Method Development and Validation for Pharmacokinetic and Tissue Distributions of Ellagic Acid Using Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS)

Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of Sample Preparation
2.2. Optimization of Chromatographic and MS/MS Conditions
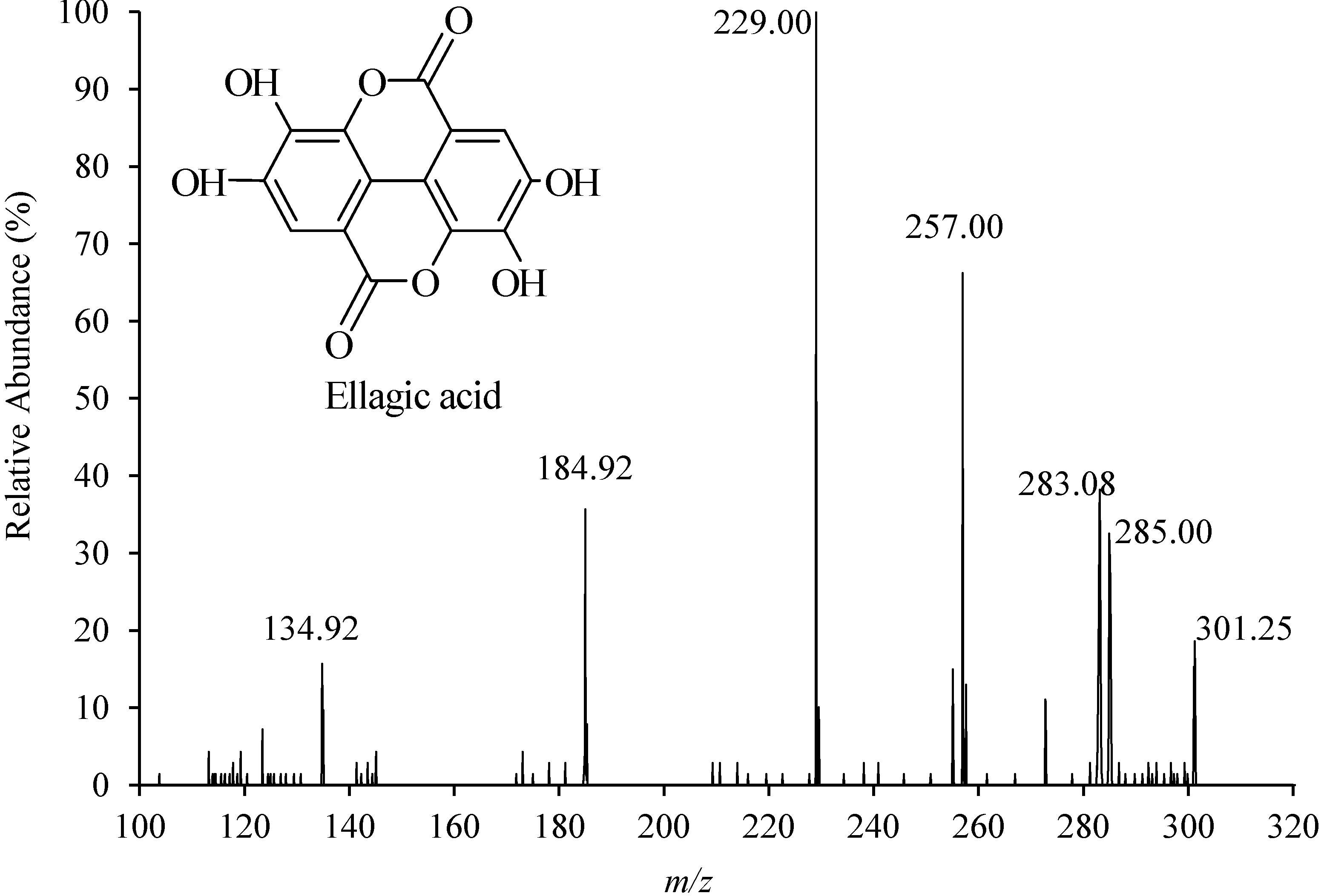
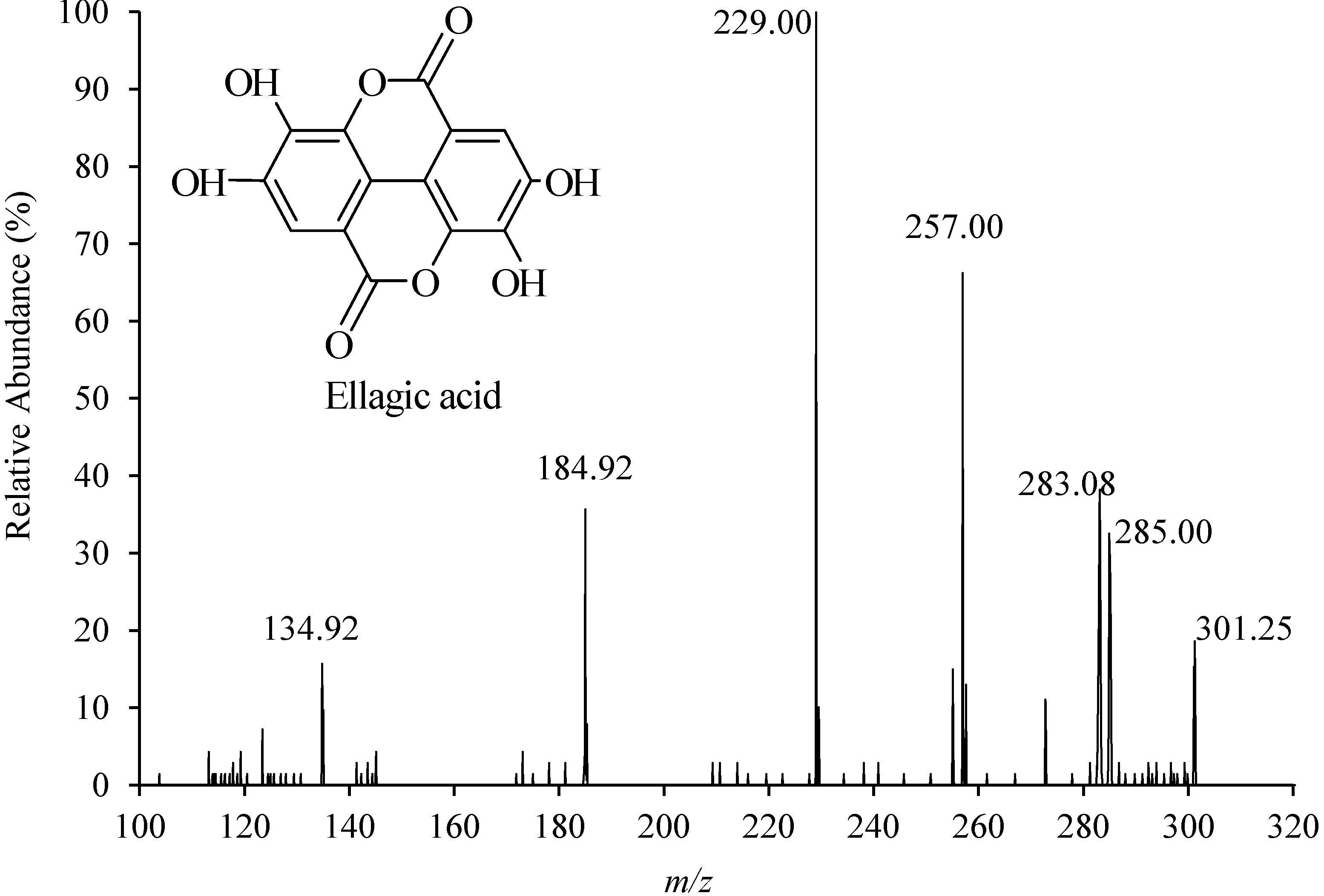
2.3. Method Validation
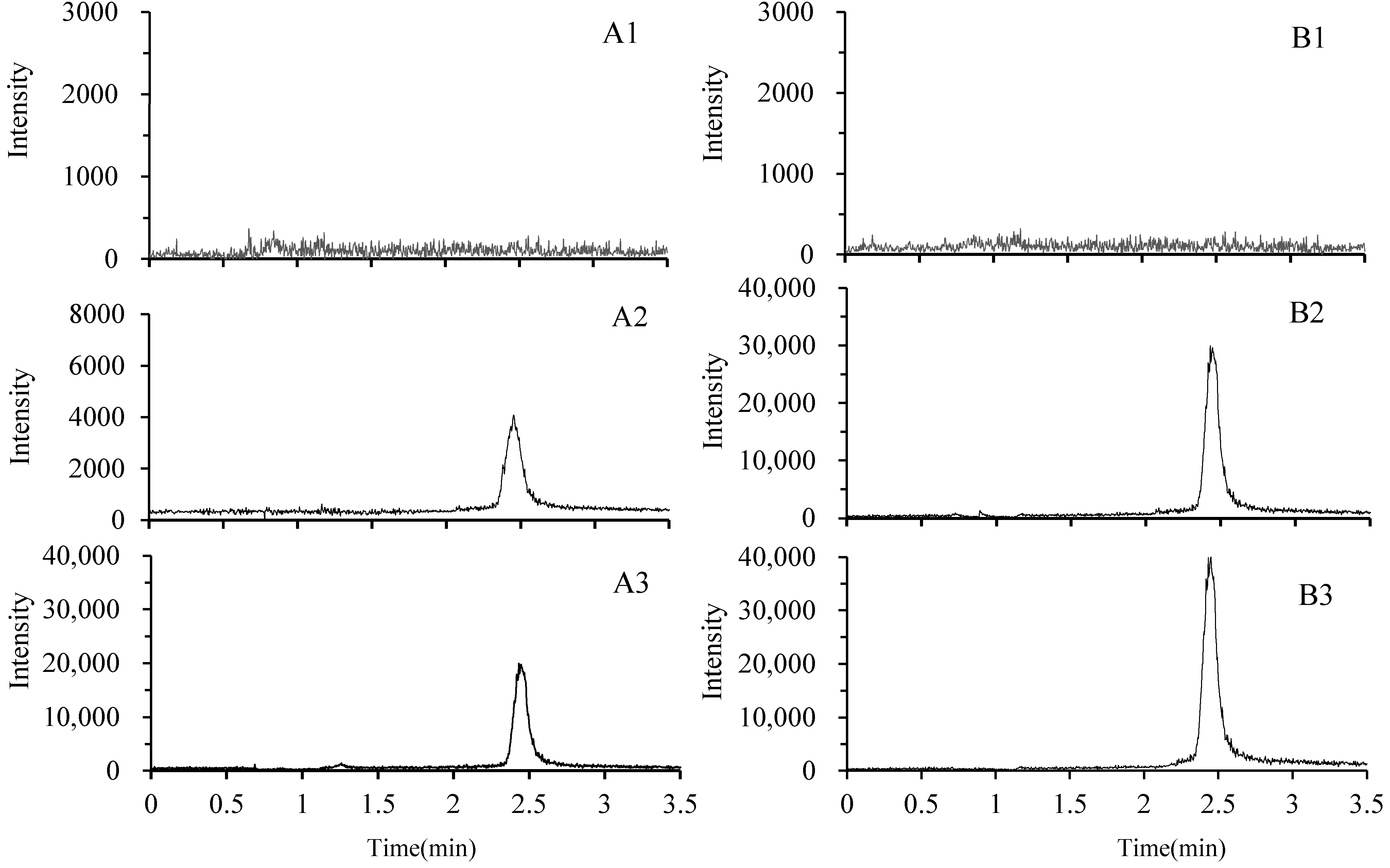
2.3.1. Selectivity and Matrix Effect
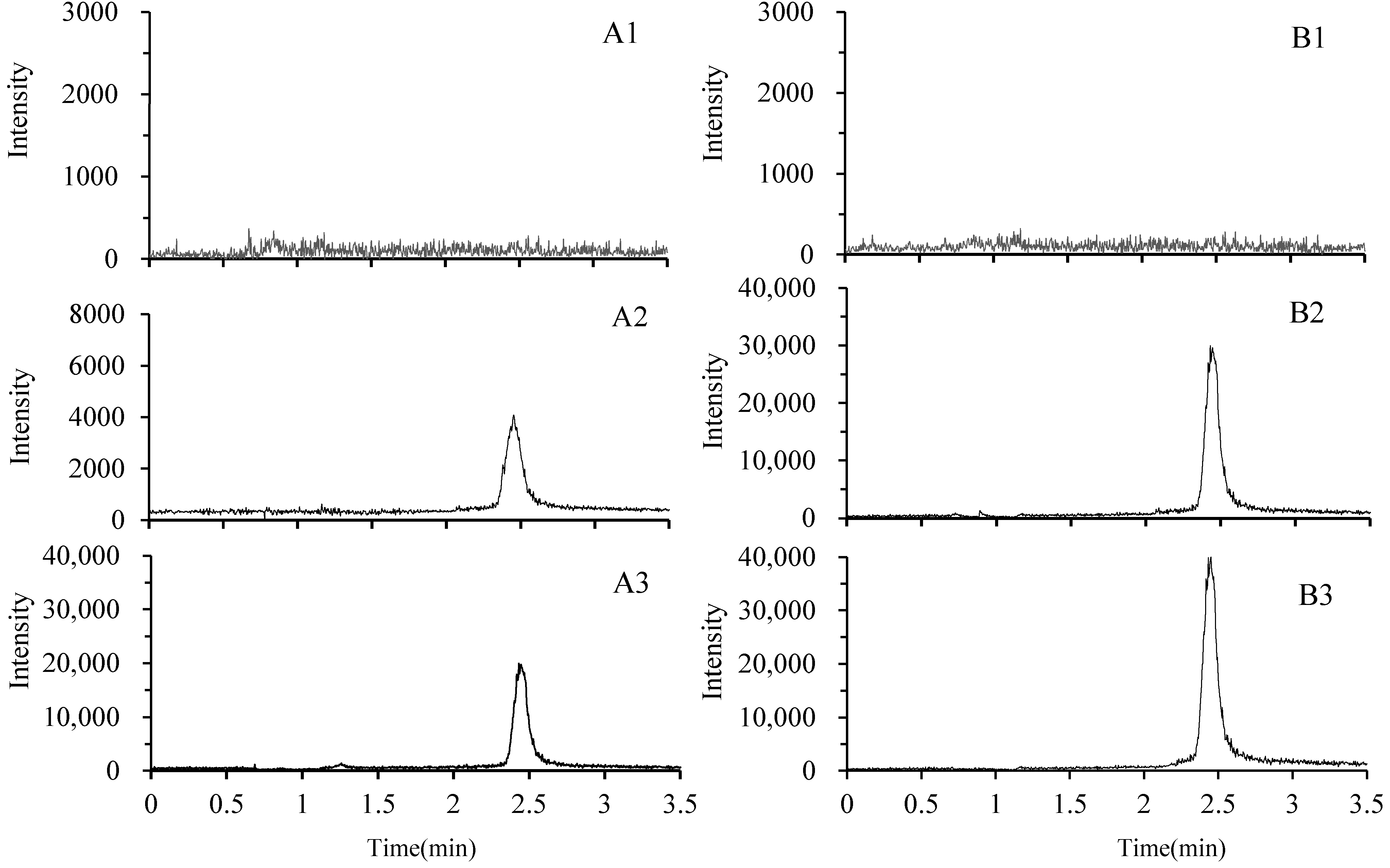
2.3.2. Linearity and the Lower Limits of Quantitation
2.3.3. Precision and Accuracy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Level ng/mL | Accuracy | Precision (RSD %) | Recovery % | Stability % | |||
|---|---|---|---|---|---|---|---|---|
| RE %, | Intraday | Inter-Day | Short Term a | Long Term b | Freeze–Thaw c | |||
| n = 5 | n = 5 | n = 15 | ||||||
