Regioselective Electrophilic Aromatic Bromination: Theoretical Analysis and Experimental Verification

Abstract
:
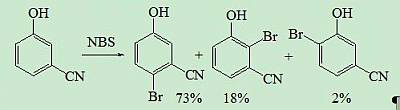{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
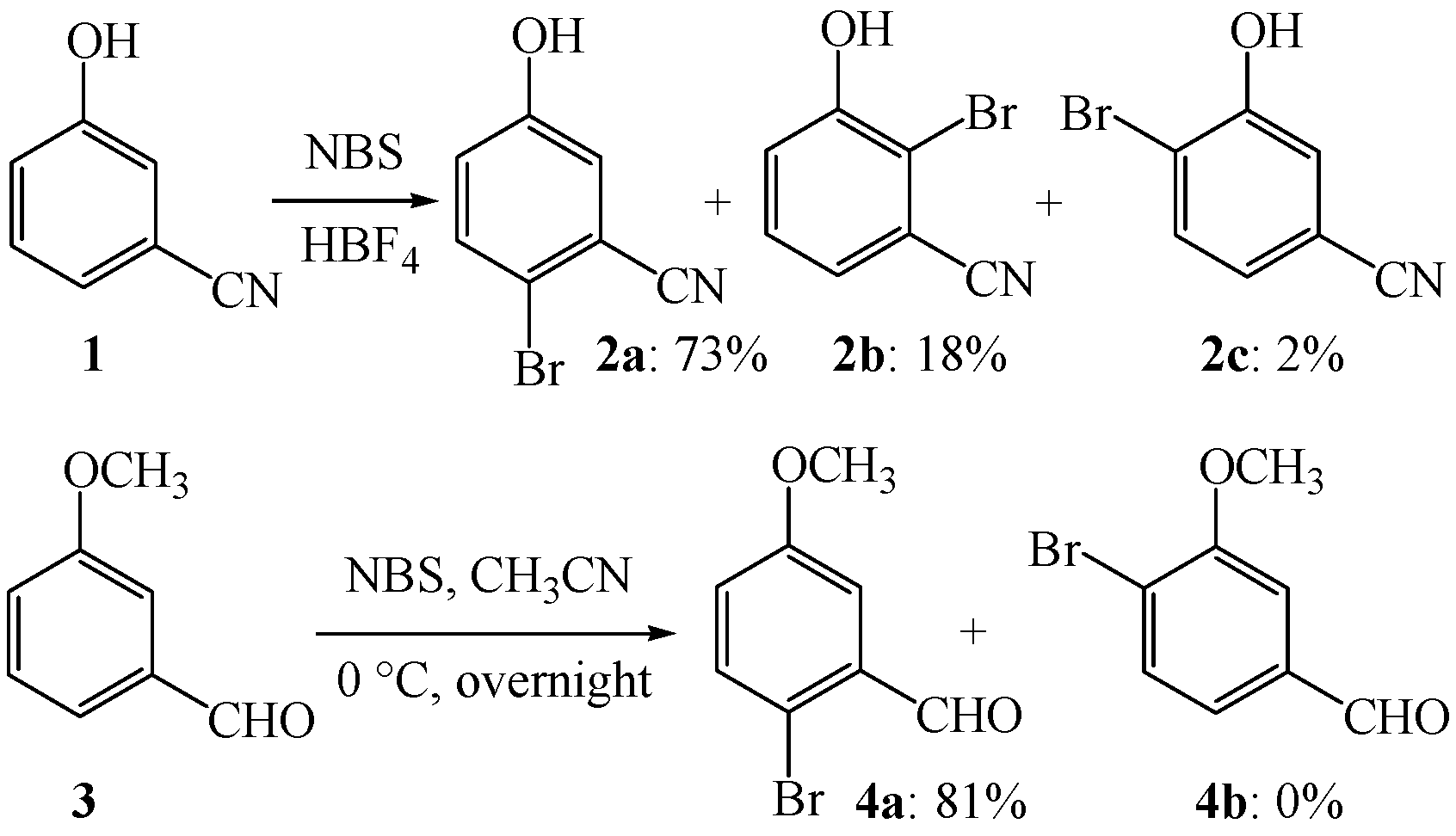{kind=link}
{kind=link}
{kind=link}
{kind=link}
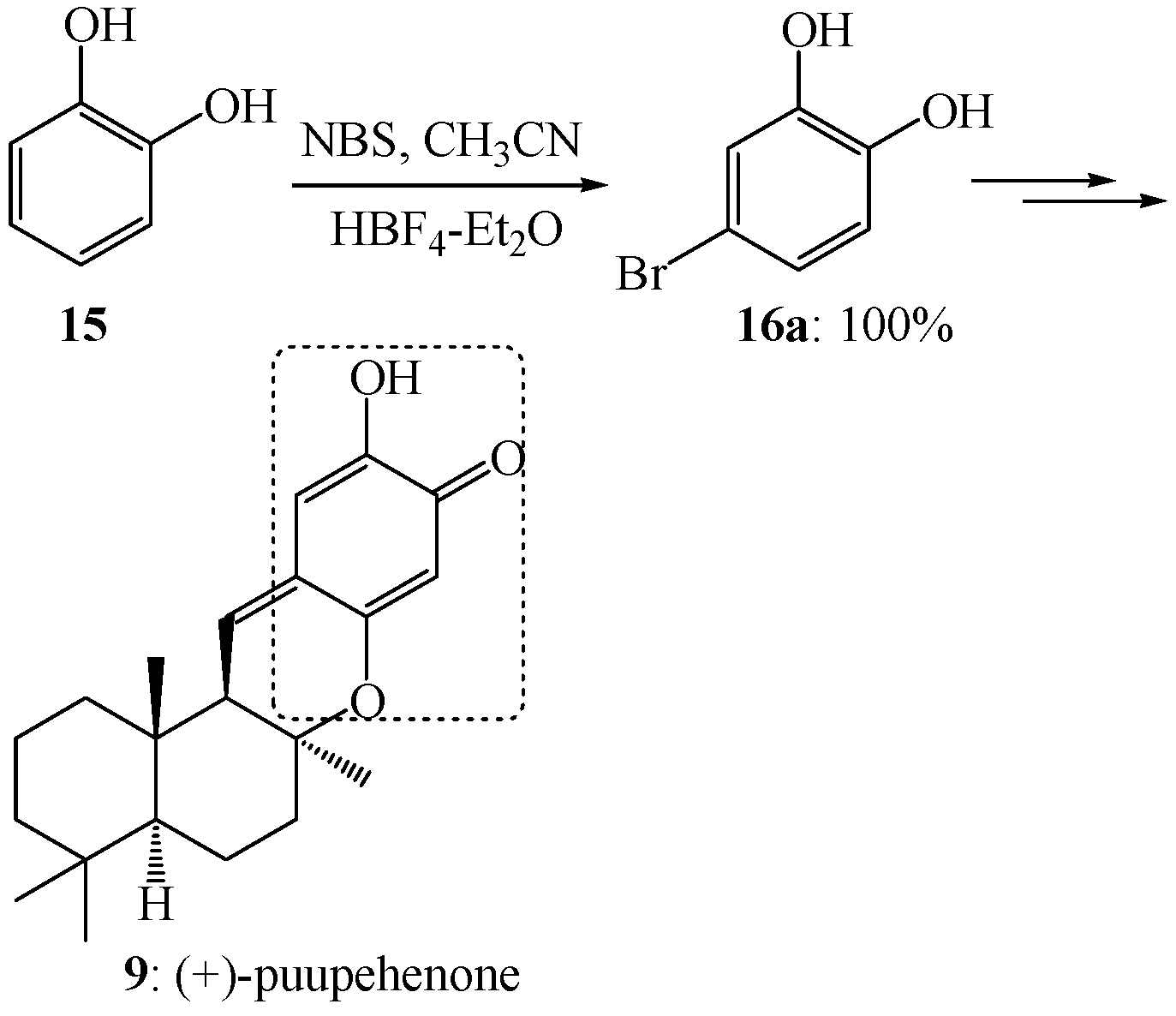{kind=link}
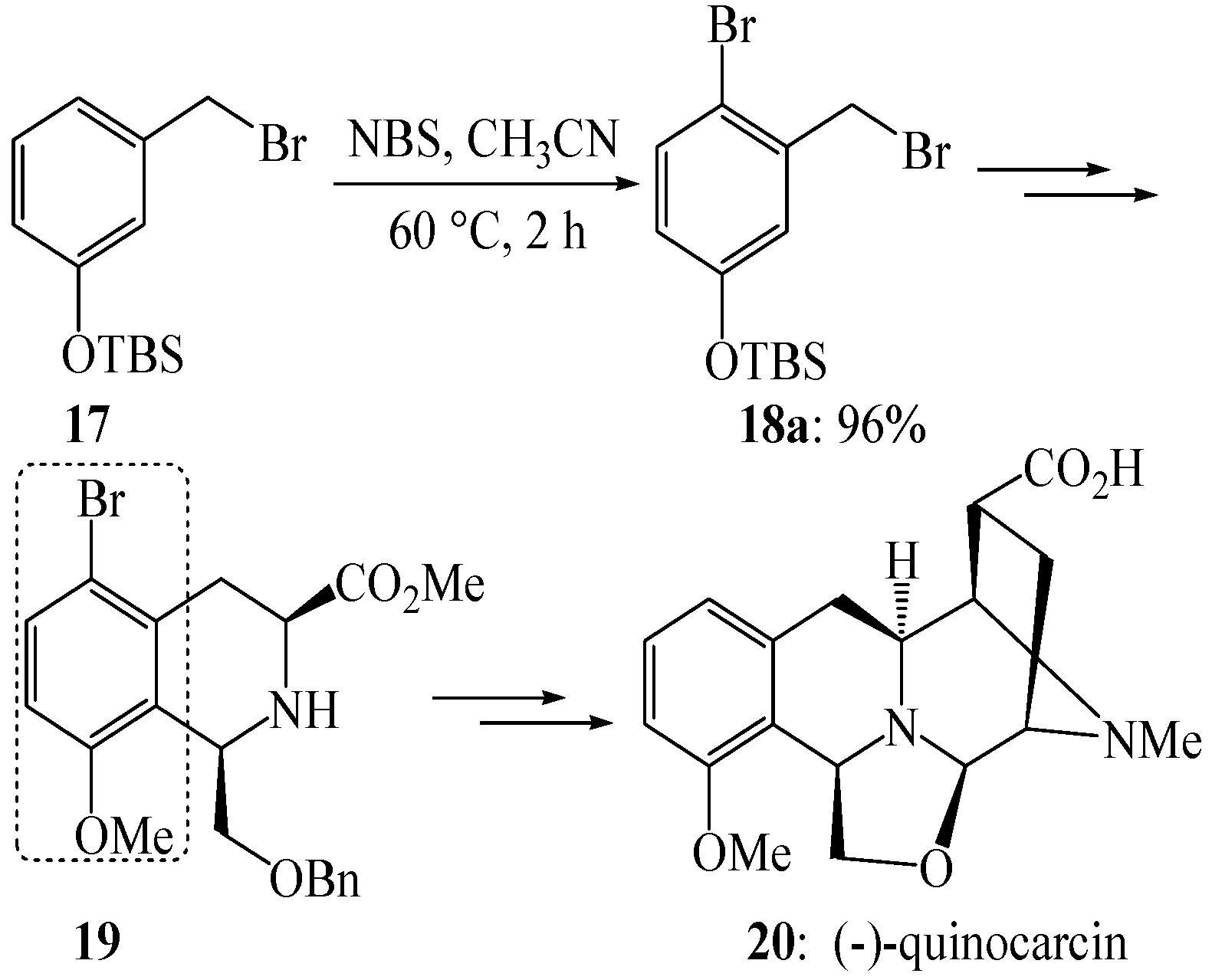{kind=link}
{kind=link}
{kind=link}
1. Introduction
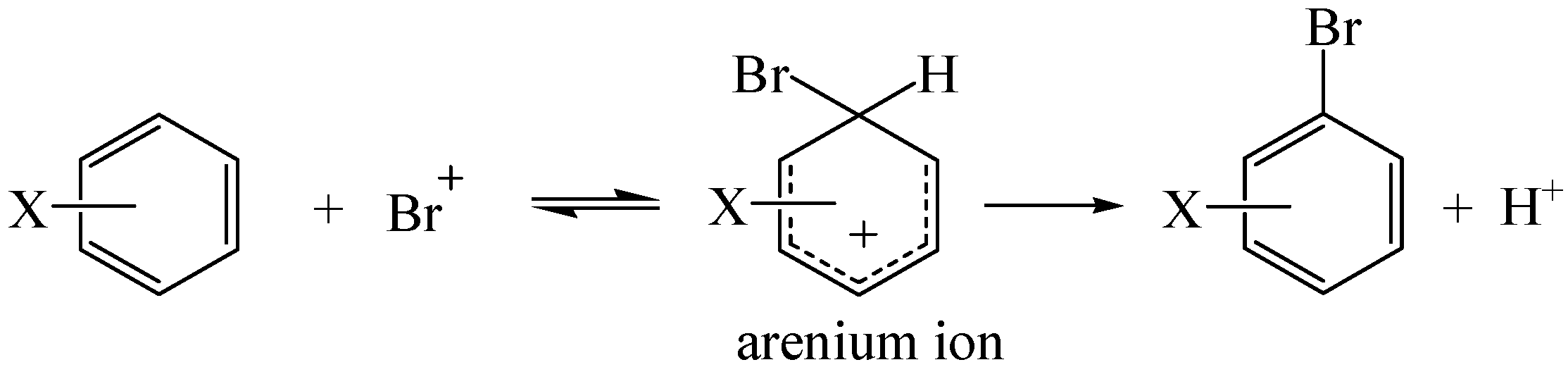
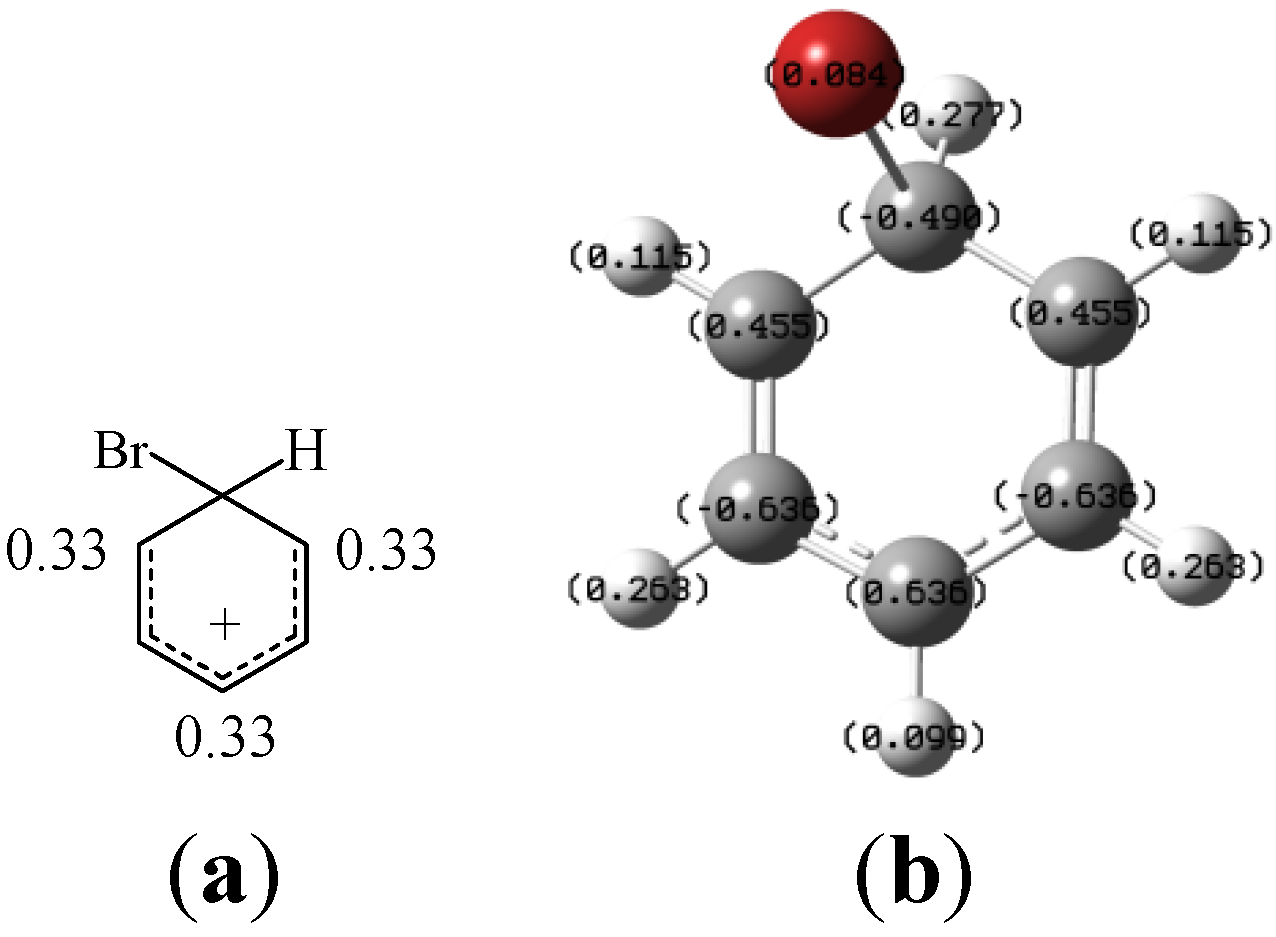
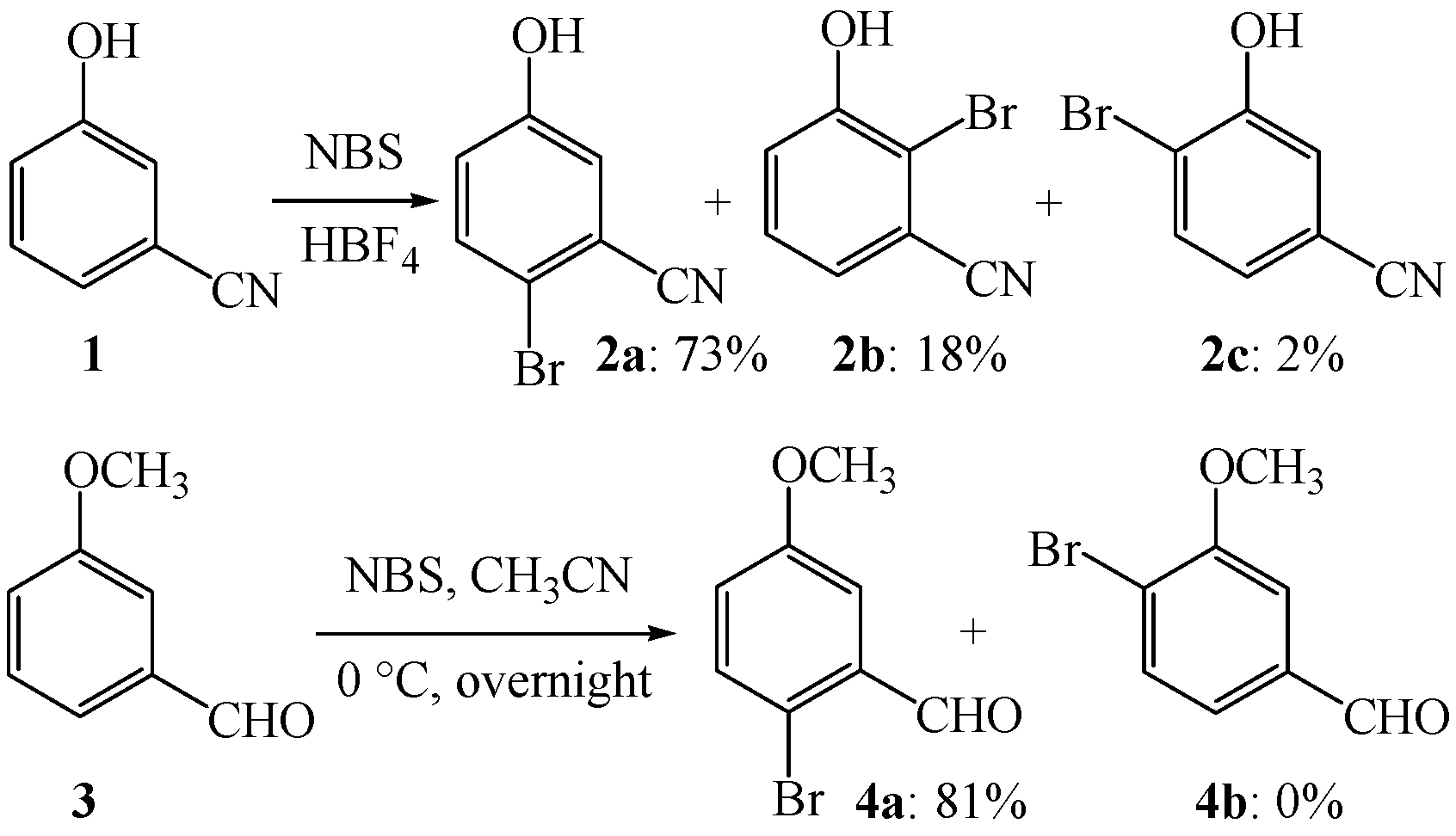
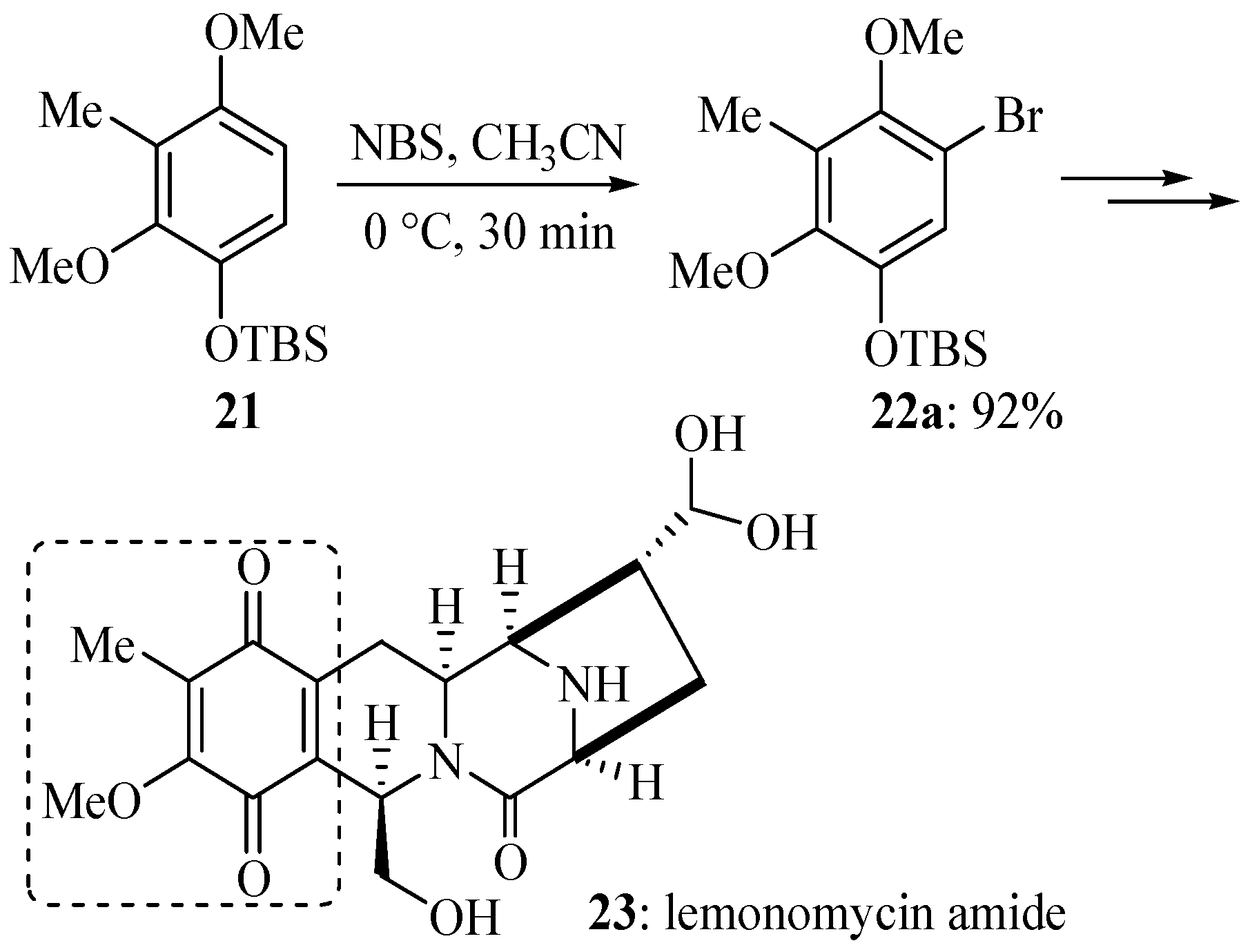
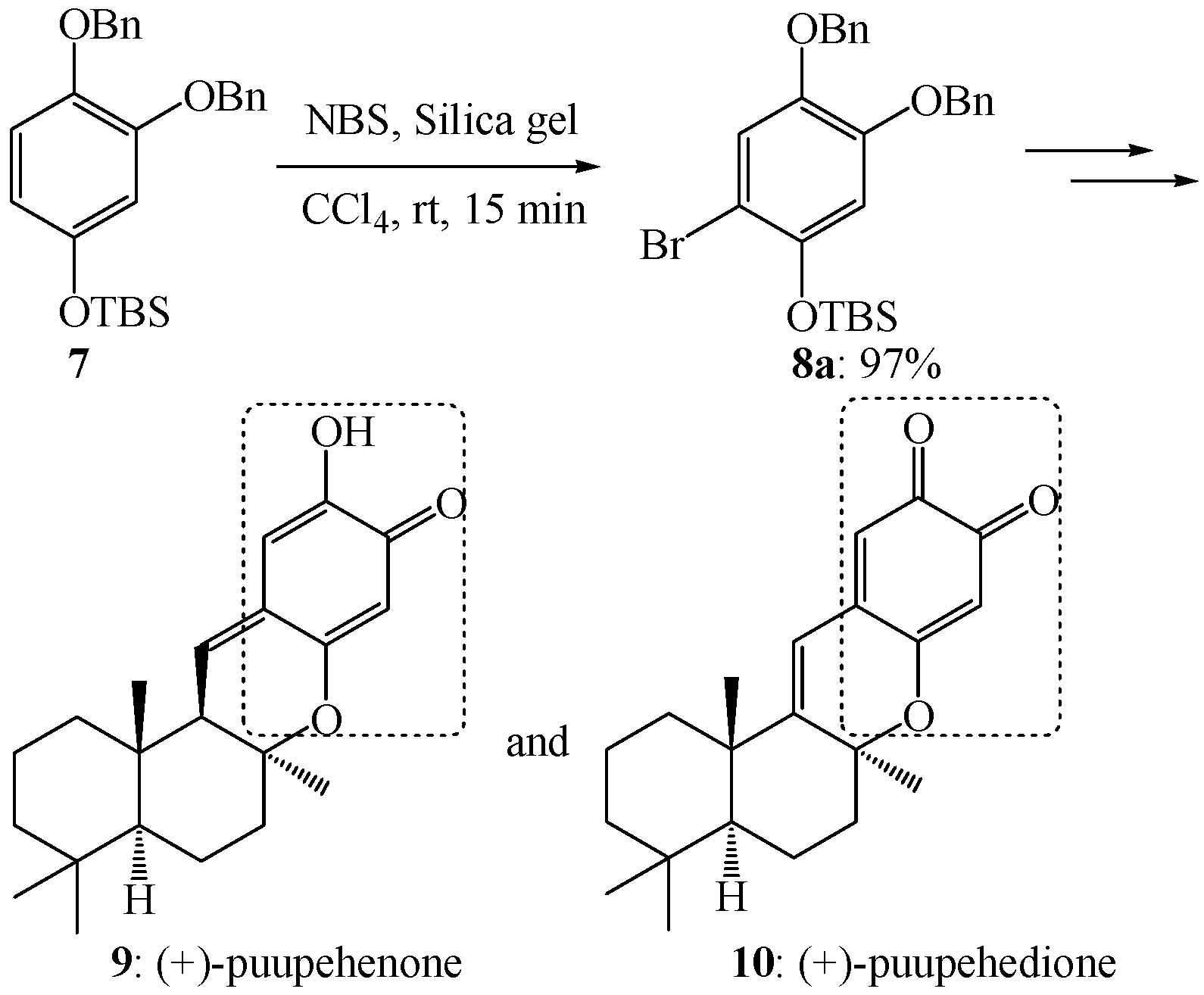
2. Results and Discussion
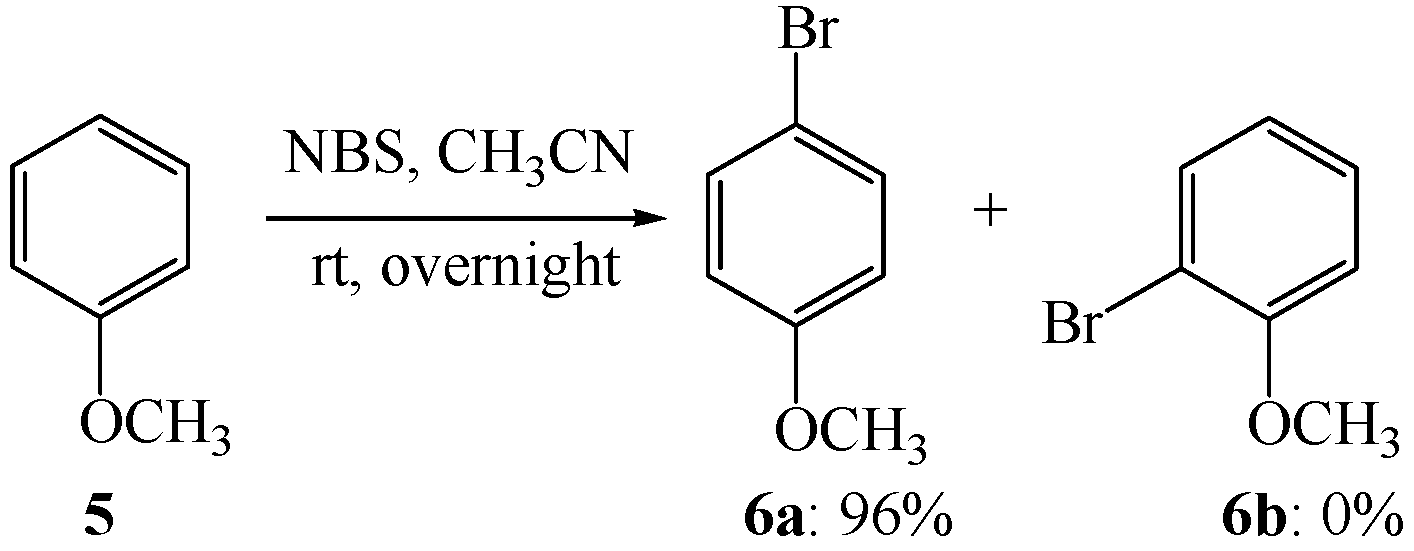
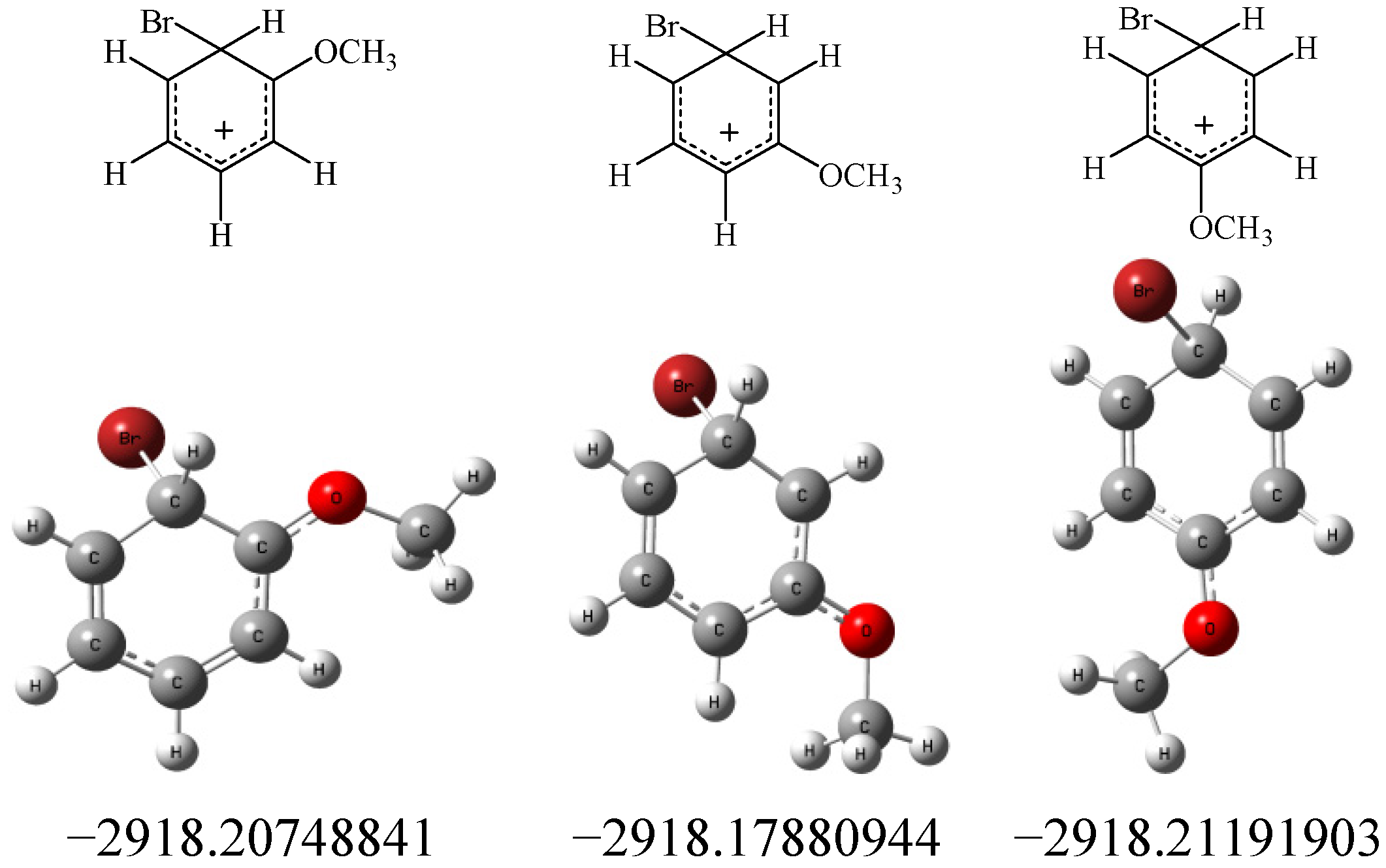
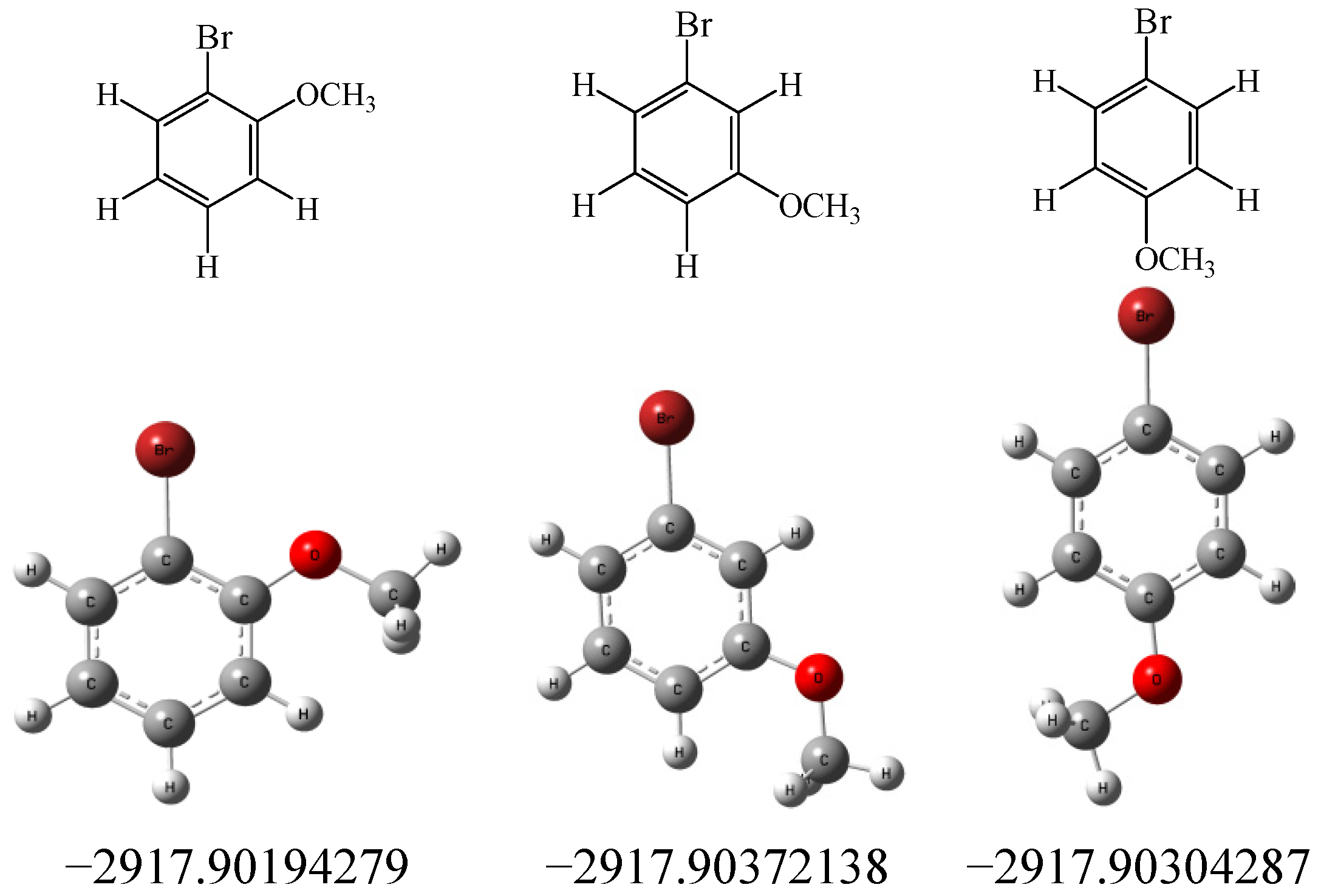

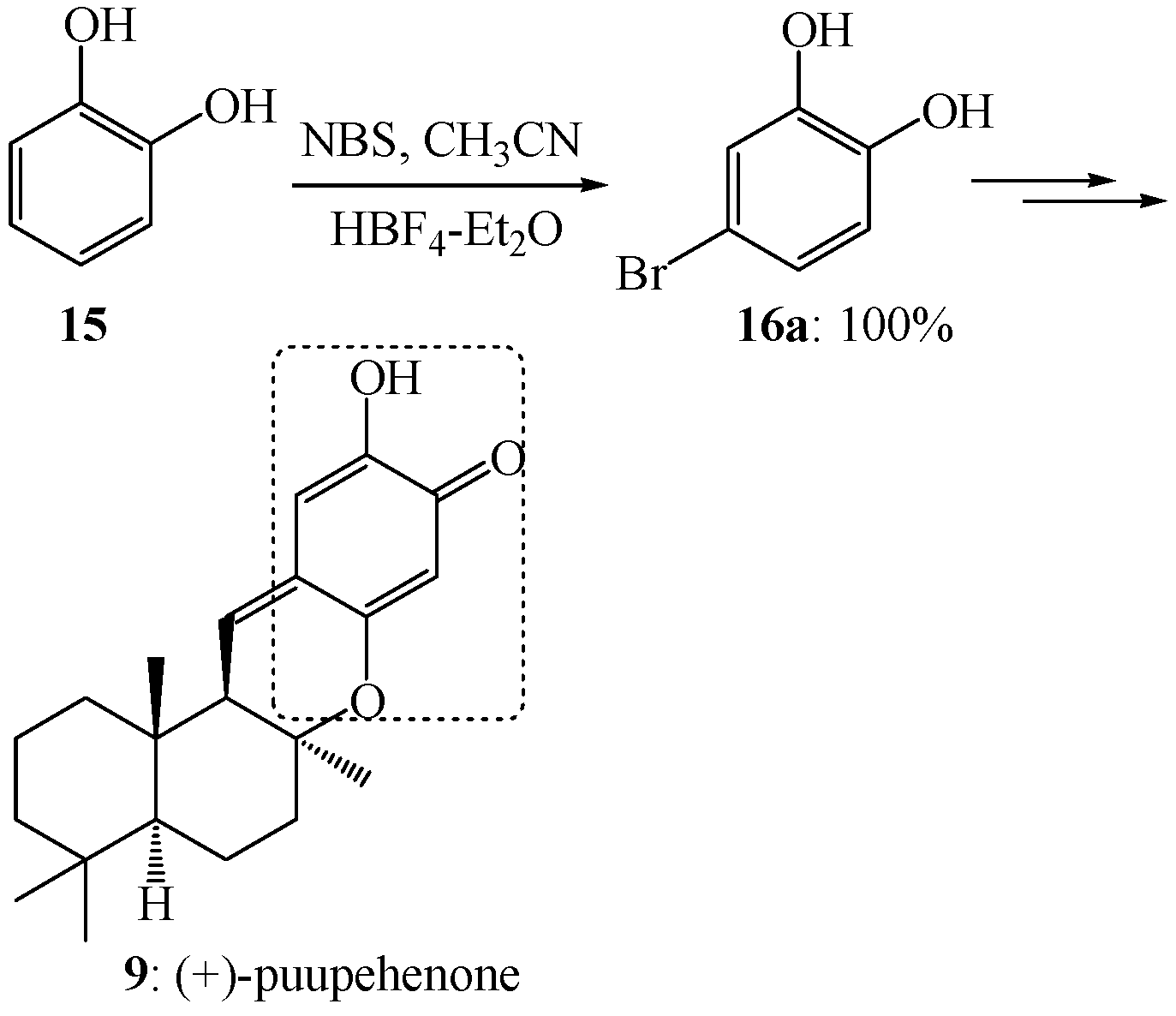
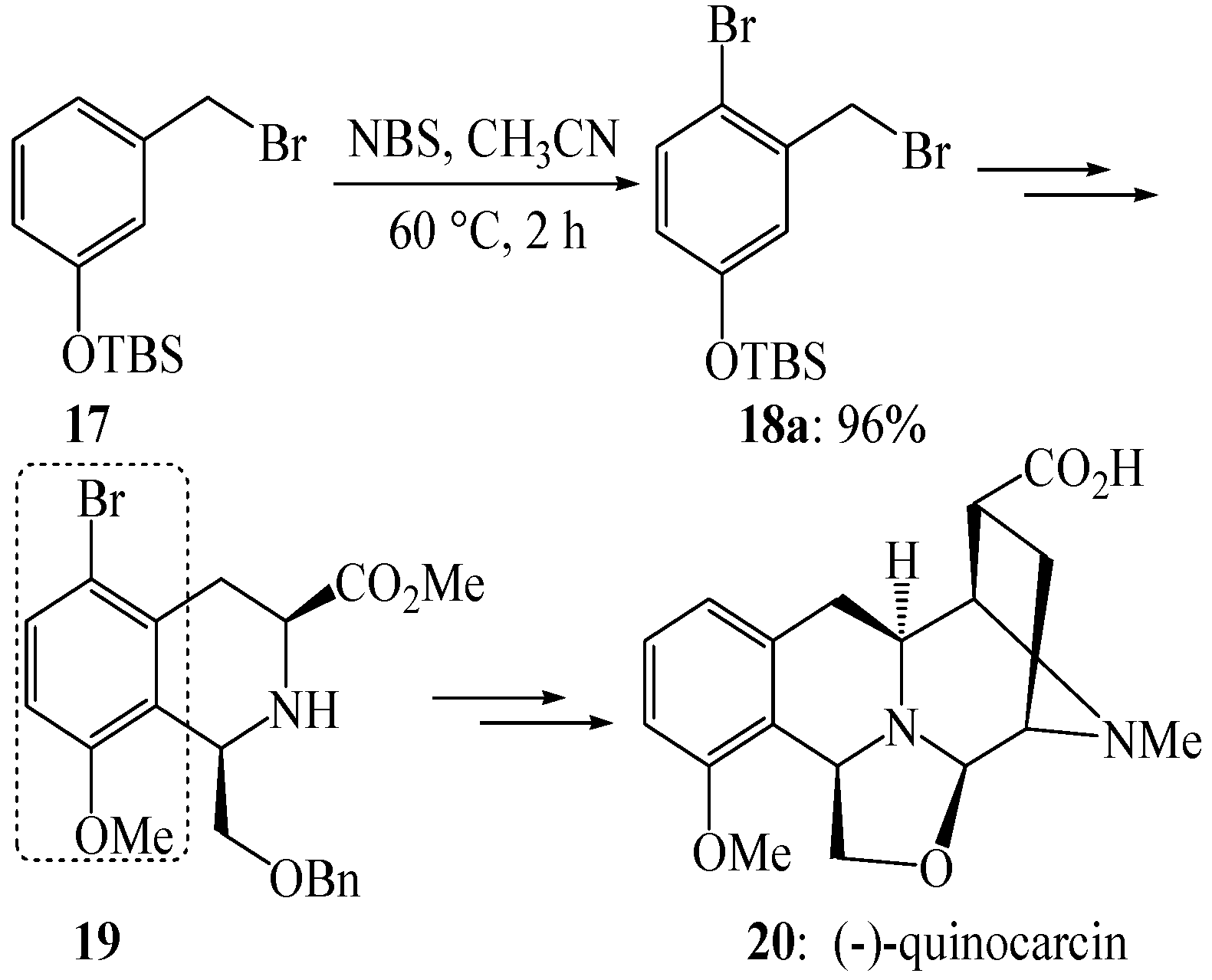
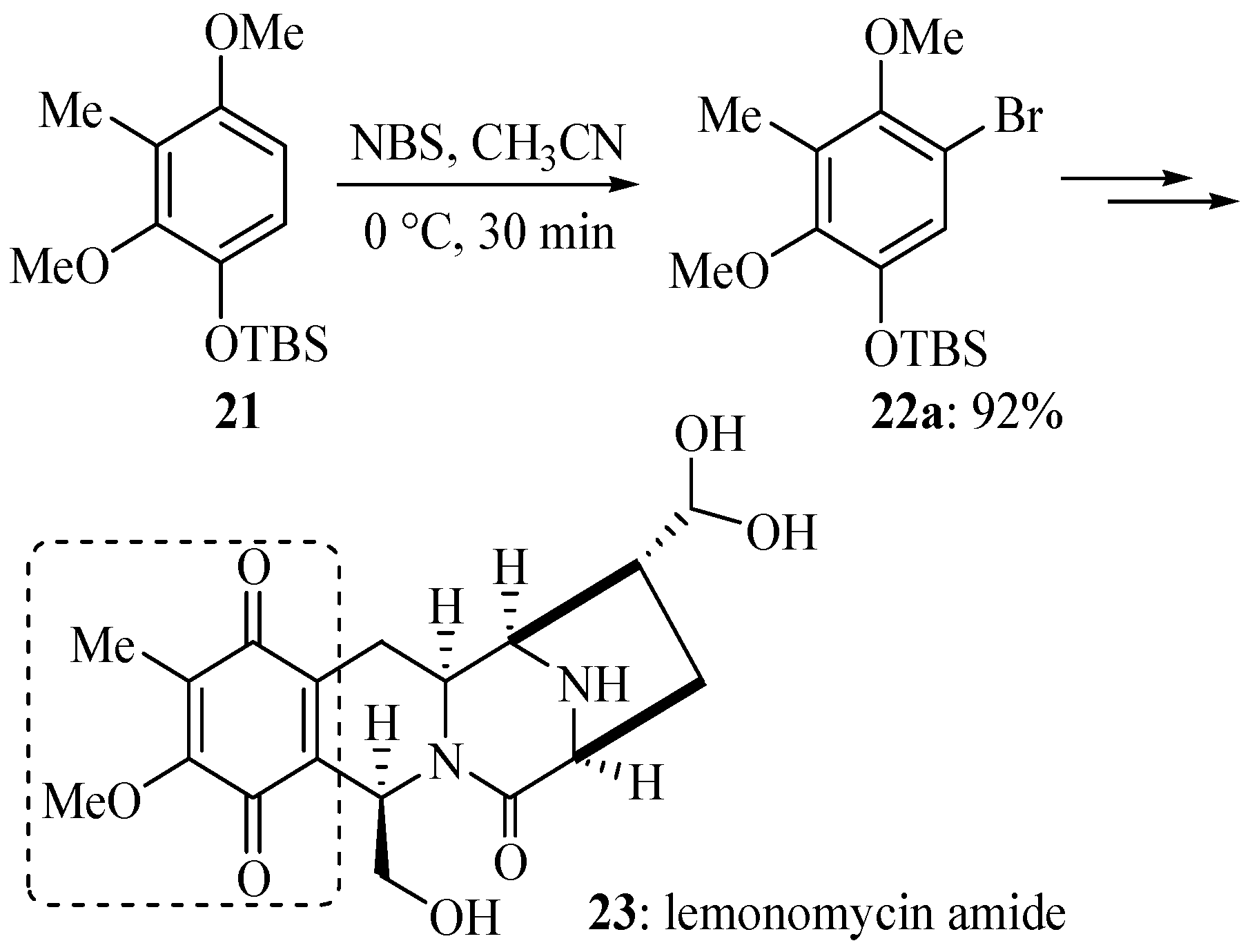

3. Experimental
3.1. General Information
3.2. Calculations
3.3. Electrophilic Aromatic Bromination of Compound 5
3.4. Electrophilic Aromatic Bromination of Compound 17
3.5. Electrophilic Aromatic Bromination of Compound 21
3.6. Electrophilic Aromatic Bromination of Compound 27a
3.7. Electrophilic Aromatic Bromination of Compound 27b
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflictts of Interest
References and Notes
