Photocatalysis for Renewable Energy Production Using PhotoFuelCells

Abstract
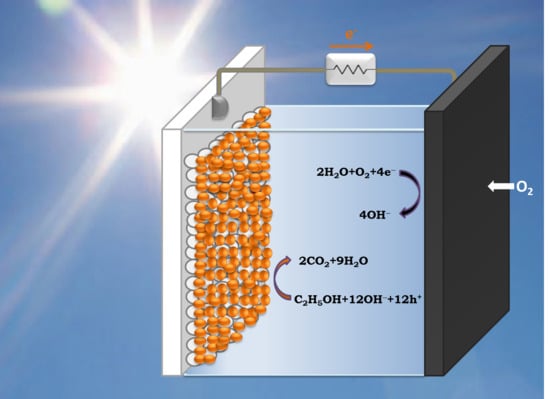
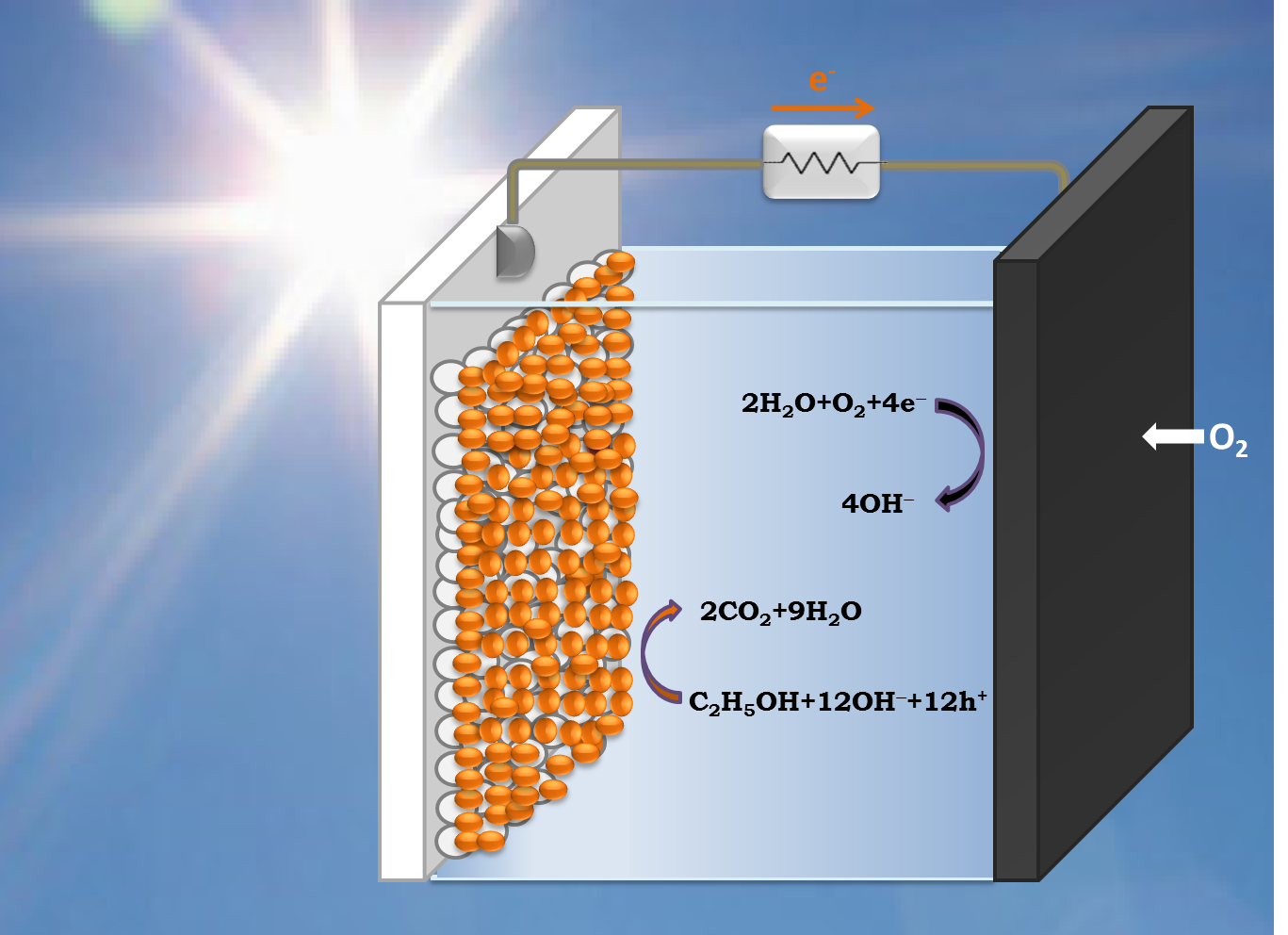
:
1. Introduction
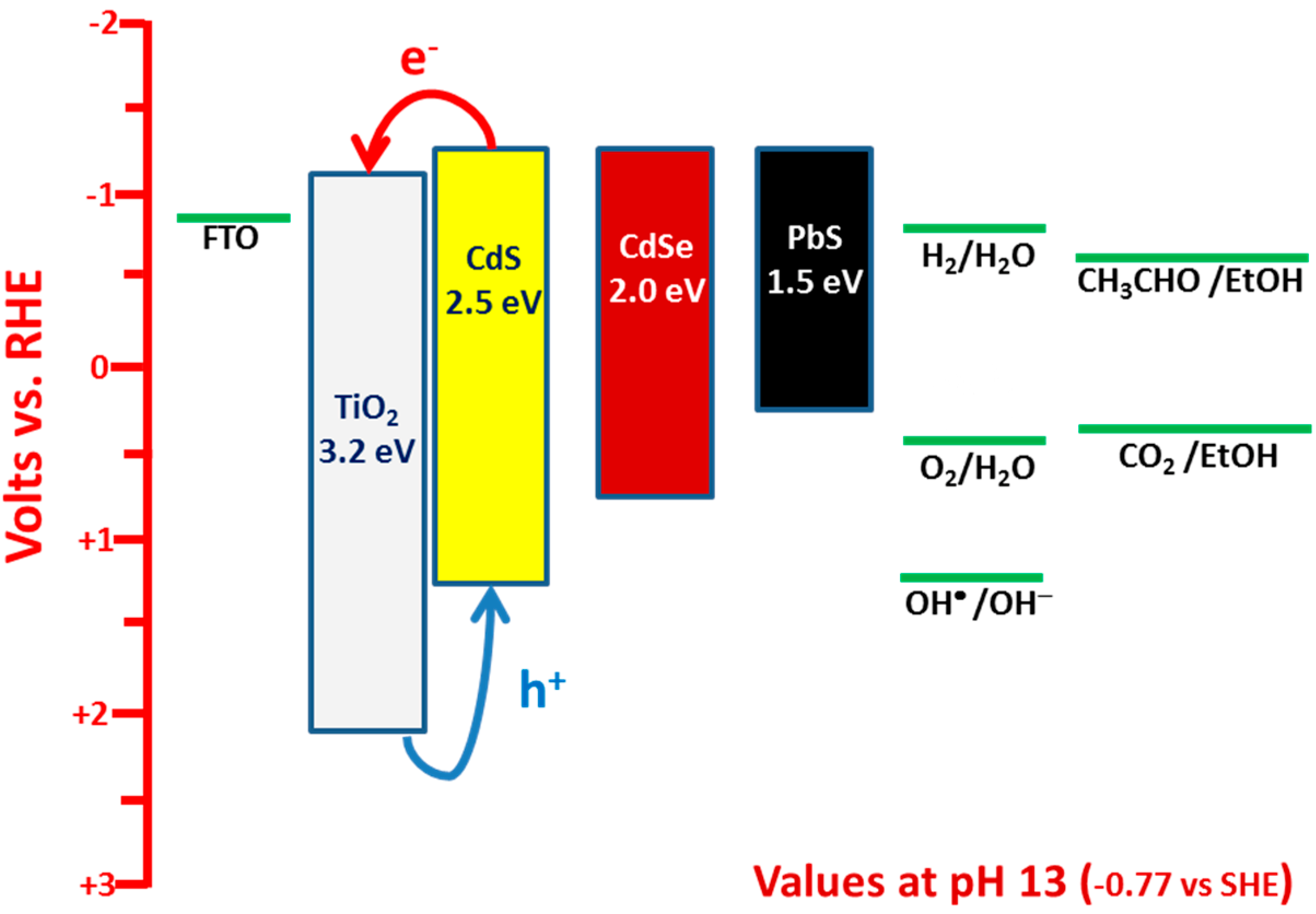
2. Model of PFC Operation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photoanode | |
|---|---|
| |
| |
| Cathode | |
| Inert environment (no O2 present) | Aerated electrolyte or cathode exposed to ambient air |
|
|
| Overall Cell Reactions (combination of anode and cathode reactions) | |
| |
| Name | Chemical Composition | Number of Electrons | ΔG0(kJ·mol−1) | E0 (V) |
|---|---|---|---|---|
| Methanol | CH3OH | 6 | −702 | 1.21 |
| Ethanol | C2H5OH | 12 | −1325 | 1.14 |
| n-Propanol | C3H7OH | 18 | −1965 | 1.13 |
| n-Butanol | C4H9OH | 24 | −2595 | 1.12 |
| n-Pentanol | C5H11OH | 30 | −3249 | 1.12 |
| Glycerol | C3H8O3 | 14 | −1656 | 1.22 |
| Sorbitol | C6H14O6 | 26 | −3084 | 1.23 |
| Acetic acid | CH3COOH | 8 | −873 | 1.13 |
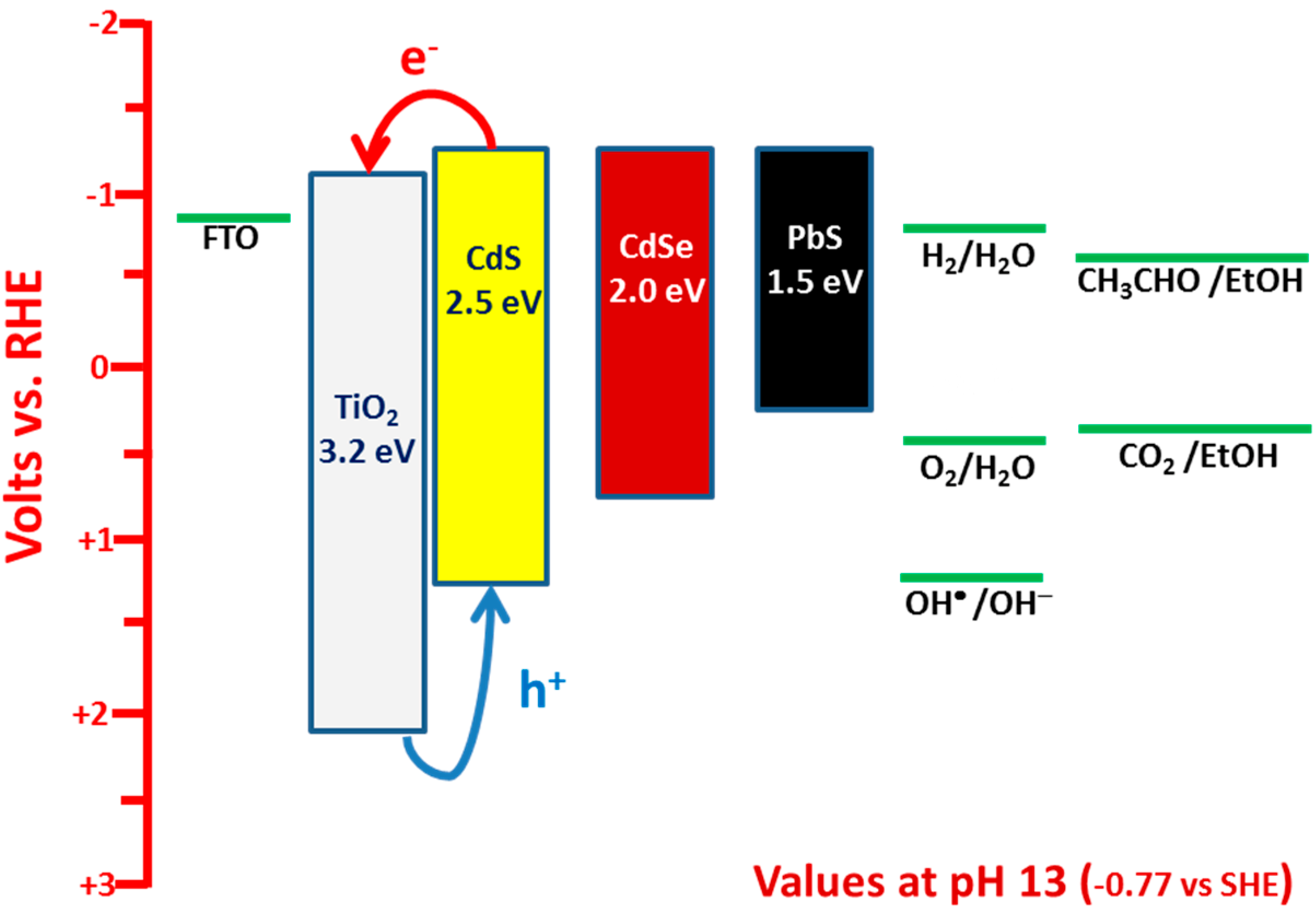
3. Results and Discussion
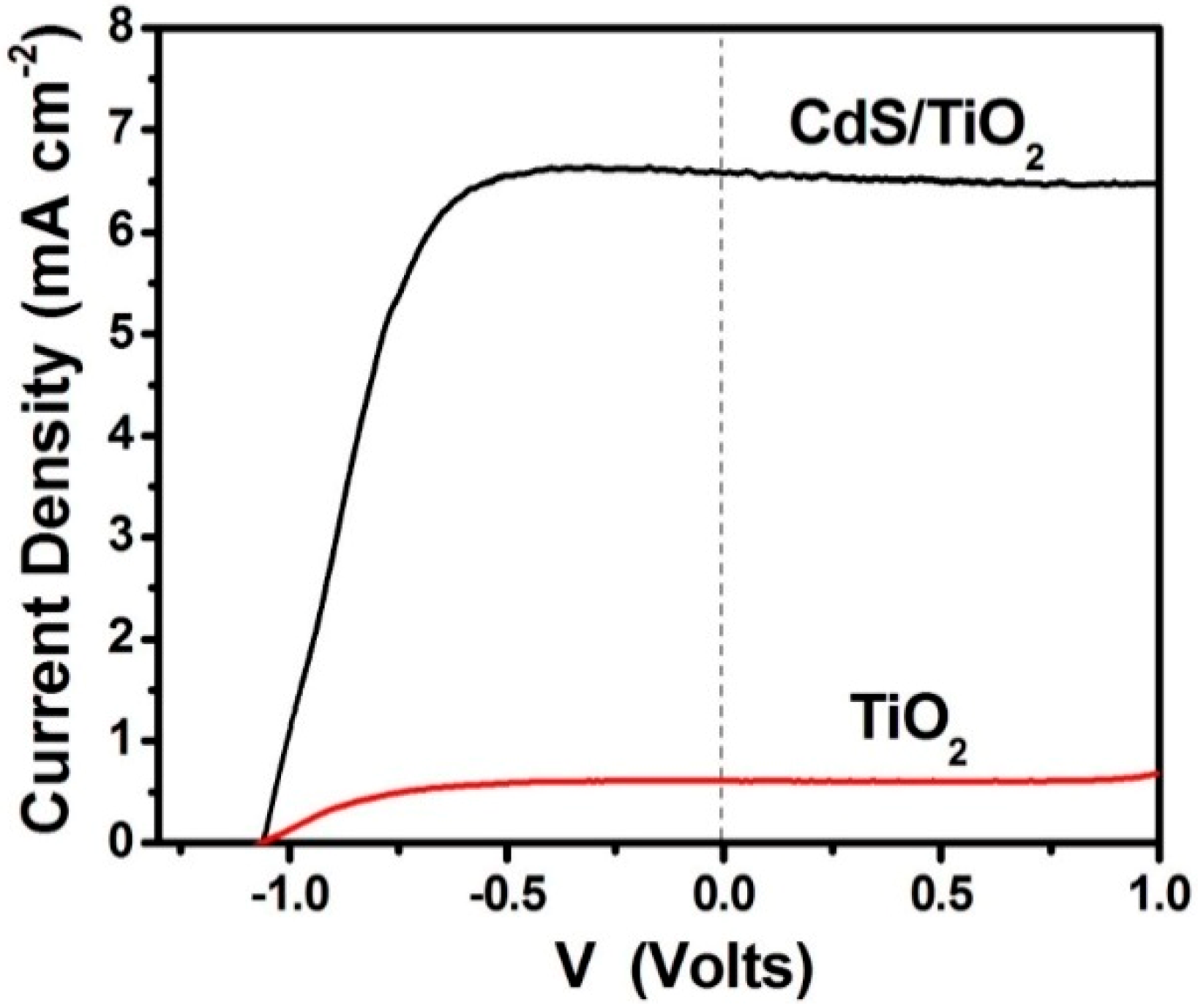
3.1. Study of PFCs Operating in the Presence of Oxygen and Producing Electricity

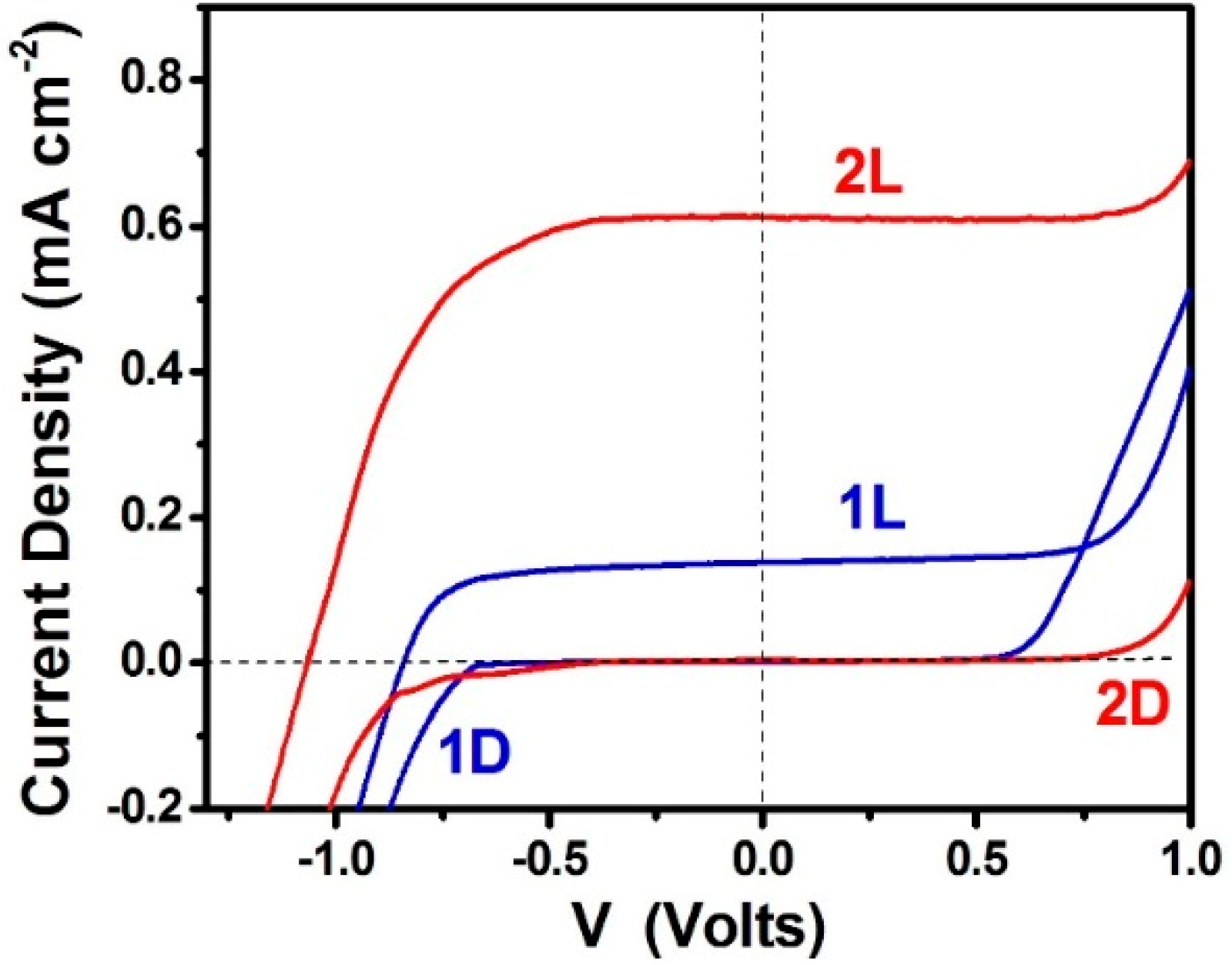
