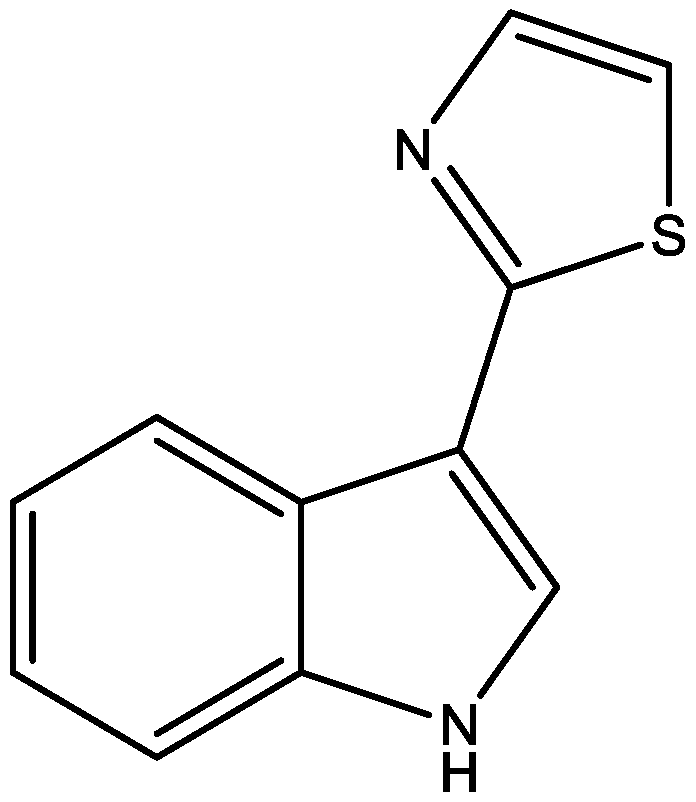
Camalexin-Induced Apoptosis in Prostate Cancer Cells Involves Alterations of Expression and Activity of Lysosomal Protease Cathepsin D

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
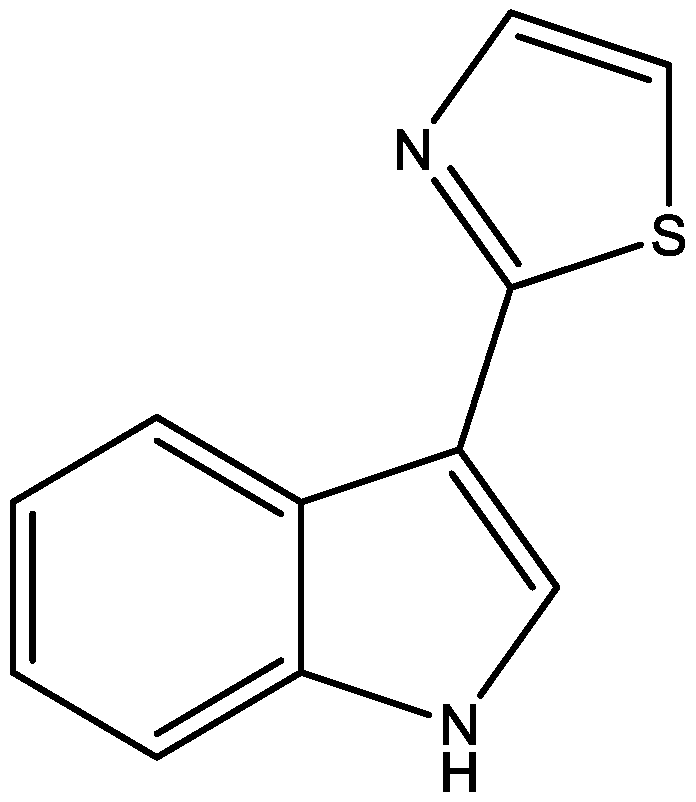
2. Results and Discussion
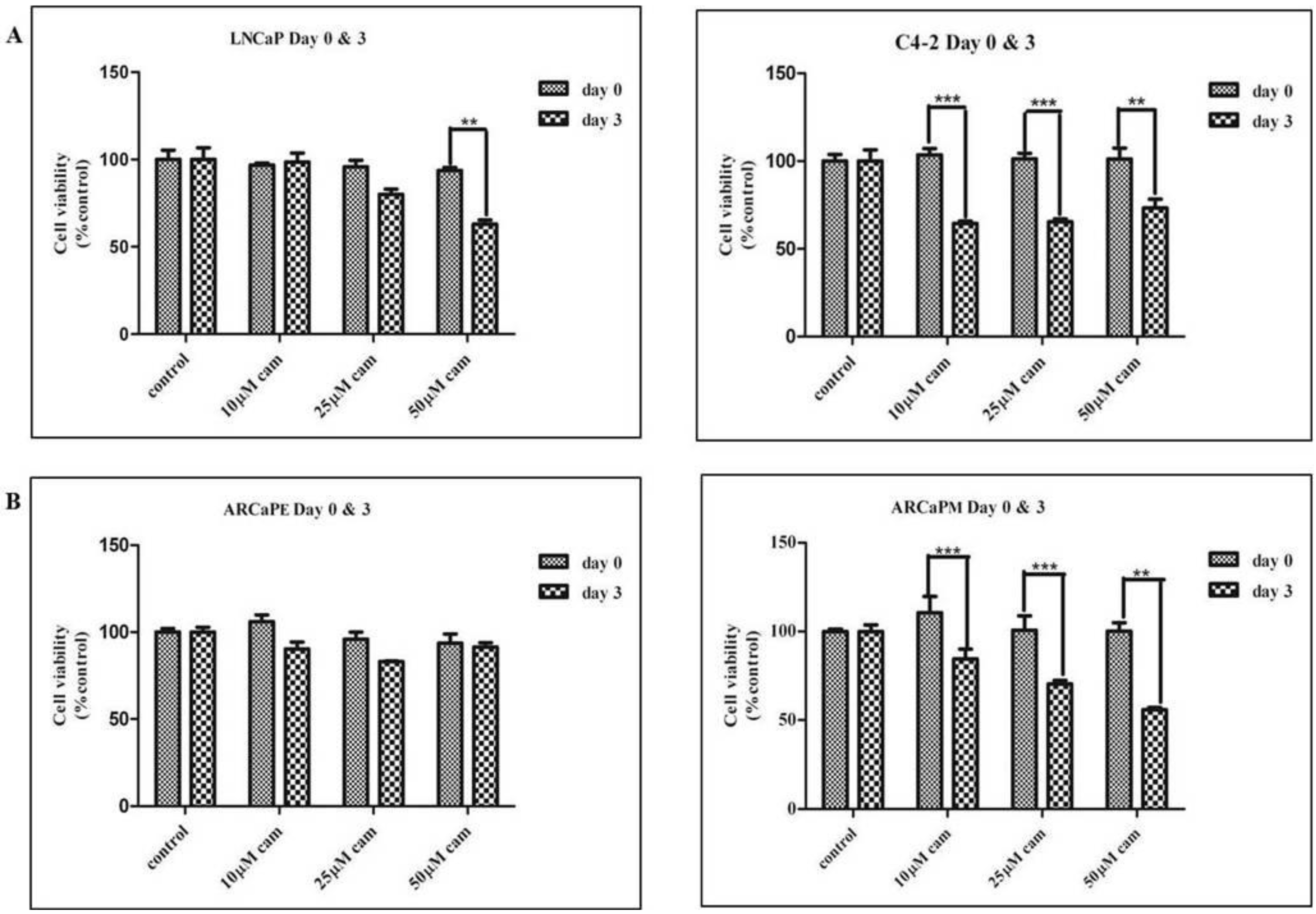
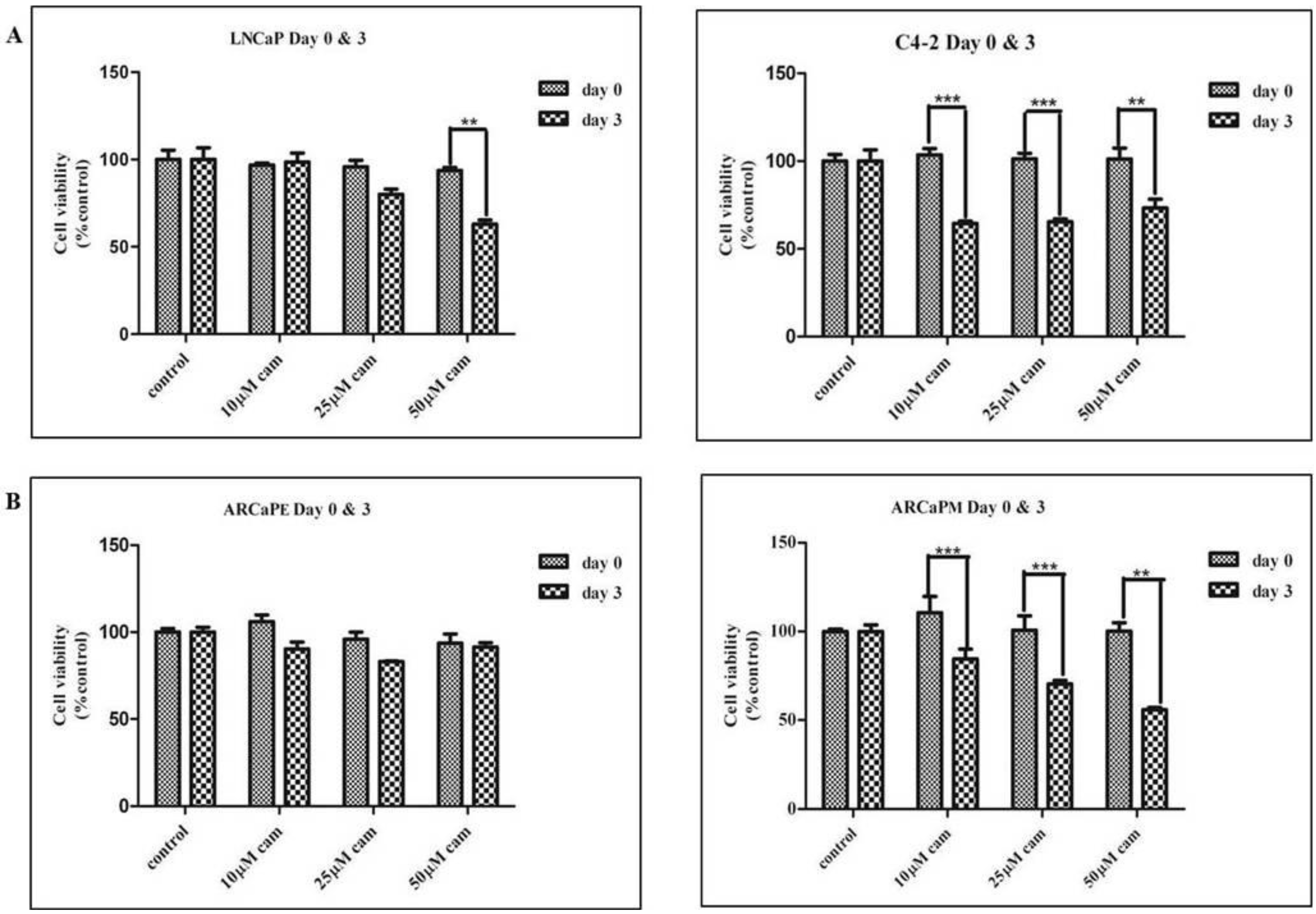
2.1. Camalexin Treatments Decreases Cell Proliferation in the More Aggressive Prostate Cancer Cells as Compared to the Lesser Aggressive Cells
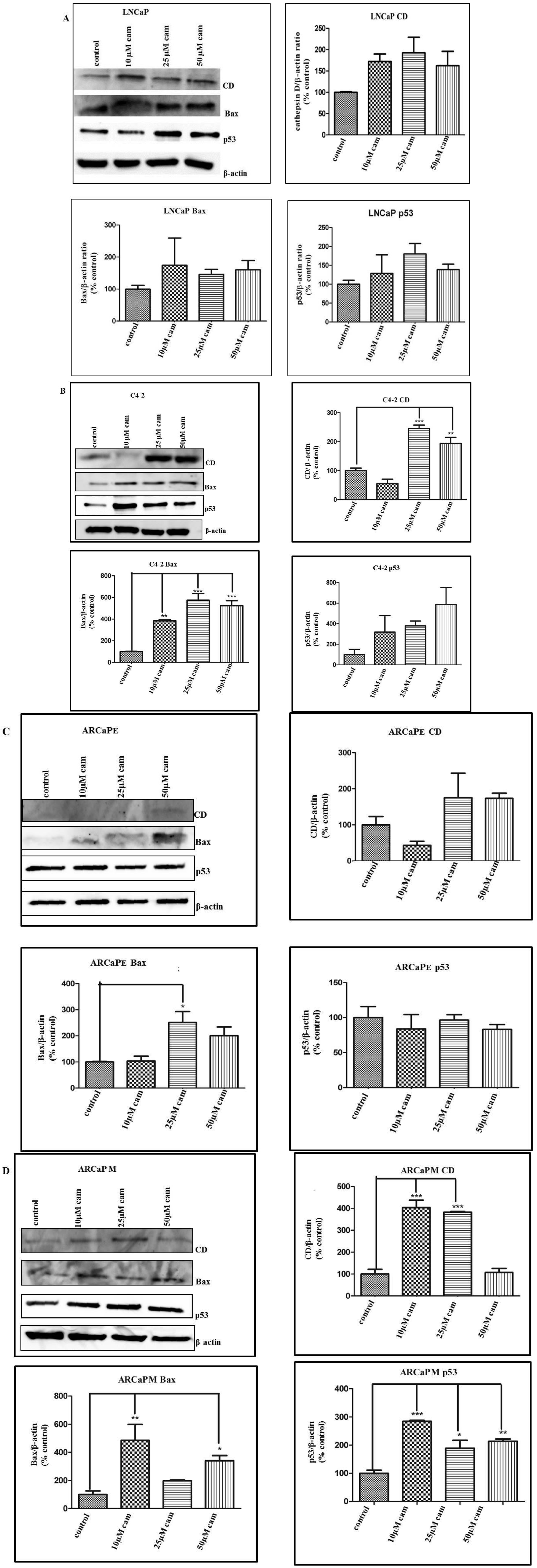
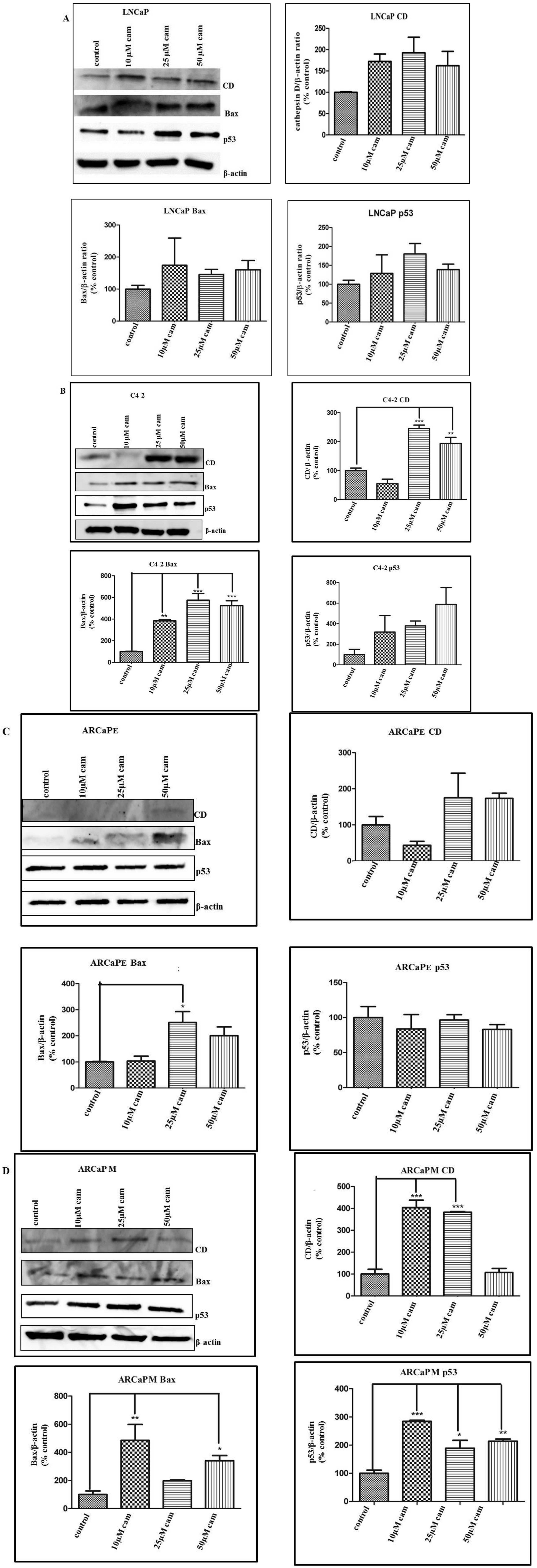
2.2. Camalexin Treatment of Prostate Cancer Cells Increases Protein Expression of the Lysosomal Protease CD, Bax and p53 Transcription Factor
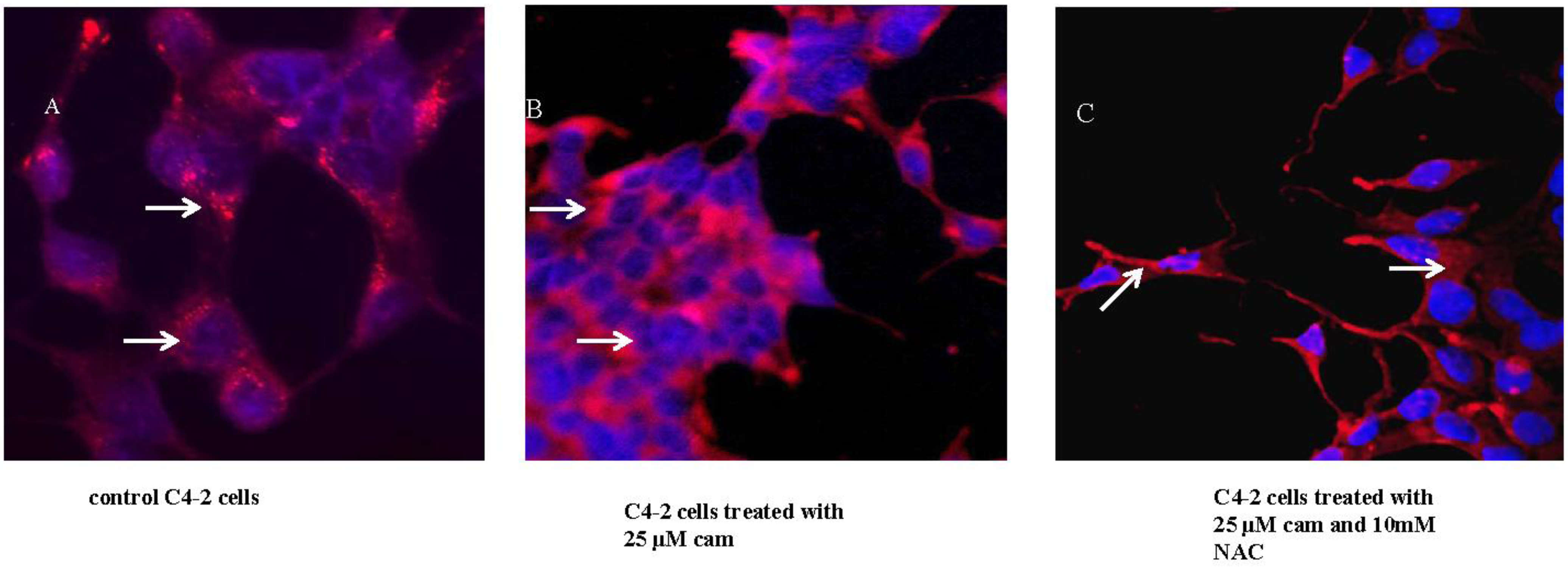
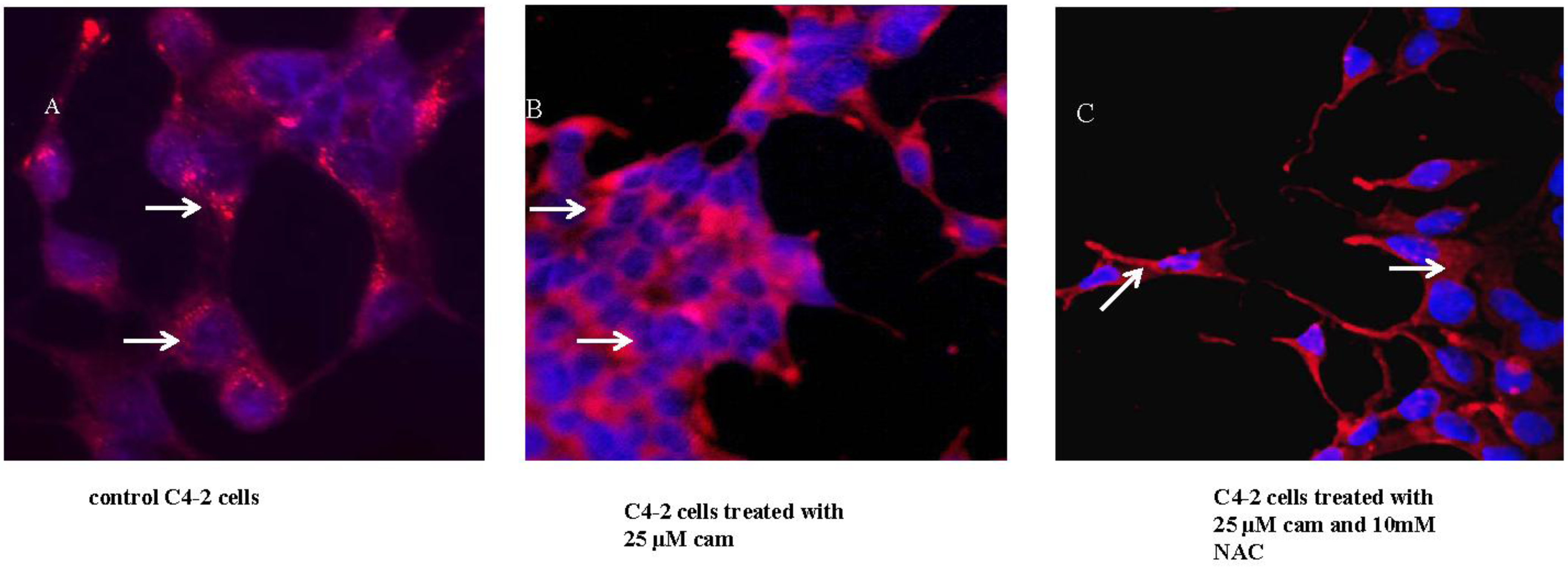
2.3. Camalexin Shifts Subcellular Localization of CD from the Lysosome to the Cytosol
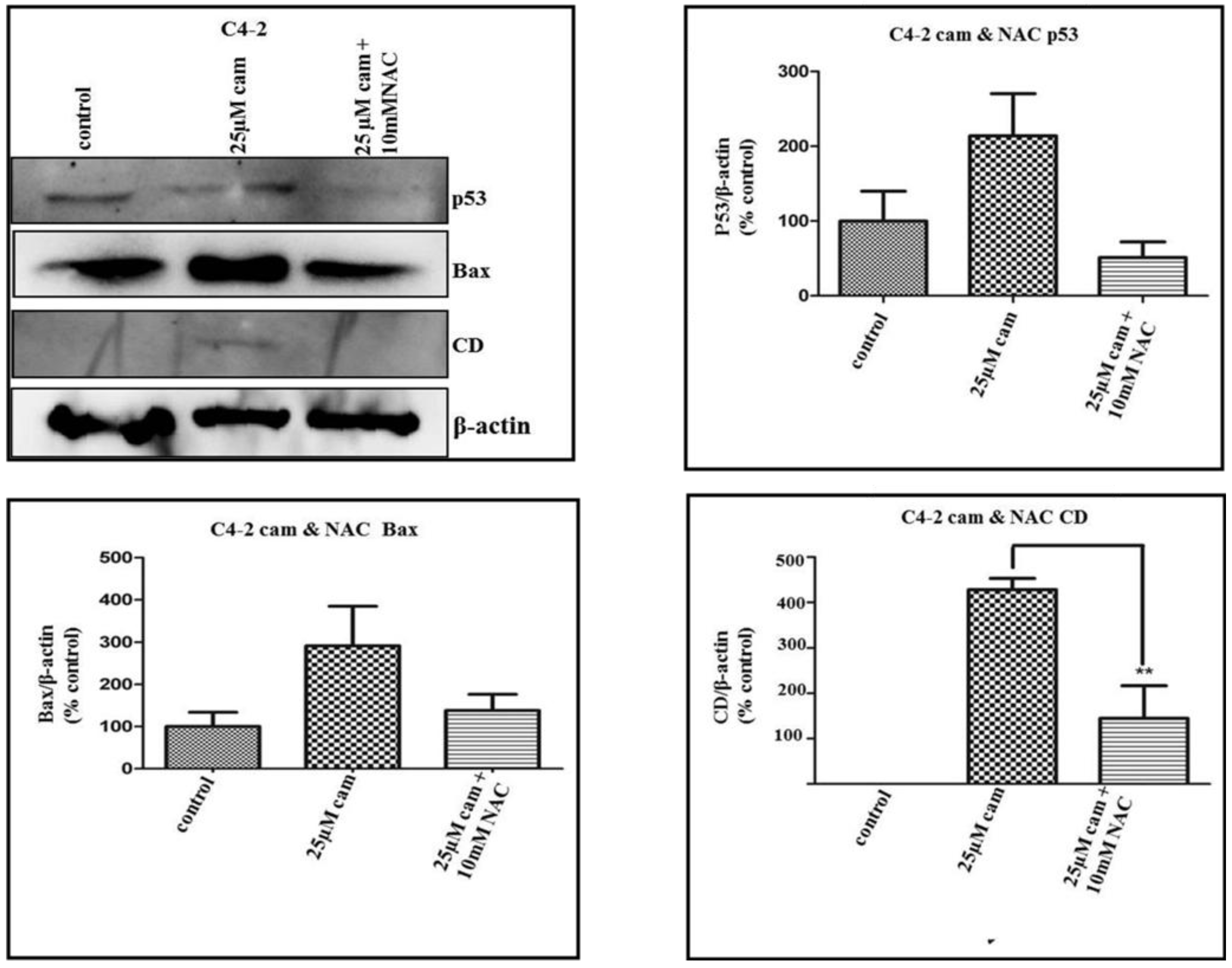
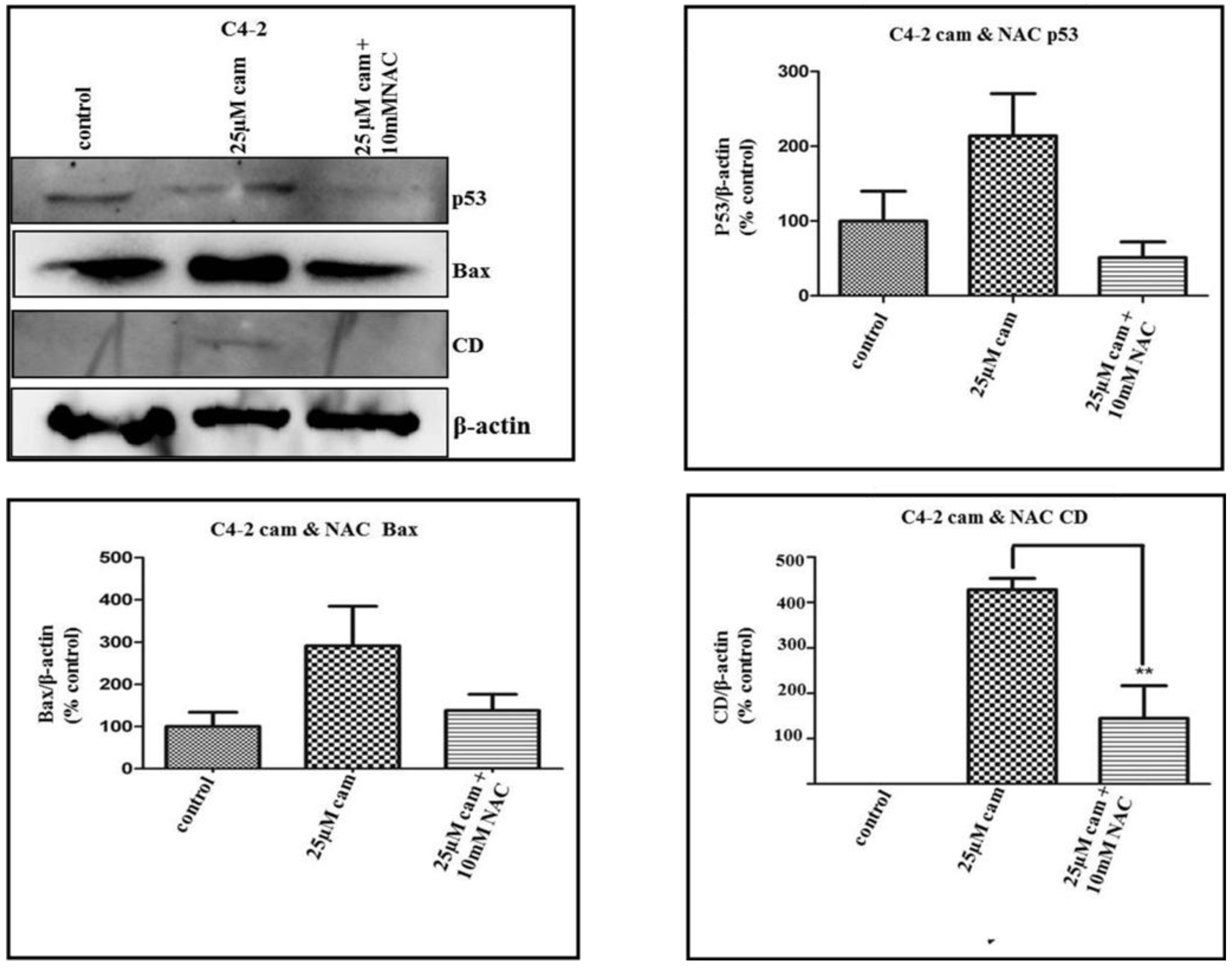
2.4. NAC Abrogates Camalexin-Mediated Increased p53, Bax and CD Protein Expression in Prostate Cancer Cells
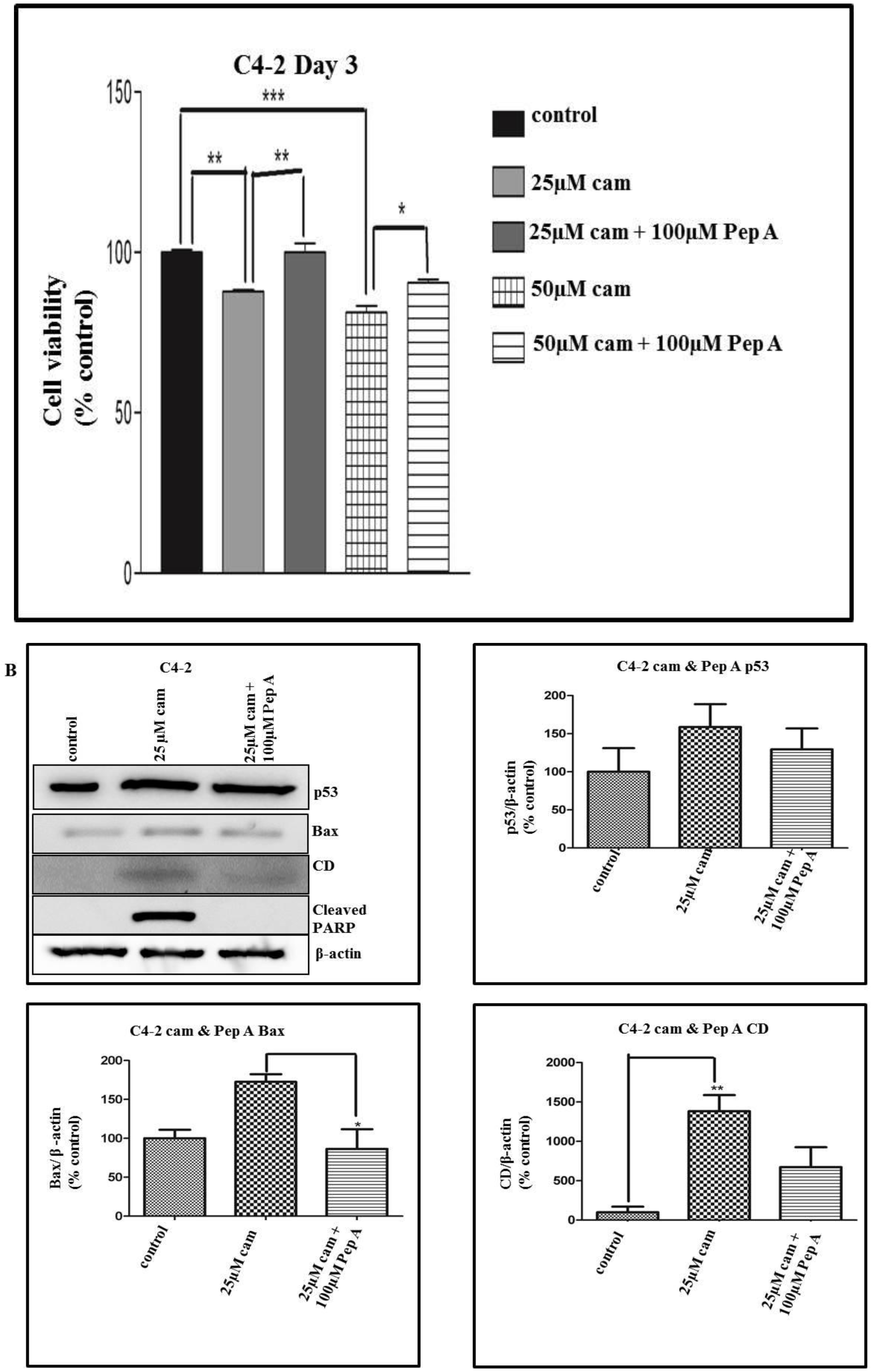
2.5. Pepstatin A, a CD Inhibitor, Antagonizes Camalexin-Mediated Decrease in Cell Viability, and Its Promotion of Pro-Apoptotic Protein Expression

2.6. Discussion
3. Experimental
3.1.Reagents and Antibodies
3.2. Cell Lines and Culture
3.3. Cell Viability Assay
3.4. Western Blot Analysis
3.5. Immunocytochemistry (ICC)
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Authors Contributions
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Thun, M.J. Cancer statistics. CA-Cancer J. Clin. 2007, 57, 43–66. [Google Scholar] [CrossRef]