- Beletskaya, I.P.; Cheprakov, A.V. The Heck reaction as a sharpening stone of palladium catalysis. Chem. Rev. 2000, 100, 3009–3066. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Non-conventional methodologies for transition-metal catalysed carbon–carbon coupling: A critical overview. Part 1: The Heck reaction. Tetrahedron 2005, 61, 11771–11835. [Google Scholar] [CrossRef]
- Phan, N.T.S.; Sluys, M.V.D.; Jones, C.W. On the nature of the active species in palladium catalyzed Mizoroki-Heck and Suzuki-Miyaura couplings–homogeneous or heterogeneous catalysis, a critical review. Adv. Synth. Catal. 2006, 348, 609–679. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Non-conventional methodologies for transition-metal catalysed carbonecarbon coupling: A critical overview. Part 2: The Suzuki reaction. Tetrahedron 2008, 64, 3047–3101. [Google Scholar]
- Martin, R.; Buchwald, S.L. Palladium-catalyzed Suzuki-Miyaura cross-coupling reactions employing sialkylbiaryl phosphine ligands. Acc. Chem. Res. 2008, 41, 1461–1473. [Google Scholar] [CrossRef]
- Phapale, V.B.; Cárdenas, D.J. Nickel-catalysed Negishi cross-coupling reactions: Scope and mechanisms. Chem. Soc. Rev. 2009, 38, 1598–1607. [Google Scholar] [CrossRef]
- Lamblin, M.; Nassar-Hardy, L.; Hierso, J.C.; Fouquet, E.; Felpin, F.X. Recyclable heterogeneous palladium catalysts in pure water: Sustainable developments in Suzuki, Heck, Sonogashira and Tsuji–Trost reactions. Adv. Synth. Catal. 2010, 352, 33–79. [Google Scholar] [CrossRef]