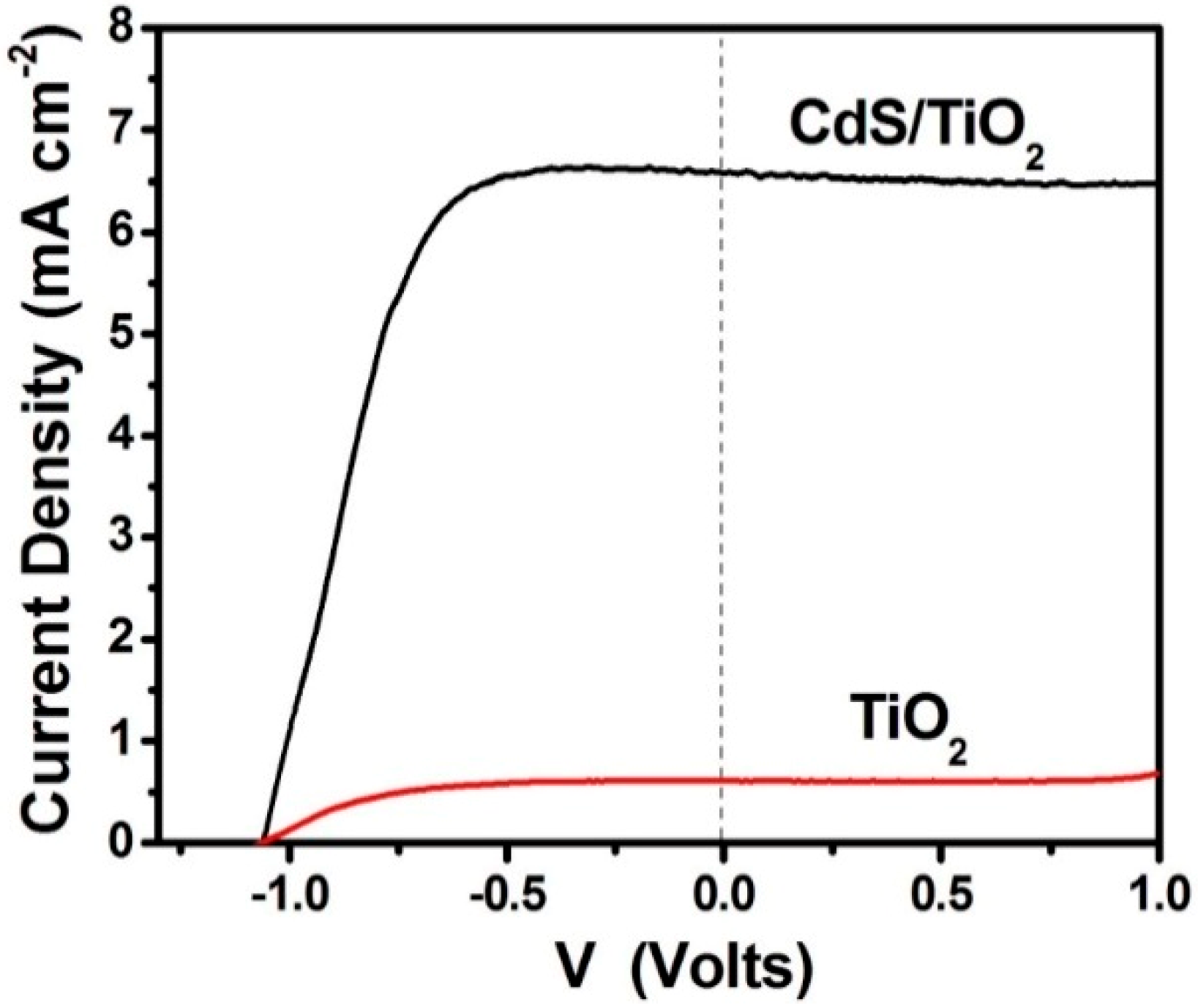
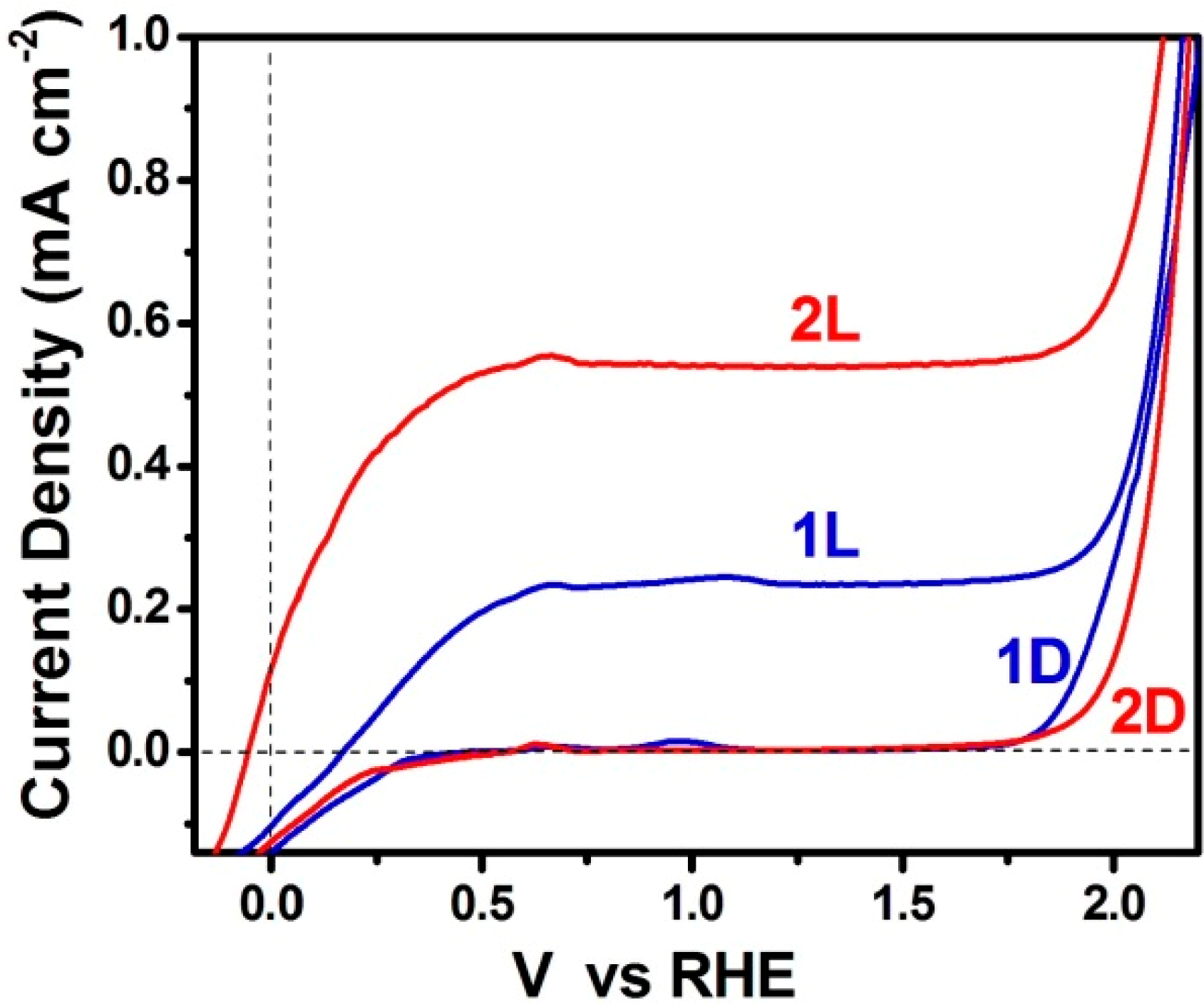
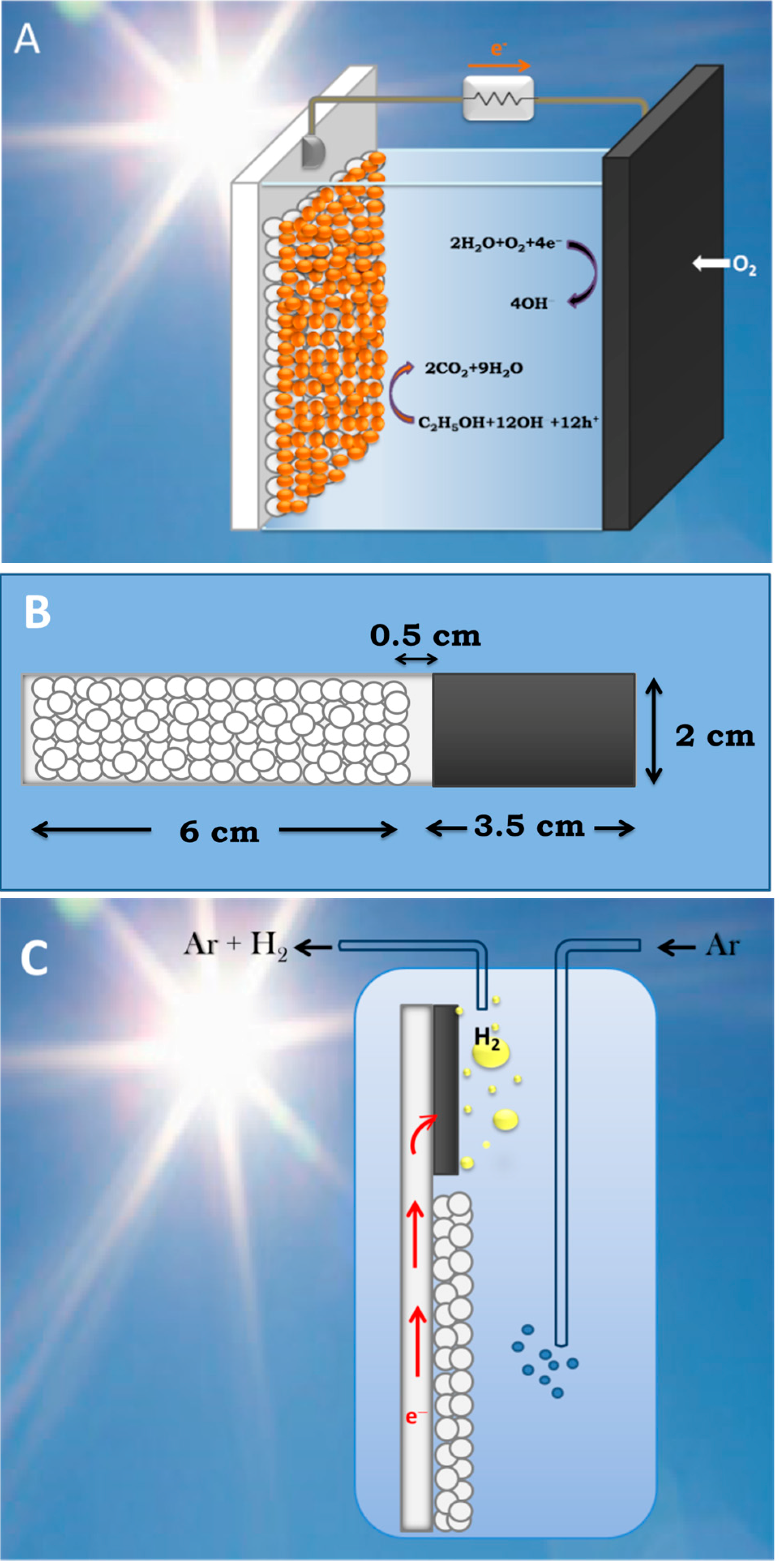
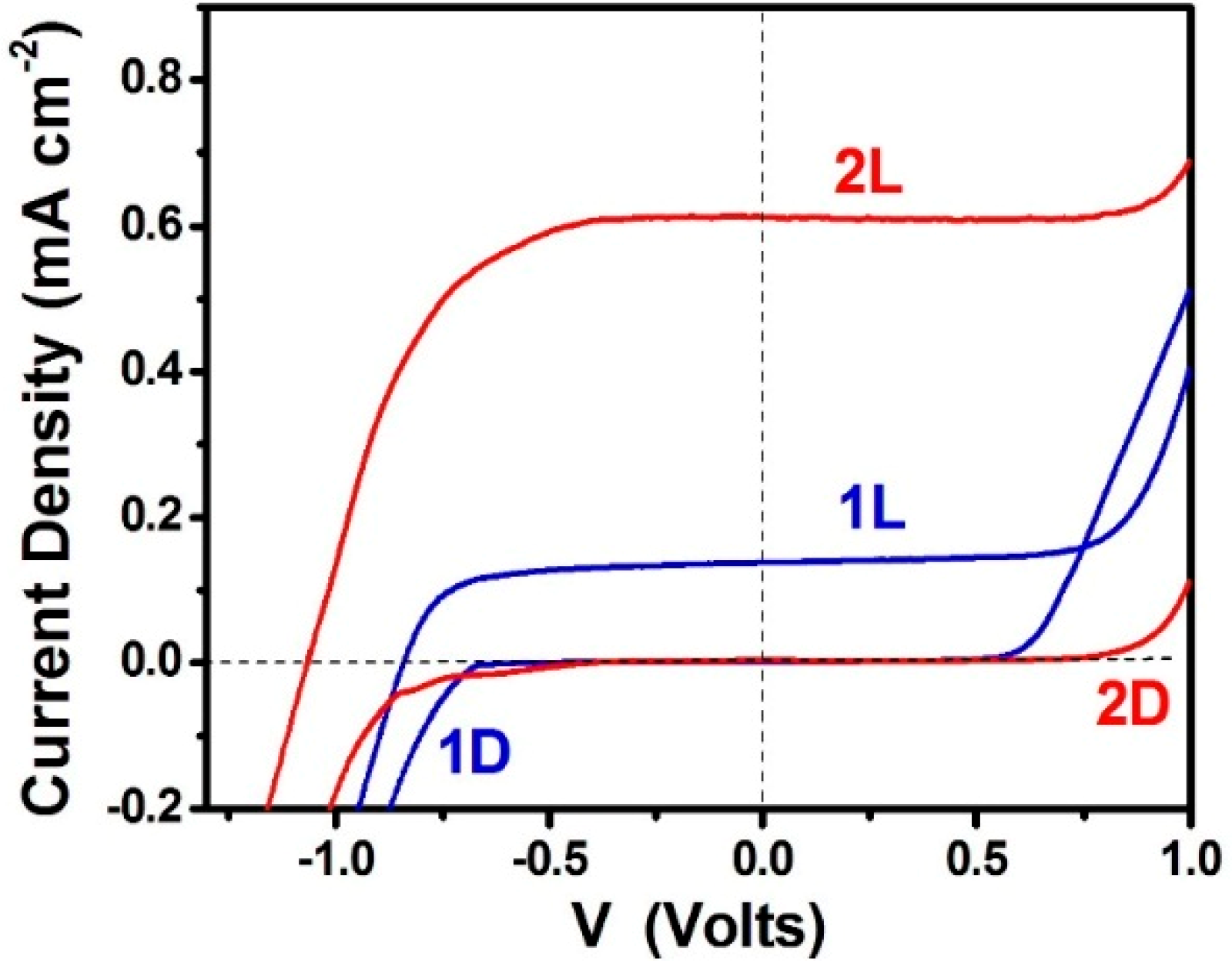
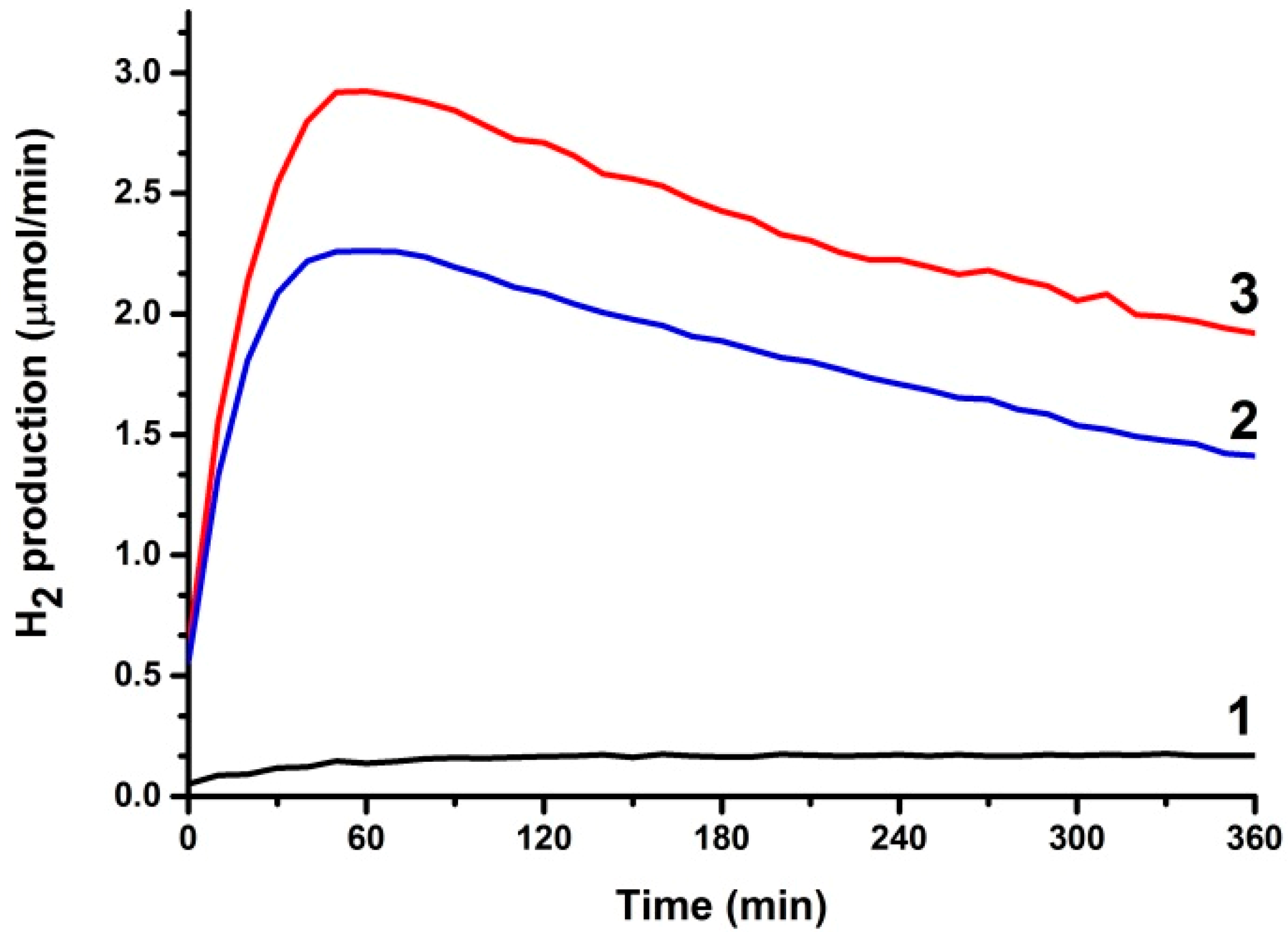
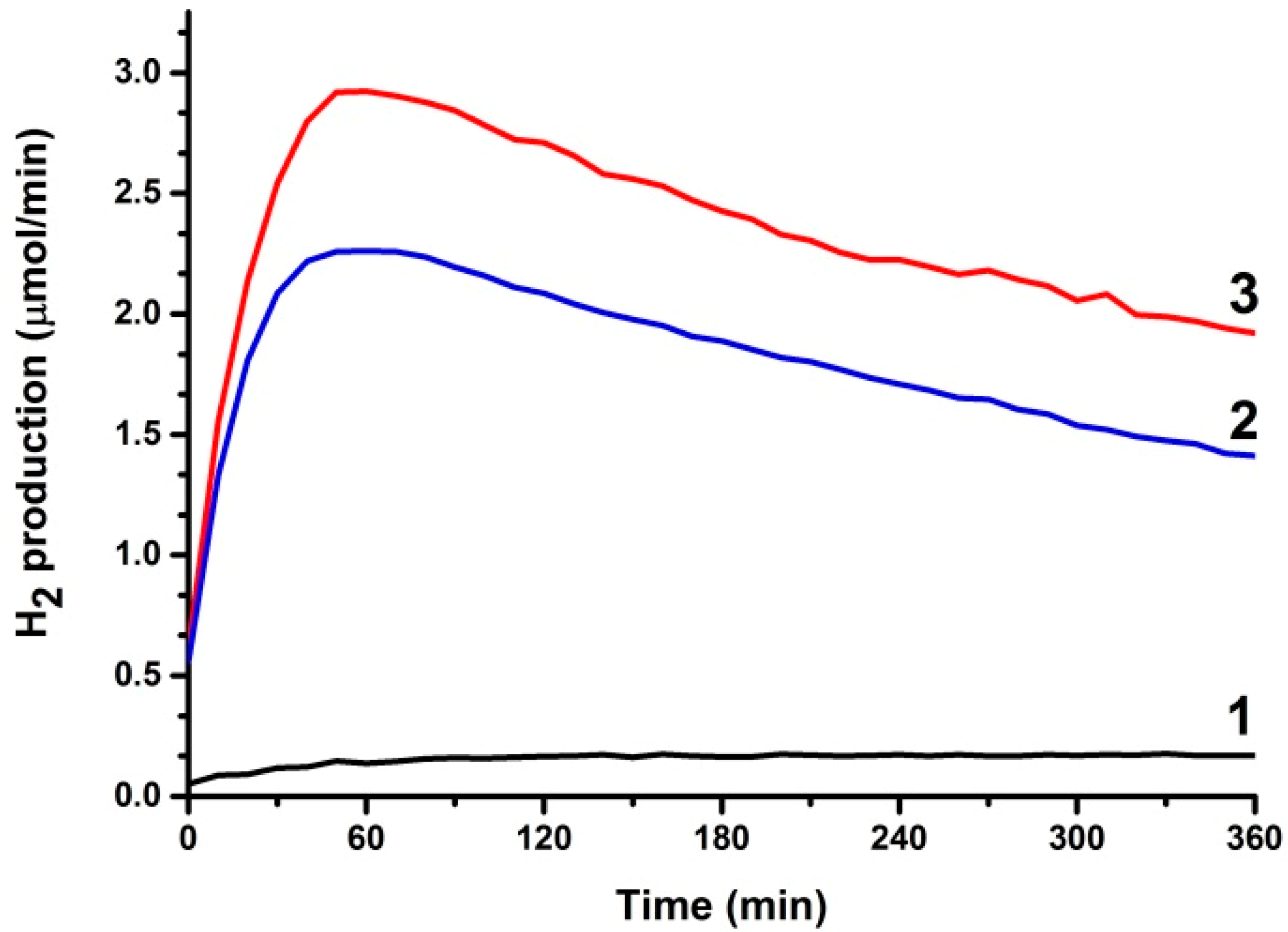
3.2. Use of PFCs for Hydrogen Production
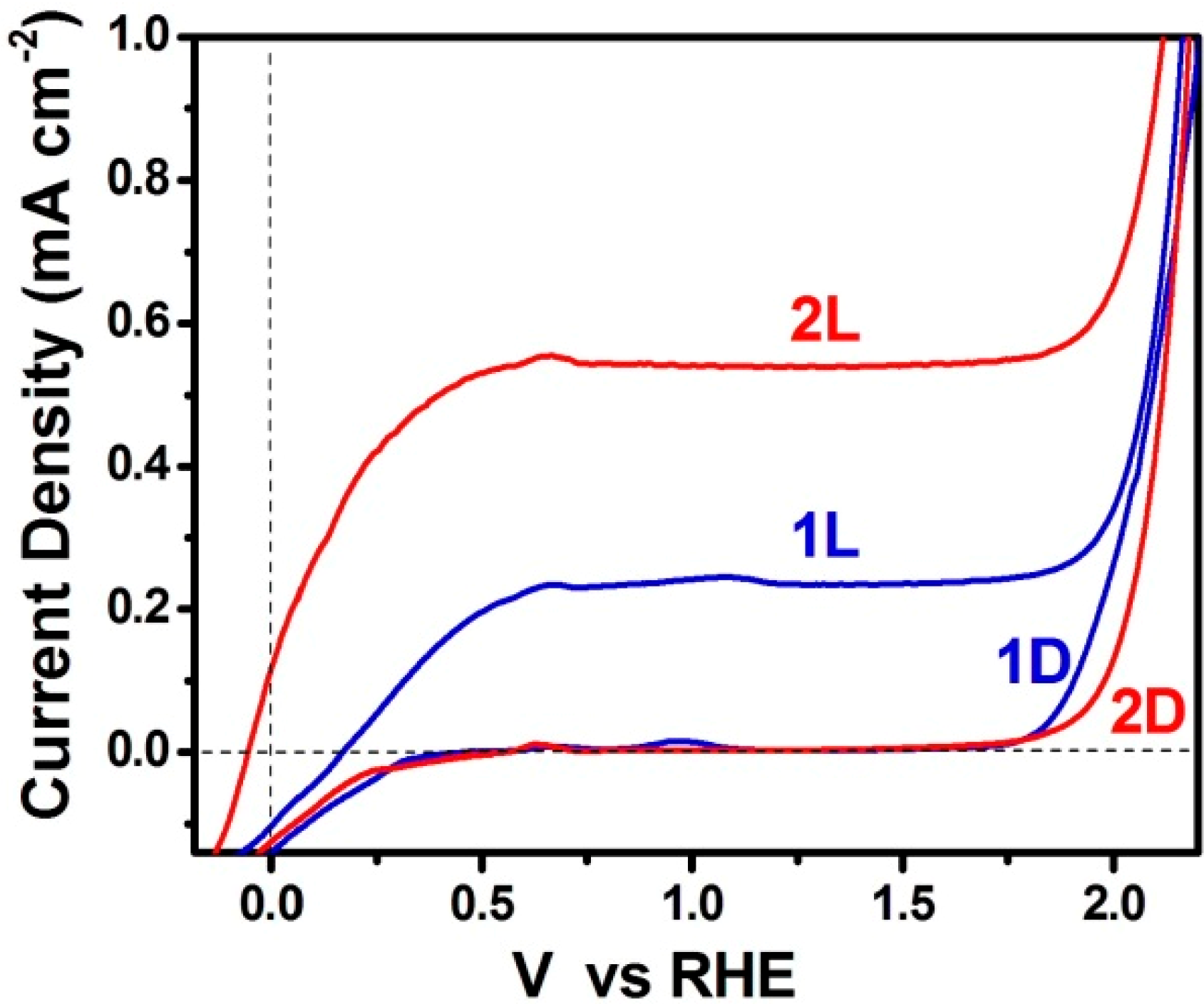
3.3. Short-Circuiting Anode and Cathode Electrode for Hydrogen Production in an Alkaline PFC. The “Photoelectrocatalytic Leaf”

4. Experimental Section
4.1. Materials
4.2. Preparation of np-TiO2 Films and Deposition of CdS by the SILAR Method
4.3. Construction of the Counter Electrode
4.4. Device (Reactor) Construction
4.5. Measurements
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kaneko, M.; Nemoto, J.; Ueno, H.; Gokan, N.; Ohnuki, K.; Horikawa, M.; Saito, R.; Shibata, T. Photoelectrochemical Reaction of Biomass and Bio-Related Compounds With Nanoporous TiO2 Film Photoanode and O2-Reducing Cathode. Electrochem. Commun. 2006, 8, 336–340. [Google Scholar] [CrossRef]
