Prodrug Strategies for Enhancing the Percutaneous Absorption of Drugs

Centre of Excellence for Pharmaceutical Sciences (PHARMACEN), North-West University, Potchefstroom 2520, South Africa
Molecules 2014, 19(12), 20780-20807; https://doi.org/10.3390/molecules191220780
Submission received: 20 October 2014
/
Revised: 3 December 2014
/
Accepted: 4 December 2014
/
Published: 12 December 2014
(This article belongs to the Special Issue Prodrugs)
Abstract
:The transdermal application of drugs has attracted increasing interest over the last decade or so, due to the advantages it offers, compared to other delivery methods. The development of an efficient means of transdermal delivery can increase drug concentrations, while reducing their systemic distribution, thereby avoiding certain limitations of oral administration. The efficient barrier function of the skin, however, limits the use of most drugs as transdermal agents. This limitation has led to the development of various strategies to enhance drug-skin permeation, including the use of penetration enhancers. This method unfortunately has certain proven disadvantages, such as the increased absorption of unwanted components, besides the drug, which may induce skin damage and irritancy. The prodrug approach to increase the skin’s permeability to drugs represents a very promising alternative to penetration enhancers. The concept involves the chemical modification of a drug into a bioreversible entity that changes both its pharmaceutical and pharmacokinetic characteristics to enhance its delivery through the skin. In this review; we report on the in vitro attempts and successes over the last decade by using the prodrug strategy for the percutaneous delivery of pharmacological molecules.
1. Introduction
The skin, being the largest organ in the human body, has various functions, including protecting the body from its external environment, from excessive water loss, friction and impact wounds, and from external stimuli that could potentially harm the body, whilst also providing sensory abilities [1]. The skin of the average adult covers approximately 1–2 m2 [2] and receives a third of the total blood circulation, thus making it an ideal route through which therapeutic agents can be administered [3]. The outermost layer of the skin, the stratum corneum (SC), is the primary layer through which most drug molecules penetrate the skin. It is a compositionally and morphologically unique biomembrane that consists of a layer of compressed, keratin-filled corneocytes, anchored in a lipid matrix. This arrangement of the corneocytes and lipids makes the SC about a thousand times less permeable to water and substances, relative to other membranes in the body [2].
The advantages of the transdermal drug delivery (TDD), compared to the oral administration route, make it a very attractive alternative. Of these advantages include better patient collaboration, control over the input kinetics, with less side effects [2]. Incorporating drugs into a TDD system helps decrease their side effects on the gastrointestinal tract, whilst it can be especially advantageous to children or infants who have trouble taking some oral formulations, due to the bitter taste, or being unable to swallow tablets [4]. Since transdermal drug administration avoids the hepatic first-pass metabolism, an improved bioavailability would occur, the administered dose would be reduced and therapy could be easily interrupted in case of a toxic effect [5].
Despite these advantages, transdermal drug delivery (TDD) has its limitations, including: (a) the skin acting as a two-way barrier prevents molecules from entering the body from the external environment, while regulating the loss of essential body fluids and electrolytes; (b) drugs requiring high-blood levels can’t be administered through the skin; (c) patch adhesives may not adhere to all skin types and may result in allergic responses; and (d) drugs or drug formulations may cause skin irritation or sensitization.
Transdermal delivery is therefore unsuitable for all drugs. For molecules to pass through the SC, they need to exhibit favorable values with regards to certain physicochemical properties, including the partition coefficient (log P), melting point, aqueous solubility, and the molecular mass and pKa of the drug. All of these properties to various degrees influence the rate of a drug’s permeation through the skin [6].
Furthermore, physiological factors, such as the skin (age, hydration, condition and metabolism), body (anatomical) location, race, gender and temperature are reported to affect the rate of drugs’ permeation through the skin [7].
In this review, the structure of the skin, physiological and physicochemical factors governing percutaneous absorption of molecules and drugs, and representatives of different therapeutic categories of agents, subjected to the prodrug strategy over the last decade, are discussed.
2. Structure of the Skin
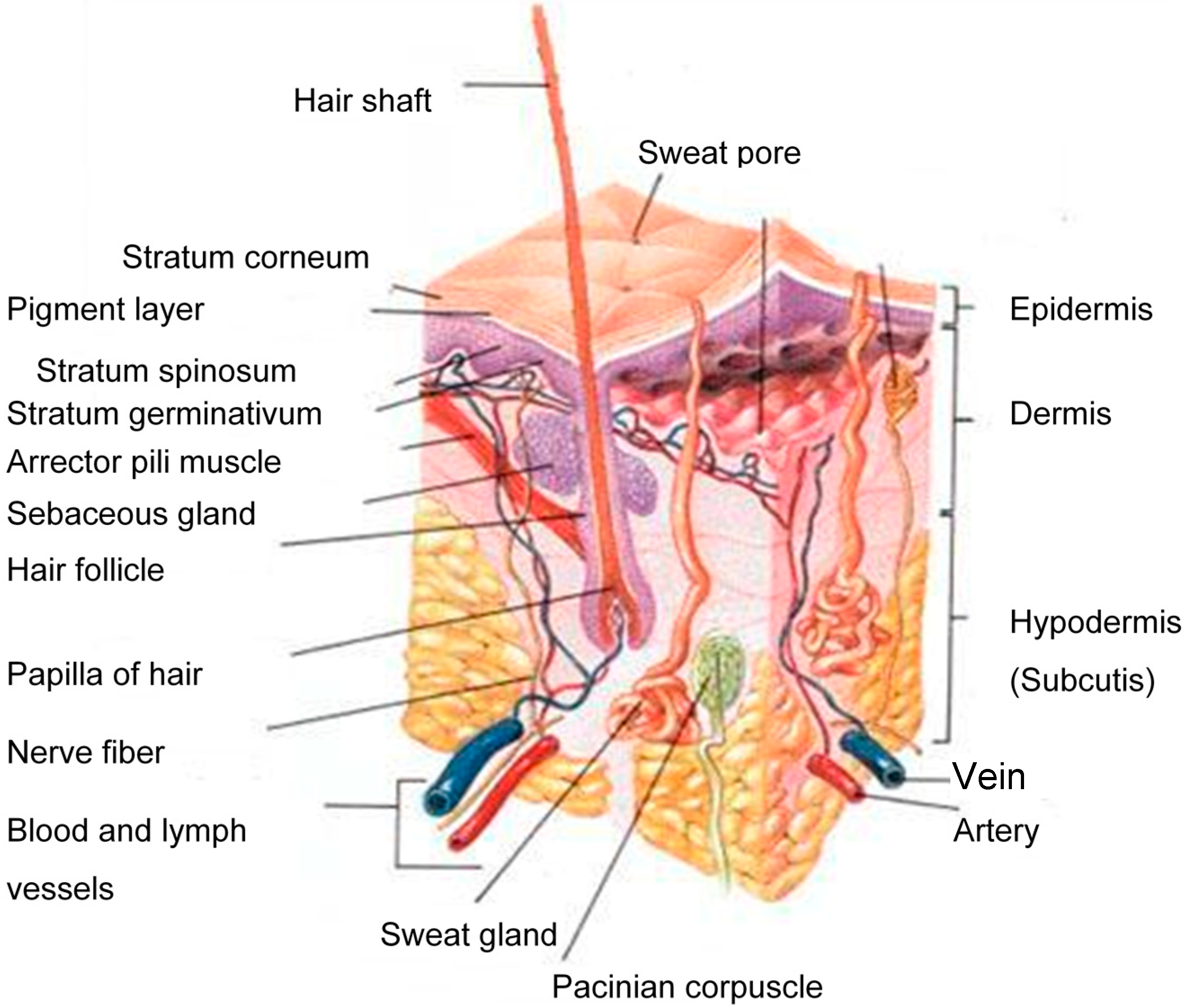
The human skin consists of three distinct layers, i.e., the stratified, avascular, cellular epidermis, the dermis and the underlying hypodermis (Figure 1) of connective tissue [8]. A therapeutic agent must cross all of these layers and enter the systemic circulation in order to exert its effect.
Figure 1.
A schematic cross-section of human skin [9].
Figure 1.
A schematic cross-section of human skin [9].
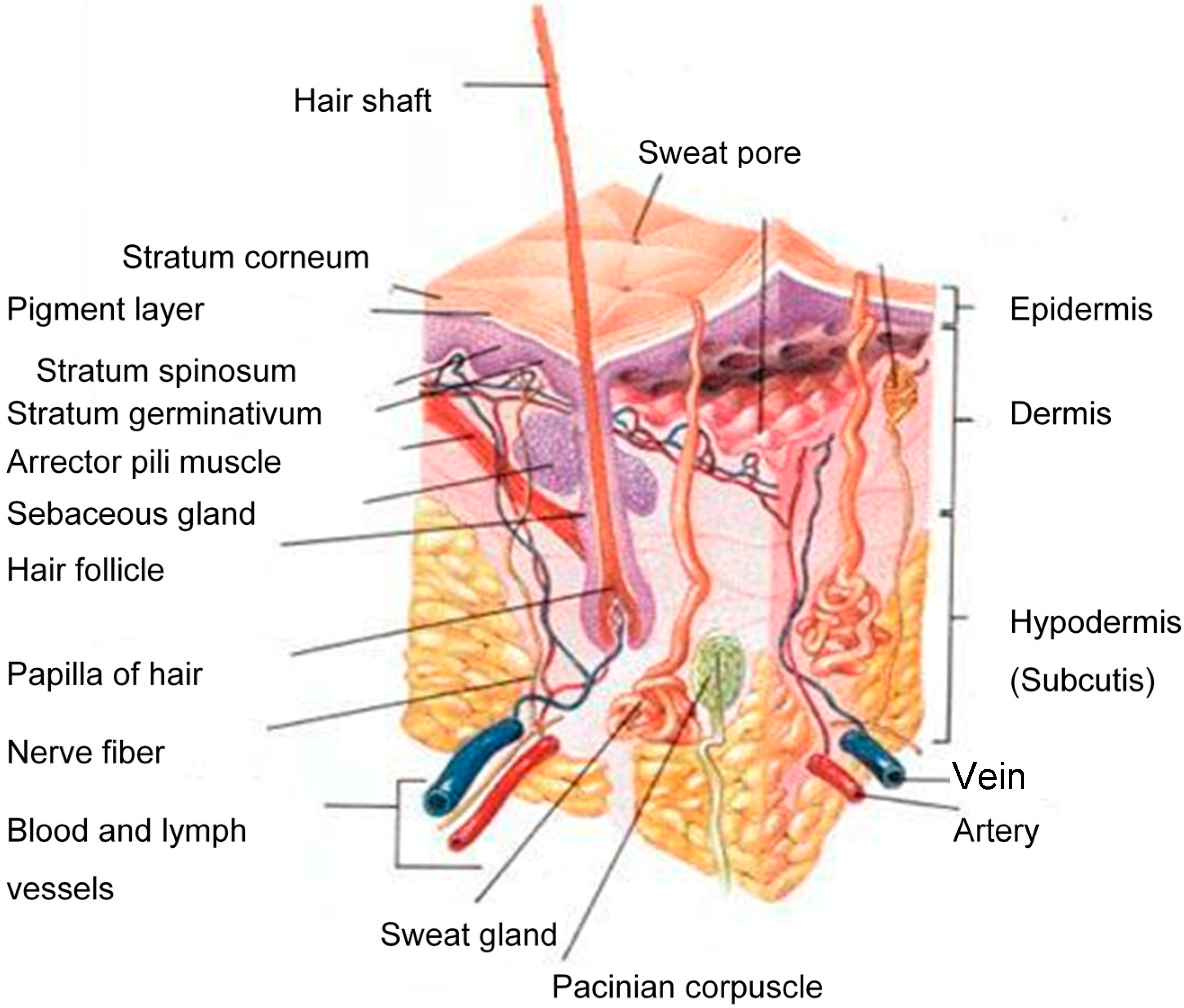
2.1. Stratum Corneum (SC)
The tissue that mainly affects drug permeation into the skin is the stratum corneum (SC), or the horny layer. The major components of the SC are the ceramides, fatty acids, cholesterol, cholesterol sulphate and sterol esters [10], which vary among individuals and body location [11] and provide the SC with its amphiphilic properties. Water plays a vital role in maintaining the SC integrity and is it also involved in the mediation of some hydrolytic enzymes’ activities.
2.2. Viable Epidermis
The epidermis is situated directly beneath the SC and is described as a complex, multiple-layered membrane. The epidermis contains no blood vessels, and must nutrients and waste products travel across this membrane in order to maintain its integrity.
2.3. Dermis
The dermis is situated between the epidermis and the underlying subcutaneous, fatty layer, i.e., the hypodermis. It is the major component of the skin and predominantly consists of collagen fibrils and elastic tissue, which provide support and flexibility to the skin. Since the dermis is very hydrophilic, hydrophilic drugs would easily pass through it, while lipophilic drugs would experience difficulties in crossing it. Structures, like lymphatic and blood vessels, nerve endings, hair follicles, sebaceous and sweat glands are found in the dermis [12]. The dermis layer has the highest blood circulation in the skin and thus regulates body temperature and removes toxins and other undesirable molecules. The dermis thus forms a “sink” condition that allows transdermal permeation to take place, due to the formed concentration gradient [13].
2.4. Hypodermis
The hypodermis, also known as the subcutaneous fatty layer, bridges the overlying dermis and the underlying body constituents. This layer is very thick and thus insolates the body and protects it against external shock.
2.5. Skin Appendages
Three main appendages are present in the skin, namely the hair follicles, and the eccrine and apocrine sweat glands. The hair follicles are found over the entire body, except in areas that take a lot of strain, such as the feet soles and the hand palms. All three appendages are situated in the dermis [14] and each has specific functions. The sebaceous glands are located in the hair follicles and secrete sebum, a mixture of fatty acids, cholesterol, triglycerides, waxes and cellular debris [15]. More lipophilic drugs hence cross this section, due to the lipophilic nature of sebum. Eccrine glands are found across most of the body and secrete sweat, a diluted salt solution with a pH ~5, and thus function as body heat regulators. The apocrine glands are located at specific areas of the body only and are usually limited to the axillae, nipples and anogenital regions, and are responsible for the odor of sweat [7].
