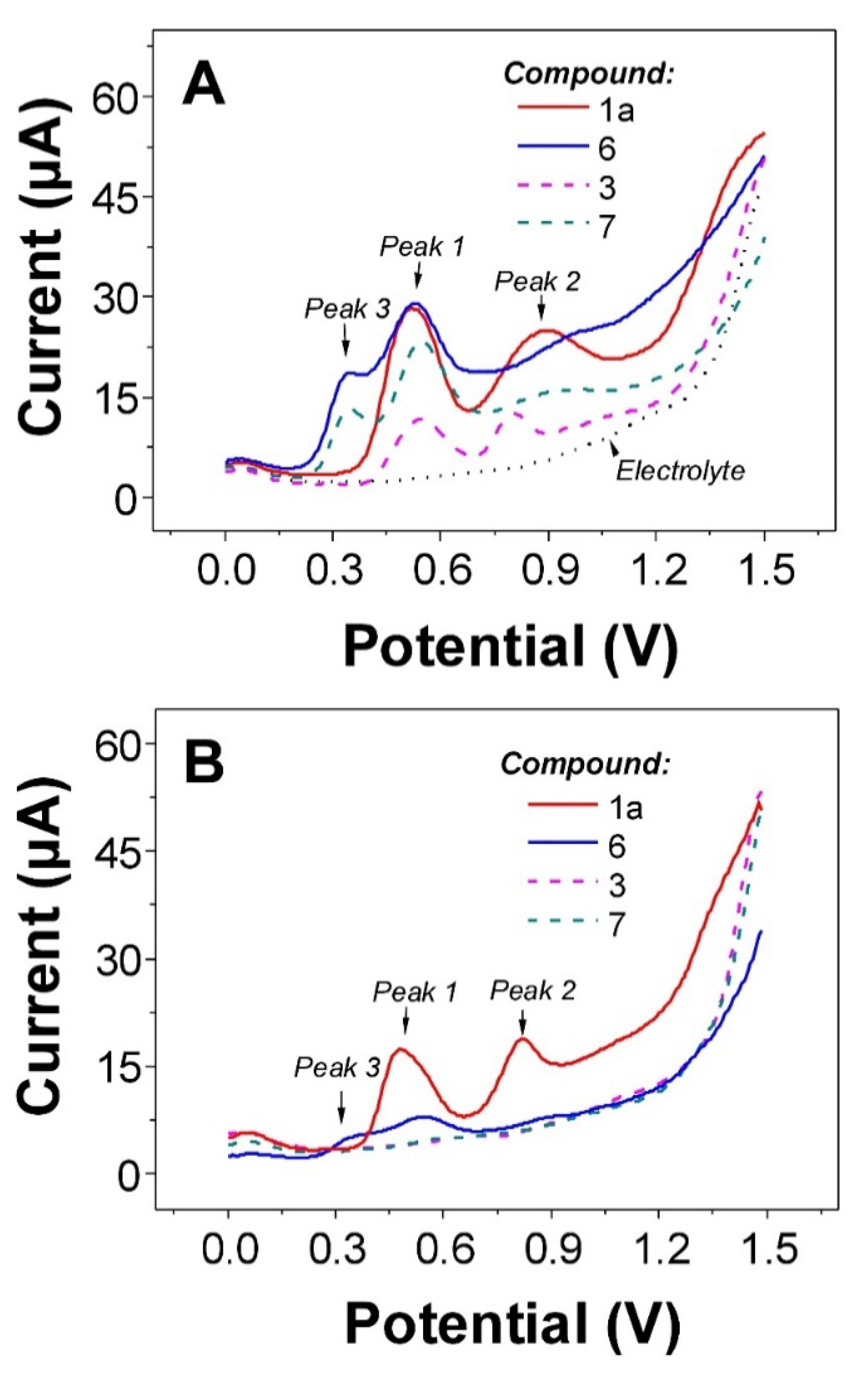
2.4. DPPH Scavenging
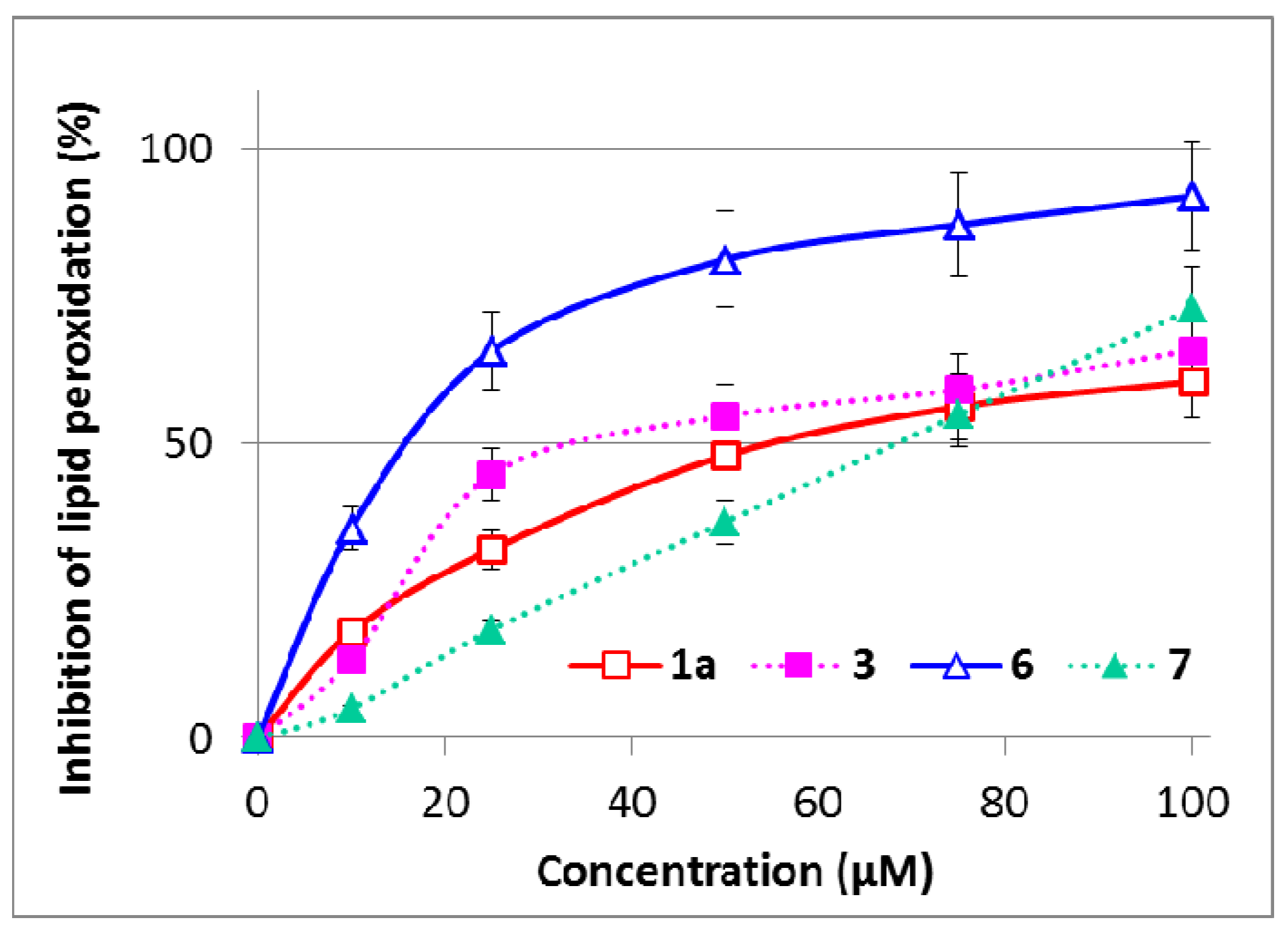
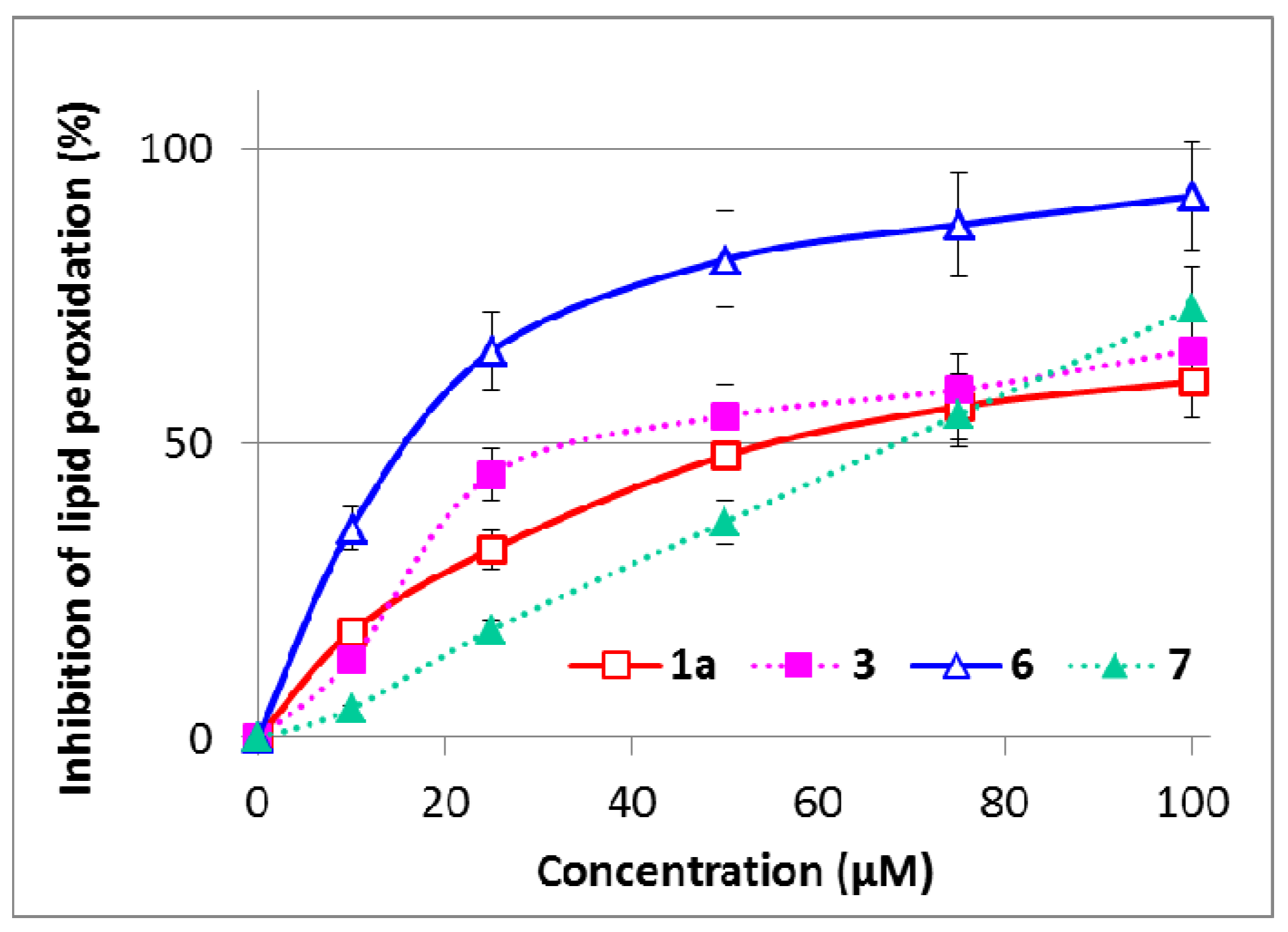
2.5. Inhibition of Microsomal Lipid Peroxidation

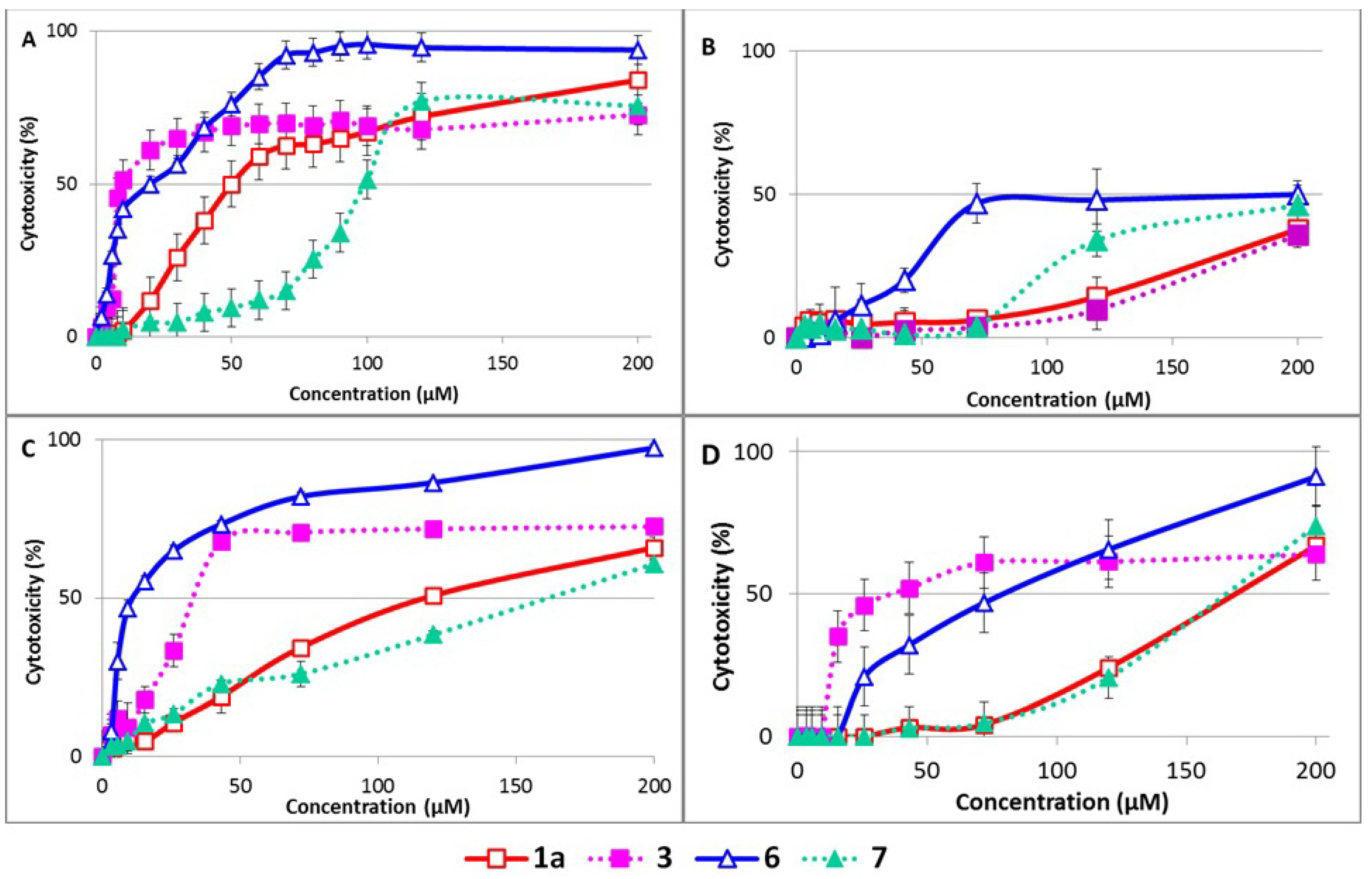
2.6. Cytotoxicity
| HUVEC | NAK | BALB/c 3T3 | HepG2 | |
|---|---|---|---|---|
| 1a | 57.8 ± 3.6 | >200 | 130.7 ± 8.0 | 165.4 ± 8.1 |
| 3 | 7.6 ± 1.9 b | >200 | 23.5 ± 5.6 b | 51.8 ± 5.9 b |
| 6 | 20.9 ± 4.6 | >200 | 11.9 ± 3.0 | 58.4 ± 9.0 |
| 7 | 98.4 ± 10.6 c | >200 | 161.4 ± 9.0 c | 172.5 ± 11.6 c |
3. Experimental
3.1. Chemicals and Reagents
3.2. General Methods
3.3. Chemistry
3.3.1. Synthesis of 12-Vinyl Dodecanedioate-23-O-Silybin B (2b)
3.3.2. General Procedure—Preparation of Diesters 3 and 4
3.3.3. Preparation of Mixed Dimer 5
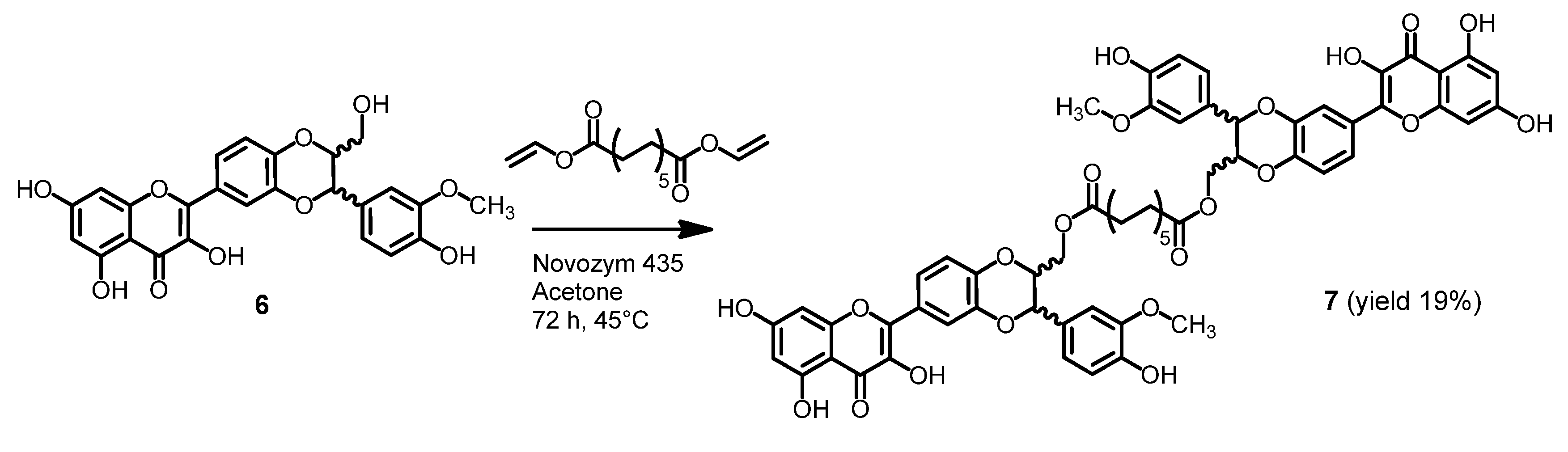
3.3.4. Preparation 2,3-Dehydrosilybin Dimer 7
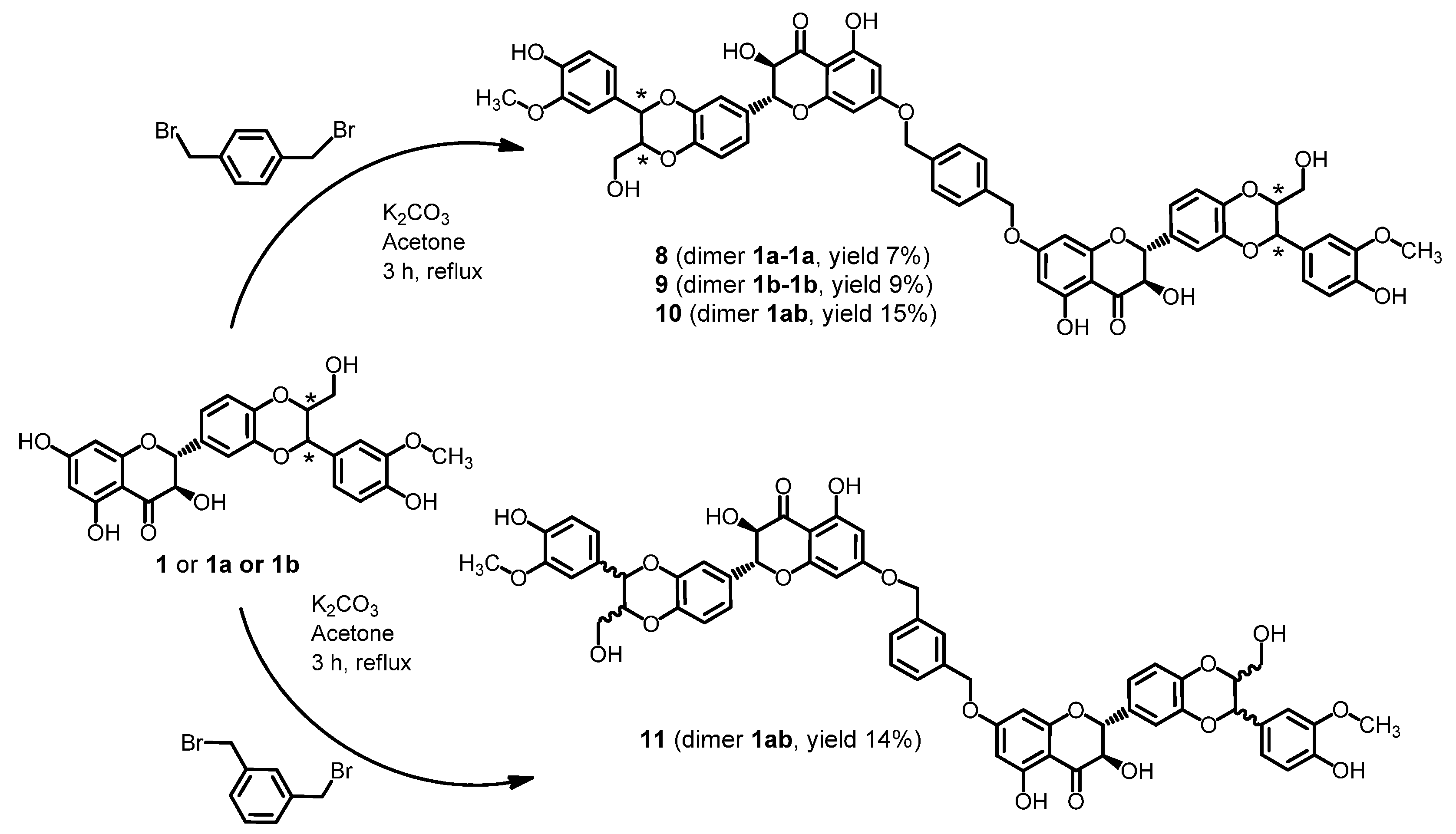
3.3.5. General Procedure: Preparation of Silybin Diethers 8, 9, 10 and 11
3.4. Stability of Dimers—HPLC Analysis
3.5. Electrochemical Measurement
3.6. DPPH Assay
3.7. Inhibition of Microsomal Lipid Peroxidation
3.8. Source of Human Tissues
3.9. Cell Cultures
3.10. Cytotoxicity Testing
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gazak, R.; Walterova, D.; Kren, V. Silybin and silymarin—New and emerging applications in medicine. Curr. Med. Chem. 2007, 14, 315–338. [Google Scholar] [CrossRef]
- Kren, V.; Gazak, R.; Purchartova, K.; Marhol, P.; Biedermann, D.; Sedmera, P. Chemoenzymatic preparative separation of silybins A and B. J. Mol. Catal. B: Enzymat. 2009, 61, 247–251. [Google Scholar]
- Gazak, R.; Trouillas, P.; Biedermann, D.; Fuksova, K.; Marhol, P.; Kuzma, M.; Kren, V. Base-catalyzed oxidation of silybin and isosilybin into 2,3-dehydro derivatives. Tetrahedron Lett. 2013, 54, 315–317. [Google Scholar] [CrossRef]