- Crawford, E.D.; Eisenberger, M.A.; McLeod, D.G. A controlled trial of leuprolide with or without flutamide in prostatic carcinoma. N. Engl. J. Med. 1989, 321, 419–420. [Google Scholar] [CrossRef]
- Beer, T.M.; El Geneidi, M.; Eilers, K.M. Docetaxel (taxotere) in the treatment of prostate cancer. Expert Rev. Anticancer Ther. 2003, 3, 261–268. [Google Scholar] [CrossRef]
- Browne, L.M.; Conn, K.L.; Ayer, W.A.; Tewari, J.P. The Camalexins: New phytoalexins produced in leaves of Camelina sativa (Cruciferae). Tetrahedron 1991, 47, 3903–3914. [Google Scholar]
- Jimenez, L.D.; Ayer, W.A.; Tewari, J.P. Phytoalexins produced in the leaves of Capsella bursa pastoris (Shepherd’s purse). Phytoprotection 1997, 78, 99–103. [Google Scholar] [CrossRef]
- Tsuji, J.; Jackson, E.P.; Gage, D.A.; Hammerschmidt, R.; Somerville, S.C. Phytoalexin accumulation in Arabidopsis Thaliana during the hypersensitive reaction to Pseudomonas syringae pv syringae. Plant. Physiol. 1992, 98, 1304–1309. [Google Scholar] [CrossRef]
- Jeandet, P.; Clément, C.; Courot, E.; Cordelier, S. Modulation of phytoalexin biosynthesis in engineered plants for disease resistance. Int. J. Mol. Sci. 2013, 14, 14136–14170. [Google Scholar] [CrossRef]
- Mezencev, R.; Galizzi, M.; Kutschy, P.; Docampo, R. Trypanosoma cruzi: Antiproliferative effect of indole phytoalexins on intracellular amastigotes in vitro. Exp. Parasitol. 2009, 122, 66–69. [Google Scholar] [CrossRef]
- Moody, C.J.; Roffey, J.R.A.; Stephens, M.A.; Stratford, I.J. Synthesis and cytotoxic activity of indole thiazoles. Anticancer Drugs 1997, 8, 489–499. [Google Scholar] [CrossRef]