3. Routes of Transdermal Delivery
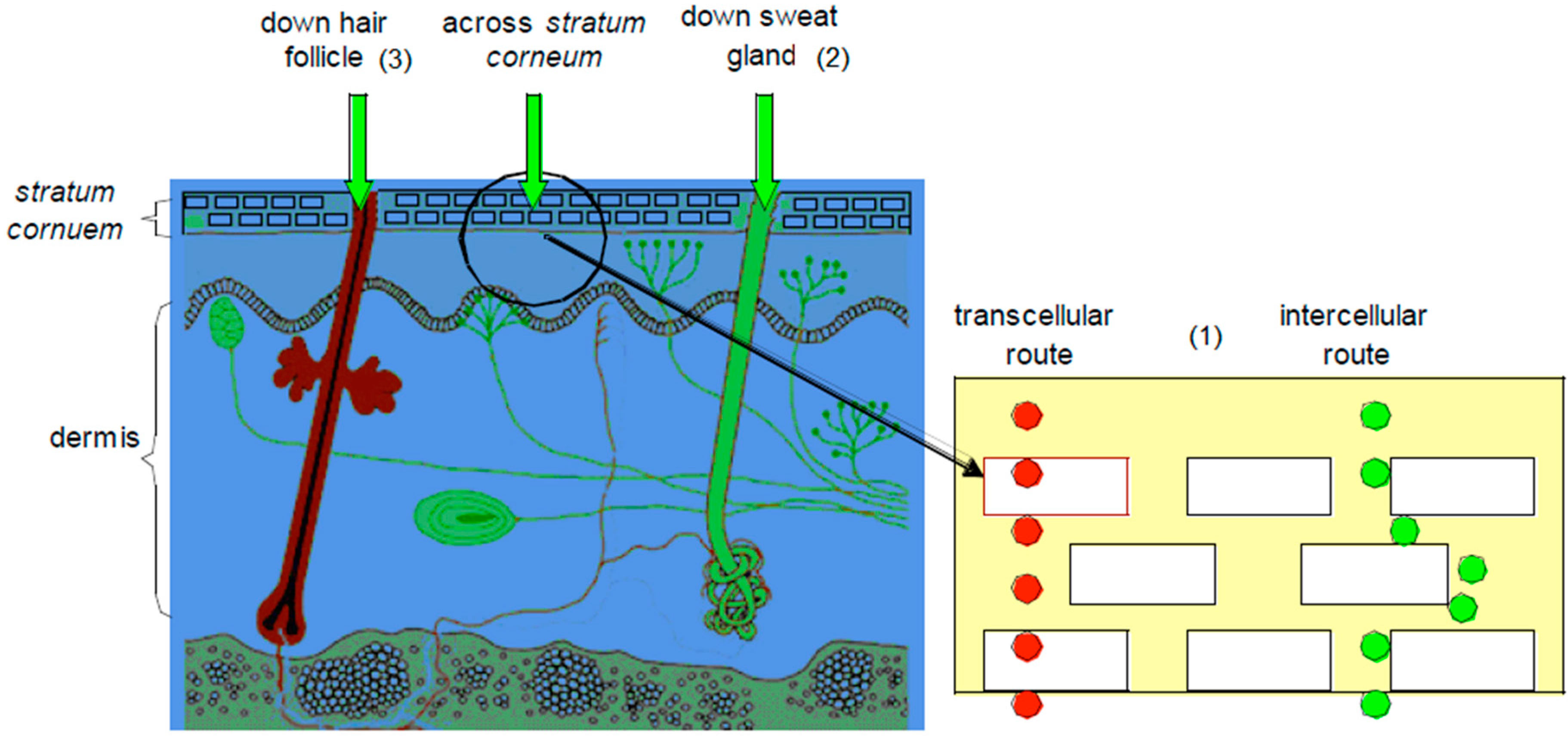
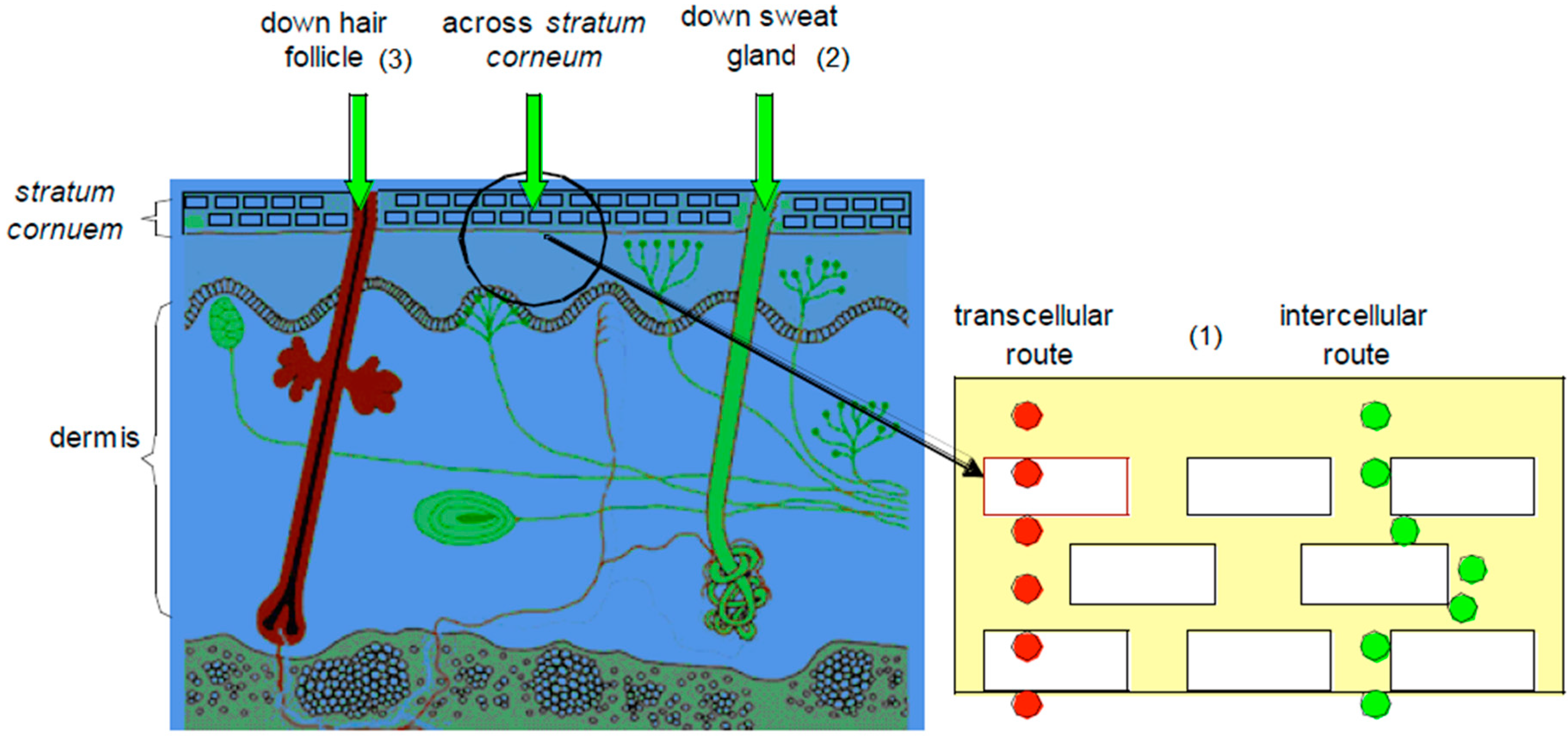
For molecules to affect the body, they must cross through the different layers of the skin to reach the systemic circulation. This can be achieved through different routes, i.e., the transepidermal routes (intra- and intercellular), the sweat glands and the hair follicles [1,7], as illustrated in Figure 2.
3.1. Transepidermal Routes
This pathway is split into the intracellular (crossing of molecule through the cells) and the intercellular (crossing of molecule in between the cells) routes.
The intracellular route, also known as the transcellular pathway, is the path through which a molecule permeates through the SC. Drug molecules that use this route move across and through the corneocytes located in the SC [1]. The keratinocytes of the SC are very hydrophilic in nature, and thus naturally, hydrophilic drugs would cross the SC with more ease. These hydrophilic keratinocytes are, however, enclosed in a multi-layered lipophilic membrane and are surrounded by a lipophilic matrix in between these cells. This lipophilic environment thus prevents hydrophilic drugs from crossing the SC [7].
The intercellular route accommodates molecules that pass in between the cells of the SC. The lipid bilayers, comprising up to 1% of the SC, are important in reducing water loss through the skin. The balance of lipid and aqueous solubility that is important for solubility in the membrane is also important for molecules traversing the intercellular route [16,17,18].
3.2. Transappendageal (Shunt Routes)
The sweat glands and hair follicles also offer pathways through which molecules can travel through the SC. Drugs that use this route normally have high molecular weights and do these charged molecules travel through the bulk of the SC with difficulty [7].
Figure 2.
A representation of transdermal delivery pathways: (1) transepidermal route (intra- and intercellular); (2) the sweat gland and (3) the hair follicle (adapted from [19]).
Figure 2.
A representation of transdermal delivery pathways: (1) transepidermal route (intra- and intercellular); (2) the sweat gland and (3) the hair follicle (adapted from [19]).
4. Factors Influencing Skin Permeation of Drugs
The transdermal permeation of drugs is significantly affected by both physiological and physicochemical factors, as discussed in the following sections.
4.1. Physiological Factors
The physiological factors that influence the absorption of molecules through a healthy skin include:
4.1.1. Skin Age
As the skin ages, some functional and structural changes take place that affect the transdermal absorption of molecules into it. Hydration plays an important role in transdermal absorption and decreases with age, as the skin loses its moisture content [20]. The enzymatic activity in the skin also reduces with age, as well as the blood flow to the skin. A lower blood flow in older people suggests that the clearance of drugs in an older SC will be slower, which in turn would negatively impact on the drug flux gradient [7].
4.1.2. Anatomical Location
The skin differs in appearance all across the body. The generalized decreasing order of absorption exists from the genitals > head and neck > trunk > arm > leg [7]. Differences in anatomical sites are therefore indicative that the location of transdermal patches, or ointment applications would affect absorption.
4.1.3. Race
Racial differences in skin function have been investigated. Weigand et al. [21] reported an increase in intracellular cohesion in black skin, while Reinertson and Wheatley [22] further demonstrated higher lipid content in black skin. Although relatively limited data are available on race, indications are that racial differences do exist among white and black skins, with regards to some anatomical and physiological functions of the skin, and that these differences could determine race related patterns in skin behavior, namely the response to irritant chemicals and the penetration of topically applied drugs.
4.1.4. Temperature
The permeation of molecules across the skin is a passive process. An increase in temperature would thus result in an increase in kinetic energy of the drug molecules, which would therefore cause the molecules to move faster through the SC. An increasing temperature would also cause structural alterations in the SC and underlying tissue and result in a faster movement of the drug through the different skin layers [7,23].
4.1.5. Skin Hydration
The water content of the skin plays a vital role in transdermal permeation. Jackson [24] found that hydration of the skin through soaking, moisturizing, or humidity, for example, causes the corneocytes in the SC to swell, which allows molecules to permeate through that layer much easier. In another study, Li and Chan [25] revealed that the use of a very hydrophilic co-solvent, like ethanol/water, had increased the permeation of anti-human immunodeficiency virus (HIV) drugs through the skin.
4.1.6. Skin Condition (Disease)
Disease compromises the natural barrier function of the skin, which affects the transdermal absorption of therapeutic agents. The rate of transdermal absorption would increase with a disease that ravages the skin, but would decrease upon self-healing of the skin [7].
4.1.7. Skin Metabolism
Enzymes in the skin are responsible for metabolism and for the elimination of drugs. For prodrugs, which have to undergo biotransformation, enzymatic activity is therefore a rate limiting factor in the transdermal absorption process [26], and is reported to be the highest in the viable epidermis of the skin [27].
4.2. Physicochemical Factors
Physicochemical factors are the primary factors that influence the transdermal absorption of drugs. For molecules designed to pass through the SC, favorable physicochemical properties are necessary [2], which include:
4.2.1. Solubility
The SC is the most important barrier to skin permeation and therefore the rate limiting factor for drugs to cross the skin. The SC is a lipophilic membrane [28] and the amount of a drug that accumulates in it bears some relationship to the solubility of that drug in some organic solvents, such as the highly lipophilic hexane. Saturated solutions better permeate through the SC, because they represent maximum thermodynamic activity [29]. An awareness of the importance of drug solubility in the SC emerged from reports during the 1980s that drugs with biphasic (water and lipid) solubility better permeated than those with high monophasic (water or lipid) solubility [17,18]. A very hydrophilic drug is unable to penetrate the skin, while a very lipophilic drug has the propensity to remain in the layers of the SC. While the SC is lipophilic in nature and favors the permeation of lipophilic drugs, the aqueous nature of the layers beneath the SC dictate that drugs should embody some hydrophilic properties to pass through them. Often, small structural changes of a drug, such as salt formation, or esterification can either enable the enhancement of aqueous, or lipid solubility [30]. Moieties, such as polyethylene glycol (PEG) that are amphiphilic in nature, show an increase in the aqueous solubility of derivatives, due to multiple hydrogen bonds that form with water molecules, resulting from an increase in chain length [31].
4.2.2. Partition Coefficient
Penetration of the SC requires that a drug partitions into the membrane. Such partitioning is an important step in the penetration of the membrane [32]. However, solubility in the membrane is the limiting physicochemical parameter. The partition coefficient is usually the key factor in determining which pathway a drug molecule would follow when passing through the SC. Hydrophilic molecules are therefore expected to pass through the intracellular routes, whereas more lipophilic molecules would use the intercellular routes. Either lipophilic or hydrophilic permeants must pass through the hydrophilic and lipophilic layers, respectively, to achieve successful transdermal delivery. In order to study and determine the partition coefficient of a drug, organic solvents are used to mimic the different phases/domains that a molecule must cross to enter the systemic circulation. The n-octanol-water partition coefficient (log P) is a good representation of the partitioning of a drug in between the lipophilic SC and the underlying hydrophilic living cells of the epidermis [33]. The log D value (the log P value at a specific pH) of a compound is usually a good indication as to whether a molecule would be a favorable candidate for transdermal permeation, or not. The lower the log P value, the more hydrophilic is the compound. Compounds with a log P lower than −1 would hence have difficulty in passing from the vehicle into the SC. Subsequently, only compounds with log P > −1 should be considered as possible candidates for transdermal delivery. Another problem arises when compounds have a log p value above 2. Such a drug is delayed in the SC, where a reservoir could be established that would create problems with achieving steady plasma concentrations within a reasonable time span [16]. Williams [7], however, reported that molecules with a log p value in the 1–3 range, had exhibited both aqueous and lipophilic properties, sufficient enough to obtain proper transdermal permeation, because they had been able to cross the lipophilic (SC) and hydrophilic layers (epidermis) of the skin. Such findings emphasize the lack of consensus among researchers with regards to the optimum partition coefficient range that is required for a drug to be efficiently delivered across the skin.
4.2.3. Molecular Mass
The skin is a compact membrane that is difficult to penetrate. Smaller molecules are therefore likely to permeate the skin at higher rates than larger molecules, hence the importance of molecular size in transdermal absorption. Idson [34] suggested an inverse relationship between molecular size and transdermal permeation. Drugs with a molecular mass (MW) between 100 and 500 Daltons were found to be suitable for transdermal transport [2,7]. A study conducted on a molecule with MW above 500 Daltons; however, also resulted in percutaneous absorption [35], suggesting that the upper limit of the MW range of a compound could indeed exceed 500 Daltons, but still efficiently permeate the skin through passive diffusion.
4.2.4. Ionization
The lipophilic nature of the SC had led to the belief that ionized drugs would be poor candidates for transdermal delivery. As a result of the complex structure of the skin, however, drugs can cross the skin via various pathways. The transcellular route is regarded as having intermediate properties, whereas the intracellular route is mainly regarded for allowing the delivery of lipophilic molecules. Ionized drugs hence cross the skin through the shunt route, but the amount of molecules that pass through that route is significantly less than unionized molecules that take the intracellular route [7].
The pH range of the viable epidermis is 7.3–7.4 and that of the SC 4.2–5.6. The drug concentration that exists in the unionized form is a function of both the dissociation constant of the drug and the pH at the absorption site [30].
From the Henderson-Hasselbach equation:
The fraction of the unionized drug is thus a function of the pH [36].
4.2.5. Hydrogen Bonding
The varied skin components (lipids, proteins, aqueous regions, enzymes, etc.) and possible drug penetrants (weak acids/bases, ionized/unionized species, neutral molecules, etc.) suggest a multitude of potential interactions between drug substances and skin tissue [7]. The formation of hydrogen bonds, or weak Van der Waals forces, would hence influence skin penetration of a permeating drug. Lag time is an important factor being influenced by drug binding to the tissue. Bound drugs to skin tissue in the SC would result in a prolonged lag time, which would translate into a delayed onset of the therapeutic action. Since the SC is mainly a hydrogen bond acceptor [37], an increase in the number of hydrogen bonding groups of the drug may inhibit its permeation across the layers of the SC [7].
4.2.6. Melting Point
Generally, organic substances with high melting points and high enthalpy of melting have lower aqueous solubility properties, because solvents can’t enter the crystalline structure of such molecules to dissolve them [38]. An indirect relationship therefore exists between the melting point and the solubility of a drug. Hadgraft et al. [39] developed a model that accurately estimates the solubility constraint (δSC) in the SC by using the melting point of the drug:
where, δSC is the solubility constraint in the SC, mp is the melting point (in K).
