Thermal Reactivity of Neutral and Oxidized Ferrocenyl-Substituted Enediynes


Abstract
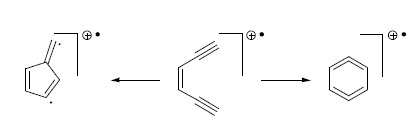
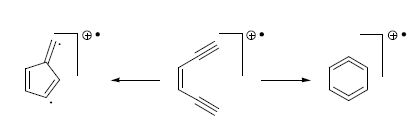
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
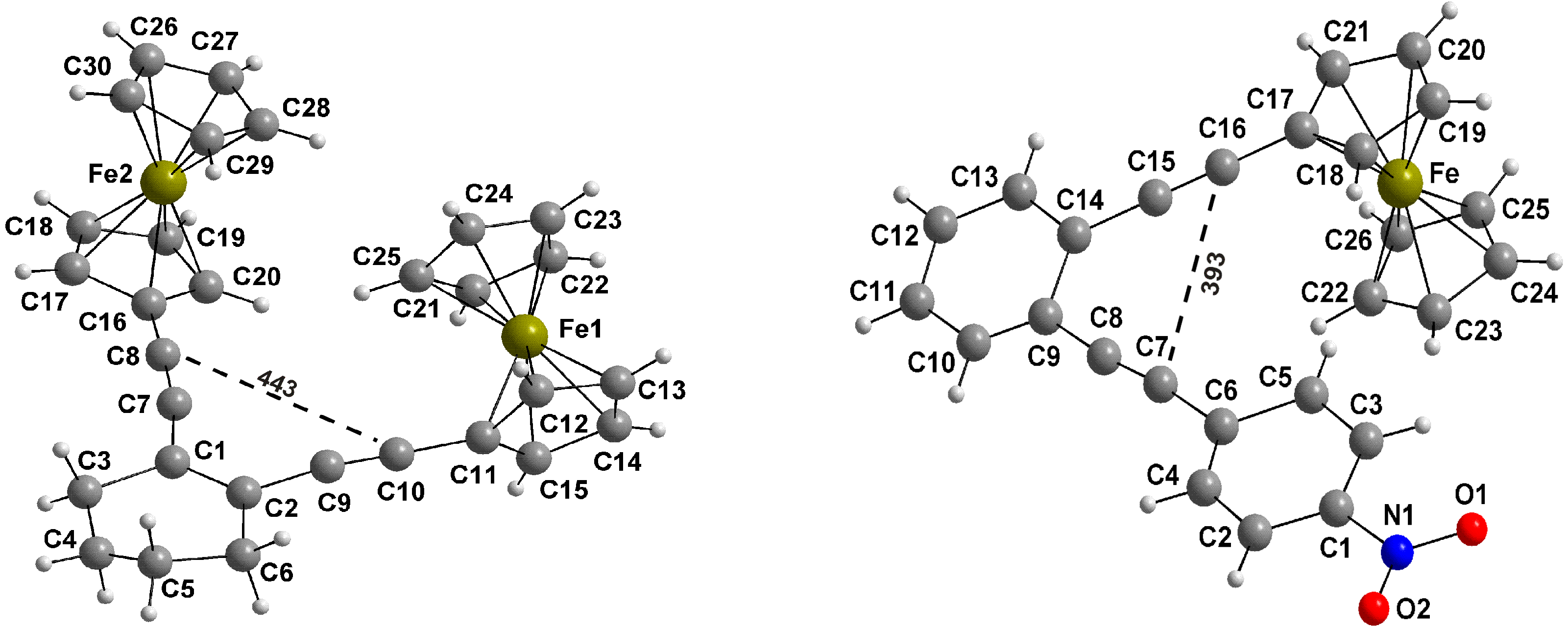
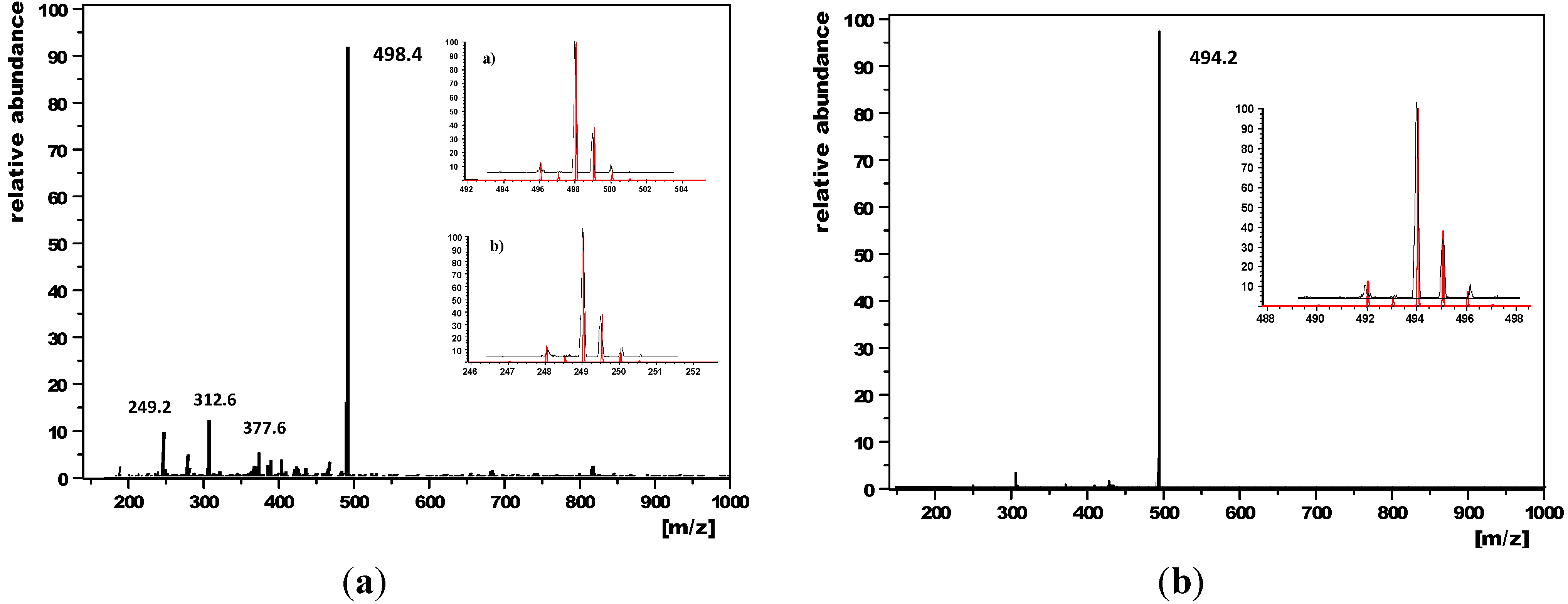
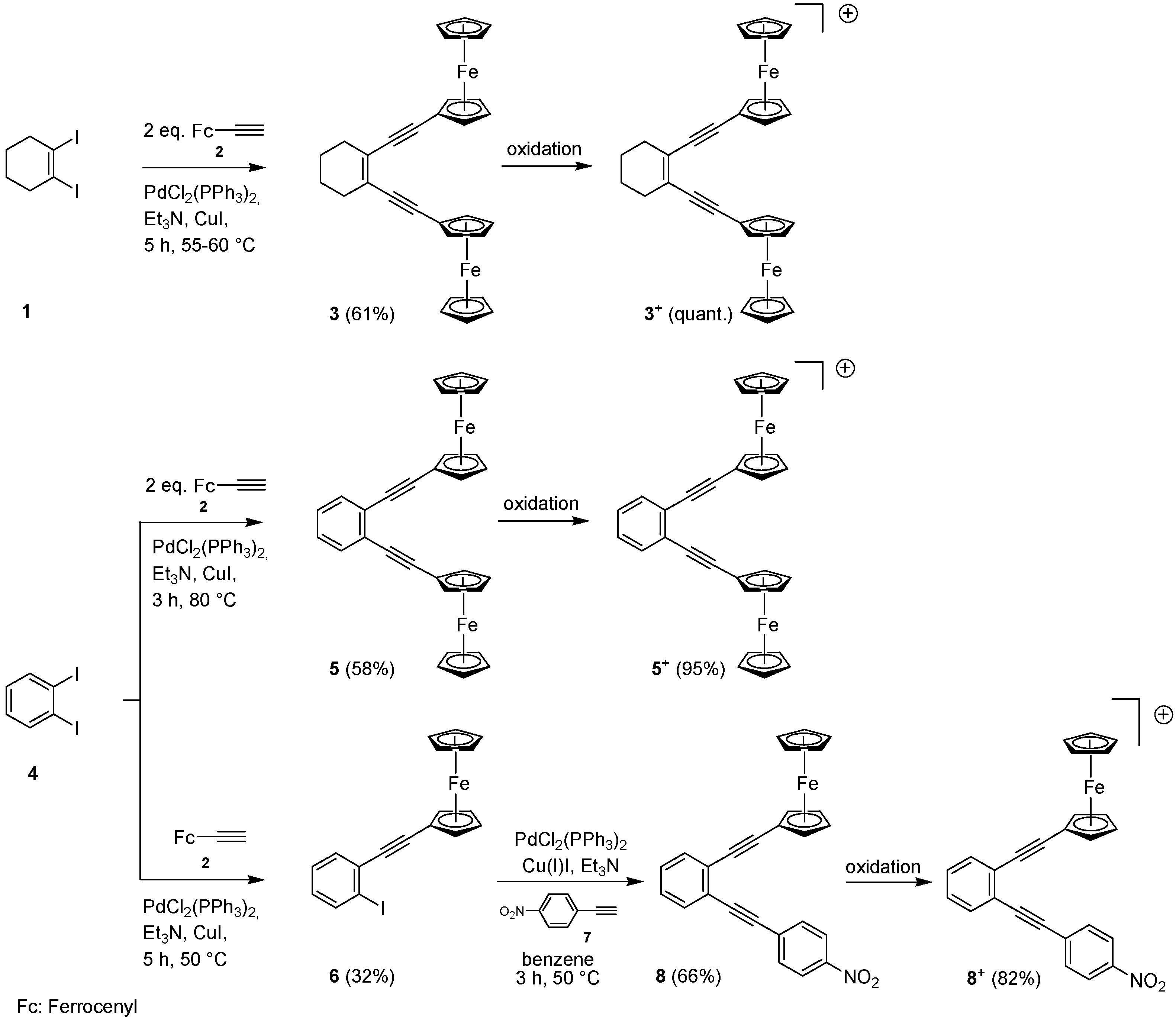
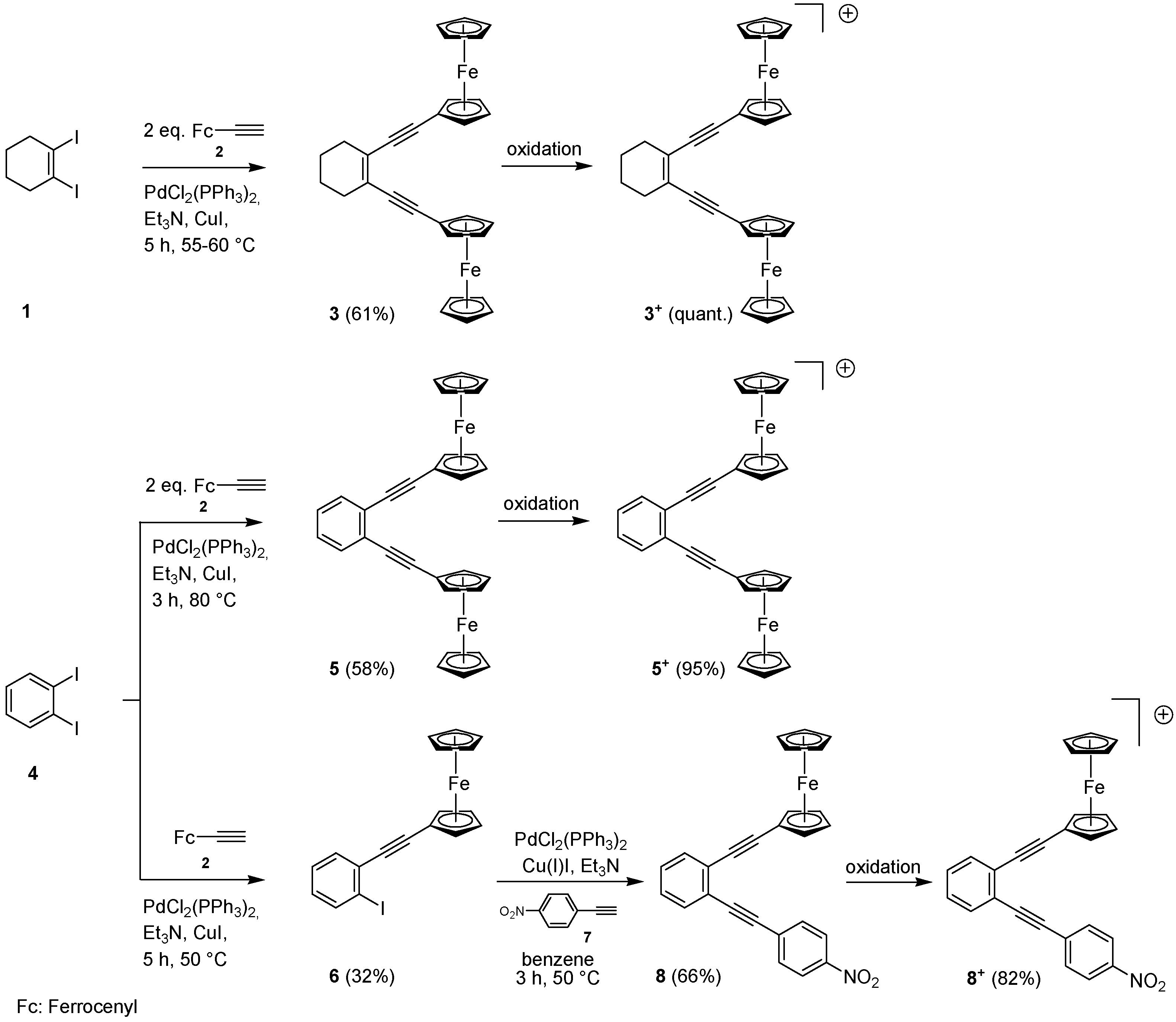
2.1. Synthesis of Enediynes 3, 5 and 8, and Their Oxidation to 3+, 5+ and 8+
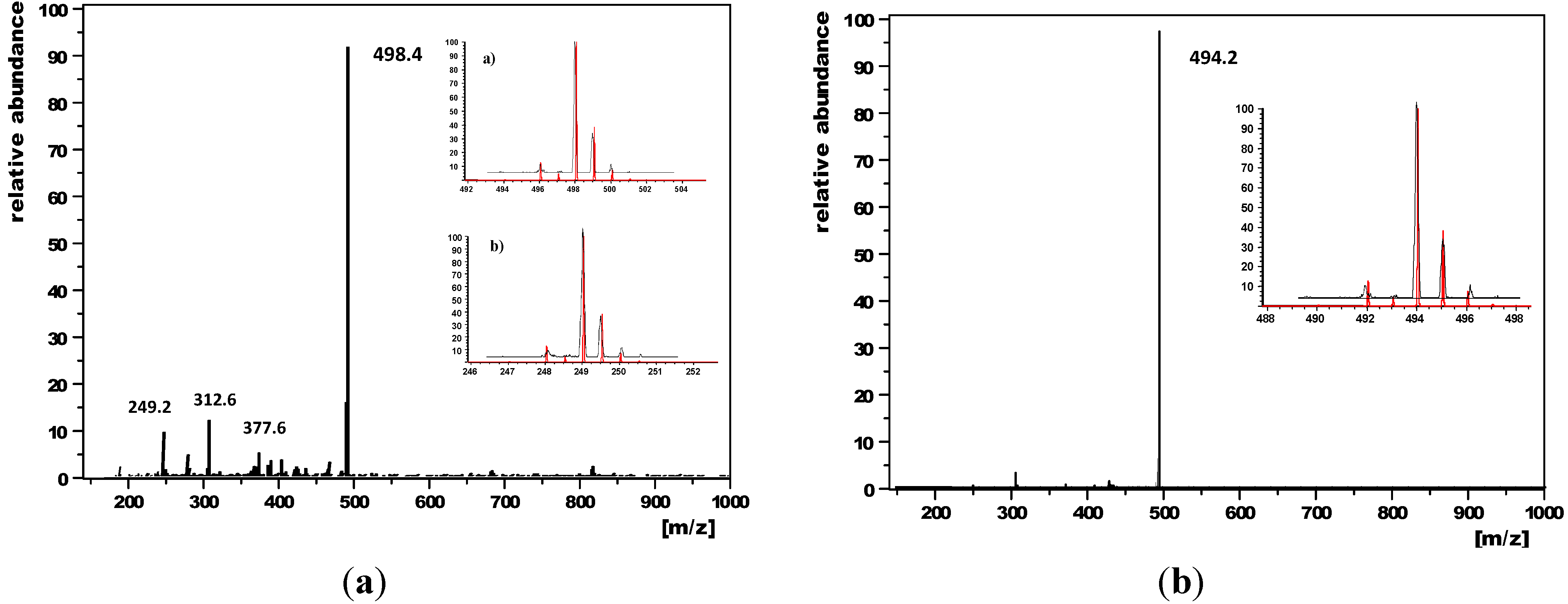
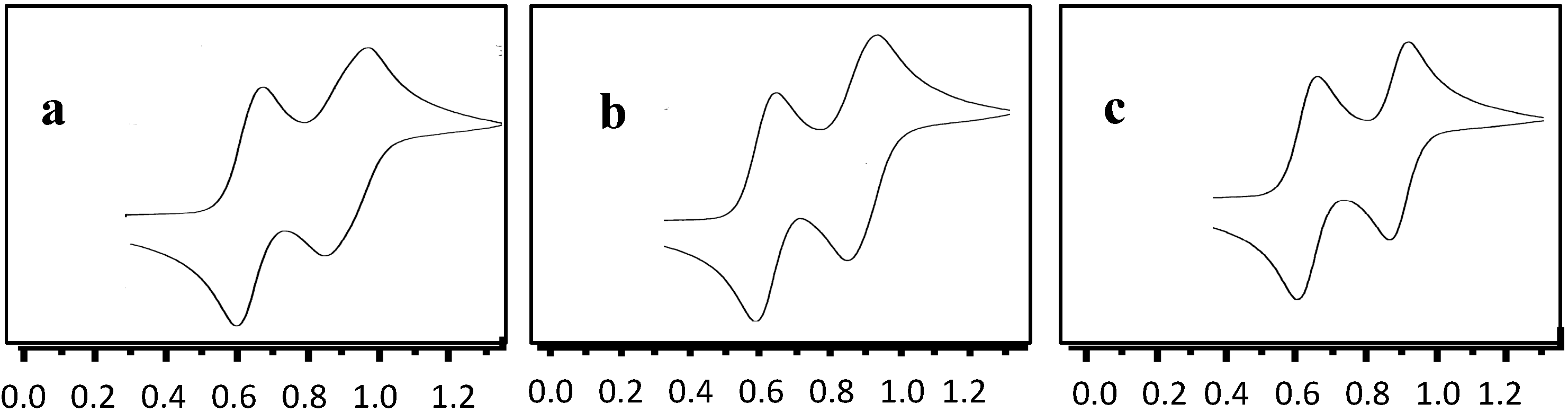
2.2. Electrochemical Properties of 3, 5 and 8

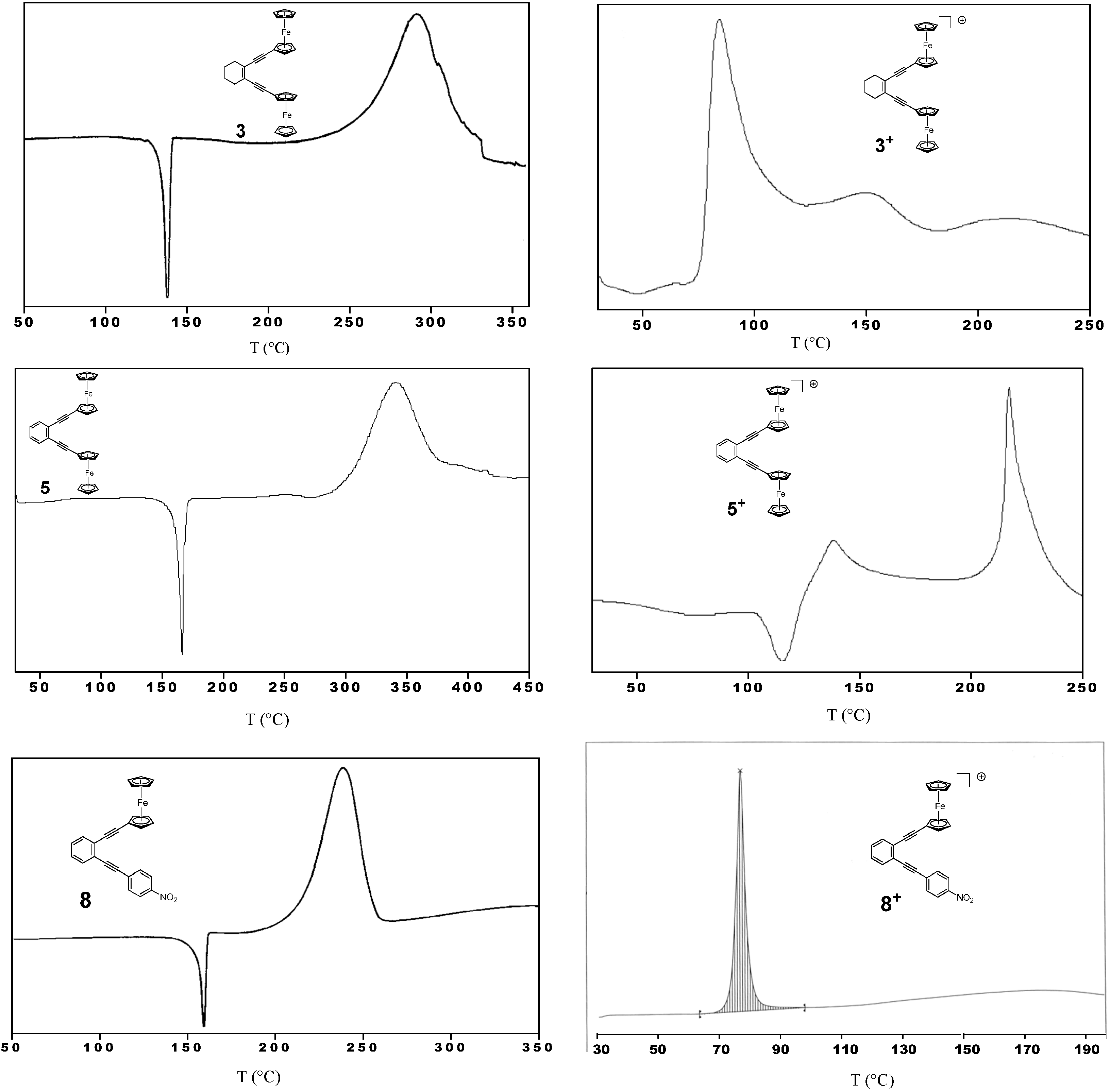
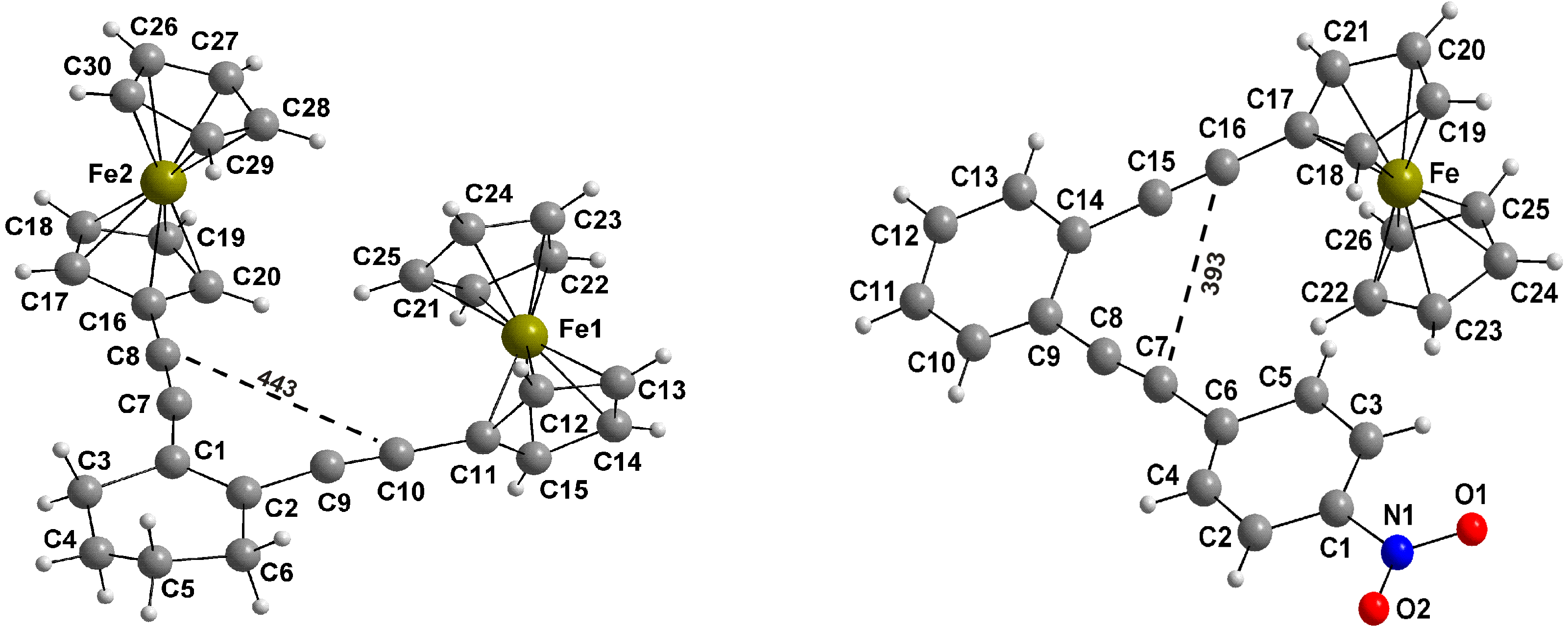
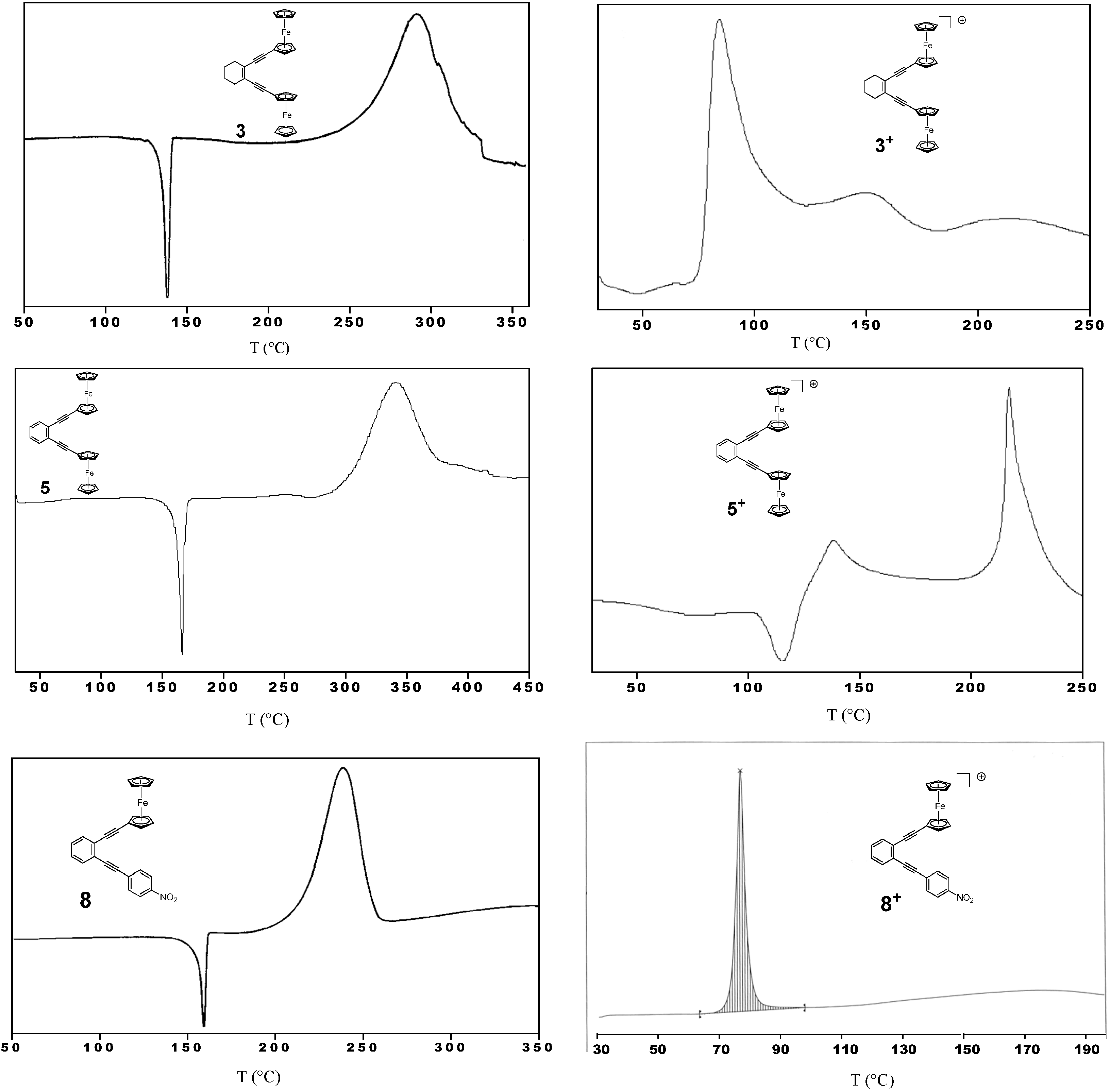
2.3. Thermal Properties
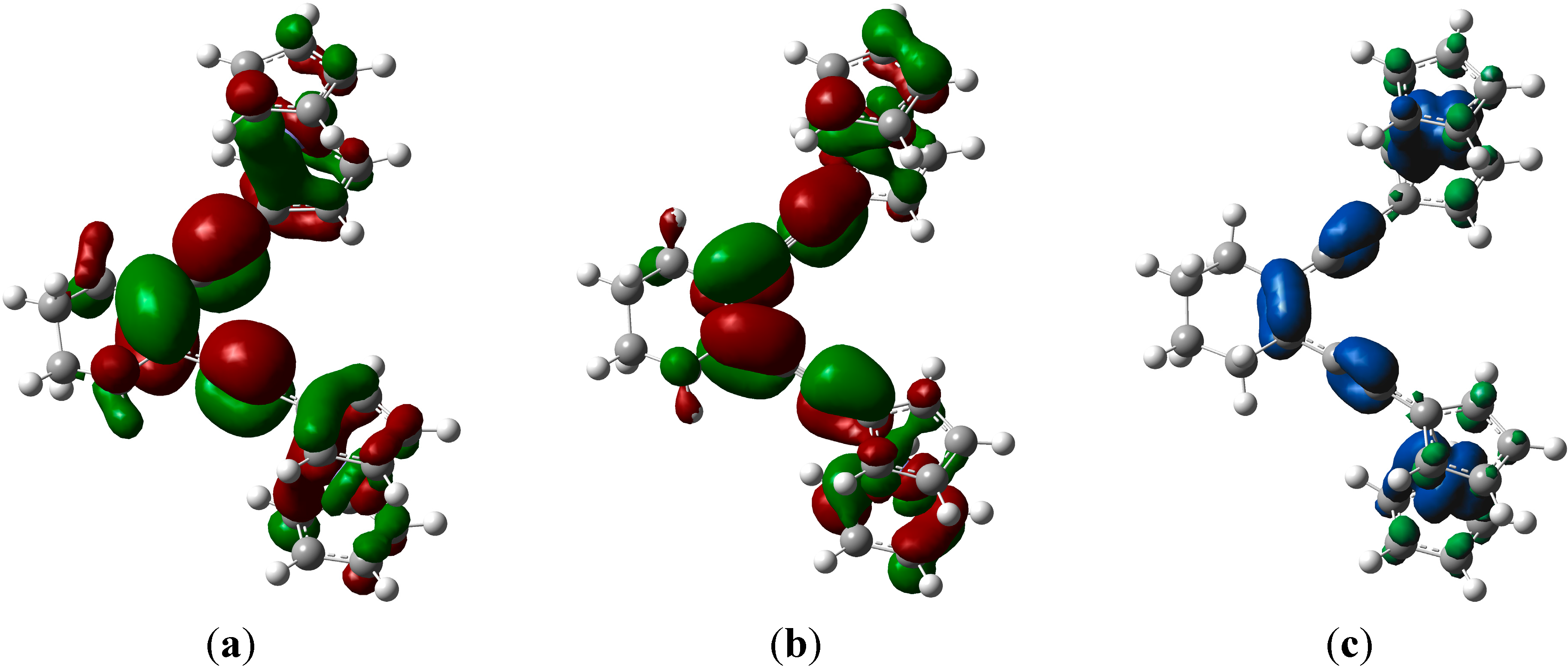
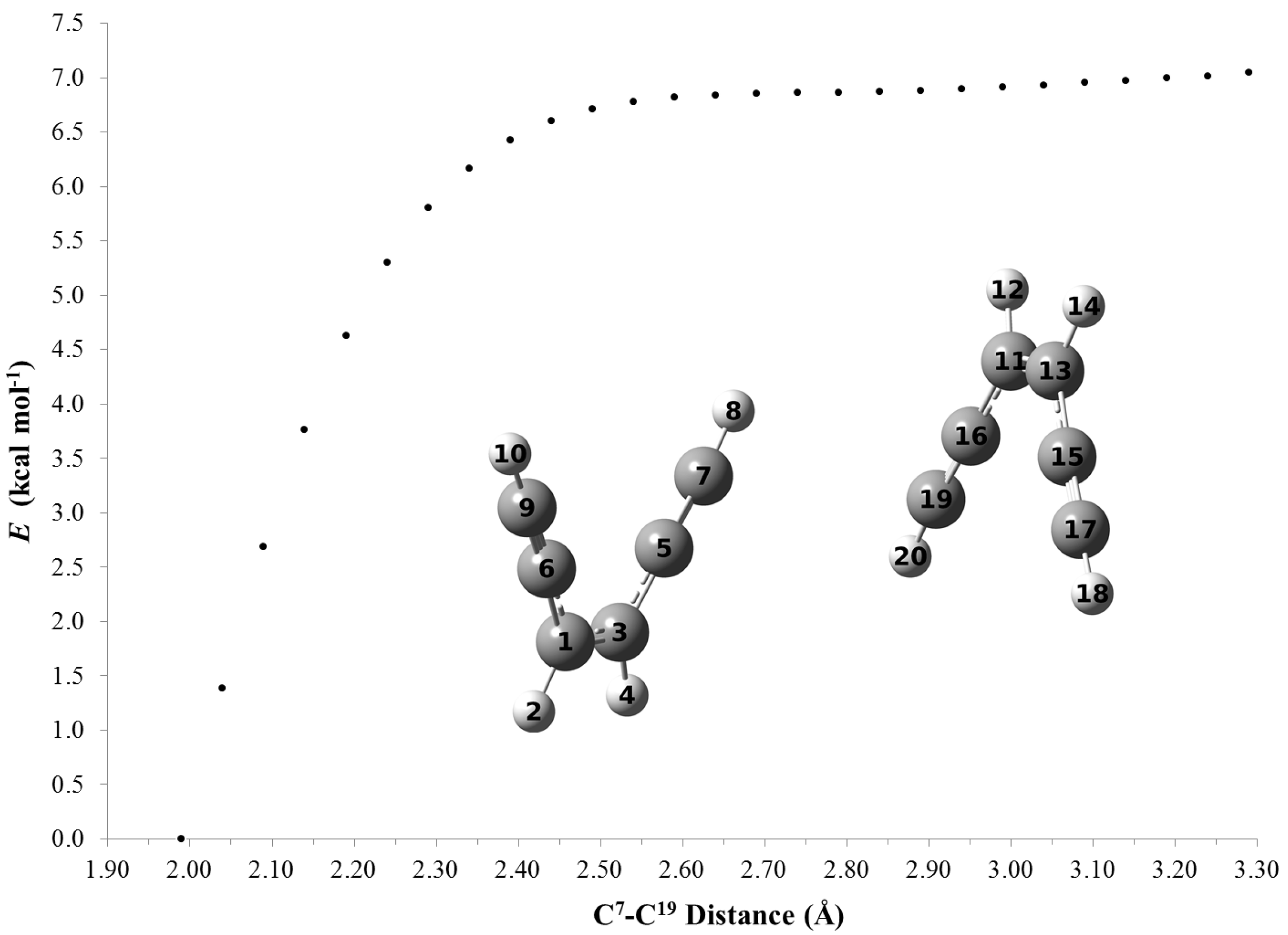
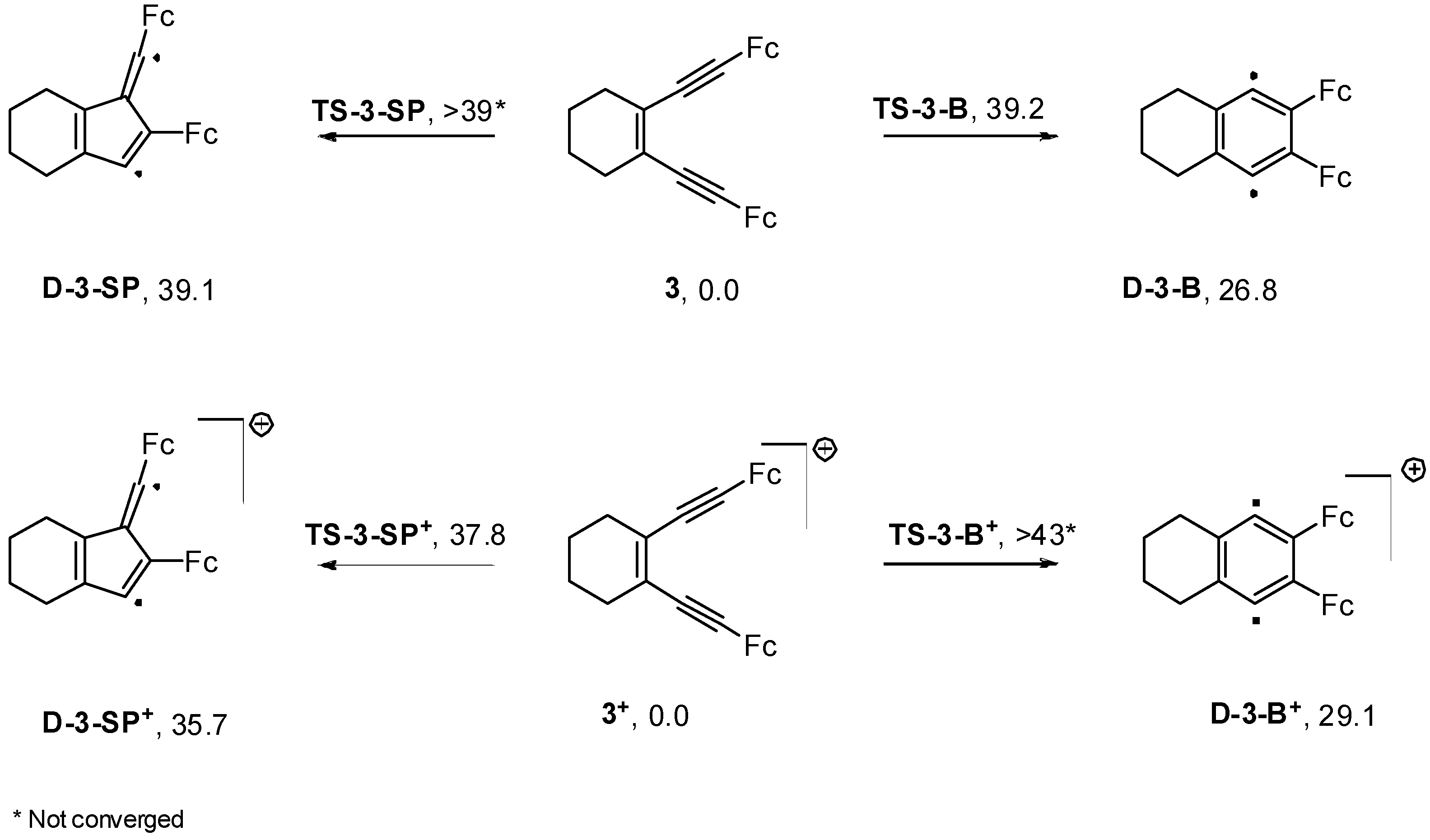
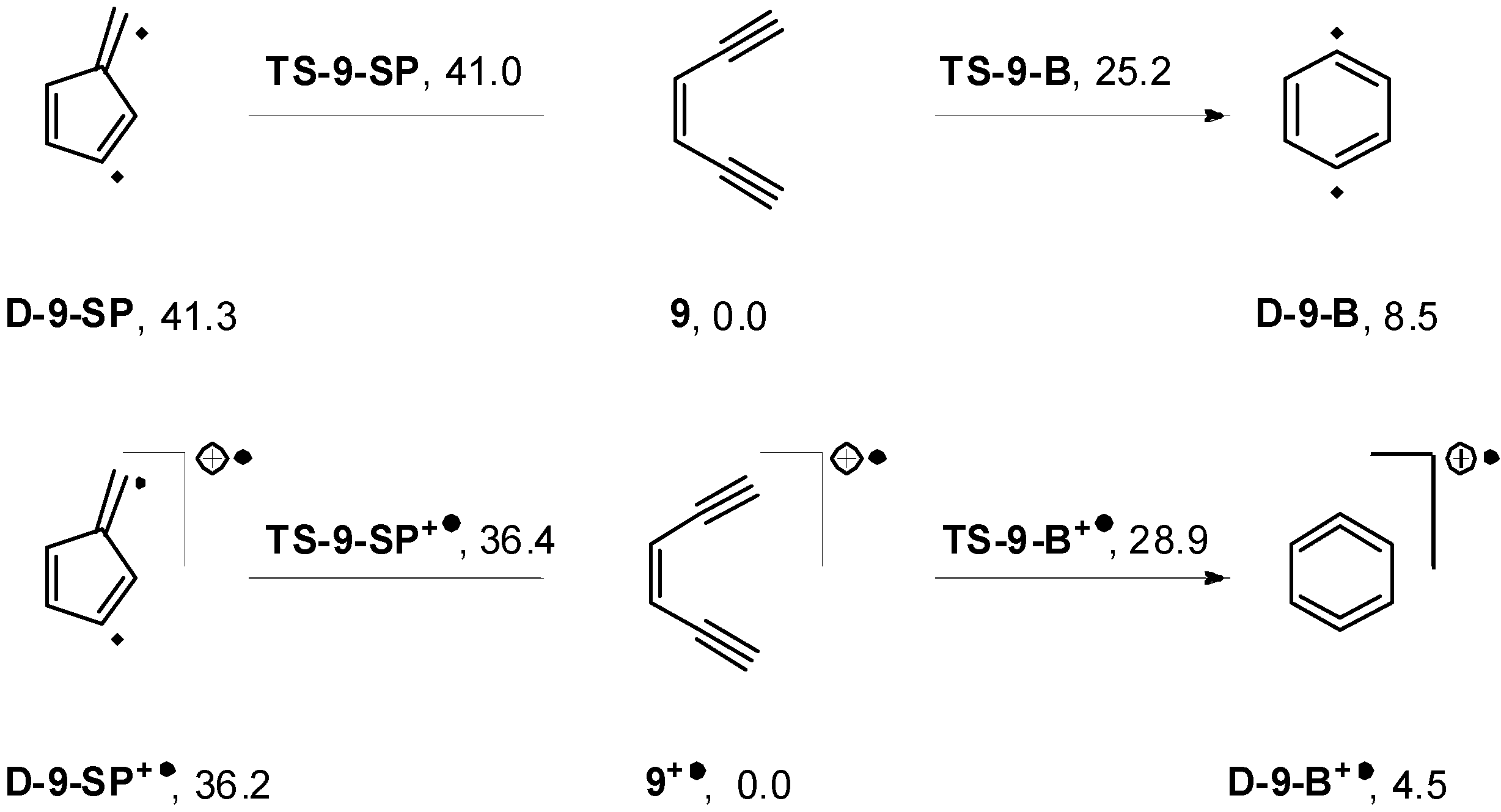
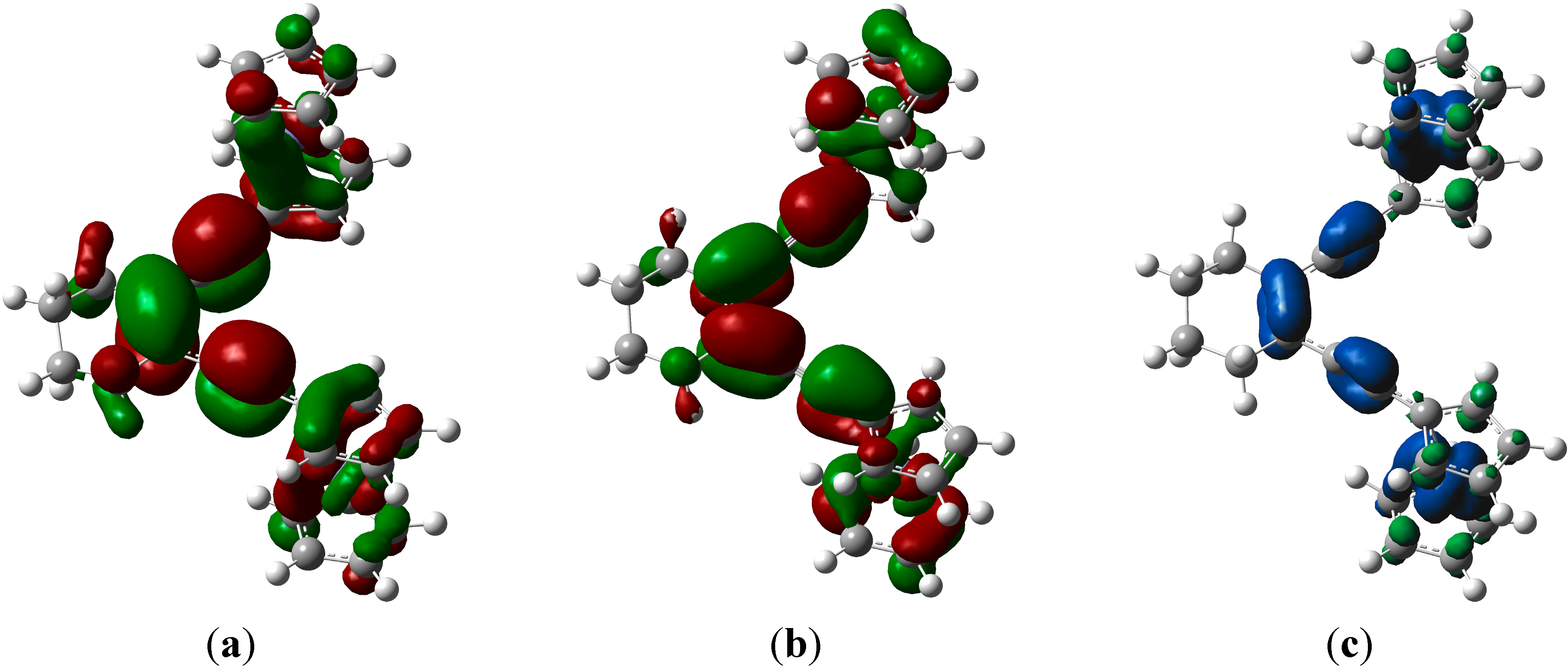
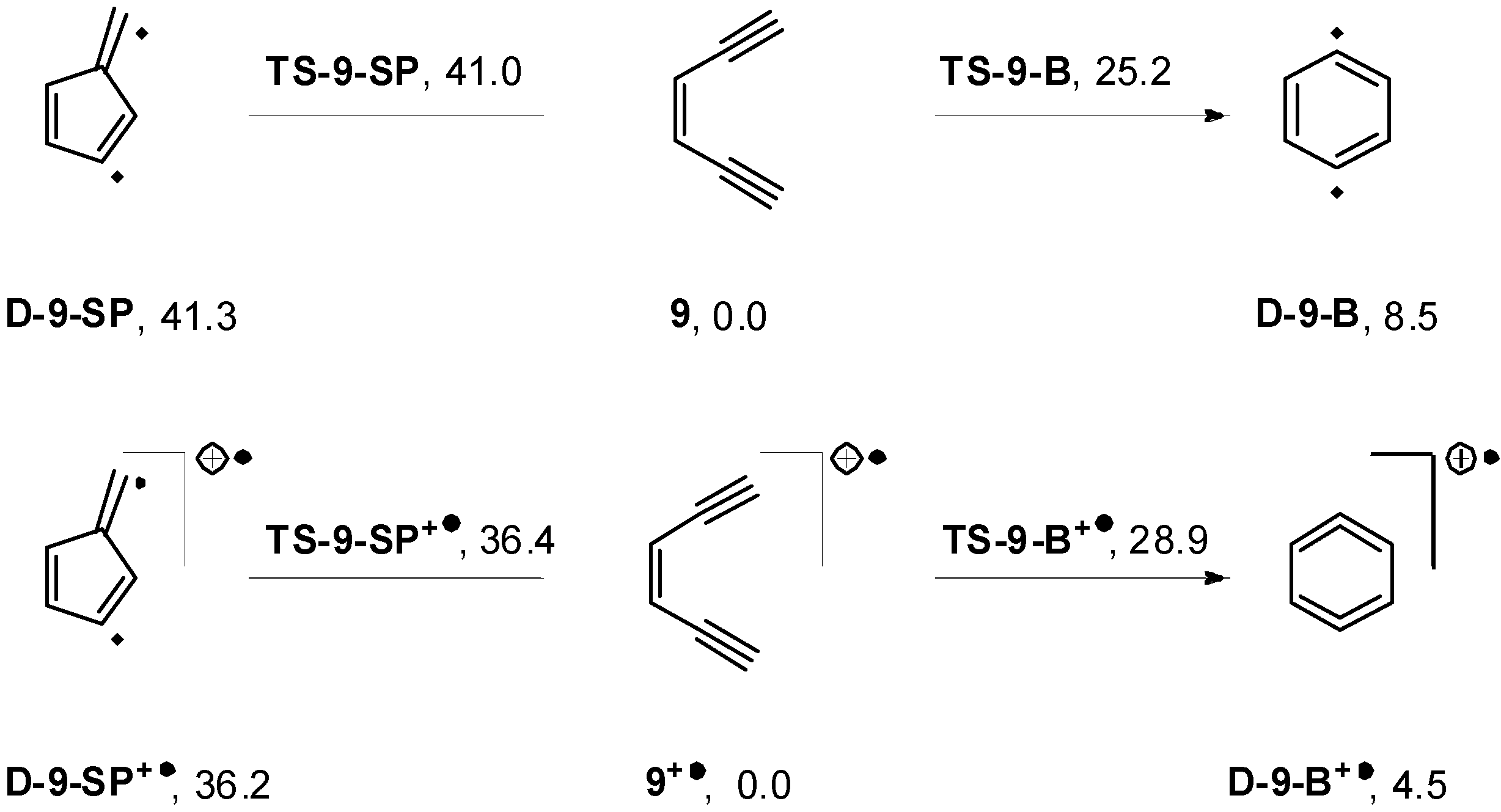
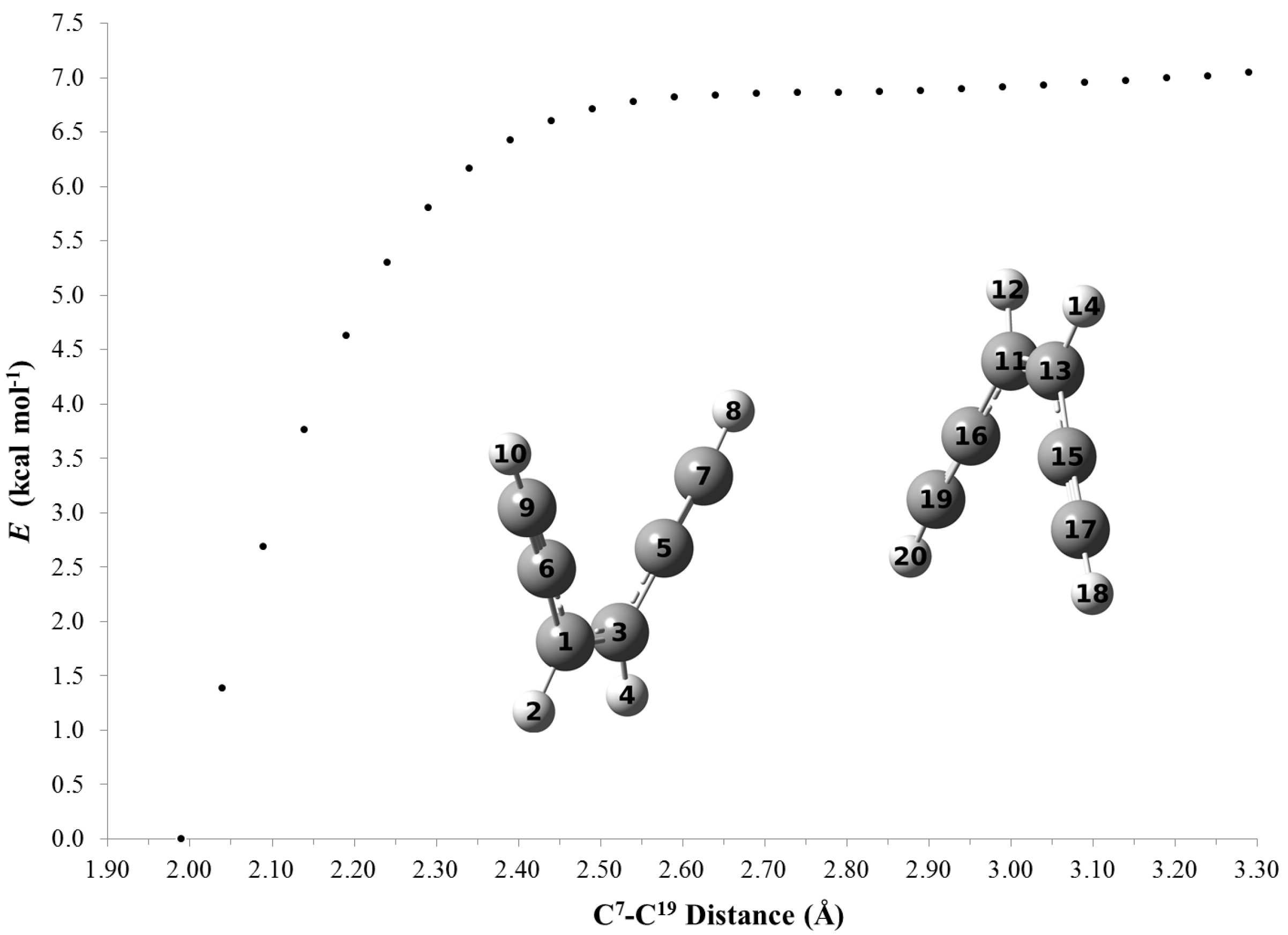
2.4. Computations


3. Experimental Section
3.1. General Information
3.2. 1,2-Bis(ferrocenylethynyl)cyclohexene (3)
3.3. Oxidation of 1,2-Bis(ferrocenylethynyl)cyclohexene 3 to the Monocation 3+
3.4. 1,2-Bis(ferrocenylethynyl)benzene (5)
3.5. Oxidation of 1,2-Bis(ferrocenylethynyl)benzene (5) to the Monocation 5+
3.6. 2-Ferrocenylethynyl-1-iodobenzene (6)
3.7. 2-(4-Nitrophenylethynyl)-1-ferrocenylethynylbenzene (8)
3.8. Oxidation of 2-(4-Nitrophenylethynyl)-1-ferrocenylethynylbenzene 8 to the Monocation 8+
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Joshi, M.C.; Rawat, D.S. Recent developments in enediyne chemistry. Chem. Biodivers. 2012, 9, 459–498. [Google Scholar]