- Smith, B.A.; Neal, C.L.; Chetram, M.; Vo, B.; Mezencev, R.; Hinton, C.; Odero-Marah, V.A. The phytoalexin camalexin mediates cytotoxicity towards aggressive prostate cancer cells via reactive oxygen species. J. Nat. Med. 2013, 67, 607–618. [Google Scholar] [CrossRef]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metast. Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Khandrika, L.; Kumar, B.; Koul, S.; Maroni, P.; Koul, H.K. Oxidative stress in prostate cancer. Cancer Lett. 2009, 28, 125–135. [Google Scholar]
- Lim, S.D.; Sun, C.; Lambeth, J.D.; Marshall, F.; Amin, M.; Chung, L.; Petros, J.A.; Arnold, R.S. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate 2005, 62, 200–207. [Google Scholar] [CrossRef]
- Gaziano, J.M.; Glynn, R.J.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Sesso, H.D.; Buring, J.E. Vitamins E and C in the prevention of prostate and total cancer in men: the Physicians' Health Study II randomized controlled trial. JAMA 2009, 301, 52–62. [Google Scholar]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug. Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Kachadourian, R.; Day, B.J. Flavonoid-induced glutathione depletion: Potential implications for cancer treatment. Free Radic. Biol. Med. 2006, 41, 65–76. [Google Scholar] [CrossRef]
- Buttk, T.M.; Sandstrom, P.A. Oxidative stress as a mediator of apoptosis. Immunol. Today 1994, 15, 7–10. [Google Scholar] [CrossRef]
- Gardner, A.M.; Xu, F.; Fady, C.; Jacoby, F.J.; Duffey, D.C.; Tu, Y.; Lichtenstein, A. Apoptotic vs. non-apoptotic cytotoxicity induced by hydrogen peroxide. Free Radic. Biol. Med. 1997, 22, 73–83. [Google Scholar] [CrossRef]