- Huber, A.; Thongphasuk, P.; Erben, G.; Lehmann, W.D.; Tuma, S.; Stremmel, W.; Chamulitrat, W. Significantly greater antioxidant anticancer activities of 2,3-dehydrosilybin than silybin. Biochim. Biophys. Acta 2008, 1780, 837–847. [Google Scholar]
- Thongphasuk, P.; Stremmel, W.; Chamulitrat, W. 2,3-dehydrosilybin is a better DNA topoisomerase I inhibitor than its parental silybin. Chemotherapy 2009, 55, 42–48. [Google Scholar] [CrossRef]
- Gazak, R.; Svobodova, A.; Psotova, J.; Sedmera, P.; Prikrylova, V.; Walterova, D.; Kren, V. Oxidised derivatives of silybin and their antiradical and antioxidant activity. Bioorg. Med. Chem. 2004, 12, 5677–5687. [Google Scholar] [CrossRef]
- Zatloukalova, M.; Kren, V.; Gazak, R.; Kubala, M.; Trouillas, P.; Ulrichova, J.; Vacek, J. Electrochemical investigation of flavonolignans and study of their interactions with DNA in the presence of Cu(II). Bioelectrochemistry 2011, 82, 117–124. [Google Scholar] [CrossRef]
- Gazak, R.; Sedmera, P.; Vrbacky, M.; Vostalova, J.; Drahota, Z.; Marhol, P.; Walterova, D.; Kren, V. Molecular mechanisms of silybin and 2,3-dehydrosilybin antiradical activity—Role of individual hydroxyl groups. Free Radic. Biol. Med. 2009, 46, 745–758. [Google Scholar] [CrossRef]
- Gazak, R.; Purchartova, K.; Marhol, P.; Zivna, L.; Sedmera, P.; Valentova, K.; Kato, N.; Matsumura, H.; Kaihatsu, K.; Kren, V. Antioxidant and antiviral activities of silybin fatty acid conjugates. Eur. J. Med. Chem. 2010, 45, 1059–1067. [Google Scholar] [CrossRef]
- Theodosiou, E.; Katsoura, M.H.; Loutrari, H.; Purchartova, K.; Kren, V.; Kolisis, F.N.; Stamatis, H. Enzymatic preparation of acylated derivatives of silybin in organic and ionic liquid media and evaluation of their antitumor proliferative activity. Biocatal. Biotrans. 2009, 27, 161–169. [Google Scholar]