- Schreiner, P.R.; Navarro-Vazquez, A.; Prall, M. Computational studies on the cyclizations of enediynes, enyne-allenes, and related polyunsaturated systems. Acc. Chem. Res. 2005, 38, 29–37. [Google Scholar]
- Poloukhtine, A.; Popik, V.V. Photoswitchable enediynes: Use of cyclopropenone as photocleavable masking group for the enediyne triple bond. Chem. Commun. 2005, 5, 617–619. [Google Scholar]
- Odedra, A.; Wu, C.J.; Pratap, T.B.; Huang, C.W.; Ran, Y.F.; Liu, R.S. Ruthenium-catalyzed aromatization of enediynes via highly regioselective nucleophilic additions on a π-alkyne functionality. A useful method for the synthesis of functionalized benzene derivatives. J. Am. Chem. Soc. 2005, 127, 3406–3412. [Google Scholar]
- Nath, M.; Pink, M.; Zaleski, J.M. Controlling both ground- and excited-State thermal barriers to Bergman cyclization with alkyne termini substitution. J. Am. Chem. Soc. 2005, 127, 478–479. [Google Scholar]
- Lewis, K.D.; Matzger, A.J. Bergman cyclization of sterically hindered substrates and observation of phenyl-shifted products. J. Am. Chem. Soc. 2005, 127, 9968–9969. [Google Scholar]
- Kovalenko, S.V.; Alabugin, I.V. Lysine–enediyne conjugates as photochemically triggered DNA double-strand cleavage agents. Chem. Commun. 2005, 1444–1446. [Google Scholar]
- Fouad, F.S.; Wright, J.M.; Plourde, G.; Purohit, A.D.; Wyatt, J.K.; El-Shafey, A.; Hynd, G.; Crasto, C.F.; Lin, Y.Q.; Jones, G.B. Synthesis and protein degradation capacity of photoactivated enediynes. J. Org. Chem. 2005, 70, 9789–9797. [Google Scholar]