Lowering the melting point of a drug would hence cause an increase in its solubility in the SC and ultimately in its permeation across the skin [40].
5. The Impact of Prodrug Strategy on the Percutaneous Absorption of Drugs
Physicochemical properties, like the lipophilicity of a drug molecule can be modified through derivatization. The fact that most drugs are only designed for oral administration emphasizes the complexity of the development of drug candidates suitable for transdermal delivery, because their physicochemical makeup has to be specifically modified to allow skin permeation. The derivatization of a drug may enhance its ability to permeate through the skin. This approach comprises the basis of the prodrug strategy, during which a drug molecule is structurally modified into derivatives, imparted with favorable physicochemical properties to facilitate its permeation, although being designed to latter yield the parent drug (active pharmaceutical ingredient) after enzymatic, or chemical release [41]. Two of the derivatization paths often used include (a) increasing lipophilicity through alkyl-chain lengthening [42], and (b) using PEGylation to bring about derivatives, such as esters, carbonates, carbamates and ether, equipped with enhanced physicochemical properties that would more readily cross the skin, compared to the parent drug [43]. This strategy could lead to an increase in a drug’s pharmacological action in the body. Several drugs, representing different categories of therapeutic agents, have been investigated over the last decade for their skin permeation ability, by employing the prodrug strategy. Typical examples of such drugs are discussed in the following sections.
5.2. Cytarabine (Anticancer)
Cytarabine (Figure 4) is an anticancer drug that is extensively used in the treatment of both acute and chronic myeloblastic leukaemia. The major impediments to a broad clinical use of cytarabine include its cell cycle (S-phase) specificity, and the rapid metabolism of the drug in plasma into its inactive metabolite, uracil arabinoside (ara-U), by the enzyme deoxycytidine deaminase [49]. A prolonged exposure of cells to cytarabine’s cytotoxic concentrations is a requirement for achieving maximum activity. In practice, it is administered either through repetitive schedules, or continuous intravenous infusion to achieve a sustained supply [50]. Patient non-compliance occurs as a result of these modes of administration being inconvenient and invasive.
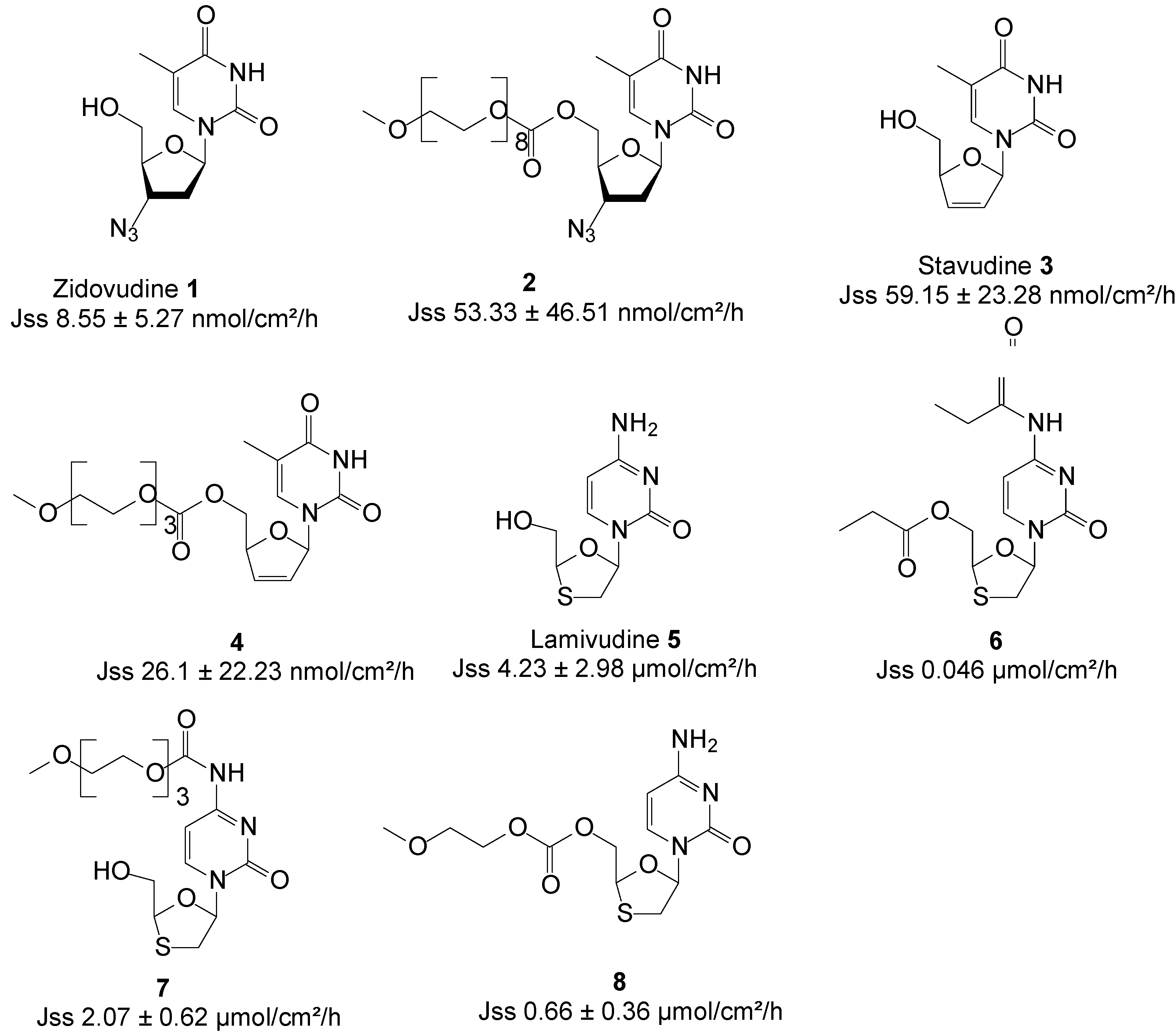
Figure 3.
Structures of ARV drugs and best skin penetrant derivatives.

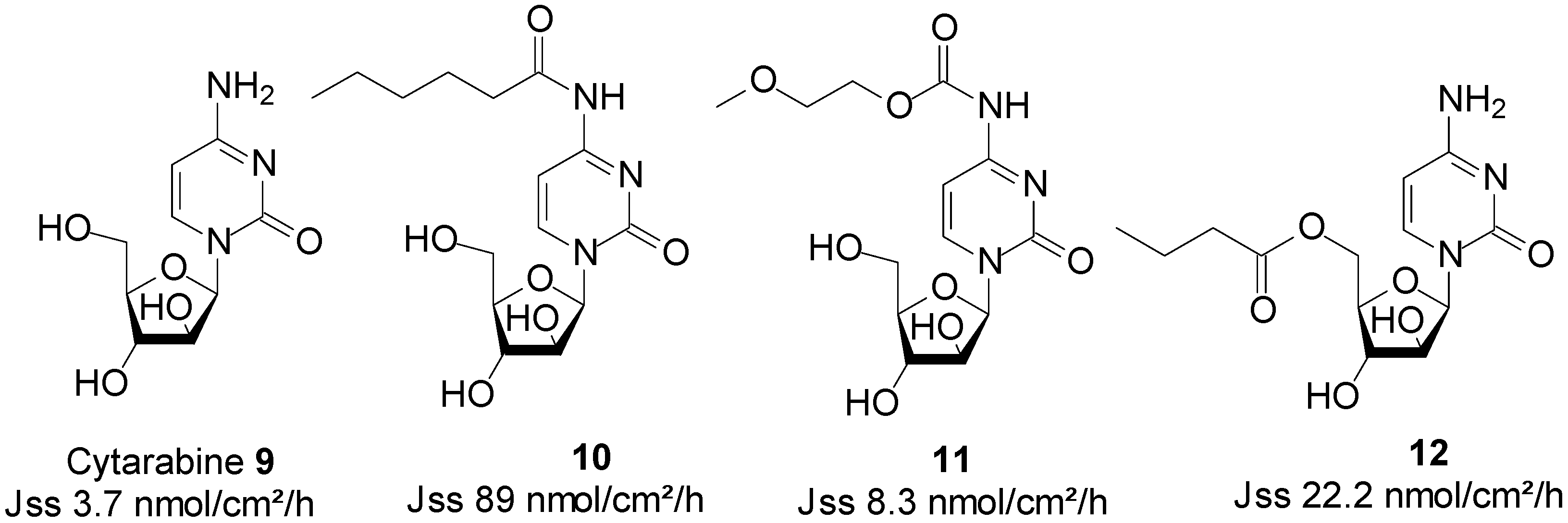
Figure 4.
Cytarabine and its best skin permeant derivatives.
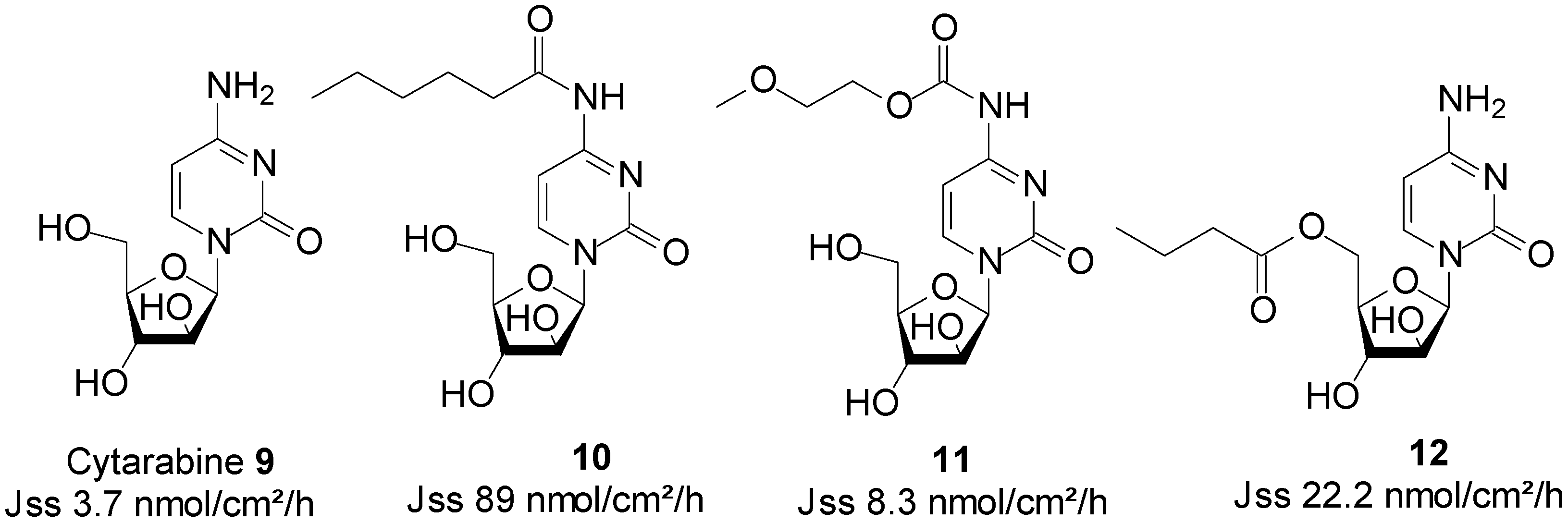
In search for a viable alternative to the intravenous route of administration of cytarabine, Legoabe et al. investigated the skin permeation ability of cytarabine 9, by employing the prodrug strategy. All Franz cell diffusion experiments were performed at saturation in aqueous PBS pH 7.4 solutions. They observed an increase in skin permeation fluxes by their series of N4-alkylamide derivatives. N-Hexyl amide 10 was found to be the best penetrant with a Jss value of 89.0 nmol/cm2/h, which was significantly higher than 3.7 nmol/cm2/h of cytarabine [51]. In another study, the n-alkyl chains were replaced by MPEG moieties, which resulted in a series of N4-MPEG carbamates. Although not significantly higher, skin penetration was enhanced, since the Jss of carbamate 11, featuring one EO, was found to be twice as high than that of cytarabine (8.3 vs. 3.7 nmol/cm2/h) [52]. The study of N-alkyl ester prodrugs of cytarabine also led to enhanced skin permeation. Indeed, N-butyl ester 12 exhibited the highest flux value of 22.2 nmol/cm2/h, which was significantly higher than that of the parent drug cytarabine (3.70 nmol/cm2/h) [53]. The structures of cytarabine and its best permeant derivatives are shown in Figure 4.
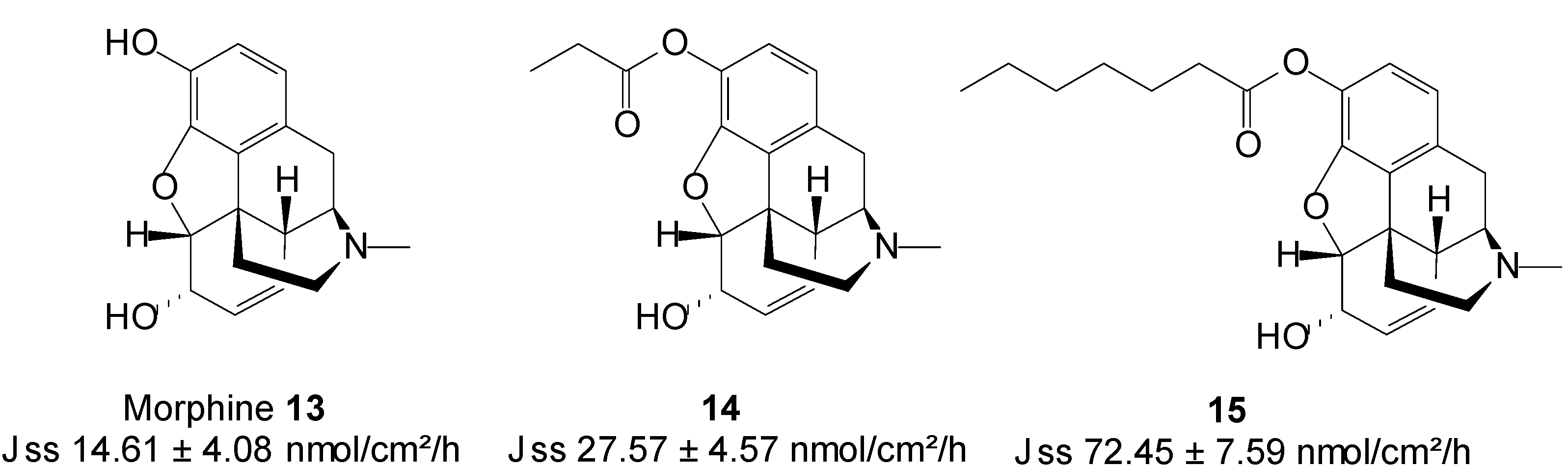
5.3. Morphine (Opioid Analgesic)
Morphine 13 is the most widely used opioid analgesic for acute and chronic pain, and is the standard against which new analgesics are measured. Morphine is inefficiently absorbed when orally administered, due to first-pass metabolism. In patients with normal renal function, the plasma half-life of morphine is 1.4~3.4 h [54].
Transdermal administration would offer a further improvement in its delivery, by enabling the continuous systemic application of morphine through intact skin, hence producing constant plasma concentrations [55]. Two N-alkyl esters (Figure 5) of morphine, viz. morphine propionate 14 and morphine enanthate 15 were synthesized as potential prodrugs for transdermal delivery. Both prodrugs were more lipophilic than their parent drug. The in vitro skin permeation of morphine and its prodrugs from citrate-phosphate buffer in pH 6 was in the order of 15 > 14 > 13. Morphine propionate and morphine enanthate therefore enhanced the transdermal delivery of morphine by 2- and 5-fold, respectively [56]. The highest flux observed with the emanate prodrug may be ascribed to the fact this compound was assessed in saturated conditions unlike the propionate 14 which was tested in dilution.
Figure 5.
Morphine and its n-alkyl ester prodrugs.