- Lianos, P. Production of Electricity and Hydrogen by Photocatalytic Degradation of Organic Wastes in a Photoelectrochemical Cell: The Concept of the Photofuelcell: A Review of a Re-Emerging Research Field. J. Hazard. Mater. 2011, 185, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Becquerel, E. Mémoire sur les effets électriques produits sous l’influence des rayons solaires. Comptes Rendus 1839, 9, 561–567. [Google Scholar]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Van de Krol, R.; Liang, Y.; Schoonman, J. Solar Hydrogen Production with Nanostructured Metal Oxides. J. Mater. Chem. 2008, 18, 2311–2320. [Google Scholar]
- Kudo, A.; Miseki, Y. Heterogeneous Photocatalyst Materials for Water Splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Gratzel, M. Photoelectrochemical Cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Bak, T.; Nowotny, J.; Rekas, M.; Sorell, C.C. Photo-Electrochemical Hydrogen Generation from Water Using Solar Energy. Materials-Related Aspects. Int. J. Hydrog. Energy 2002, 27, 991–1022. [Google Scholar] [CrossRef]
- Antoniadou, M.; Sfaelou, S.; Dracopoulos, V.; Lianos, P. Platinum-Free Photoelectrochemical Water Splitting. Catal. Commun. 2014, 43, 72–74. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Antoniadou, M.; Kondarides, D.I.; Lianos, P. Aldol Condensation Products During Photocatalytic Oxidation of Ethanol in a Photoelectrochemical Cell. Appl. Catal. B 2010, 100, 124–132. [Google Scholar] [CrossRef]