- Theodosiou, E.; Loutrari, H.; Stamatis, H.; Roussos, C.; Kolisis, F.N. Biocatalytic synthesis and antitumor activities of novel silybin acylated derivatives with dicarboxylic acids. New Biotechnol. 2011, 28, 342–348. [Google Scholar] [CrossRef]
- Monti, D.; Gazak, R.; Marhol, P.; Biedermann, D.; Purchartova, K.; Fedrigo, M.; Riva, S.; Kren, V. Enzymatic kinetic resolution of silybin diastereoisomers. J. Nat. Prod. 2010, 73, 613–629. [Google Scholar] [CrossRef]
- Chen, X.Y.; Zenger, K.; Lupp, A.; Kling, B.; Heilmann, J.; Fleck, C.; Kraus, B.; Decker, M. Tacrine-silibinin codrug shows neuro- and hepato protective effects in vitro and pro-cognitive and hepatoprotective effects in vivo. J. Med. Chem. 2012, 55, 5231–5242. [Google Scholar] [CrossRef]
- Zarrelli, A.; Romanucci, V.; Greca, M.D.; de Napoli, L.; Previtera, L.; di Fabio, G. New silybin scaffold for chemical diversification: Synthesis of novel 23-phosphodiester silybin conjugates. Synlett 2013, 24, 45–48. [Google Scholar]
- Gazak, R.; Sedmera, P.; Marzorati, M.; Riva, S.; Kren, V. Laccase-mediated dimerization of the flavonolignan silybin. J. Mol. Catal. B 2008, 50, 87–92. [Google Scholar] [CrossRef]
- Trouillas, P.; Marsal, P.; Svobodova, A.; Vostalova, J.; Gazak, R.; Hrbac, J.; Sedmera, P.; Kren, V.; Lazzaroni, R.; Duroux, J.L.; et al. Mechanism of the antioxidant action of silybin and 2,3-dehydrosilybin flavonolignans: A joint experimental and theoretical study. J. Phys. Chem. A 2008, 112, 1054–1063. [Google Scholar] [CrossRef]
- Pliskova, M.; Vondracek, J.; Kren, V.; Gazak, R.; Sedmera, P.; Walterova, D.; Psotova, J.; Simanek, V.; Machala, M. Effects of silymarin flavonolignans and synthetic silybin derivatives on estrogen and aryl hydrocarbon receptor activation. Toxicology 2005, 215, 80–89. [Google Scholar] [CrossRef]