- Bhattacharyya, S.; Pink, M.; Baik, M.H.; Zaleski, J.M. A unique approach to metal-induced Bergman cyclization: Long-range enediyne activation by ligand-to-metal charge transfer. Angew. Chem. Int. Ed. Engl. 2005, 44, 592–595. [Google Scholar]
- Basak, A.; Roy, S.K.; Mandal, S. Activation of macrocyclic enediynes by transannular cyclization. Angew. Chem. Int. Ed. Engl. 2005, 44, 132–135. [Google Scholar]
- Banfi, L.; Basso, A.; Guanti, G.; Riva, R. Design and synthesis of heterocycle fused enediyne prodrugs activable at will. ARKIVOC 2006, 2006, 261–275. [Google Scholar] [CrossRef]
- Kar, M.; Basak, A. Design, synthesis, and biological activity of unnatural enediynes and related analogues equipped with pH-dependent or phototriggering devices. Chem. Rev. 2007, 107, 2861–2890. [Google Scholar]
- Gredičak, M.; Jerić, I. Enediyne compounds—New promises in anticancer therapy. Acta Pharm. 2007, 57, 133–150. [Google Scholar]
- Liang, Z.-X. Complexity and simplicity in the biosynthesis of enediyne natural products. Nat. Prod. Rep. 2010, 27, 499–528. [Google Scholar]
- Perrin, C.L.; Reyes-Rodríguez, G.J. Reactivity of nucleophiles toward a p-benzyne derived from an enediyne. J. Phys. Org. Chem. 2013, 26, 206–210. [Google Scholar]
- Krupička, M.; Sander, W.; Marx, D. Mechanical manipulation of chemical reactions: Reactivity switching of Bergman cyclizations. J. Phys. Chem. Lett. 2014, 5, 905–909. [Google Scholar]
- Jones, R.R.; Bergman, R.G. p-Benzyne. Generation as an intermediate in a thermal isomerization reaction and trapping evidence for the 1,4-benzenediyl structure. J. Am. Chem. Soc. 1972, 94, 660–661. [Google Scholar]