- Antoniadou, M.; Panagiotopoulou, P.; Kondarides, D.I.; Lianos, P. Photocatalysis and Photoelectrocatalysis Using Nanocrystalline Titania Alone or Combined with Pt, RuO2 or NiO Co-Catalysts. J. Appl. Electrochem. 2012, 42, 737–743. [Google Scholar] [CrossRef]
- Antoniadou, M.; Kondarides, D.I.; Dionysiou, D.D.; Lianos, P. Quantum Dot Sensitized Titania Applicable as Photoanode in Photoactivated Fuel Cells. J. Phys. Chem. C 2012, 116, 16901–16909. [Google Scholar] [CrossRef]
- Sfaelou, S.; Sygellou, L.; Dracopoulos, V.; Travlos, A.; Lianos, P. Effect of the Nature of Cadmium Salts on the Effectiveness of CdS SILAR Deposition and Its Consequences on the Performance of Sensitized Solar Cells. J. Phys. Chem. C 2014, 118, 22873–22880. [Google Scholar] [CrossRef]
- Yu, H.; Irie, H.; Hashimoto, K. Conduction Band Energy Level Control of Titanium Dioxide: Toward an Efficient Visible-Light-Sensitive Photocatalyst. J. Am. Chem. Soc. 2010, 132, 6898–6899. [Google Scholar] [CrossRef] [PubMed]