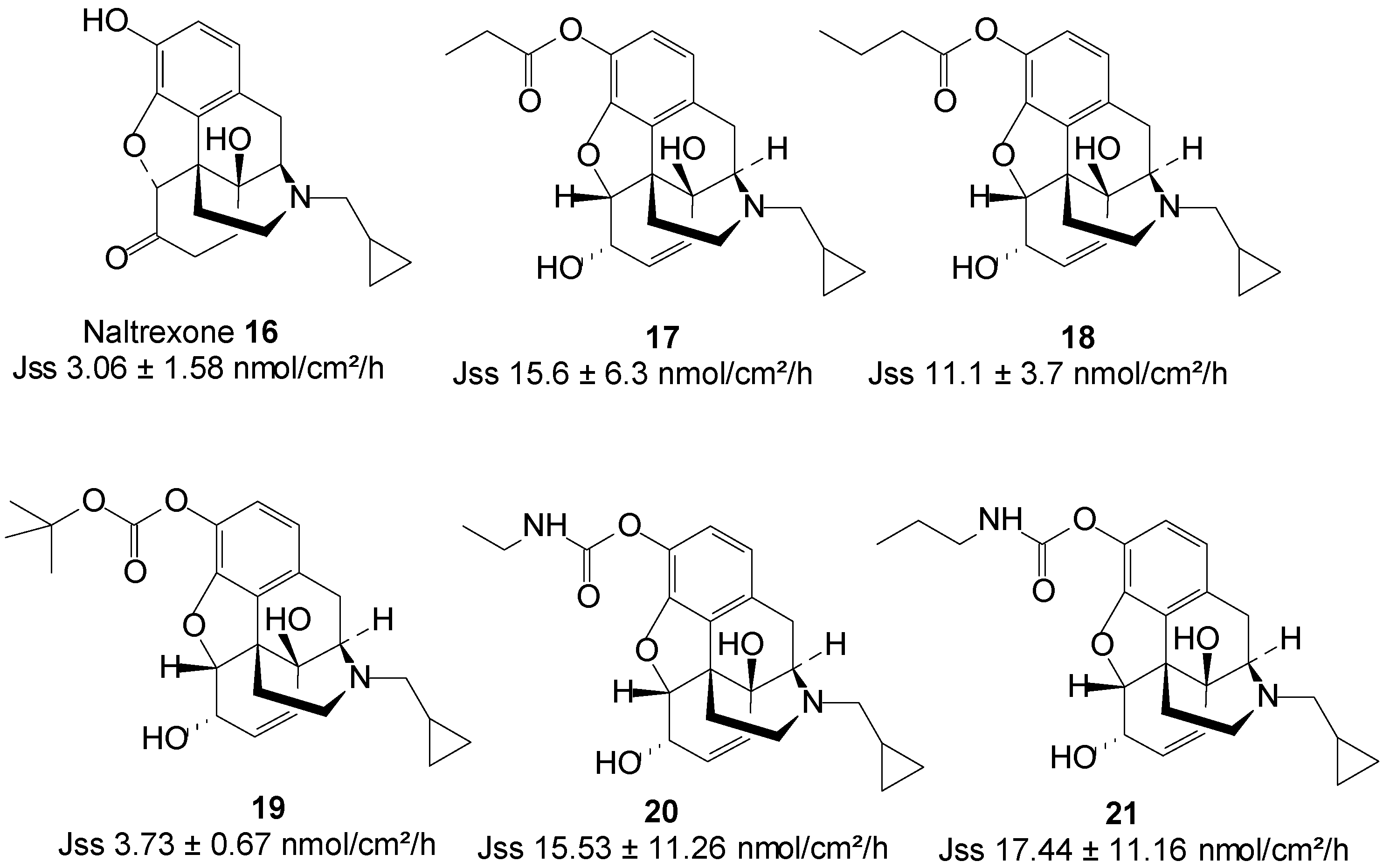
5.4. Naltrexone (Opioid Antagonist)
Naltrexone (NTX), an opioid antagonist, is used for alcohol dependence and opioid addiction [57]. In maintenance therapy for alcohol abstinence, the NTX dose must be given regularly to maintain a minimum effective plasma drug concentration. Standard drug regimens for chronic therapy create non-compliance problems and require high motivation of NTX patients [58]. Sustained release formulations would be extremely useful in maintenance therapy and may result in the improvement of the therapeutic effectiveness of NTX. Additionally, a non-oral delivery route for NTX could reduce oral NTX related gastrointestinal side effects, including nausea, abdominal pain and vomiting [41,59]. NTX bioavailability would also be increased via a delivery route that bypasses first-pass metabolism, as this drug is found to undergo extensive first-pass metabolism with a resultant low oral bioavailability of a mere 5%–40% [60].
In search for a non-oral delivery route for NTX 16, Stinchcomb and co-workers assessed the human skin permeation of straight-chain alkyl ester, and branched-chain alkyl ester and carbonate prodrugs of naltrexone alongside the parent drug from mineral oil in saturated conditions. All the straight-chain ester prodrugs enhanced the skin permeation of NTX. The acetate 17 and propionate 18 were the best permeants with Jmax values 15.6 ± 6.3 and 11.1 ± 3.7 nmol/cm2/h, respectively, higher than 2.5 ± 1.5 nmol/cm2/h of NTX [61].
Outcomes of branched-chain prodrugs were disappointing as most derivatives displayed significantly lower transdermal fluxes than parent NTX. The best penetrant was found to be tert-butyl carbonate 19 (Figure 6), having a steady-state flux (Jss) value of 3.73 ± 0.67, which was similar to 3.06 ± 1.58 nmol/cm2/h of the parent NTX [62]. Thus, no skin permeation enhancement was achieved in this study.
Figure 6.
Naltrexone and its best skin penetrant prodrugs.
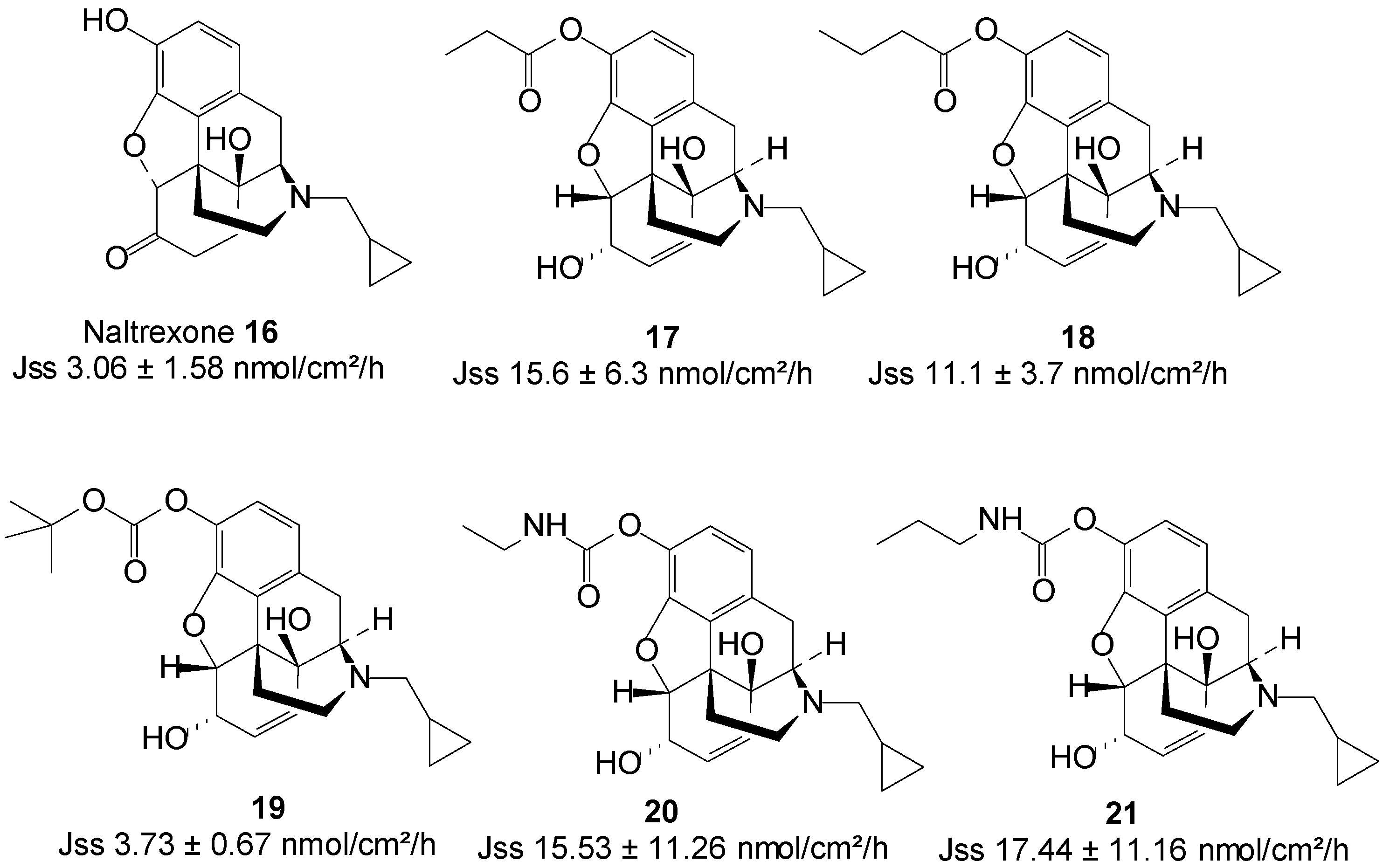
Guided by the findings of previous studies, they further investigated the human skin permeation of 3-O-alkyl carbamate prodrugs of NTX. Two series of prodrugs viz. straight-chain N-monoalkyl and branched-chain N,N-dialkyl carbamates (Figure 6) were synthesized. Here again, all the straight-chain carbamate prodrugs enhanced the skin permeation of NTX while the branched-chain derivatives did not. Interestingly, like in the case of the straight-chain esters, the N-ethyl 20 and N-propyl 21 carbamate prodrugs were the best penetrants, enhancing the permeation of NTX up to 3-fold [63]. The results of these three studies confirm that the nature and type of promoiety are crucial to obtaining maximum skin permeation of a drug using the prodrug strategy.
5.5. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
Non-steroidal anti-inflammatory drugs (NSAIDs) are also known as non-steroidal anti-inflammatory agents/analgesics (NSAIAs), or non-steroidal anti-inflammatory medicines (NSAIMs). This group of medications has analgesic (pain reducing) and antipyretic (fever reducing) effects. In higher doses, they also have anti-inflammatory effects by reducing swelling.
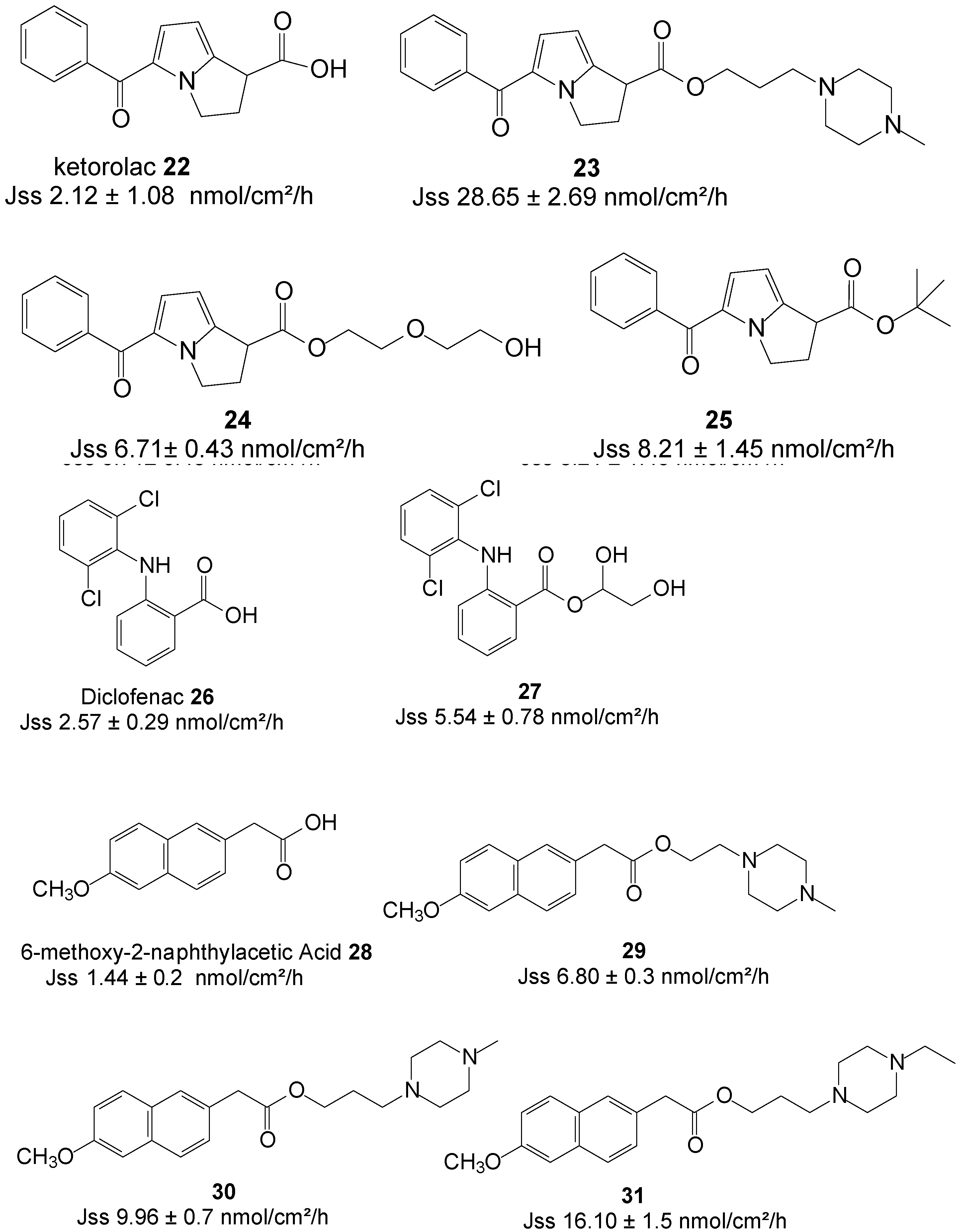
Ketorolac is an NSAID agent with powerful analgesic and low anti-inflammatory activities, and is widely used in the management of both moderate and severe pain [64]. Although the oral bioavailability of ketorolac is reported to be 90%, with very low hepatic first-pass elimination, its biological half-life of 4–6 h requires frequent administration to maintain a therapeutic effect [65]. The long-term use of currently available dosage forms of ketorolac results in gastrointestinal ulceration and acute renal failure [66]. The transdermal delivery route is regarded as a viable alternative for the safe delivery of ketorolac.
Quandil et al. [67] synthesized a series of piperazinylalkyl ester prodrugs of ketorolac 22 and assessed their potential for permeating dorsal rat skin alongside the parent drug in PBS. The study resulted in a significant increase in skin permeation of the prodrugs. The best permeation rate was achieved with ester 23 (Figure 7) with Jss values of 44.60 ± 12.14 nmol/cm2/h and 24.15 ± 3.50 nmol/cm2/h at pH 5 and 7.4, respectively. These values were significantly higher than those ketorolac; 28.65 ± 2.69 and 2.12 ± 1.08 nmol/cm2/h at pH 5 and 7.4, respectively. Skin permeation was thus enhanced in the study.
Figure 7.
Structures of NSAIDs and their best permeant ester prodrugs.

Polyoxyethylene glycol ester prodrugs of ketorolac were also synthesized and their ability to permeate human skin in vitro was also assessed. Gel B with the composition of carbomer (1%), triethanolamine (1%), distilled water (83%) and the ester prodrug 24 (15%) (Figure 7), featuring two EO units, was found the best permeant with a Jss value of 6.71 ± 0.43 nmol/cm2/h, slightly higher than 4.1 nmol/cm2/h of gel A containing ketorolac (15%) instead [68].