- Lockhart, T.P.; Comita, P.B.; Bergman, R.G. Kinetic evidence for the formation of discrete 1,4-dehydrobenzene intermediates. Trapping by inter- and intramolecular hydrogen atom transfer and observation of high-temperature CIDNP. J. Am. Chem. Soc. 1981, 103, 4082–4090. [Google Scholar]
- Lockhart, T.P.; Bergman, R.G. Evidence for the reactive spin state of 1,4-dehydrobenzenes. J. Am. Chem. Soc. 1981, 103, 4091–4096. [Google Scholar]
- Lindh, R.; Persson, B.J. Ab initio study of the Bergman reaction: The autoaromatization of hex-3-ene-1,5-diyne. J. Am. Chem. Soc. 1994, 116, 4963–4969. [Google Scholar]
- Warner, B.P.; Millar, S.P.; Broene, R.D.; Buchwald, S.L. Controlled acceleration and inhibition of Bergman cyclization by metal chlorides. Science 1995, 269, 814–816. [Google Scholar]
- König, B.; Hollnagel, H.; Ahrens, B.; Jones, P.G. Activation of macrocyclicbiaryl-enediynes by metal ion coordination. Angew. Chem. Int. Ed. Engl. 1995, 34, 2538–2540. [Google Scholar]
- König, B.; Pitsch, W.; Thondorf, I. Synthesis, structure, and reactivity of enediyne macrocycles. J. Org. Chem. 1996, 61, 4258–4261. [Google Scholar]
- McPhee, M.M.; Kerwin, S.M. Synthesis and metal ion binding studies of enediyne—Containing crown ethers. J. Org. Chem. 1996, 61, 9385–9393. [Google Scholar]
- Basak, A.; Shain, J.C. Synthesis and thermal behavior of a novel diazaenediyne and its copper(II) complex. Tetrahedron Lett. 1998, 39, 1623–1624. [Google Scholar]
- Basak, A.; Shain, J.C. Synthesis and thermal reactivity of a novel macrocyclicenediyne and its copper(II) complex. Tetrahedron Lett. 1998, 39, 3029–3030. [Google Scholar]