- Berube, G. Natural and synthetic biologically active dimeric molecules: Anticancer agents, anti-hiv agents, steroid derivatives and opioid antagonists. Curr. Med. Chem. 2006, 13, 131–154. [Google Scholar] [CrossRef]
- Mott, B.T.; Tripathi, A.; Siegler, M.A.; Moore, C.D.; Sullivan, D.J.; Posner, G.H. Synthesis and antimalarial efficacy of two-carbon-linked, artemisinin-derived trioxane dimers in combination with known antimalarial drugs. J. Med. Chem. 2013, 56, 2630–2641. [Google Scholar] [CrossRef]
- Jenett-Siems, K.; Kohler, I.; Kraft, C.; Pertz, H.H.; Kren, V.; Fiserova, A.; Kuzma, M.; Ulrichova, J.; Bienzle, U.; Eich, E. In vitro antiplasmodial activities of semisynthetic N,N'-spacer-linked oligomeric ergolines. Bioorg. Med. Chem. 2004, 12, 817–824. [Google Scholar] [CrossRef]
- Chan, K.F.; Zhao, Y.Z.; Burkett, B.A.; Wong, I.L.K.; Chow, L.M.C.; Chan, T.H. Flavonoid dimers as bivalent modulators for P-glycoprotein-based multidrug resistance: Synthetic apigenin homodimers linked with defined-length poly(ethylene glycol) spacers increase drug retention and enhance chemosensitivity in resistant cancer cells. J. Med. Chem. 2006, 49, 6742–6759. [Google Scholar] [CrossRef]
- Dolle, C.; Magrone, P.; Riva, S.; Ambrosi, M.; Fratini, E.; Peruzzi, N.; Lo Nostro, P. Symmetric and asymmetric bolaamphiphiles from ascorbic acid. J. Phys. Chem. B 2011, 115, 11638–11649. [Google Scholar]
- Chebil, L.; Humeau, C.; Falcimaigne, A.; Engasser, J.M.; Ghoul, M. Enzymatic acylation of flavonoids. Process Biochem. 2006, 41, 2237–2251. [Google Scholar]
- Magrone, P.; Cavallo, F.; Panzeri, W.; Passarella, D.; Riva, S. Exploiting enzymatic regioselectivity: A facile methodology for the synthesis of polyhydroxylated hybrid compounds. Org. Biomol. Chem. 2010, 8, 5583–5590. [Google Scholar] [CrossRef]