More recently, Liu et al. investigated the impact of ester promoieties on the transdermal delivery of ketorolac. Different n-alkyl and aryl ester prodrugs of ketorolac were synthesized and their permeation across nude mouse skin evaluated in a 30% ethanol/pH 7.4 Buffer. Ketorolac tert-butyl ester 25 (Figure 7) exhibited the highest penetration of all compounds and tested with a flux of 8.21 ± 1.45 nmol/cm2/h, which represented an enhancement ratio of 2.5, compared to ketorolac (3.24 ± 0.25 nmol/cm2/h) [69].
The transdermal delivery of diclofenac 26 was also evaluated by using the prodrug strategy. Methanol, ethylene glycol, glycerol and 1,3-propylene glycol esters were synthesized and the In vitro fluxes across human epidermal membrane determined using saturated aqueous solutions (vehicle, PBS pH 7.4). The glycerol ester prodrug 27 was the best permeant of all with a flux of 5.54 ± 0.78 nmol/cm2/h, which was almost 2-fold higher that of diclofenac itself (2.57 ± 0.29 nmol/cm2/h) [70]. Diclofenac displayed higher permeation flux across hairless rat skin than its prodrugs from hydroxy-functionalized polyacrylate patches. This inferiority in fluxes of the prodrugs has been ascribed as probably due to strong interaction between the ester prodrugs and the ester type adhesives [71].
Furthermore, another NSAID which has recently been investigated is nabumetone. Piperazinylalkyl ester prodrugs (Figure 7) of its active metabolite, 6-methoxy-2-naphthylacetic acid 28 were synthesized and evaluated in vitro from saturated PBS solutions for the purpose of percutaneous drug delivery. Rat dorsal skin was used in Franz diffusion cells. The 4-ethylpiperazinyl-1-propyl ester prodrug 31 showed a 10-fold higher flux in permeating the skin compared to the parent 28 (16.10 vs. 1.44 nmol/cm2/h) [72]. Thus, enhancement of percutaneous absorption was achieved in this study by using the prodrug strategy.
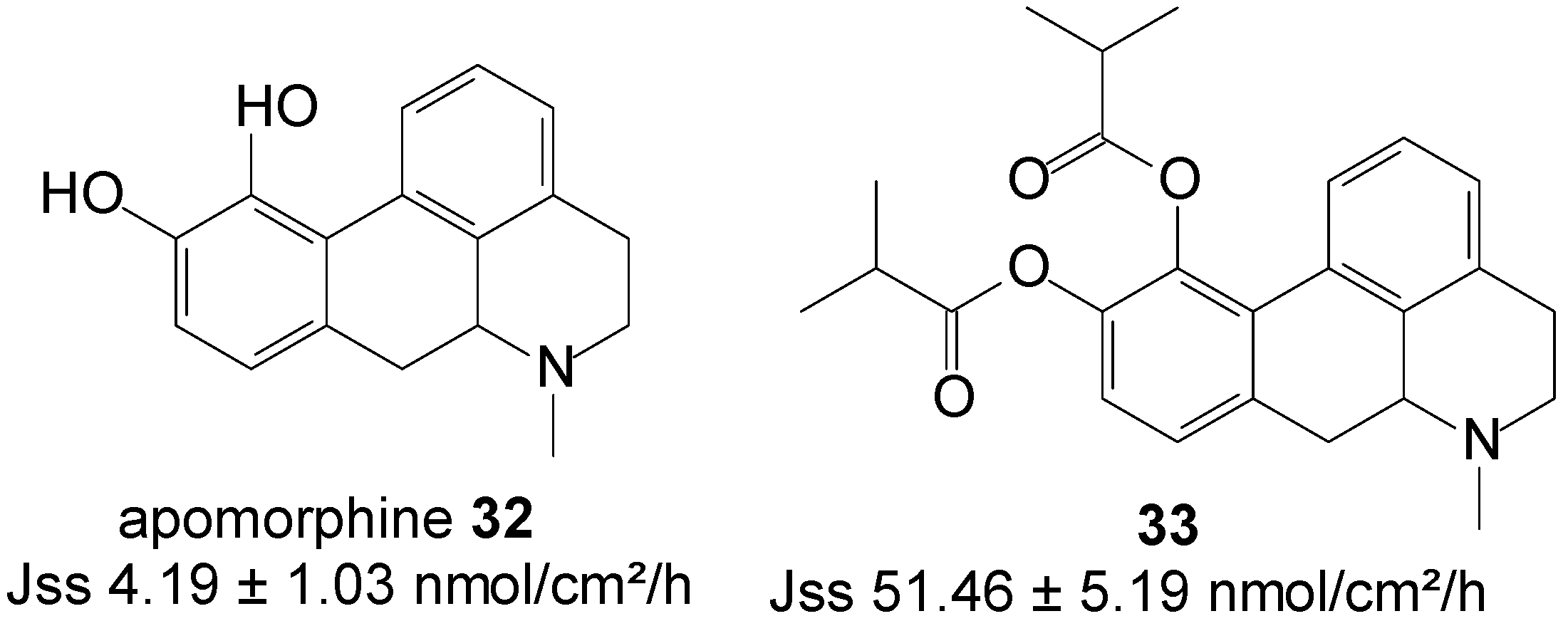
5.6. Apomorphine (Anti-Parkinsonian)
Apomorphine 32 is a classic mixed type of dopamine D1 and D2 receptor agonists, used in the therapy of Parkinson’s disease [73]. Orally administered apomorphine in advanced Parkinson’s disease patients is unsuccessful, due to its high dose requirement as a result of metabolic constraints and the first-pass metabolic effect. High oral doses of apomorphine can furthermore cause gastrointestinal complications, associated with nephrotoxicity [74]. Apomorphine is most commonly administered through repeated subcutaneous infusions or injections, which invariably result in the appearance of subcutaneous nodules. The transdermal delivery of apomorphine could be an ideal alternative route of administration, due to the advantages of eliminating the first-pass metabolism, a reduction in gastrointestinal side effects, and sustained release to resolve its short half-life (32 min).
Diester prodrugs of apomorphine viz. diacetyl apomorphine and diisobutyryl apomorphine were synthesized and their in vitro permeation across nude mouse skin assessed from saturated deionized solutions. Significantly higher skin permeation was achieved during this study with diisobutyryl apomorphine ester 33 (Figure 8). This prodrug displayed a Jss flux of 51.46 ± 5.19 nmol/cm2/h, which exceeded the flux of apomorphine base (4.19 ± 1.03 nmol/cm2/h) by 10-fold [75].
Figure 8.
Apomorphine, its diester prodrug, and their respective skin permeation fluxes.

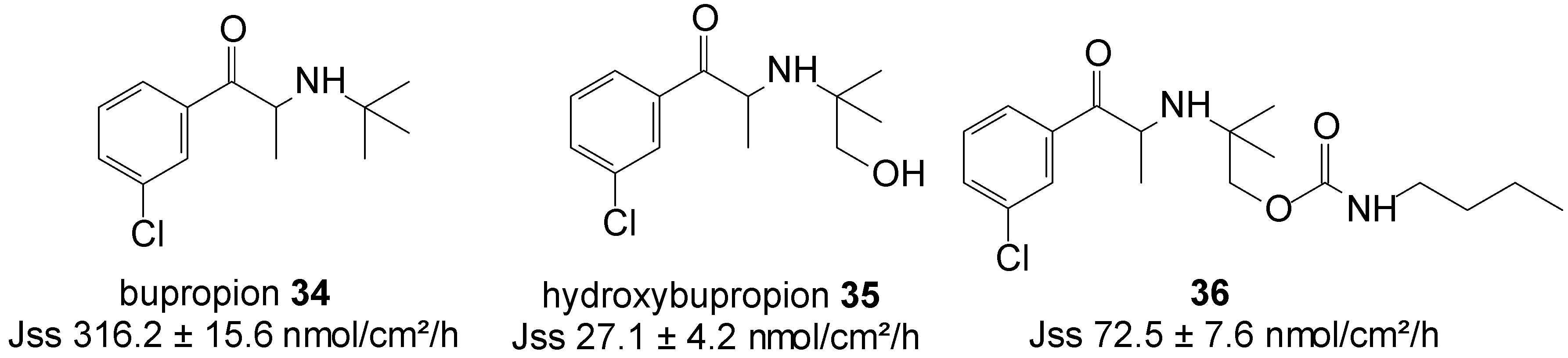
5.7. Bupropion (Anti-Depressant)
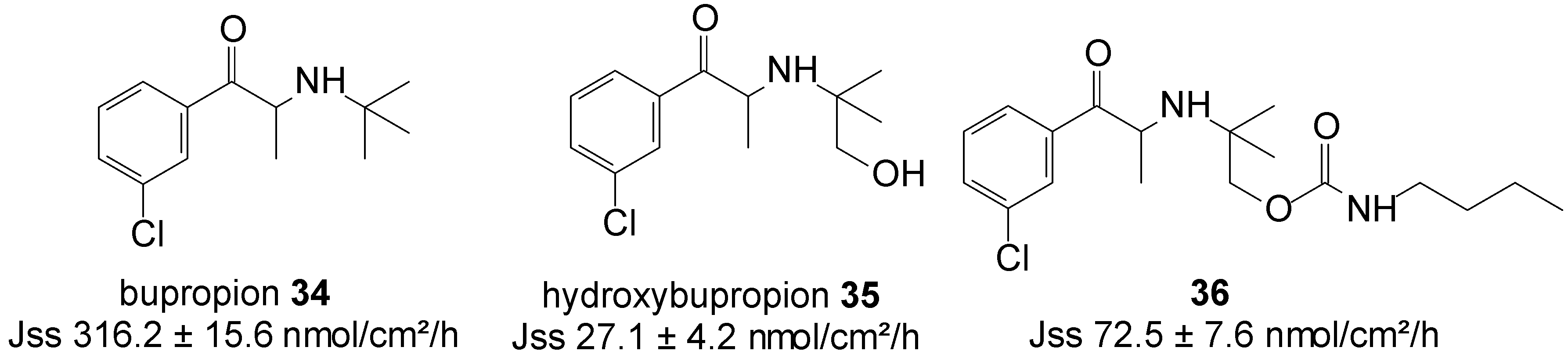
Bupropion (BUP) 34 is an aminoketone that is used as an antidepressant and non-nicotine aid to smoking cessation [76]. In addition to its side effects, such as nausea and vomiting, even cases of seizures, either due to BUP overdoses or unintentional exposure, are reported [77]. To curb the dose related risk of seizures, associated with the high peak concentration of the drug following oral administration, BUP HCl is administered in split doses. Alternatively, the transdermal delivery of BUP would reduce its dose related side effects, increase its bioavailability by reducing its metabolism and the first-pass metabolic effect, and allow for the easier removal of drug input, while maintaining therapeutic concentrations for long periods, with non-invasive zero-order release.
Carbamate prodrug 36 of hydroxybupropion 35 (Figure 9) was synthesized and the in vitro percutaneous absorption through excised human skin assessed, alongside bupropion 34 and 35 from saturated mineral oil. The in vitro, mean steady-state flux of 34 was significantly higher than that of hydroxybupropion 35 (316.2 ± 15.6 vs. 27 ± 4 nmol/cm2/h). Although bupropion traversed human skin at sufficient rates to achieve the required plasma levels, however, it was found chemically unstable and hygroscopic, and unsuitable for transdermal formulation. Contrary, although hydroxybupropion was found stable, its transport across skin was much slower. Alternatively, prodrug 36 was found to be stable, whilst also providing a 2.7-fold increase in the transdermal flux of hydroxybupropion 35 (72.5 ± 7.6 vs. 27 ± 4 nmol/cm2/h) across human skin in vitro. Carbamate 36 was hence regarded as a viable option for the transdermal delivery of hydroxybupropion [78].
Figure 9.
Bupropion, hydroxypropion and the carbamate prodrug of hydroxypropion.
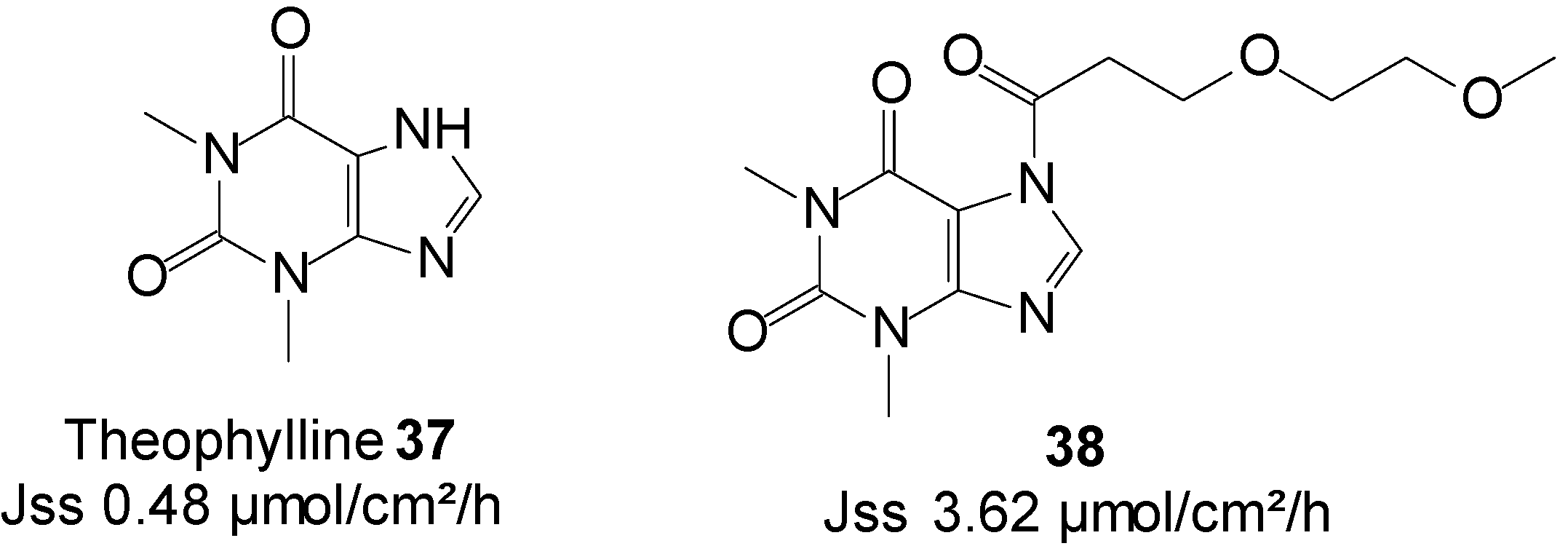
5.8. Theophylline (Cardiovascular)
Theophylline is a methylxanthine drug that is intravenously administered in therapies for respiratory diseases, such as asthma. The use of theophylline is complicated by its interaction with various drugs, mainly cimetidine and phenytoin. Because it has a narrow therapeutic index, it must be monitored through direct measurement of serum theophylline to avoid high levels related toxicity. Seizures can also occur in severe cases of toxicity. The topical delivery of theophylline would as a result appear as a credible route to reduce its risk of high dose related toxicity and other possible side effects, including nausea, diarrhea, increased heart rate, arrhythmias and central nervous system excitation (headaches, insomnia, irritability, dizziness and lightheadedness).
Theophylline was the first amide/imide for which prodrugs were synthesized and were characterized for their ability to enhance the topical delivery of the parent drug from isopropyl myristate (IPM) vehicle [79,80].
More recently, Majumdar et al. [81] investigated prodrugs of theophylline 37 by incorporating ethyleneoxy groups in the promoiety. The study resulted in an enhanced skin permeation rate of the drug. Prodrug 38 (Figure 10), featuring two EO units, achieved the highest delivery of total species through hairless mouse skin from IPM/PBS saturated solutions, which was more than seven times that of the parent, theophylline (3.62 vs. 0.48 µmol/cm2/h).
Figure 10.
Theophylline and its best skin penetrant amide prodrug.