- Forrest, V.J.; Kang, Y.H.; McClain, D.E.; Robinson, D.H.; Ramakrishnan, N. Oxidative stress- induced apoptosis prevented by trolox. Free Radic. Biol. Med. 1994, 16, 675–684. [Google Scholar] [CrossRef]
- Kagedal, K.; Johansson, A.C.; Johansson, U.; Heimlich, G.; Roberg, K.; Wang, N.S.; Jürgensmeier, J.M.; Ollinger, K. Lysosomal membrane permeabilization during apoptosis-involvement of Bax? Int. J. Exp. Pathol. 2005, 86, 309–321. [Google Scholar] [CrossRef]
- Mathiasen, I.S.; Jaattela, M. Triggering caspase independent cell death to combat cancer. Trends Mol. Med. 2002, 8, 212–220. [Google Scholar] [CrossRef]
- Roberg, K.; Ollinger, K. Oxidative stress causes relocation of the lysosomal enzyme cathepsin D with ensuing apoptosis in neonatal rat cardiomyocytes. Am. J. Pathol. 1998, 152, 1151–1156. [Google Scholar]
- Kagedal, K.; Zhao, M.; Svensson, I.; Brunk, U. Sphingosine induced apoptosis is dependent on lysosomal proteases. Biochem. J. 2001, 359, 335–343. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Deussing, J.; Miyoshi, H.; Bronk, S.F.; Svingen, P.A.; Peters, C.; Kaufmann, S.H.; Gores, G.J. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Invest. 2000, 106, 1127–1137. [Google Scholar] [CrossRef]
- Yuan, X.M.; Li, W.; Dalen, H.; Lotem, J.; Kama, R.; Sachs, L.; Brunk, U.T. Lysosomal destabilization in p53-induced apoptosis. Proc. Natl. Acad. Sci. USA 2002, 99, 6286–6291. [Google Scholar]
- Deiss, L.P.; Galinka, H.; Berissi, H.; Cohen, O.; Kimchi, A. Cathepsin D protease mediates programmed cell death induced by interferon gamma, Fas/APO-1 and TNF-alpha. EMBO J. 1996, 15, 3861–3870. [Google Scholar]