- Zatloukalova, M.; Enache, T.A.; Kren, V.; Ulrichova, J.; Vacek, J.; Oliveira-Brett, A.M. Effect of 3-O-galloyl substitution on the electrochemical oxidation of quercetin and silybin galloyl esters at glassy carbon electrode. Electroanalysis 2013, 25, 1621–1627. [Google Scholar] [CrossRef]
- Vacek, J.; Zatloukalova, M.; Desmier, T.; Nezhodova, V.; Hrbac, J.; Kubala, M.; Kren, V.; Ulrichova, J.; Trouillas, P. Antioxidant, metal-binding and DNA-damaging properties of flavonolignans: A joint experimental and computational highlight based on 7-O-galloylsilybin. Chem. Biol. Interact. 2013, 205, 173–180. [Google Scholar] [CrossRef]
- Valentova, K.; Vidlar, A.; Zatloukalova, M.; Stuchlik, M.; Vacek, J.; Simanek, V.; Ulrichova, J. Biosafety and antioxidant effects of a beverage containing silymarin and arginine. A pilot, human intervention cross-over trial. Food Chem. Toxicol. 2013, 56, 178–183. [Google Scholar] [CrossRef]
- Wang, F.; Huang, K.X.; Yang, L.X.; Gong, J.X.; Tao, Q.F.; Li, H.B.; Zhao, Y.; Zeng, S.; Wu, X.M.; Stockigt, J.; et al. Preparation of C-23 esterified silybin derivatives and evaluation of their lipid peroxidation inhibitory and DNA protective properties. Bioorg. Med. Chem. 2009, 17, 6380–6389. [Google Scholar] [CrossRef]
- Abourashed, E.A.; Mikell, J.R.; Khan, I.A. Bioconversion of silybin to phase I and II microbial metabolites with retained antioxidant activity. Bioorg. Med. Chem. 2012, 20, 2784–2788. [Google Scholar] [CrossRef]
- Zhu, L.F.; Xu, M.; Zhu, H.T.; Wang, D.; Yang, S.X.; Yang, C.R.; Zhang, Y.J. New flavan-3-ol dimer from green tea produced from camellia taliensis in the Ai-Lao Mountains of Southwest China. J. Agric. Food Chem. 2012, 60, 12170–12176. [Google Scholar] [CrossRef]