- Antoniadou, M.; Lianos, P. Near Ultraviolet and Visible Light Photoelectrochemical Degradation of Organic Substances Producing Electricity and Hydrogen. J. Photochem. Photobiol. A 2009, 204, 69–74. [Google Scholar] [CrossRef]
- Antoniadou, M.; Lianos, P. Production of Electricity by Photoelectrochemical Oxidation of Ethanol in a PhotoFuelCell. Appl. Catal. B 2010, 99, 307–313. [Google Scholar] [CrossRef]
- Sfaelou, S.; Antoniadou, M.; Trakakis, G.; Dracopoulos, V.; Tasis, D.; Parthenios, J.; Galiotis, C.; Papagelis, K.; Lianos, P. Buckypaper as Pt-Free Cathode Electrode in Photoactivated Fuel Cells. Electrochim. Acta 2012, 80, 399–404. [Google Scholar] [CrossRef]
- Balis, N.; Dracopoulos, V.; Antoniadou, M.; Lianos, P. One-Step Electrodeposition of Polypyrrole Applied as Oxygen Reduction Electrocatalyst in Photoactivated Fuel Cells. Electrochim. Acta 2012, 70, 338–343. [Google Scholar] [CrossRef]
- Fujishima, A.; Kato, T.; Maekawa, E.; Honda, K. Mechanism of the Current Doubling Effect. I. The ZnO Photoanode in Aqueous Solution of Sodium Formate. Bull. Chem. Soc. Jpn. 1981, 54, 1671–1674. [Google Scholar] [CrossRef]