- Basak, A.; Shain, J.C.; Khamrai, U.K.; Rudra, K.R.; Basak, A. Nitrogen substituted cyclic enediynes: Synthesis, thermal reactivity and complexation with metal ions. J. Chem. Soc. Perkin Trans. 1 2000, 1955–1964. [Google Scholar]
- Maier, M.E. Design of enediyne prodrugs. Synlett 1995, 13–26. [Google Scholar]
- Grissom, J.W.; Gunawardena, G.U.; Klingberg, D.; Huang, D. The chemistry of enediynes, enyne-allenes and related compounds. Tetrahedron 1996, 52, 6453–6513. [Google Scholar]
- Benites, P.J.; Rawat, D.S.; Zaleski, J.M. Metalloenediynes: Ligand field control of thermal Bergman cyclization reactions. J. Am. Chem. Soc. 2000, 122, 7208–7217. [Google Scholar]
- Maier, M.E.; Greiner, B. Synthesis and reactivity of a p-methoxyphenyl-substituted enediyne. A case of electronic influence on the rate of the Bergman cycloaromatization. Liebigs Ann. Chem. 1992, 855–861. [Google Scholar]
- Nicolaou, K.C.; Liu, A.; Zeng, Z.; McComb, S. Redox-controlled Bergman cycloaromatizations. Designed enediynes with DNA-cleaving properties and antitumor activity. J. Am. Chem. Soc. 1992, 114, 9279–9282. [Google Scholar]
- Semmelhack, M.F.; Neu, T.; Foubelo, F. Arene 1,4-diradical formation from o-dialkynylarenes. Tetrahedron Lett. 1992, 33, 3277–3280. [Google Scholar]
- Semmelhack, M.F.; Neu, T. Arene 1,4-diradical formation from o-dialkynylarenes. J. Org. Chem. 1994, 59, 5038–5047. [Google Scholar]
- Schmittel, M.; Kiau, S. Polar effects in the transition state of the Bergman cyclization. Chem. Lett. 1995, 953–954. [Google Scholar]