- Lu, Y.R.; Foo, L.Y. Antioxidant and radical scavenging activities of polyphenols from apple pomace. Food Chem. 2000, 68, 81–85. [Google Scholar] [CrossRef]
- Fujisawa, S.; Atsumi, T.; Ishihara, M.; Kadoma, Y. Cytotoxicity, ROS-generation activity and radical-scavenging activity of curcumin and related compounds. Anticancer Res. 2004, 24, 563–569. [Google Scholar]
- Osman, A.M. Multiple pathways of the reaction of 2,2-diphenyl-1-picrylhydrazyl radical (DPPH center dot) with (+)-catechin: Evidence for the formation of a covalent adduct between DPPH center dot and the oxidized form of the polyphenol. Biochem. Bioph. Res. Commun. 2011, 412, 473–478. [Google Scholar] [CrossRef]
- Kawabata, J.; Okamoto, Y.; Kodama, A.; Makimoto, T.; Kasai, T. Oxidative dimers produced from protocatechuic and gallic esters in the DPPH radical scavenging reaction. J. Agric. Food Chem. 2002, 50, 5468–5471. [Google Scholar]
- Yamaguchi, H.; Ishii, E.; Tashiro, K.; Miyazaki, S. Role of umbilical vein endothelial cells in hematopoiesis. Leukemia Lymphoma 1998, 31, 61–69. [Google Scholar] [CrossRef]
- Rasmussen, C.; Thomas-Virnig, C.; Allen-Hoffmann, B.L. Classical human epidermal keratinocyte cell culture. In Methods in Molecular Biology; Randell, S.H., Fulcher, M.L., Eds.; Humana Press: Clifton, NJ, USA, 2013; Volume 945, pp. 161–175. [Google Scholar]
- Mersch-Sundermann, V.; Knasmuller, S.; Wu, X.-J.; Darroudi, F.; Kassie, F. Use of a human-derivedliver cell line for the detection of cytoprotective, antigenotoxic and cogenotoxic agents. Toxicology 2004, 198, 329–340. [Google Scholar] [CrossRef]
- Pham, A.; Bortolazzo, A.; White, J.B. Rapid dimerization of quercetin through an oxidative mechanism in the presence of serum albumin decreases its ability to induce cytotoxicity in MDA-MB-231 cells. Biochem. Bioph. Res. Commun. 2012, 427, 415–420. [Google Scholar] [CrossRef]