| plasma | 50 | −5.3 | 5.7 | 7.2 | 103.3 ± 6.4 | 93.2 ± 3.6 | 92.6 ± 6.4 | 90.5 ± 7.8 |
| 200 | 3.2 | 4.1 | 3.6 | 95.3 ± 3.2 | 94.7 ± 5.2 | 93.6 ± 6.2 | 92.6 ± 8.0 | |
| 800 | −2.8 | 3.0 | 4.0 | 98.2 ± 4.6 | 94.9 ± 4.5 | 94.4 ± 5.7 | 93.2 ± 6.9 | |
| liver | 50 | −3.5 | 4.5 | 6.8 | 90.4 ± 8.7 | 92.6 ± 3.6 | 89.6 ± 8.7 | 90.4 ± 7.4 |
| 200 | −1.8 | 4.1 | 4.3 | 94.5 ± 5.2 | 93.7 ± 6.5 | 93.3 ± 6.3 | 91.3 ± 6.9 | |
| 800 | 4.6 | 3.2 | 4.7 | 96.8 ± 4.7 | 95.1 ± 5.0 | 91.5 ± 5.7 | 92.4 ± 3.6 | |
2.3.4. Recovery and Stability
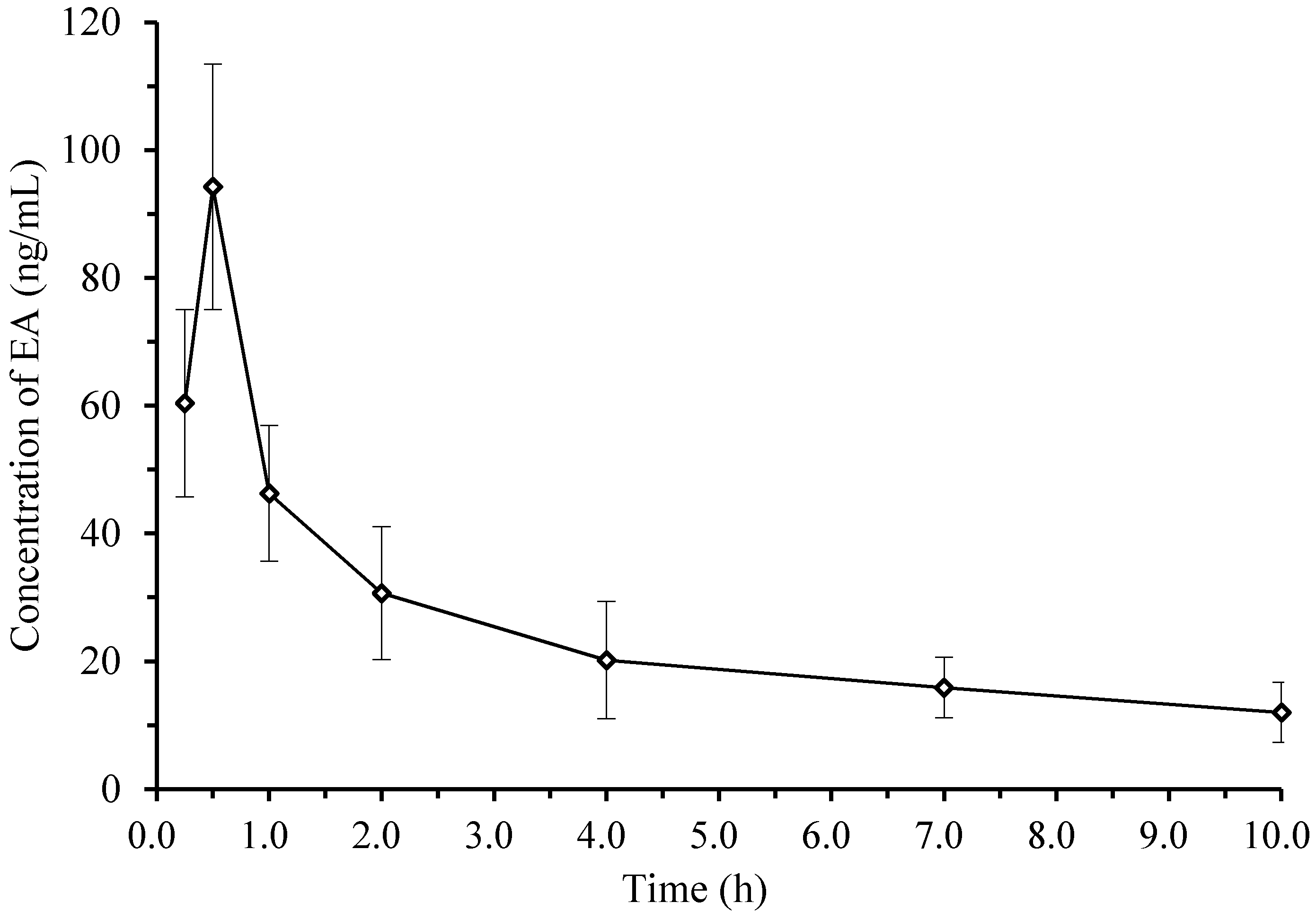
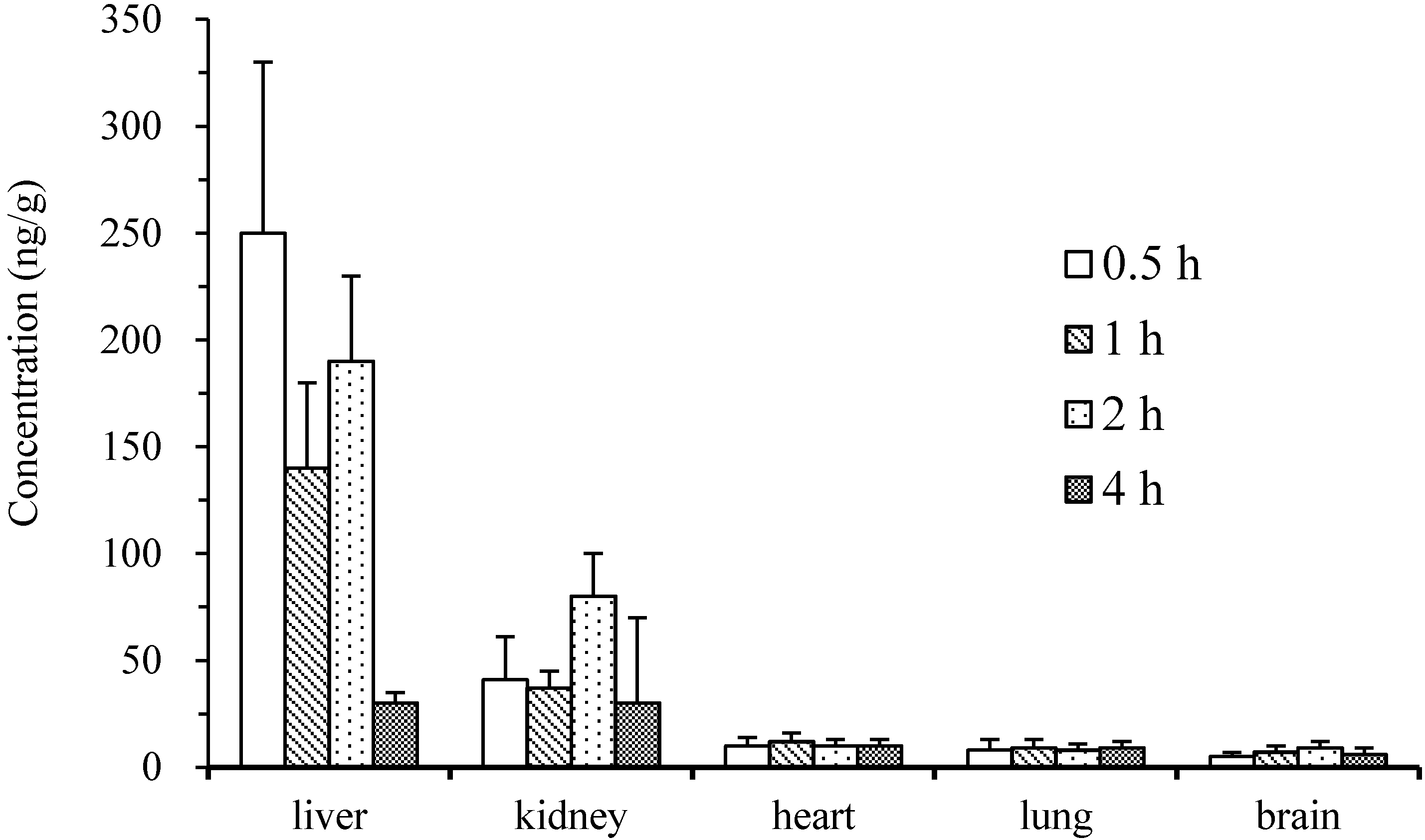
2.4. Pharmacokinetics and Tissue Distribution

| Parameters | Unit | Mean ± SD |
|---|---|---|
| t1/2α | h | 0.25 ± 0.02 |
| t1/2β | h | 6.86 ± 0.05 |
| CL/F | L/h/kg | 109.3 ± 11.7 |
| AUC0–t | ng/mL × h | 252.0 ± 17.0 |
| AUC0–∞ | ng/mL × h | 457.2 ± 34.0 |