- Wu, G.S.; Saftig, P.; Peters, C.; El-Deiry, W.S. Potential role for cathepsin D in p53-dependent tumor suppression and chemosensitivity. Oncogene 1998, 16, 2177–2183. [Google Scholar]
- Ollinger, K. Inhibition of cathepsin D prevents free-radical induced apoptosis in rat cardiomyocytes. Arch. Biochem. Biophys. 2000, 373, 346–351. [Google Scholar] [CrossRef]
- Roberg, K. Relocalization of cathepsin D and cytochrome c early in apoptosis revealed by immunoelectron microscopy. Lab. Invest. 2001, 81, 149–158. [Google Scholar] [CrossRef]
- Kagedal, K.; Johansson, U.; Ollinger, K. The lysosomal protease cathepsin D mediates apoptosis induced by oxidative stress. FASEB J. 2001, 15, 1592–1594. [Google Scholar]
- Takuma, K.; Kiriu, M.; Mori, K.; Lee, E.; Enomoto, R.; Baba, A.; Matsuda, T. Roles of cathepsins in reperfusion-induced apoptosis in cultured astrocytes. Neurochem. Int. 2003, 42, 153–159. [Google Scholar] [CrossRef]
- Roberg, K.; Kagedal, K.; Ollinger, K. Microinjection of cathepsin d induces caspase-dependent apoptosis in fibroblasts. Am. J. Pathol. 2002, 161, 89–96. [Google Scholar] [CrossRef]
- Heinrich, M.; Neumeyer, J.; Jakob, M.; Hallas, C.; Tchikov, V.; Winoto-Morbach, S.; Wickel, M.; Schneider-Brachert, W.; Trauzold, A.; Hethke, A.; et al. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3activation. Cell Death Differ. 2004, 11, 550–563. [Google Scholar] [CrossRef]
- Bidere, N.; Lorenzo, H.K.; Carmona, S.; Laforge, M.; Harpe, F.; Dumont, C.; Senik, A. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J. Biol. Chem. 2003, 278, 31401–31411. [Google Scholar]