- Narayanan, R. Recent advances in noble metal nanocatalysts for Suzuki and Heck cross-coupling reactions. Molecules 2010, 15, 2124–2138. [Google Scholar] [CrossRef]
- Lennox, A.J.J.; Lloyd-Jones, G.C. Transmetalation in the Suzuki–Miyaura coupling: The fork in the trail. Angew. Chem. Int. Ed. 2013, 52, 7362–7370. [Google Scholar] [CrossRef]
- Pellissier, H.; Santelli, M. The use of arynes in organic synthesis. Tetrahedron 2003, 59, 701–730. [Google Scholar] [CrossRef]
- Tadross, P.M.; Stoltz, B.M. A comprehensive history of arynes in natural product total synthesis. Chem. Rev. 2012, 112, 3550–3577. [Google Scholar] [CrossRef]
- Smith, K.; James, D.M.; Matthews, I.; Bye, M.R. Selective para-bromination of phenols via a regenerable polymer-bound tetraalkylammonium tribromide. J. Chem. Soc. Perkin Trans. 1 1992, 1992, 1877–1878. [Google Scholar]
- Ghiaci, M.; Sedaghat, M.E.; Ranjbari, S.; Gil, A. Regioselective bromination of organic substrates by LDH-CO32−-Br− promoted by V2O5-H2O2. Appl. Catal. A: Gen. 2010, 384, 18–26. [Google Scholar] [CrossRef]
- Wortel, T.M.; Oudijn, D.; Vleugel, C.J.; Roeiofsen, D.P.; Bekkum, H.V. Selective bromination of halobenzenes using zeolite catalysts. J. Catal. 1979, 60, 110–120. [Google Scholar] [CrossRef]