| K10 | 1/h | 0.54 ± 0.04 |
| K12 | 1/h | 1.90 ± 0.19 |
| K21 | 1/h | 0.47 ± 0.05 |
| Ka | 1/h | 14.52 ± 1.87 |
| Cmax | ng/mL | 93.6 ± 31.0 |
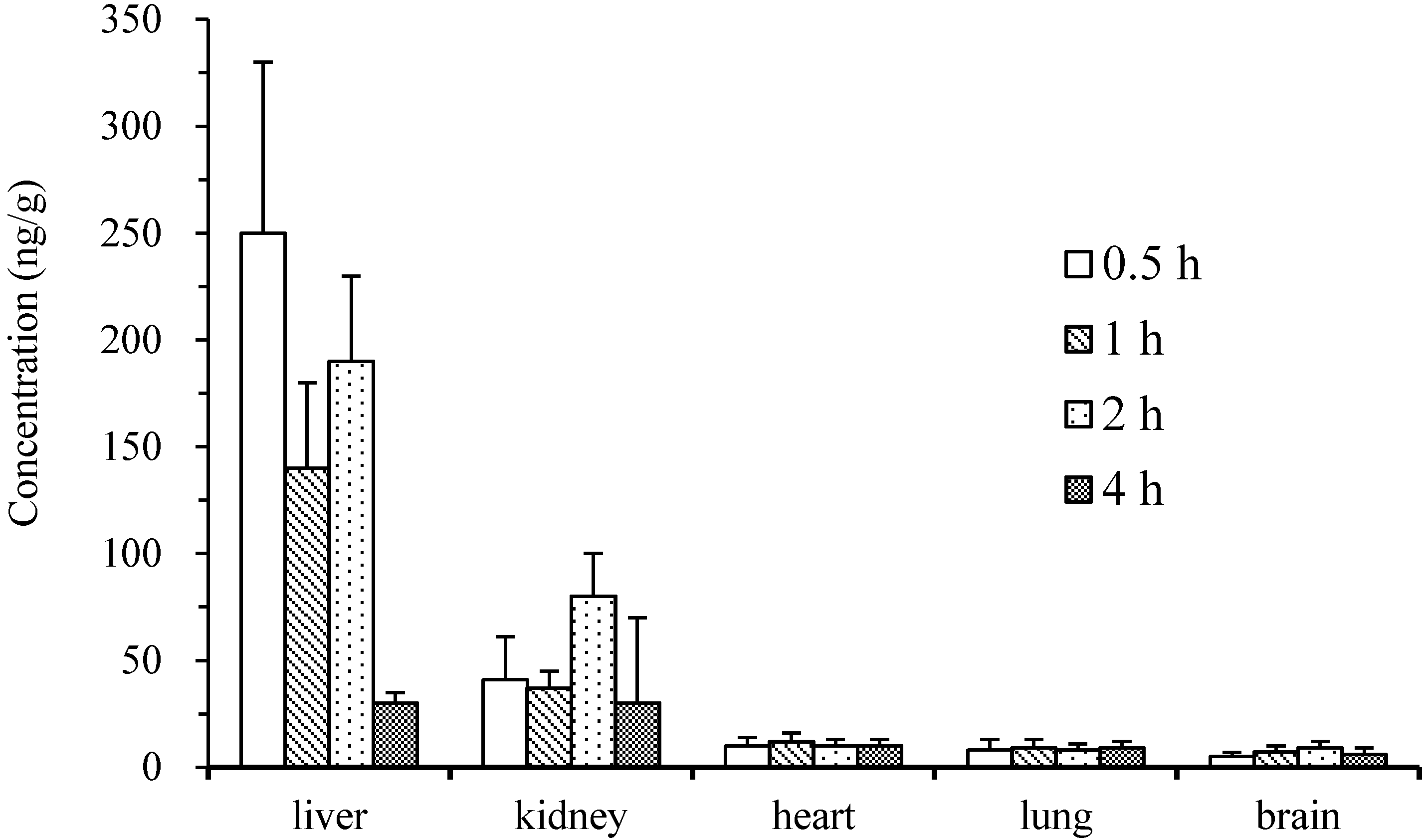
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Drug Administration and Sample Preparation
3.3. Solid Phase Extraction
3.4. Instrumentation for LC and MS
3.5. Method Validation
3.5.1. Preparation of Standard Solutions, Plasma and Tissue Calibrators, and Quality Controls
3.5.2. Specificity and Matrix Effect
3.5.3. Linearity, Accuracy, and Precision
3.5.4. Recovery
3.5.5. Stability
3.5.6. Data Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Larrosa, M.; García-Conesa, M.T.; Espín, J.C.; Tomás-Barberán, F.A. Ellagitannins, ellagic acid and vascular health. Mol. Asp. Med. 2010, 31, 513–539. [Google Scholar] [CrossRef]