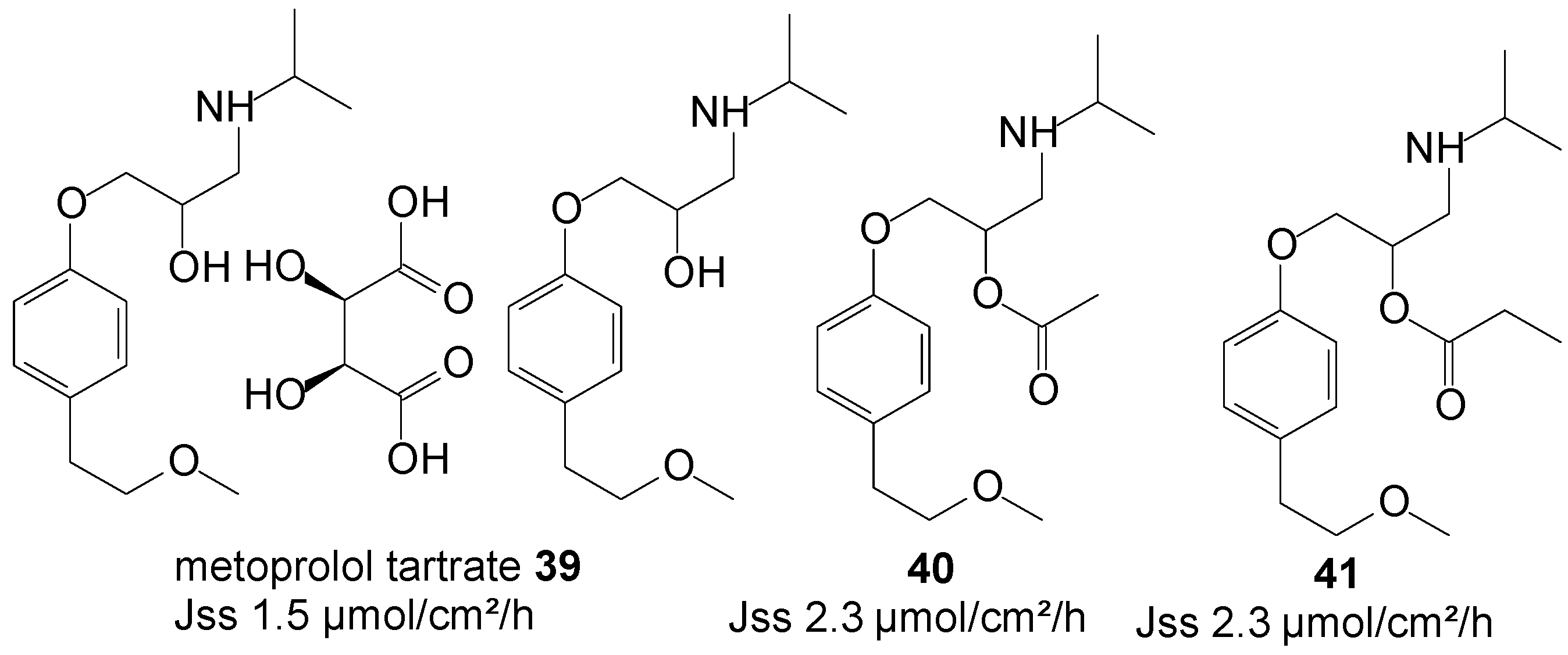
5.9. Metoprolol Tartrate (β-Adrenergic Blocker)
Metoprolol tartrate, a β-selective adrenergic blocking agent, is a well-established drug for the treatment of angina pectoris, myocardial infarction, congestive heart failure and hypertension. Metoprolol reduces blood pressure by reducing cardiac output by slowing the heart rate, and is it a useful first line therapy in the treatment of mild to moderate essential hypertension. Its efficacy is, however, reduced by its extensive first-pass metabolism following oral administration. The transdermal route, by virtue of its capability to avoid the hepatic first-pass metabolic effect, is expected to achieve higher systemic bioavailability.
Figure 11.
Metoprolol and its ester prodrugs.

Anroop et al. [82] conducted a comparative skin permeability study of metoprolol tartrate 39 and its ester prodrugs through passive permeation from 20% ethanol-acetate buffer pH 4 unsaturated solutions. The study resulted in both the acetate 40 and propionate 41 prodrugs (Figure 11) permeating the skin with similar flux, higher than the parent drug (2.3 vs. 1.5 µmol/cm2/h). A marginal skin permeation enhancement was thus achieved using the prodrug approach in this study.
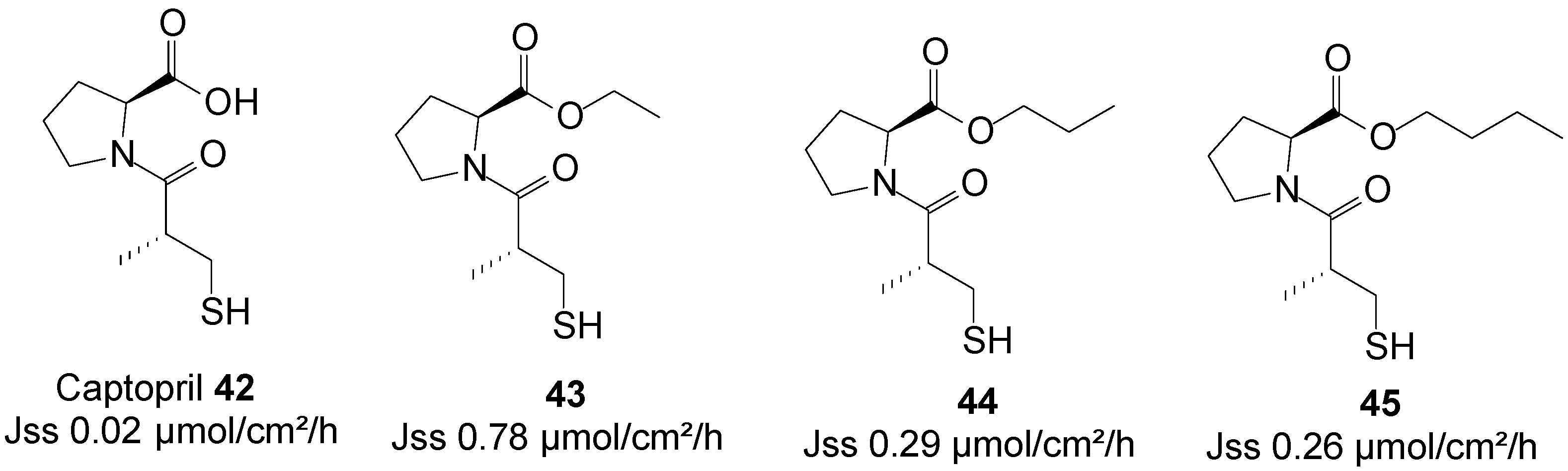
5.10. Captopril (Anti-Hypertensive)
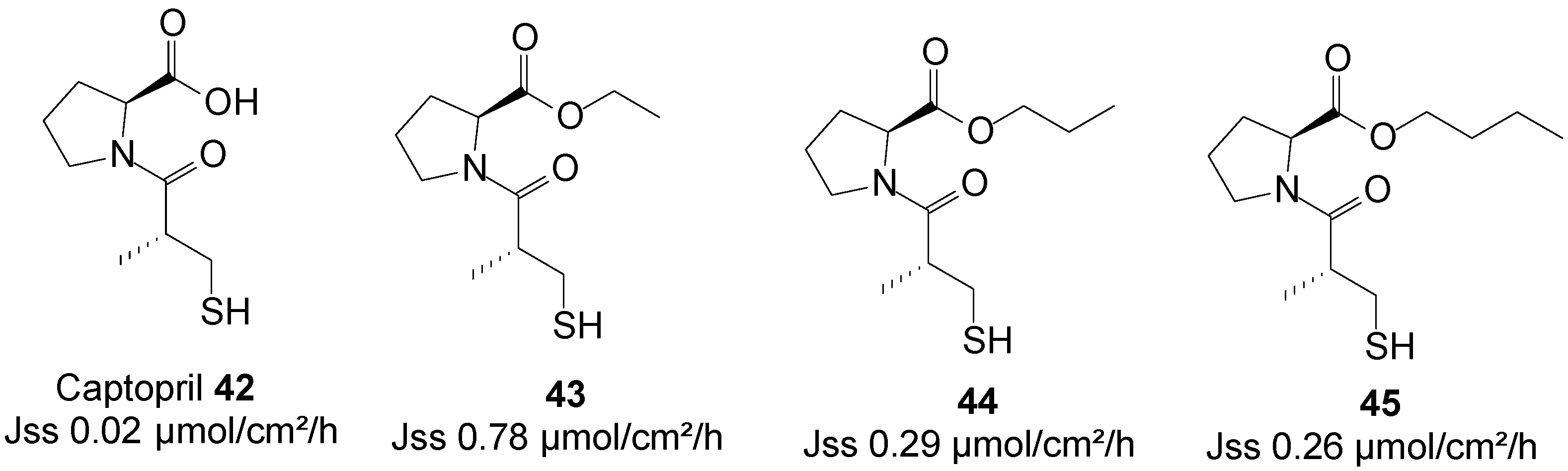
Captopril is an angiotensin-converting enzyme inhibitor, primarily used for the treatment of hypertension. The use of captopril is commonly associated with rash and taste disturbances (metallic, or loss of taste). It also has a relatively poor pharmacokinetic profile, including poor oral bioavailability and a short half-life. Subsequently, it must be administered two or three times per day to reach therapeutic levels, which counteracts patient compliance. Transdermal drug delivery therefore has the potential to act as a viable alternative administration route for this drug by increasing its patient compliance profile.
A series of n-alkyl carboxyl ester prodrugs of captopril 42 were synthesized and were subjected to percutaneous absorption in Franz diffusion cell experiments from saturated PBS pH 7.4 solutions using porcine skin. Captopril and its prodrugs crossed the skin relatively freely. The highest skin permeation fluxes were observed for the ethyl 43, propyl 44 and butyl 45 ester prodrugs (Figure 12). Adoption of the prodrug strategy hence resulted in an increase in skin permeation of the parent captopril [83].
Figure 12.
Captopril and its best n-alkyl ester prodrugs.
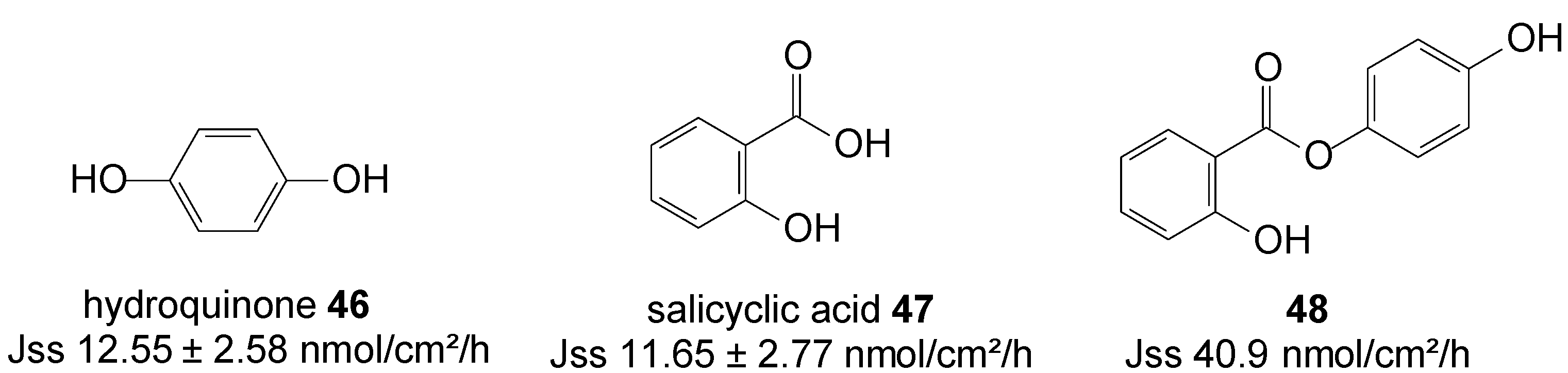
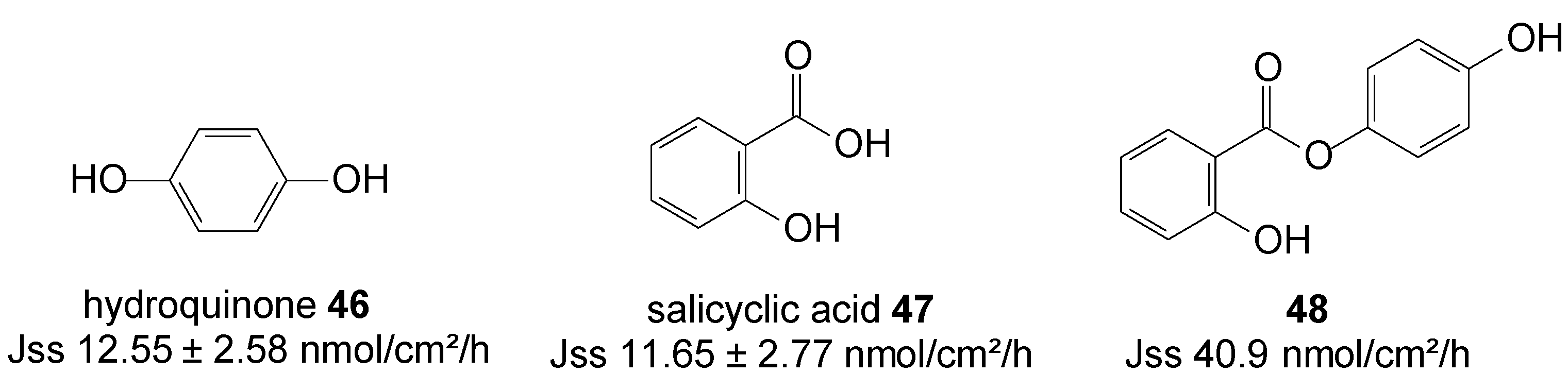
5.11. Hydroquinone (Anti-Melasma)
Hydroquinone 46 and salicylic acid 47 are drugs for treating melasma through the mechanisms of tyrosinase inhibition and chemical peeling, respectively. However, their frequency use is associated with skin irritation, which has limited use of both drugs. Salicylic acid is drug for peeling also known as an ultraviolet B sunscreen while hydroquinone is anti-melasma agent [84]. HQ in combination with peeling agents offers a synergistic therapy for treating melasma [85]. HQ is as a hydrophilic molecule with limited skin permeability and also poses some problems with storage instability and skin toxicity such as contact dermatitis and allergic dermatitis. To improve the efficacy and safety of melasma therapy, intense efforts has been devoted to enhance drug absorption and reduce toxic risk.
In order to promote cutaneous delivery of 46 and 47 for treating melasma, and to take advantage of the synergistic activity of skin lightening and chemical peeling combined in one unique entity, prodrugs of hydroquinone and salicylic acid were synthesized via covalent bonding (Figure 13) and their permeation through pig skin was assessed from 20% PEG400/PBC pH 7.4 saturated solutions. The 4-hydroxyphenyl 2-hydroxybenzoate prodrug or co-drug 48 permeated the skin with significantly higher flux compared to the individual parent drugs [86].
Figure 13.
Structures of hydroquinone, salicylic acid and their best skin permeant ester co-drug 48.
Figure 13.
Structures of hydroquinone, salicylic acid and their best skin permeant ester co-drug 48.
5.12. Haloperidol (Anti-Schizophrenic)
Haloperidol 49 continues to be a useful agent in the treatment of schizophrenia. The monthly intra-muscular administration of 49 in decanoate ester form proves to be valuable in reducing the problems of confused or agitated patients forgetting (or refusing) to take their medication. However, drawbacks associated to the administration of this ester by the intra-muscular route include local reactions and tenderness at the site of injection [87]. Furthermore, parenteral depot preparations require a healthcare professional to administer them, and short of removing the depot site by surgery, it is not possible to terminate treatment once it has commenced. Haloperidol delivered transdermally, and therefore avoiding the GI route, could among others be used to combat nausea and vomiting associated with opiate therapy in palliative care.