- De la Vega, F.; Sasson, Y. Selective liquid-phase bromination of toluene catalysed by zeolites. Zeolites 1989, 9, 418–422. [Google Scholar] [CrossRef]
- De la Vega, F.; Sasson, Y. Selective para-bromination of toluene catalysed by Na-Y zeolite in the presence of an epoxide. J. Chem. Soc. Chem. Commun. 1989, 653–653. [Google Scholar] [CrossRef]
- De la Vega, F.; Sasson, Y. Selectivity in the liquid-phase bromination of aromatics catalysed by zeolites. Zeolites 1991, 11, 617–621. [Google Scholar] [CrossRef]
- De la Vega, F.; Sasson, Y. Highly selective bromination of toluene in a bromine-oxirane-zeolite system. Zeolites 1993, 13, 341–347. [Google Scholar] [CrossRef]
- Smith, K.; Bahzad, D. Highly efficient para-selective bromination of simple aromatic substrates by means of bromine and a reusable zeolite. Chem. Commun. 1996, 1996, 467–468. [Google Scholar] [CrossRef]
- Smith, K.; He, P.; Taylor, A. Selective para-bromination of phenyl acetate under the control of zeolites, bases, acetic anhydride or metal acetates in the liquid phase. Green Chem. 1999, 1, 35–38. [Google Scholar] [CrossRef]
- Smith, K.; El-Hiti, G.A.; Hammond, M.E.W.; Bahzad, D.; Li, Z.; Siquet, C. Highly efficient and selective electrophilic and free radical catalytic bromination reactions of simple aromatic compounds in the presence of reusable zeolites. J. Chem. Soc. Perkin Trans. 1 2000, 2000, 2745–2752. [Google Scholar]