- Larrosa, M.; García-Conesa, M.T.; Espín, J.C.; Tomás-Barberán, F.A. Bioavailability and metabolism of ellagic acid and ellagitannins. In Flavonoids and Related Compounds: Bioavailability and Function, 1st ed.; Spencer, J.P.E., Crozier, A., Eds.; CRC Press Inc: Boca Raton, FL, USA, 2012; pp. 183–195. [Google Scholar]
- Aguilera-Carbo, A.; Augur, C.; Prado-Barragan, L.A.; Favela-Torres, E.; Aguilar, C.N. Microbial production of ellagic acid and biodegradation of ellagitannins. Appl. Microbiol. Biot. 2008, 78, 189–199. [Google Scholar] [CrossRef]
- Doyle, B.; Griffiths, L.A. The metabolism of ellagic acid in the rat. Xenobiotica 1980, 10, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Boukharta, M.; Jalbert, G.; Castonguay, A. Biodistribution of ellagic acid and dose-related inhibition of lung tumorigenesis in A/J mice. Nutr. Cancer 1992, 18, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Lei, F.; Xing, D.M.; Xiang, L.; Zhao, Y.N.; Wang, W.; Zhang, L.J.; Du, L.J. Pharmacokinetic study of ellagic acid in rat after oral administration of pomegranate leaf extract. J. Chromatogr. B 2003, 796, 189–194. [Google Scholar] [CrossRef]
- Seeram, N.P.; Lee, R.; Heber, D. Bioavailability of ellagic acid in human plasma after consumption of ellagitannins from pomegranate (Punica granatum L.) juice. Clin. Chim. Acta 2004, 348, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Stoner, G.D.; Sardo, C.; Apseloff, G.; Mullet, D.; Wargo, W.; Pound, V.; Singh, A.; Sanders, J.; Aziz, R.; Casto, B.; et al. Pharmacokinetics of anthocyanins and ellagic acid in healthy volunteers fed freeze-dried black raspberries daily for 7 days. J. Clin. Pharmacol. 2005, 45, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Espín, J.C.; Larrosa, M.; García-Conesa, M.T.; Tomás-Barberán, F.A. Biological significance of urolithins, the gut