- Xu, J.; Wang, R.; Xie, Z.H.; Odero-Marah, V.; Pathak, S.; Multani, A.; Chung, L.W.; Zhau, H.E. Prostate cancer metastasis: Role of the host microenvironment in promoting epithelial to mesenchymal transition and increased bone and adrenal gland metastasis. Prostate 2006, 66, 1664–1673. [Google Scholar] [CrossRef]
- Odero-Marah, V.A.; Wang, R.; Chu, G.; Zayzafoon, M.; Xu, J.; Shi, C.; Marshall, F.F.; Zhau, H.E.; Chung, L.W. Receptor activator of NF-KappaB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Res. 2008, 18, 858–870. [Google Scholar] [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane Permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Stova, V.; Turk, V.; Turk, B. Lysosomal cysteine cathepsins: signaling pathways in apoptosis. Biol. Chem. 2007, 388, 555–560. [Google Scholar]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes and oxidative stress in aging and apoptosis. Biochem. Biophys. Acta 2008, 1780, 1291–1303. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T.; Gustafsson, B.; Brunk, U.T. Lysosomal labilization. IUBMB Life 2006, 58, 531–539. [Google Scholar] [CrossRef]
- Antunes, F.; Cadenas, E.; Brunk, U.T. Apoptosis induced by exposure to low steady state concentration of H2O2 is a consequence of lysosomal rupture. Biochem. J. 2001, 356, 549–555. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Leist, M.; Gores, G.J. Lysosomes in cell death. Oncogene 2004, 23, 2881–2890. [Google Scholar] [CrossRef]
- Gimenez-Bonafe, P.; Tortosa, A.; Perez-Thomas, R. Overcoming drug resistance by enhancing apoptosis by tumor cells. Curr. Cancer Drug Targets. 2009, 9, 320–340. [Google Scholar] [CrossRef]