Thus, a series of prodrugs including ethyl, propyl, butyl, octyl and decyl O-acyl esters of haloperidol were synthesized and their permeation across guinea pig skin were evaluated from aqueous PBS pH 7.4 saturated solutions. Cetrimide was included in the receptor phase as solubilizing agent but did not significantly alter the barrier properties of the membranes. As the prodrug ester chain length increased, the flux of the prodrugs decreased. The ethyl ester progrug 50 (Figure 14) was the best penetrant among all prodrugs. However, the permeation of the parent drug, 49, across guinea pig skin was found to be greater than all its derivatives. Thus, no percutaneous absorption was enhanced by adopting the prodrug strategy in this study [88].
Figure 14.
Structures of haloperidol and its ethyl ester prodrug.

The physiochemical data, together with the skin permeation fluxes of all parent drugs and their best skin penetrant prodrugs, are summarized in Table 1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Physiochemical data and skin permeation fluxes of all parent drugs and best percutaneous penetrant derivatives.
| Drug/Prodrugs | Therapeutic Category | MW (g/mol) | Sw (mM) PBS pH 7.4 | Log D n-octanol/PBS pH 7.4 | Flux (nmol/cm2h) | Vehicle/Donor Phase Condition | Skin Penetration Enhancement | Ref. |
|---|---|---|---|---|---|---|---|---|
| Zidovudine 1 * | ARVs | 267.2 | 95.4 | −0.7 ± 0.04 | 8.55 ± 5.27 | PBS pH 7.4/saturated | moderate | [44] |
| 2 | 677.7 | 104.8 | −1.1 ± 0.05 | 53.33 ± 46.51 | ||||
| Stavudine 3 * | 224.2 | 380.2 | −1.1 ± 0.04 | 59.15 ± 23.28 | no | [45] | ||
| 4 | 414.4 | nd | −0.7 ± 0.04 | 26.1 ± 22.23 | ||||
| Lamivudine 5 * | 229.3 | 82 | −0.83 ± 0.06 | 4.23 ± 2.98 a | no | [46] | ||
| 6 | 285.32 | 18.0 | 1.9 | 0.046 a | ||||
| 7 | 331.34 | 25.8 | −0.7 ± 0.01 | 2.07± 0.62 a | [47] | |||
| 8 | 375.40 | nd | nd | 0.66± 0.36 a | ||||
| Cytarabine 9 * | Anticancer | 243.2 | 738.8 | −1.9 ± 0.02 | 3.7 | PBS pH7.4/saturated | highly significant | [50] |
| 10 | 341.4 | 14.53 | 0.91 ± 0.05 | 89.0 | ||||
| 11 | 345.3 | 230 | −1.2 ± 0.03 | 8.3 | moderate | [51] | ||
| 12 | 313.3 | 48.46 | −0.26 ± 0.00 | 22.2 | significant | [52] | ||
| Morphine 13 * | Opioid Analgesic | 285.3 | >100 | –1.13 ± 0.28 | 14.61 ± 4.08 | PBC/unsaturated | [56] | |
| 14 | 341.4 | 30.1 | 1.75 ± 0.02 | 27.57 ± 4.57 | ||||
| 15 | 397.5 | 3.97 | nd | 72.45 ± 7.59 | PBC/saturated | significant | ||
| Naltrexone 16 * | Opioid Antagonist | 341 | 0.26 h | nd | 2.5 ± 1.5 | mineral oil/saturated | moderate | [61] |
| 17 | 383 | 2.04 h | 15.6 ± 6.3 | |||||
| 18 | 397 | 4.41 h | 11.1 ± 3.7 | |||||
| 16 * | 341 | 0.24 h | 3.06 ± 1.58 | no | [62] | |||
| 19 | 441 | 5.6 h | 3.73 ± 0.67 | |||||
| 16 * | 341 | 0.24 h | 3.68 ± 2.37 | moderate | [63] | |||
| 20 | 412 | 1.01 h | 15.53 ± 11.26 | |||||
| 21 | 425 | 0.96 h | 17.44 ± 11.16 | moderate | ||||
| Ketorolac 22 * | NSAIDs | 255.27 | 70.87 | –0.83 ± 0.08 | 2.12 ± 1.08 | PBS pH 7.4/saturated | significant | [66] |
| 23 | 395.50 | 35.64 | 2.15 ± 0.09 | 24.15 ± 3.50 | ||||
| Ketorolac 22 * | 255.27 | 19.1 | 1.14 ± 0.01 | 4.1 | gel A | marginal | [67] | |
| 24 | 343.37 | 28.3 | 2.13 ± 0.02 | 6.71 ± 0.43 | gel B | |||
| Ketorolac 22 * | 255.27 | 8205.2 b | nd | 3.24 ± 0.25 | 30% ethanol in PBC/saturated | moderate | [68] | |
| 25 | 311.2 | 172.8 b | nd | 8.21 ± 1.45 | ||||
| Diclofenac 26 * | 282.13 | 0.0041 c | 2.97 e | 2.57 ± 0.29 | PBS pH 7.4/saturated | moderate | [69] | |
| 27 | 340.21 | 0.662 d | 1.57 f | 5.54 ± 0.78 | ||||
| 6-Meo-2-naphthylacetic acid 28 | 216.24 | 63.34 | 0.23 ± 0.02 | 1.44 ± 0.2 | PBS pH 7.4/saturated | moderate | [71] | |
| 29 | 342.4 | 21.58 | 0.23 ± 0.01 | 6.80 ± 0.3 | ||||
| 30 | 356.5 | 20.09 | 0.22 ± 0.02 | 9.96 ± 0.7 | moderate | |||
| 31 | 370.5 | 6.20 | 0.53 ± 0.08 | 16.10 ± 1.5 | significant | |||
| Apomorphine 32 * | Anti-Parkinsonian | 267.3 | nd | 2.0 ± 0.1 g | 4.19 ± 1.03 | Water/saturated | significant | [72] |
| 33 | 407.51 | 1.6 ± 0.03 g | 51.46 ± 5.19 | |||||
| Bupropion 34 * | Antidepressant | 239.75 | 54.2 h | nd | 316.2 ± 15.6 | mineral oil/saturated | no | [77] |
| 35 | 255.74 | 3.9 h | 27.1 ± 4.2 | moderate | ||||
| 36 | 354.88 | 16.4 h | 72.5 ± 7.6 | |||||
| Theophylline 37 * | Cardiovascular agent | 180.17 | 0.3 i | −2.14 j | 0.48 a | IPM/saturated | moderate | [80] |
| 38 | 326 | 2.6 i | −1.52 j | 3.62 a | ||||
| Metoprolol tartrate 39 * | β-Adrenergic blocker | 684.82 | >1000 k | nd | 1.5 ± 0.17 a | 20% ethanol-acetate buffer pH 4/unsaturated | marginal | [81] |
| 40 | 309.41 | 8.3 k | 2.3 ± 0.17 a | |||||
| 41 | 323.43 | 5.9 k | 2.3 ± 0.17 a | |||||
| Captopril 42 * | Anti-hypertensive | 217.29 | 66.7 | 0.02 a | PBS pH 7.4/saturated | highly significant | [82] | |
| 43 | 245.29 | 35.6 | 0.78 a | |||||
| 44 | 259.29 | 18.3 | 0.29 a | significant | ||||
| 45 | 273.29 | 18.0 | 0.26 a | significant | ||||
| Hydroquinone 46 * | Anti-melasma | 110.11 | >50,000 l | 1.63 ± 0.01 | 12.55 ± 2.58 | 20% PEG400/PBC pH 7.4/saturated | moderate | [85] |
| 48 | 230.22 | 2425.4 l | 3.77 ±0.05 | 40.86 ± 9.31 | ||||
| Haloperidol 49 * | anti-schizophrenic | 375.86 | 60.2 m | nd | 2.136 ± 0.316 | PBS/saturated | no | [87] |
| 50 | 417.90 | 10.8 m | 0.664 ± 0.123 |
*: Parent drugs; a: flux expressed in µmol/cm2 h; b: Solubility in 30% ethanol; c: Solubility in 5% PG (propylene glycol) in PBS pH 2.5; d: Solubility in 5% PG in water; e: Partition coefficient (octanol/5% PG PBS pH 2.5); f: Partition coefficient (octanol/5% PG water); g: Partition coefficient (octanol/water); h: Solubility in mineral oil; i: Solubility in IPM (isopropyl myristate), j partition coefficient (IMP/PBS pH 4.0); k: Solubility in pH 4 acetate buffer; Gel A: carbomer (1%), triethanolamine (15%), 22 (15%), distilled water (83%); Gel B: carbomer (1%), triethanolamine (15%), 24 (15%), distilled water (83%); nd (not determined); PBC (phosphate buffer citrated); l: Solubility (nmol/mL) in 20% PEG400/PBS pH 7.4 buffer; m: Solubility (nmol/mL) in PBS.
The majority of derivatives listed in Table 1 demonstrated enhanced skin permeation fluxes in comparison with their respective parent drugs, regardless of the vehicle used. This is a further indicative that the vehicle-type is not a primary factor dictating percutaneous absorption of drugs, as previously reported [89]. The compounds also had molecular weights and partition coefficients in the recommended ranges (Sections 5.2.2. and 5.2.3.), which is in accordance early reports that these two factors are keys to the enhancement of skin permeation of drugs [89].
6. Conclusions
Research studies in recent years support the prodrug strategy as a viable approach which may be utilized to enhance skin permeation of therapeutic agents. However, in the design of a derivative that is intended to enter the systemic circulation through the skin, it is a prerequisite to ensure that the key physicochemical parameters of that compound, such as molecular weight and partition coefficient, are within the recommended ranges. These parameters are dependent of the promoiety linked to the parent scaffold. Therefore, the choice of the promoiety appears crucial in order to obtain optimum delivery percutaneous delivery of drug using the prodrug strategy.
Acknowledgments
The author thanks the North-West University and the National Research Foundation (NRF) for financial support.
Disclaimer
Any opinion, findings and conclusions, or recommendations expressed in this material are those of the author and therefore the NRF does not accept any liability in regard thereto.
Conflicts of Interest
The author has no conflict of interest to declare.
References
- Barry, B.W. Novel mechanisms and devices to enable successful transdermal drug delivery. Eur. J. Pharm. Sci. 2001, 14, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Kalia, Y.N.; Guy, R.H. Transdermal drug delivery: overcoming the skin’s barrier function. Pharm. Sci. Technol. Today 2000, 3, 318–325. [Google Scholar] [CrossRef]
- Chien, Y.W. Advances in transdermal systemic medication. In Transdermal Controlled Systemic Medications; Chien, Y.W., Ed.; Marcel Dekker: New York, NY, USA, 1987; pp. 1–22. [Google Scholar]
- Schiffman, S.S.; Zervakis, J.; Shaio, E.; Heald, A.E. Effect of nucleoside analogs zidovudine, didanosine, stavudine, and lamivudine on the sense of taste. Nutrition 1999, 15, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Mitragotri, S. In situ determination of partition and diffusion coefficients in the lipid bilayers of the stratum corneum. Pharm. Res. 2000, 17, 1026–1029. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.; Poulson, R.T. Absorption of drugs through the skin. In Handbook of Experimental Pharmacology: Concepts in Biomedical Pharmacology; Brodie, B.B., Gillette, J.R., Eds.; Springer Verslag: New York, NY, USA, 1971; Part 1; pp. 103–162. [Google Scholar]
- Williams, A.C. Transdermal and Topical Drug Delivery: From Theory to Clinical Practice; Pharmaceutical Press: London, UK, 2003; pp. 1–242. [Google Scholar]
- Ranade, V.V. Drug delivery systems. 6. Transdermal drug delivery. J. Clin. Pharmacol. 1991, 31, 401–418. [Google Scholar] [PubMed]
- Kadam, A.S.; Ratnaparkhi, M.P.; Chaudhary, S.P. Transdermal drug delivery: An overview. Int. J. Res. Dev. Pharm. Life Sci. 2014, 3, 1042–1053. [Google Scholar]
- Wertz, P.W.; Miethke, M.C.; Long, S.A.; Strauss, J.S.; Downing, D.T. The composition of the ceramides from human stratum corneum and from comedones. J. Investig. Dermatol. 1985, 84, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Lampe, M.A.; Burlingame, A.L.; Whitney, J.; William, M.L.; Brown, B.E.; Roitman, E.; Elias, P.M. Human stratum corneum lipids: Characterization and regional variations. J. Lipid Res. 1983, 24, 120–130. [Google Scholar]
- Schaefer, H.; Hensby, C. Skin permeability and models of percutaneous absorption. In Skin Pharmacology and Toxicology: Recent Advances; Galli, C.L., Hensby, C.N., Marinovich, M., Eds.; Plenum: New York, NY, USA, 1990; pp. 77–83. [Google Scholar]
- Cross, S.E.; Roberts, M.S. Subcutaneous absorption kinetics of interferon and other solutes. J. Pharm. Pharmacol. 1993, 45, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.A.A.; Savin, J.A.; Dahl, M.V. Clinical Dermatology, 2nd ed.; Blackwell Science: London, UK, 1995. [Google Scholar]
- Montaga, W. The skin. Sci. Am. 1965, 212, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Guy, R.H.; Hadgraft, J. Structure-activity correlations in percutaneous absorption. In Percutaneous Absorption: Mechanisms-Methodology-Drug Delivery; Bronaugh, R.L., Maibach, H.I., Eds.; Marcel Dekker: New York, NY, USA, 1989; pp. 95–109. [Google Scholar]
- Sloan, K.B.; Koch, S.A.M.; Siver, K.G. Mannich base derivatives of theophylline and 5-fluorouracil: Synthesis, properties and topical delivery characteristics. Int. J. Pharm. 1984, 21, 251–264. [Google Scholar] [CrossRef]
- Sloan, K.B. Prodrugs for dermal delivery. Adv. Drug Deliv. Rev. 1989, 3, 67–10. [Google Scholar] [CrossRef]
- Morrow, D.I.J.; McCarron, P.A.; Woolfson, A.D.; Donnelly, R.F. Innovative strategies for enhancing topical and transdermal drug delivery. Open Drug Deliv. J. 2007, 1, 36–59. [Google Scholar] [CrossRef]
- Potts, R.O.; Buras, E.M.; Chrisman, D.A. Changes with age in moisture content of human skin. J. Investig. Dermatol. 1984, 82, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Weigand, D.A.; Haygood, C.; Gaylor, J.R. Cell layers and density of Negro and Caucasian stratum corneum. J. Investig. Dermatol. 1974, 62, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Reinertson, R.P.; Wheatley, V.R. Studies on the chemical composition of human epidermal lipids. J. Investig. Dermatol. 1959, 32, 49–59. [Google Scholar] [PubMed]
- Sun, Y. Skin absorption enhancement by physical means: Heat, ultrasound and electricity. In Transdermal and Topical Drug Delivery Systems; Gosh, T.K., Pfister, W.R., Yum, S.I., Eds.; Interpharm Press: Buffalo Grove, IL, USA, 1997; pp. 327–355. [Google Scholar]
- Jackson, E.W. Toxicological aspects of percutaneous absorption. In Skin Permeation, Fundamentals and Application; Zats, J.L., Ed.; Allured Publishing Corporation: Wheaton, IL, USA, 1993; pp. 177–192. [Google Scholar]
- Li, X.; Chan, W.K. Transport, metabolism and elimination mechanisms of anti-HIV agents. Adv. Drug Deliv. Rev. 1999, 39, 81–103. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Sathyan, G.; Hwang, S.S. Clinical assessment of transdermal drug delivery systems. In Transdermal and Topical Drug Delivery Systems; Gosh, T.K., Pfister, W.R., Yum, S.I., Eds.; Interpharm Press: Buffalo Grove, IL, USA, 1997; pp. 215–248. [Google Scholar]
- Tojo, K. The prediction of transdermal permeation: Mathematical models. In Transdermal and Topical Drug Delivery Systems; Gosh, T.K., Pfister, W.R., Yum, S.I., Eds.; Interpharm Press: Buffalo Grove, IL, USA, 1997; pp. 113–138. [Google Scholar]
- Flynn, G.L. Mechanism of percutaneous absorption from physicochemical evidence. In Percutaneous Absorption: Mechanism-Methodology-Drug Delivery, 2nd ed.; Bronaugh, R.L., Maibach, H.I., Eds.; Marcel Dekker: New York, NY, USA, 1989; pp. 27–51. [Google Scholar]
- Higuchi, T. Physical chemical analysis of percutaneous absorption process from creams and ointments. J. Soc. Cosmet. Chem. 1960, 11, 85–97. [Google Scholar]
- Abdou, H.M. Dissolution, Bioavailability and Bioequivalence; Mack Publishing Company: Easton, PA, USA, 1989; pp. 52–73. [Google Scholar]
- Solomons, T.W. Organic Chemistry, 6th ed.; Wiley, J. & Sons Inc.: New York, NY, USA, 1996; pp. 452–453. [Google Scholar]
- Hadgraft, J.; Wolff, M. Physicochemical and pharmacokinetic parameters affecting percutaneous absorption. In Dermal and Transdermal Drug Delivery: New Insights and Perspectives, Proceedings of the 2nd International Symposium of the International Association for Pharmaceutical Technology, Frankfurt, Germany, 11–13 November 1991; Gurny, R., Teuber, A., Eds.; Wissenschaftliche Verlagsgesellschaft: Stuttgart, Germany, 1993. [Google Scholar]
- Tenjarla, S.N.; Puranajoti, P.; Kasina, R.; Mandal, T. Terbutaline transdermal delivery: Preformulation studies and limitations of in vitro predictive parameters. J. Pharm. Pharmacol. 1996, 48, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Idson, B. Percutaneous absorption. J. Pharm. Sci. 1975, 64, 901–924. [Google Scholar] [CrossRef]
- Billich, A.; Vyplel, H.; Grassberger, M.; Schmook, F.P.; Steck, A.; Stuetz, A. Novel cyclosporin derivatives featuring enhanced skin penetration despite increased molecular weight. Bioorg. Med. Chem. 2005, 13, 3157–3167. [Google Scholar] [CrossRef] [PubMed]
- Barry, B.W. Dermatological formulations. In Percutaneous Absorption: Mechanisms-Methodology-Drug Delivery; Bronaugh, R.L., Maibach, H.I., Eds.; Marcel Dekker: New York, NY, USA, 1983; pp. 160–172. [Google Scholar]
- Pugh, W.J.; Roberts, M.S.; Hadgraft, J. Epidermal permeability: Penetrant structure relationships. 3. The effect of hydrogen bonding interactions and molecular size on diffusion through the stratum corneum. Int. J. Pharm. 1996, 138, 149–165. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Hadgraft, J.; Cordes, G.; Wolff, M. Prediction of the transdermal delivery of β-blockers. In Die Haut als Transportorgan Für Arzneistoffe; Rietbrock, N., Ed.; Steinkopff Verslag: Darmstadt, Germany, 1990; pp. 133–143. [Google Scholar]
- Stott, R.W.; Williams, A.C.; Barry, B.W. Transdermal delivery from eutectic systems: Enhanced permeation of a model drug, ibuprofen. J. Control. Release 1998, 50, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.B. Drugs and the pharmaceutical sciences. In Prodrugs: Topical and Ocular Drug Delivery; Sloan, K.B., Ed.; Marcel Dekker Inc.: New York, NY, USA, 1992; pp. 221–297. [Google Scholar]
- Waranis, R.P.; Sloan, K.B. The effect of vehicle and prodrug properties and their interactions on the delivery of 6-mercaptopurine through skin: Bisacyloxymethyl-6-mercaptopurine prodrugs. J. Pharm. Sci. 1987, 76, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Pasut, G. PEGylation: Successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Global Report: UNAIDS Report on the Global AIDS epidemic. 2013. Available online: http://www.unaids.org/en/resources/campaigns/globalreport2013/globalreport/ (accessed on 20 October 2014).