- Antoniadou, M.; Daskalaki, V.M.; Balis, N.; Kondarides, D.I.; Kordulis, Ch.; Lianos, P. Photocatalysis and Photoelectrocatalysis Using (CdS-ZnS)/TiO2 Combined Photocatalysts. Appl. Catal. B 2011, 107, 188–196. [Google Scholar] [CrossRef]
- Selli, E.; Chiarello, G.L.; Quartarone, E.; Mustarelli, P.; Rossetti, I.; Forni, L. A Photocatalytic Water Splitting Device for Separate Hydrogen and Oxygen Evolution. Chem. Commun. 2007, 5022–5024. [Google Scholar]
- Horiuchi, Y.; Toyao, T.; Takeuchi, M.; Matsuoka, M.; Anpo, M. Recent Advances in Visible-Light-Responsive Photocatalysts for Hydrogen Production and Solar Energy Conversion—From Semiconducting TiO2 to MOF/PCP Photocatalysts. Phys. Chem. Chem. Phys. 2013, 15, 13243–13253. [Google Scholar] [CrossRef] [PubMed]
- Antoniadou, M.; Sfaelou, S.; Lianos, P. Quantum Dot Sensitized Titania for Photo-Fuel-Cell and for Water Splitting Operation in the Presence of Sacrificial Agents. Chem. Eng. J. 2014, 254, 245–251. [Google Scholar] [CrossRef]
- Nomikos, G.N.; Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Kinetic and Mechanistic Study of the Photocatalytic Reforming of Methanol Over Pt/TiO2 Catalyst. Appl. Catal. B 2014, 146, 249–257. [Google Scholar] [CrossRef]