- Alabugin, I.V.; Manoharan, M.; Kovalenko, S.V. Tuning rate of the Bergman cyclization of benzannelated enediynes with ortho substituents. Org. Lett. 2002, 4, 1119–1122. [Google Scholar]
- Alabugin, I.V.; Manoharan, M. Reactant destabilization in the Bergman cyclization and rational design of light- and pH-activated enediynes. J. Phys. Chem. A 2003, 107, 3363–3371. [Google Scholar]
- Schmittel, M.; Viola, G.; Dall’Acqua, F.; Morbach, G. A novel concept to activate enediynes for DNA cleavage. Chem. Commun. 2003, 646–647. [Google Scholar]
- Schmittel, M.; Röck, M. Reaction of enol cation radicals in the presence of nucleophiles. Chem. Ber. 1992, 125, 1611–1620. [Google Scholar]
- Schmittel, M.; von Seggern, H. Electron-transfer catalyzed reactions 3. Ketene diene [4+2] cycloaddition products via cation radical initiated Diels-Alder reaction or vinylcyclobutanone rearrangement. J. Am. Chem. Soc. 1993, 115, 2165–2177. [Google Scholar]
- Rodriguez, J.G.; Onate, A.; Martin-Villamil, R.M.; Fonseca, J. A practical synthesis of ethynylferrocene from ferrocenecarboxaldehyde: Structure of 1,4-diferrocenyl-1,3-butadiyne. J. Organomet. Chem. 1996, 513, 71–76. [Google Scholar]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. Convenient synthesis of acetylenes. Catalytic substitutions of acetylenic hydrogen with bromo alkenes, iodoarenes, and bromopyridines. Tetrahedron Lett. 1975, 50, 4467–4470. [Google Scholar]
- De Meijere, A.; Meyer, F.E. Fine feathers make fine birds: The Heck reaction in modern garb. Angew. Chem. Int. Ed. Engl. 1994, 33, 2379–2411. [Google Scholar]