microbial ellagic acid-derived metabolites: The evidence so far. Evid. Based Complement. Altern. Med. 2013, 2013. [Google Scholar] [CrossRef]
- Smart, R.C.; Huang, M.T.; Chang, R.L.; Sayer, J.M.; Jerina, D.M.; Conney, A.H. Disposition of the naturally occurring antimutagenic plant phenol, ellagic acid, and its synthetic derivatives, 3-O-decylellagic acid and 3,3'-di-O-methylellagic acid in mice. Carcinogenesis 1986, 7, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Mattila, P.; Kumpulainen, J. Determination of free and total phenolic acids in plant-derived foods by HPLC with diode-array detection. J. Agric. Food Chem. 2002, 50, 3660–3667. [Google Scholar] [CrossRef] [PubMed]
- Krishna, V.N.; Meyyanathan, S.N.; Gowramma, B.; Babu, B.; Elango, K. Development and validation of LC-APCI-MS method for the estimation of ellagic acid in fresh and processed fruit products. J. Appl. Pharm. Sci. 2013, 3, 8–12. [Google Scholar]
- Whitley, A.C.; Stoner, G.D.; Darby, M.V.; Walle, T. Intestinal epithelial cell accumulation of the cancer preventive polyphenol ellagic acid-extensive binding to protein and DNA. Biochem. Pharmacol. 2003, 66, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Murugan, V.; Mukherjee, K.; Maiti, K.; Mukherjee, P.K. Enhanced oral bioavailability and antioxidant profiles of ellagic acid by phospholipids. J. Agric. Food Chem. 2009, 57, 4559–4565. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Hu, L.; Zhang, M.; Sun, J.; Qiu, Y.; Rui, J.; Yang, X. A sensitive UPLC-MS/MS method for simultaneous determination of eleven bioactive components of Tong-Xie-Yao-Fang decoction in rat biological matrices. J. Chromatogr. B 2014, 944, 90–100. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds ellagic acid are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, L.; Yin, P.; Ma, C.; Liu, Y. Method Development and Validation for Pharmacokinetic and Tissue Distributions of Ellagic Acid Using Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS). Molecules 2014, 19, 18923-18935. https://doi.org/10.3390/molecules191118923
Yan L, Yin P, Ma C, Liu Y. Method Development and Validation for Pharmacokinetic and Tissue Distributions of Ellagic Acid Using Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS). Molecules. 2014; 19(11):18923-18935. https://doi.org/10.3390/molecules191118923
Chicago/Turabian StyleYan, Linlin, Peipei Yin, Chao Ma, and Yujun Liu. 2014. "Method Development and Validation for Pharmacokinetic and Tissue Distributions of Ellagic Acid Using Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS)" Molecules 19, no. 11: 18923-18935. https://doi.org/10.3390/molecules191118923