- Narender, N.; Mohan, K.V.V.K.; Reddy, R.V.; Srinivasu, P.; Kulkarni, S.J.; Raghavan, K.V. Liquid phase bromination of phenols using potassium bromide and hydrogen peroxide over zeolites. J. Mol. Catal. A: Chem. 2003, 192, 73–77. [Google Scholar] [CrossRef]
- Mistry, A.G.; Smith, K. A superior synthetic method for the bromination of indoles and benzimidazoles. Tetrahedron Lett. 1986, 27, 1051–1054. [Google Scholar] [CrossRef]
- Smith, K.; James, D.M.; Mistry, A.G.; Bye, M.R.; Faulkner, D.J. A new method for bromination of carbazoles, β-carbolines and iminodibenzyls by use of N-bromosuccinimide and silica gel. Tetrahedron 1992, 48, 7479–7488. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Li, J.H. An efficient copper-catalysed aerobic oxybromination of arenes in water. Green Chem. 2010, 12, 2124–2126. [Google Scholar] [CrossRef]
- Roy, S.C.; Guin, C.; Rana, K.K.; Maiti, G. An efficient chemo and regioselective oxidative nuclear bromination of activated aromatic compounds using lithium bromide and ceric ammonium nitrate. Tetrahedron Lett. 2001, 42, 6941–6942. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, B.V.S.; Reddy, P.S.R.; Basak, A.K.; Narsaiah, A.V. Efficient halogenation of aromatic systems using N-halosuccinimides in ionic liquids. Adv. Synth. Catal. 2004, 346, 77–82. [Google Scholar] [CrossRef]