- Figueiredo, P.; Elhabiri, M.; Toki, K.; Saito, N.; Dangles, O.; Brouillard, R. New aspects of anthocyanin complexation. Intramolecular copigmentation as a means for colour loss? Phytochemistry 1996, 41, 301–308. [Google Scholar] [CrossRef]
- Di Meo, F.; Garcia, J.C.S.; Dangles, O.; Trouillas, P. Highlights on anthocyanin pigmentation and copigmentation: A matter of flavonoid π-stacking complexation to be described by dft-d. J. Chem. Theory Comput. 2012, 8, 2034–2043. [Google Scholar] [CrossRef]
- Nave, F.; Bras, N.F.; Cruz, L.; Teixeira, N.; Mateus, N.; Ramos, M.J.; di Meo, F.; Trouillas, P.; Dangles, O.; de Freitas, V. Influence of a flavan-3-ol substituent on the affinity of anthocyanins (pigments) toward vinylcatechin dimers and proanthocyanidins (copigments). J. Phys. Chem. B 2012, 116, 14089–14099. [Google Scholar] [CrossRef]
- Velu, S.S.; di Meo, F.; Trouillas, P.; Sancho-Garcia, J.C.; Weber, J.F. Regio- and stereocontrolled synthesis of oligostilbenoids: Theoretical highlights at the supramolecular level. J. Nat. Prod. 2013, 76, 538–546. [Google Scholar] [CrossRef]
- Gazak, R.; Marhol, P.; Purchartova, K.; Monti, D.; Biedermann, D.; Riva, S.; Cvak, L.; Kren, V. Large-scale separation of silybin diastereoisomers using lipases. Process Biochem. 2010, 45, 1657–1663. [Google Scholar] [CrossRef]
- Zatloukalova, M.; Orolinova, E.; Kubala, M.; Hrbac, J.; Vacek, J. Electrochemical determination of transmembrane protein Na+/K+-ATPase and its cytoplasmic loop C45. Electroanalysis 2012, 24, 1758–1765. [Google Scholar]
- Joyeux, M.; Mortier, F.; Fleurentin, J. Screening of antiradical, antilipoperoxidant and hepatoprotective effects of 9 plant-extracts used in caribbean folk medicine. Phytother. Res. 1995, 9, 228–230. [Google Scholar] [CrossRef]