- N’Da, D.D.; Breytenbach, J.C. Synthesis of methoxypoly(ethylene glycol) carbonate prodrugs of zidovudine and penetration through human skin in vitro. J. Pharm. Pharmacol. 2009a, 61, 721–731. [Google Scholar] [CrossRef]
- N’Da, D.D.; Breytenbach, J.C.; Legoabe, L.J.; Breytenbach, W.J. Synthesis and in vitro transdermal penetration of methoxypoly(ethylene glycol) carbonate derivatives of stavudine. Med. Chem. 2009, 5, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Gerber, M.; Breytenbach, J.C.; du Plessis, J. Transdermal penetration of zalcitabine, lamivudine and synthesised N-acyl lamivudine esters. Int. J. Pharm. 2008, 351, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Van Heerden, J.; Breytenbach, J.C.; N’Da, D.D.; du Plessis, J.; Breytenbach, W.J.; du Preez, J.L. Synthesis and in vitro transdermal penetration of methoxypoly(ethylene glycol) carbonate and carbamate derivatives of lamivudine (3TC). Med. Chem. 2010, 6, 91–99. [Google Scholar]
- Rustum, Y.M.; Raymakers, R.A. 1-β-arabinofuranosylcytosine in therapy of leukemia: Preclinical and clinical overview. Pharmacol. Ther. 1992, 56, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Hamada, A.; Kawaguchi, T.; Nakano, M. Clinical pharmacokinetics of cytarabine formulations. Clin. Pharmacokinet. 2002, 41, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Legoabe, L.J.; Breytenbach, J.C.; N’Da, D.D.; Breytenbach, W.J. In-vitro transdermal penetration of cytarabine and its N4-alkylamide derivatives. J. Pharm. Pharmacol. 2010, 62, 756–761. [Google Scholar] [PubMed]
- Legoabe, L.J.; N’Da, D.D.; Breytenbach, J.C.; du Preez, J.L.; du Plessis, J. Synthesis and transdermal permeation of novel N4-methoxypoly(ethylene glycol) carbamates of cytarabine. Drug Dev. Ind. Pharm. 2010, 36, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Legoabe, L.J.; Breytenbach, J.C.; N’Da, D.D.; Breytenbach, W.J. Transdermal absorption of cytarabine and its 5’-O- alkyl ester derivatives. Med. Chem. 2010, 6, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Lugo, R.A.; Kern, S.E. Clinical pharmacokinetics of morphine. J. Pain Palliat. Care Pharmacother. 2002, 16, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Grond, S.; Radbruch, L.; Lehmann, K.A. Clinical pharmacokinetics of transdermal opioids. Clin. Pharmacokinet. 2000, 38, 59–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Sung, K.C.; Huang, J.F.; Yeh, C.H.; Fang, J.Y. Ester prodrugs of morphine improve transdermal drug delivery: A mechanistic study. J. Pharm. Pharmacol. 2007, 59, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Terenius, L. Rational treatment of addiction. Curr. Opin. Chem. Biol. 1998, 2, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, J.L.; Sullivan, M.A.; Church, S.H.; Seracini, A.; Collins, E.; Kleber, H.D.; Nunes, E.V. Behavioral naltrexone therapy: an integrated treatment for opiate dependence. J. Subst. Abuse Treat. 2002, 23, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Kranzler, H.R.; Lowe, V.M.; Kirk, J.V. Naltrexone vs. nefazodone for treatment of alcohol dependence: A placebo controlled trial. Neuropyschopharmacology 2000, 22, 493–503. [Google Scholar] [CrossRef]
- Wall, M.; Brine, D.R.; Perez-Reyes, M. Metabolism and disposition of naltrexone in man after oral and intravenous administration. Drug Metab. Dispos. 1981, 9, 369–375. [Google Scholar]
- Stinchcomb, A.I.; Swaan, P.W.; Ekabo, O.; Harris, K.K.; Browe, J.; dana c. Hammell, D.C.; Cooperman, T.A.; Pearsall, M. Straight-chain naltrexone ester prodrugs: Diffusion and concurrent esterase biotransformation in human skin. J. Pharm. Sci. 2002, 91, 2571–2578. [Google Scholar] [CrossRef] [PubMed]
- Vaddi, H.K.; Hamad, M.O.; Chen, J.; Banks, S.L.; Crooks, P.A.; Stinchcomb, A.L. Human skin permeation of branched-chain 3-O-alkyl ester and carbonate prodrugs of naltrexone. Pharm. Res. 2005, 22, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Vaddi, H.K.; Banks, S.L.; Chen, J.; Hammell, D.C.; Crooks, P.A.; Stinchcomb, A.L. Human skin permeation of 3-O-alkyl carbamate prodrugs of naltrexone. J. Pharm. Sci. 2009, 98, 2611–2625. [Google Scholar] [CrossRef] [PubMed]
- Kokki, H. Nonsteroidal anti-inflammatory drugs for postoperative pain: A focus on children. Paediatr. Drugs 2003, 5, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Buckley, M.M.; Brogden, R.N. Ketorolac: A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs 1990, 39, 86–109. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, D.I. Minimizing the adverse effects of ketorolac. Drug Saf. 2000, 22, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Qandil, A.; Al-Nabulsi, S.; Al-Taani, B.; Tashtoush, B. Synthesis of piperazinylalkyl ester prodrugs of ketorolac and their in vitro evaluation for transdermal delivery. Drug Dev. Ind. Pharm. 2008, 34, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Puglia, C.; Filosa, R.; Peduto, A.; de Caprariis, P.; Rizza, L.; Bonina, F.; Blasi, P. Evaluation of alternative strategies to optimize ketorolac transdermal delivery. AAPS PharmSciTech 2006, 7, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.S.; Hsieh, P.W.; Aljuffali, I.A.; Lin, Y.K.; Chang, S.H.; Wang, J.J.; Fang, J.Y. The impact of ester promoieties on the transdermal delivery of ketorolac. J. Pharm. Sci. 2014, 103, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Lobo, S.; Li, H.; Farhan, N.; Yan, G. Evaluation of diclofenac prodrugs for enhancing transdermal delivery. Drug Dev. Ind. Pharm. 2014, 40, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Lobo, S.; Li, H. Can diclofenac ester prodrug promote direct penetration across the skin? J. Chem. Pharm. Res. 2014, 6, 2701–2713. [Google Scholar]
- Pawar, V.; Thosani, R.; Kanhed, A.; Giridhar, R.; Yadav, M.R. Potential of piperazinylalkylester prodrugs of 6-methoxy-2-Naphthylacetic Acid (6-MNA) for percutaneous drug delivery. AAPS PharmSciTech 2014, 2014, 1–10. [Google Scholar]
- Subramony, J.A. Apomorphine in dopaminergic therapy. Mol. Pharm. 2006, 3, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Danhof, M.; van der Geest, R.; van Laar, T.; Boddé, H.E. An integrated pharmacokinetic-pharmacodynamic approach to optimization of rapomorphine delivery in Parkinson’s disease. Adv. Drug Deliv. Rev. 1998, 33, 203–263. [Google Scholar] [CrossRef]
- Liu, K.S.; Sung, K.C.; Al-Suwayeh, S.A.; Ku, M.C.; Chu, C.C.; Wang, J.J.; Fang, J.Y. Enhancement of transdermal apomorphine delivery with a diester prodrug strategy. Eur. J. Pharm. Biopharm. 2011, 78, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.; Berkel, H.J.; Parish, R.C.; Simar, M.R.; Syed, A.; Bocchini, J.A., Jr.; Wilson, J.T.; Manno, J.E. Single-dose pharmacokinetics of bupropion in adolescents: Effects of smoking status and gender. J. Clin. Pharmacol. 2001, 41, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, G.; Velez, L.I.; Keyes, D.C. Intentional bupropion overdoses. J. Emerg. Med. 2004, 27, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Kiptoo, P.K.; Paudel, K.S.; Hammell, D.C.; Pinninti, R.R.; Chen, J.; Crooks, P.A.; Stinchcomb, A.L. Transdermal delivery of bupropion and its active metabolite, hydroxybupropion: A prodrug strategy as an alternative approach. J. Pharm. Sci. 2009, 98, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.B.; Bodor, N. Hydroxymethyl and acyloxymethyl prodrugs of theophylline: Enhanced delivery of polar drugs through skin. Int. J. Pharm. 1982, 12, 299–313. [Google Scholar] [CrossRef]
- Kerr, D.; Roberts, W.; Tebbett, I.; Sloan, K.B. 7-Alkylcarbonyloxymethyl prodrugs of theophylline: Topical delivery of theophylline. Int. J. Pharm. 1998, 167, 37–48. [Google Scholar] [CrossRef]
- Majumdar, S.; Mueller-Spaeth, M.; Sloan, K.B. Prodrugs of theophylline incorporating ethyleneoxy groups in the promoiety: Synthesis, characterization, and transdermal delivery. AAPS PharmSciTech 2012, 13, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Anroop, B.; Ghosh, B.; Parcha, V.; Khanam, J. Comparative skin permeability of metoprolol tartrate and its ester prodrugs by passive permeation and iontophoresis. Asian J. Pharm. Sci. 2008, 3, 47–57. [Google Scholar]
- Moss, G.P.; Gullick, D.R.; Cox, P.A.; Alexander, C.; Ingram, M.J.; Smart, J.D.; Pugh, W.J. Design, synthesis and characterization of captopril prodrugs for enhanced percutaneous absorption. J. Pharm. Pharmacol. 2006, 58, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.D.; Sung, J.H.; Lee, D.W.; Kim, J.S.; Jeon, E.M.; Kim, D.D.; Kim, D.W.; Kim, J.O.; Piao, M.G.; Li, D.X.; et al. Evaluation of physicochemical properties, skin permeation and accumulation profiles of salicylic acid amide prodrugs as sunscreen agent. Int. J. Pharm. 2011, 419, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Gladstone, H.B.; Nguyen, S.L.; Williams, R.; Ottomeyer, T.; Wortzman, M.; Jeffers, M.; Moy, R.L. Efficacy of hydroquinone cream (USP 4%) used alone or in combination with salicylic acid peels in improving photodamage on the neck and upper chest. Dermatol. Surg. 2000, 26, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-W.; Aljuffali, I.A.; Fang, C.-L.; Chang, S.-H.; Fang, J.-Y. Hydroquinone-salicylic acid conjugates as novel anti-melasma actives show superior skin targeting compared to the parent drugs. J. Dermatol. Sci. 2014, 76, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Hamann, G.L.; Egan, T.M.; Wells, B.G.; Grimmig, J.E. Injection site reactions after intramuscular administration of haloperidol decanoate 100 mg/mL. J. Clin. Psychiatry 1990, 51, 502–504. [Google Scholar] [PubMed]
- Morris, A.P.; Brain, K.R.; Heard, C.M. Skin permeation and ex vivo skin metabolism of O-acyl haloperidol ester prodrugs. Int. J. Pharm. 2009, 367, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Juntunen, J.; Majumdar, S.; Sloan, K.B. The effect of water solubility of solutes on their flux through human skin in vitro: A prodrug database integrated into the extended Flynn database. Int. J. Pharm. 2008, 351, 92–103. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
N'Da, D.D. Prodrug Strategies for Enhancing the Percutaneous Absorption of Drugs. Molecules 2014, 19, 20780-20807. https://doi.org/10.3390/molecules191220780
AMA Style
N'Da DD. Prodrug Strategies for Enhancing the Percutaneous Absorption of Drugs. Molecules. 2014; 19(12):20780-20807. https://doi.org/10.3390/molecules191220780
Chicago/Turabian StyleN'Da, David D. 2014. "Prodrug Strategies for Enhancing the Percutaneous Absorption of Drugs" Molecules 19, no. 12: 20780-20807. https://doi.org/10.3390/molecules191220780