- Pla, D.; Albericio, F.; Alvarez, M. Regioselective monobromination of free and protected phenols. Eur. J. Org. Chem. 2007, 1921–1924. [Google Scholar]
- Majetich, G.; Hicks, R.; Reister, S. Electrophilic aromatic bromination using bromodimethylsulfonium bromide generated in situ. J. Org. Chem. 1997, 62, 4321–4326. [Google Scholar] [CrossRef]
- Chassaing, C.; Haudrechy, A.; Langlois, Y. 1,3-Dibromo-5,5-dimethyihydantoin, a useful reagent for aromatic bromination. Tetrahedron Lett. 1997, 38, 4415–4416. [Google Scholar] [CrossRef]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry; Springer: New York, NY, USA, 2007. [Google Scholar]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry; McGraw-Hill: New York, NY, USA, 2007. [Google Scholar]
- Smith, W.B. Ab initio studies of the bromination of benzene. J. Phys. Org. Chem. 2003, 16, 34–39. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Mathew, T.; Hoole, D.; Esteves, P.M.; Wang, Q.; Rasul, G.; Olah, G.A. N-Halosuccinimide/BF3-H2O, efficient electrophilic halogenating systems for aromatics. J. Am. Chem. Soc. 2004, 126, 15770–15776. [Google Scholar]
- Galabov, B.; Koleva, G.; Schaefer, H.F.; Schleyer, P.V.R. Electrophile affinity: Quantifying reactivity for the bromination of arenas. J. Org. Chem. 2010, 75, 2813–2819. [Google Scholar] [CrossRef]