- Rossi, R.; Carpita, A.; Bellina, F. Palladium- and/or copper-mediated cross-coupling reactions between 1-alkynes and vinyl, aryl, 1 alkynyl, 1,2-propadienyl, propargyl and allylic halides or related compounds. A review. Org. Prep. Proc. Int. 1995, 27, 127–160. [Google Scholar]
- Heck, R.F. Palladium Reagents in Organic Syntheses; Academic Press: London, UK, 1985. [Google Scholar]
- Voigt, K.; von Zezschwitz, P.; Rosauer, K.; Lansky, A.; Adams, A.; Reiser, O.; de Meijere, A. The twofold Heck reaction on 1,2-dihalocycloalkenes and subsequent 6π-electrocyclization of the resulting (E,Z,E)-1,3,5-hexatrienes. A new formal {2+2+2}-assembly of six-membered rings. Eur. J. Org. Chem. 1998, 1521–1534. [Google Scholar]
- Plevyak, J.E.; Dickerson, J.E.; Heck, R.F. Selective palladium-catalyzed vinylic substitutions with bromoiodo aromatics. J. Org. Chem. 1979, 44, 4078–4080. [Google Scholar]
- Takahashi, S.; Kuroyama, Y.; Sonogashira, K.; Hagihara, N. A convenient synthesis of ethynylarenes and diethynylarenes. Synthesis 1980, 627–630. [Google Scholar]
- Thorand, S.; Krause, N. Improved procedures for the palladium-catalyzed coupling of terminal alkynes with aryl bromides (Sonogashira coupling). J. Org. Chem. 1998, 63, 8551–8553. [Google Scholar]
- Diallo, A.K.; Absalon, C.; Ruiz, J.; Astruc, D. Ferrocenyl-terminated redox stars: Synthesis and electrostatic effects in mixed-valence stabilization. J. Am. Chem. Soc. 2011, 133, 629–641. [Google Scholar]
- Prall, M.; Wittkopp, A.; Schreiner, P.R. Can Fulvenes Form from Enediynes? A Systematic high-level computational study on parent and benzannelated enediyne and enyne–allene cyclizations. J. Phys. Chem. A 2001, 105, 9265–9274. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Engels, B.; Lennartz, C.; Hanrath, M.; Schmittel, M.; Strittmatter, M. A theoretical and experimental study on the regioselectivity of biradical cyclizations in enyne-allenes: Influence of substituents on the switch from the Myers-Saito to the novel C2-C6 cyclization. Angew. Chem. Int. Ed. Engl. 1998, 37, 1960–1963. [Google Scholar]
- Stahl, F.; Moran, D.; Schleyer, P.V.; Prall, M.; Schreiner, P.R. Aromaticity of the Bergman, Myers-Saito, Schmittel, and directly related cyclizations of enediynes. J. Org. Chem. 2002, 67, 1453–1461. [Google Scholar]
- Wenthold, P.G.; Lipton, M.A. A density functional molecular orbital study of the C2–C7 and C2–C6 cyclization pathways of 1,2,4-heptatrien-6-ynes. The role of benzannulation. J. Am. Chem. Soc. 2000, 122, 9265–9270. [Google Scholar]
- Vavilala, C.; Byrne, N.; Kraml, C.M.; Ho, D.M.; Pascal, R.A. Thermal C1–C5 diradical cyclization of enediynes. J. Am. Chem. Soc. 2008, 130, 13549–13551. [Google Scholar]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cinar, M.E.; Morbach, G.; Schmittel, M. Thermal Reactivity of Neutral and Oxidized Ferrocenyl-Substituted Enediynes. Molecules 2014, 19, 18399-18413. https://doi.org/10.3390/molecules191118399
Cinar ME, Morbach G, Schmittel M. Thermal Reactivity of Neutral and Oxidized Ferrocenyl-Substituted Enediynes. Molecules. 2014; 19(11):18399-18413. https://doi.org/10.3390/molecules191118399
Chicago/Turabian StyleCinar, Mehmet Emin, Guido Morbach, and Michael Schmittel. 2014. "Thermal Reactivity of Neutral and Oxidized Ferrocenyl-Substituted Enediynes" Molecules 19, no. 11: 18399-18413. https://doi.org/10.3390/molecules191118399