- Kroemer, G.; Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer. 2005, 5, 886–897. [Google Scholar] [CrossRef]
- Li, W.; Yuan, X.; Nordgren, G.; Dalen, H.; Dubowchik, G.M.; Firestone, R.A.; Brunk, U.T. Induction of cell death by the lysosomotropic detergent MSDH. FEBS Lett. 2000, 470, 35–39. [Google Scholar] [CrossRef]
- Groth-Pederson, L.; Jaattela, M. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett. 2013, 332, 265–274. [Google Scholar] [CrossRef]
- Mezencev, R.; Updegrove, T.; Kutschy, P.; Repovska, M.; McDonald, J.F. Camalexin induces apoptosis in Jurkat T-leukemia cells by increased concentration of reactive oxygen species and activation of caspases-8 and -9. J. Nat. Med. 2011, 65, 488–499. [Google Scholar] [CrossRef]
- Fehrenbacher, N.; Jaattela, M. Lysosomes as targets for cancer therapy. Cancer Res. 2005, 65, 2993–2995. [Google Scholar]
- Rosse, T.; Olivier, R.; Monney, L.; Rager, M.; Conus, S.; Fellay, I.; Jansen, B.; Borner, C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature 1998, 391, 496–499. [Google Scholar] [CrossRef]
- Eskes, R.; Antonsson, B.; Osen-Sand, A.; Montessuit, S.; Richter, C.; Sadoul, R.; Mazzei, G.; Nichols, A.; Martinou, J.C. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J. Cell. Biol. 1998, 143, 217–224. [Google Scholar] [CrossRef]
- Johansson, A.C.; Appelqvist, H.; Nilsson, C.; Kagedal, K.; Roberg, K.; Ollinger, K. Regulation of apoptosis-associated lysosomal membrane Permeabilization. Apoptosis 2010, 15, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Werneburg, N.W.; Guciarddi, M.E.; Brunk, S.F.; Kaufmann, S.H.; Gores, G.J. Tumor necrosis factor-related apoptosis inducing ligand activates a lysosomal pathway of apoptosis that is regulated by Bcl-2 proteins. J. Biol. Chem. 2007, 282, 28960–28970. [Google Scholar]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, A.