- Kong, J.; Galabov, B.; Koleva, G.; Zou, J.J.; Schaefer, H.F., III; Schleyer, P.V.R. The inherent competition between addition and substitution reaction of Br2 with benzene and arenas. Angew. Chem. Int. Ed. 2011, 50, 6809–6813. [Google Scholar] [CrossRef]
- Brown, J.J.; Cockroft, S.L. Aromatic reactivity revealed: Beyond resonance theory and frontier orbitals. Chem. Sci. 2013, 4, 1772–1780. [Google Scholar] [CrossRef]
- Ansell, M.F.; Dewar, M.J.S. Aromatic substitution. Nature 1955, 175, 982–983. [Google Scholar] [CrossRef]
- Esteves, P.M.; Carneiro, J.W.M.; Cardoso, S.P.; Barbosa, A.G.H.; Laali, K.K.; Rasul, G.; Prakash, G.K.S.; Olah, G.A. Unified mechanistic concept of electrophilic aromatic nitration: Convergence of computational results and experimental data. J. Am. Chem. Soc. 2003, 125, 4836–4849. [Google Scholar] [CrossRef]
- Hubig, S.M.; Kochi, J.K. Direct observation of the wheland intermediate in electrophilic aromatic substitution. Reversible formation of nitrosoarenium cations. J. Am. Chem. Soc. 2000, 122, 8279–8288. [Google Scholar] [CrossRef]
- In the case of a highly reactive electrophile with a highly reactive aromatic reactant, the formation of the electrophile may be the rate-determining step, in which the transition state comes early on the reaction coordinate and resembles the aromatic reactant. The reaction profile may be related to the fundamental electronic characteristics of the aromatic reactant.