- Chiarello, G.L.; Dozzi, M.V.; Scavini, M.; Grunwaldt, J.-D.; Selli, E. One Step Flame-Made Fluorinated Pt/TiO2 Photocatalysts for Hydrogen Production. Appl. Catal. B 2014, 160–161, 144–151. [Google Scholar] [CrossRef]
- Ismail, A.A.; Bahnemann, D.W. Photochemical Splitting of Water for Hydrogen Production by Photocatalysis: A Review. Sol. Energy Mater. Sol. Cells 2014, 128, 85–101. [Google Scholar] [CrossRef]
- Puskelova, J.; Michal, R.; Caplovicova, M.; Antoniadou, M.; Caplovic, L.; Plesch, G.; Lianos, P. Hydrogen Production by Photocatalytic Ethanol Reforming Using Eu- and S-Doped Anatase. Appl. Surf. Sci. 2014, 305, 665–669. [Google Scholar] [CrossRef]
- Nicolau, Y.F. Solution Deposition of Thin Solid Compound Films by a Successive Ionic-Layer Adsorption and Reaction Process. Appl. Surf. Sci. 1985, 22–23, 1061–1074. [Google Scholar] [CrossRef]
- Trevisan, R.; Rodenas, P.; Gonzalez-Pedro, V.; Sima, C.; Sanchez, R.S.; Barea, E.M.; Mora-Sero, I.; Fabregat-Santiago, F.; Gimenez, S. Harnessing Infrared Photons for Photoelectrochemical Hydrogen Generation. A PbS Quantum Dot Based “Quasi-Artificial Leaf”. J. Phys. Chem. Lett. 2013, 4, 141–146. [Google Scholar] [CrossRef]