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Oda, E.; Ohk, R.; Murasaw, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokin, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef]
- Repnik, U.; Stoka, V.; Turk, V.; Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochem. Biophys. Acta 2012, 1824, 22–33. [Google Scholar]
- Sample Availability: Samples of the compound camalexin, were obtained from Roman Mezencev, and can also be obtained commercially from AfferChem Inc., New Brunswick, NJ.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Smith, B.; Randle, D.; Mezencev, R.; Thomas, L.; Hinton, C.; Odero-Marah, V. Camalexin-Induced Apoptosis in Prostate Cancer Cells Involves Alterations of Expression and Activity of Lysosomal Protease Cathepsin D. Molecules 2014, 19, 3988-4005. https://doi.org/10.3390/molecules19043988
Smith B, Randle D, Mezencev R, Thomas L, Hinton C, Odero-Marah V. Camalexin-Induced Apoptosis in Prostate Cancer Cells Involves Alterations of Expression and Activity of Lysosomal Protease Cathepsin D. Molecules. 2014; 19(4):3988-4005. https://doi.org/10.3390/molecules19043988
Chicago/Turabian StyleSmith, Basil, Diandra Randle, Roman Mezencev, LeeShawn Thomas, Cimona Hinton, and Valerie Odero-Marah. 2014. "Camalexin-Induced Apoptosis in Prostate Cancer Cells Involves Alterations of Expression and Activity of Lysosomal Protease Cathepsin D" Molecules 19, no. 4: 3988-4005. https://doi.org/10.3390/molecules19043988