- Hammond, G.S. A correlation of reaction rates. J. Am. Chem. Soc. 1955, 77, 334–338. [Google Scholar] [CrossRef]
- Holleman, A.F. Some factors influencing substitution in the benzene ring. Chem. Rev. 1924, 1, 187–230. [Google Scholar] [CrossRef]
- Oberhauser, T. A new bromination method for phenols and anisoles: NBS/HBF4-Et2O in CH3CN. J. Org. Chem. 1997, 62, 4504–4506. [Google Scholar] [CrossRef]
- Carreno, M.C.; Ruano, J.L.G.; Sanz, G.; Toledo, M.A.; Urbano, A. N-Bromosuccinimide in acetonitrile: A mild and regiospecific nuclear brominating reagent for methoxybenzenes and naphthalenes. J. Org. Chem. 1995, 60, 5328–5331. [Google Scholar] [CrossRef]
- Zysman-Colman, E.; Arias, K.; Siegel, J.S. Synthesis of arybromides from arenas and N-bromosuccinimde (NBS) in acetonitrile-A convenient method for aromatic bromination. Can. J. Chem. 2009, 87, 440–447. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Barrero, A.F.; Alvarez-Manzaneda, E.J.; Chahboun, R. Enantiospecific synthesis of (+)-puupehenone from (−)-sclareol and protocatechualdehyde. Tetrahedron Lett. 1997, 38, 2325–2328. [Google Scholar] [CrossRef]
- Barrero, A.F.; Alvarez-Manzaneda, E.J.; Chahboun, R.; Armstrong, V. Synthesis and antitumor activity of puupehedione and related compounds. Tetrahedron 1999, 55, 15181–15208. [Google Scholar] [CrossRef]
- Alvarez-Manzaneda, E.J.; Chahboun, R.; Perez, I.B.; Cabrera, E.; Alvarez, E.; Alvarez-Manzaneda, R. First enantiospecific synthesis of the antitumor marine sponge metabolite (−)-15-oxopuupehenol from (−)-sclareol. Org. Lett. 2005, 7, 1477–1480. [Google Scholar]
- Quideau, S.; Ledon, M.; Lamidey, A.M. Enantiospecific synthesis of the antituberculosis marine sponge metabolite (+)-puupehenone. The arenol oxidative activation route. Org. Lett. 2002, 4, 3975–3978. [Google Scholar] [CrossRef]
- Wu, Y.C.; Liu, L.; Li, H.J.; Wang, D.; Chen, Y.J. Skraup-Doebner-von Miller quinoline synthesis revisited: Reversal of the regiochemistry for γ-aryl-β,γ-unsaturated α-ketoesters. J. Org. Chem. 2006, 71, 6592–6595. [Google Scholar] [CrossRef]
- Wu, Y.C.; Chen, Y.J.; Li, H.J.; Zou, X.M.; Hu, F.Z.; Yang, H.Z. Synthesis of trifluoromethyl-promoted functional pyrazolo[1,5-a]pyrimidine and pyrazolo[5,1-d][1,2,3,5]tetrazine-4(3H)-ones. J. Fluor. Chem. 2006, 127, 409–416. [Google Scholar] [CrossRef]
- Wu, Y.C.; Liu, L.; Wang, D.; Chen, Y.J. Efficient synthesis of 3-arylaminopyrroline-2-ones by the tandem reaction of anilines and β,γ-unsaturated α-ketoesters. J. Heterocycl. Chem. 2006, 43, 949–955. [Google Scholar] [CrossRef]
- Wu, Y.C.; Liu, L.; Liu, Y.L.; Wang, D.; Chen, Y.J. TFA-mediated tandem Friedel-Crafts alkylation/cyclization/hydrogen transfer process for the synthesis of flavylium compounds. J. Org. Chem. 2007, 72, 9383–9386. [Google Scholar] [CrossRef]
- Wu, Y.C.; Li, H.J.; Liu, L.; Wang, D.; Yang, H.Z.; Chen, Y.J. Efficient construction of pyrazolo[1,5-a]pyrimidine scaffold and its exploration as a new heterocyclic fluorescent platform. J. Fluor. 2008, 18, 357–363. [Google Scholar] [CrossRef]
- Wu, Y.C.; Li, H.J.; Yang, H.Z. A sensitive and highly selective fluorescent sensor for In3+. Org. Biomol. Chem. 2010, 8, 3394–3397. [Google Scholar] [CrossRef]
- Wu, Y.C.; Li, H.J.; Liu, L.; Demoulin, N.; Liu, Z.; Wang, D.; Chen, Y.J. Facile synthesis of spiropyrans from chromene hemiacetal esters and bifunctional nucleophiles. Synlett 2011, 2011, 1573–1578. [Google Scholar] [CrossRef]
- Wu, Y.C.; Li, H.J.; Liu, L.; Demoulin, N.; Liu, Z.; Wang, D.; Chen, Y.J. Hafnium triflate as an efficient catalyst for direct Friedel-Crafts reactions of chromene hemiacetals. Adv. Synth. Catal. 2011, 353, 907–912. [Google Scholar] [CrossRef]
- Wu, Y.C.; Li, H.J.; Liu, L.; Liu, Z.; Wang, D.; Chen, Y.J. Cascade reaction of β,γ-unsaturated α-ketoesters with phenols in trityl chloride/TFA system. Highly selective synthesis of 4-aryl-2H-chromenes and their applications. Org. Biomol. Chem. 2011, 9, 2868–2877. [Google Scholar] [CrossRef]
- Wu, Y.C.; Liron, M.; Zhu, J.P. Asymmetric total synthesis of (−)-quinocarcin. J. Am. Chem. Soc. 2008, 130, 7148–7152. [Google Scholar] [CrossRef]
- Wu, Y.C.; Bernadat, G.; Masson, G.; Couturier, C.; Schlama, T.; Zhu, J.P. Synthetic studies on (−)-lemonomycin: An efficient asymmetric synthesis of lemonomycinone amide. J. Org. Chem. 2009, 74, 2046–2052. [Google Scholar]
- Wu, Y.C.; Zhu, J.P. Asymmetric total syntheses of (−)-renieramycin M and G and (−)-jorumycin using aziridine as a lynchpin. Org. Lett. 2009, 11, 5558–5561. [Google Scholar] [CrossRef]
- Besler, B.H.; Merz, K.M., Jr.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, H.-J.; Wu, Y.-C.; Dai, J.-H.; Song, Y.; Cheng, R.; Qiao, Y. Regioselective Electrophilic Aromatic Bromination: Theoretical Analysis and Experimental Verification. Molecules 2014, 19, 3401-3416. https://doi.org/10.3390/molecules19033401
Li H-J, Wu Y-C, Dai J-H, Song Y, Cheng R, Qiao Y. Regioselective Electrophilic Aromatic Bromination: Theoretical Analysis and Experimental Verification. Molecules. 2014; 19(3):3401-3416. https://doi.org/10.3390/molecules19033401
Chicago/Turabian StyleLi, Hui-Jing, Yan-Chao Wu, Jian-Hong Dai, Yan Song, Runjiao Cheng, and Yuanyuan Qiao. 2014. "Regioselective Electrophilic Aromatic Bromination: Theoretical Analysis and Experimental Verification" Molecules 19, no. 3: 3401-3416. https://doi.org/10.3390/molecules19033401