- Strataki, N.; Antoniadou, M.; Dracopoulos, V.; Lianos, P. Visible-Light Photocatalytic Hydrogen Production from Ethanol-Water Mixtures Using a Pt-CdS-TiO2 Photocatalyst. Catal. Today 2010, 151, 53–57. [Google Scholar] [CrossRef]
- Daskalaki, V.M.; Antoniadou, M.; Li Puma, G.; Kondarides, D.I.; Lianos, P. Solar Light-Responsive Pt/CdS/TiO2 Photocatalysts for Hydrogen Production and Simultaneous Degradation of Inorganic or Organic Sacrificial Agents in Wastewater. Environ. Sci. Technol. 2010, 44, 7200–7205. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michal, R.; Sfaelou, S.; Lianos, P. Photocatalysis for Renewable Energy Production Using PhotoFuelCells. Molecules 2014, 19, 19732-19750. https://doi.org/10.3390/molecules191219732
Michal R, Sfaelou S, Lianos P. Photocatalysis for Renewable Energy Production Using PhotoFuelCells. Molecules. 2014; 19(12):19732-19750. https://doi.org/10.3390/molecules191219732
Chicago/Turabian StyleMichal, Robert, Stavroula Sfaelou, and Panagiotis Lianos. 2014. "Photocatalysis for Renewable Energy Production Using PhotoFuelCells" Molecules 19, no. 12: 19732-19750. https://doi.org/10.3390/molecules191219732