- Siekevitz, P.; Sidney, P.C.; Nathan, O.K. Preparation of microsomes and submicrosomal fractions: Mammalian. In Methodsin Enzymology; Academic Press: Waltham, MA, USA, 1962; Volume 5, pp. 61–68. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Council for International Organizations of Medical Sciences. International ethical guidelines for biomedical research involving human subjects. Bull. Med. Ethics 2002, 182, 17–23. [Google Scholar]
- Gazak, R.; Valentova, K.; Fuksova, K.; Marhol, P.; Kuzma, M.; Medina, M.A.; Oborna, I.; Ulrichova, J.; Kren, V. Synthesis and antiangiogenic activity of new silybin galloyl esters. J. Med. Chem. 2011, 54, 7397–7407. [Google Scholar] [CrossRef]
- Atanasova, G.; Jans, R.; Zhelev, N.; Mitev, V.; Poumay, Y. Effects of the cyclin-dependent kinase inhibitor CYC202 (R-roscovitine) on the physiology of cultured human keratinocytes. Biochem. Pharmacol. 2005, 70, 824–836. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the new compounds are available in miligram amounts from the authors at the Institute of Microbiology, Prague, Czech Republic.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vavříková, E.; Vacek, J.; Valentová, K.; Marhol, P.; Ulrichová, J.; Kuzma, M.; Křen, V. Chemo-Enzymatic Synthesis of Silybin and 2,3-Dehydrosilybin Dimers. Molecules 2014, 19, 4115-4134. https://doi.org/10.3390/molecules19044115
Vavříková E, Vacek J, Valentová K, Marhol P, Ulrichová J, Kuzma M, Křen V. Chemo-Enzymatic Synthesis of Silybin and 2,3-Dehydrosilybin Dimers. Molecules. 2014; 19(4):4115-4134. https://doi.org/10.3390/molecules19044115
Chicago/Turabian StyleVavříková, Eva, Jan Vacek, Kateřina Valentová, Petr Marhol, Jitka Ulrichová, Marek Kuzma, and Vladimír Křen. 2014. "Chemo-Enzymatic Synthesis of Silybin and 2,3-Dehydrosilybin Dimers" Molecules 19, no. 4: 4115-4134. https://doi.org/10.3390/molecules19044115