Quality Evaluation of Pharmaceutical Formulations Containing Hydrochlorothiazide

Abstract
:1. Introduction
2. Results and Discussion
2.1. Quality Control Tests

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test | Specifications | Lab 1 (Ref) | Lab 2 (Gen) | Lab 3 (Sim) | Lab 4 (Sim) | Lab 5 (Comp) | Lab 6 (Comp) |
|---|---|---|---|---|---|---|---|
| Identification | Identification by TLC | Approved | Approved | Approved | Approved | Approved | Approved |
| Hardness | Minimum 30 N | >32 N | >45 N | >55 N | >30 N | not applicable | not applicable |
| Disintegration | Disintegrates in water at 37 °C within 30 min | <1 min | <1 min | <1 min | <1 min | <1 min | <1 min |
| Weight | −7.5% < X < +7.5% | Approved | Approved | Approved | Approved | Approved | Reproved |
| Friability | Maximum 1.5% | 0.04% | 0.06% | 0.006% | 0.11% | not applicable | not applicable |
| Uniformity of dosage units | 85.0 < X < 115.0% VR, RSD < 6.0% | Average = 220.75% RSD = 1.12% | Average = 243.76% RSD = 2.76% | Average = 203.85% RSD = 1.1% | Average = 172.75% RSD = 1.35% | Average = 183.60% RSD = 3.13% | Average = 198.85% RSD = 3.7% |
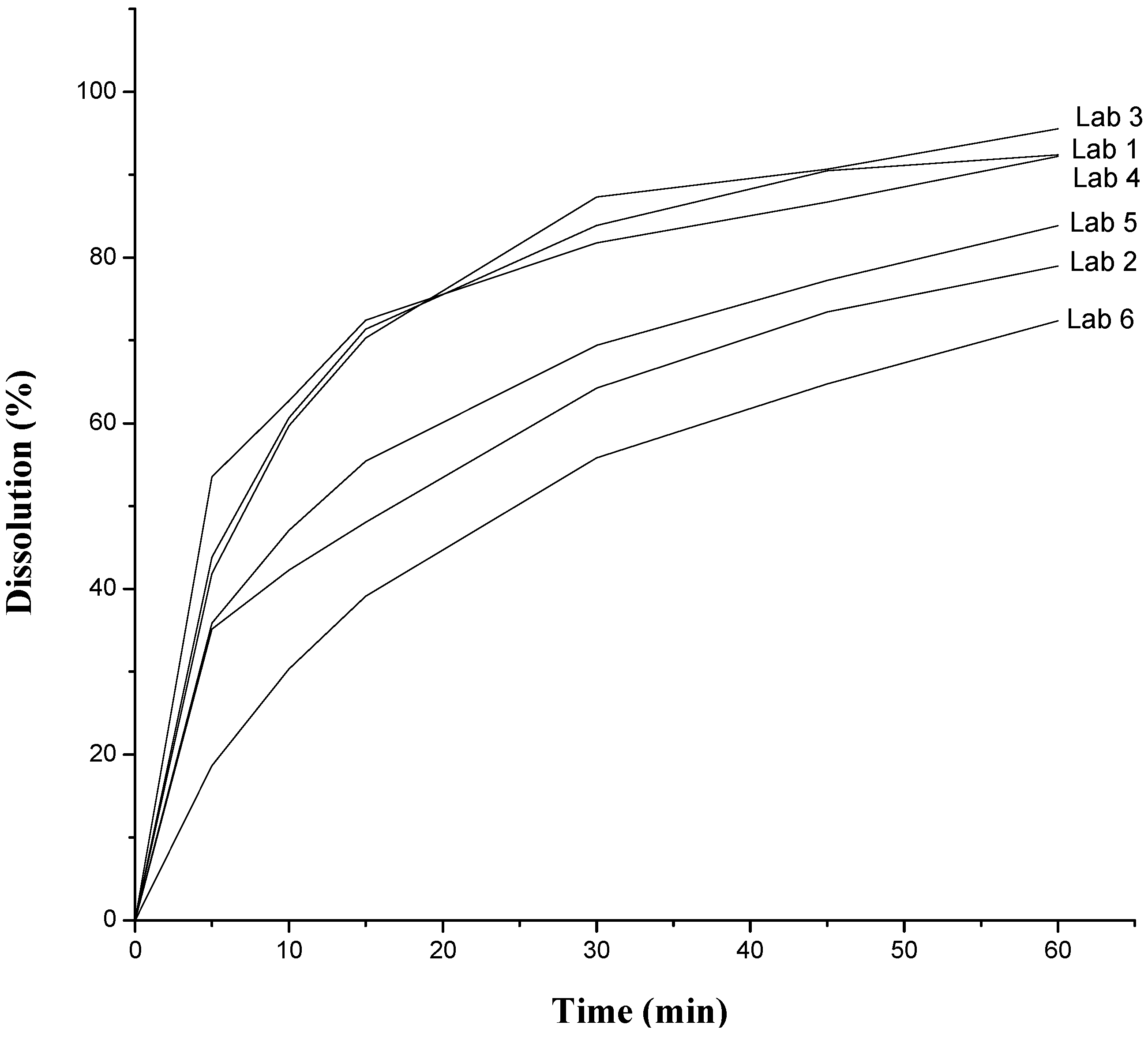
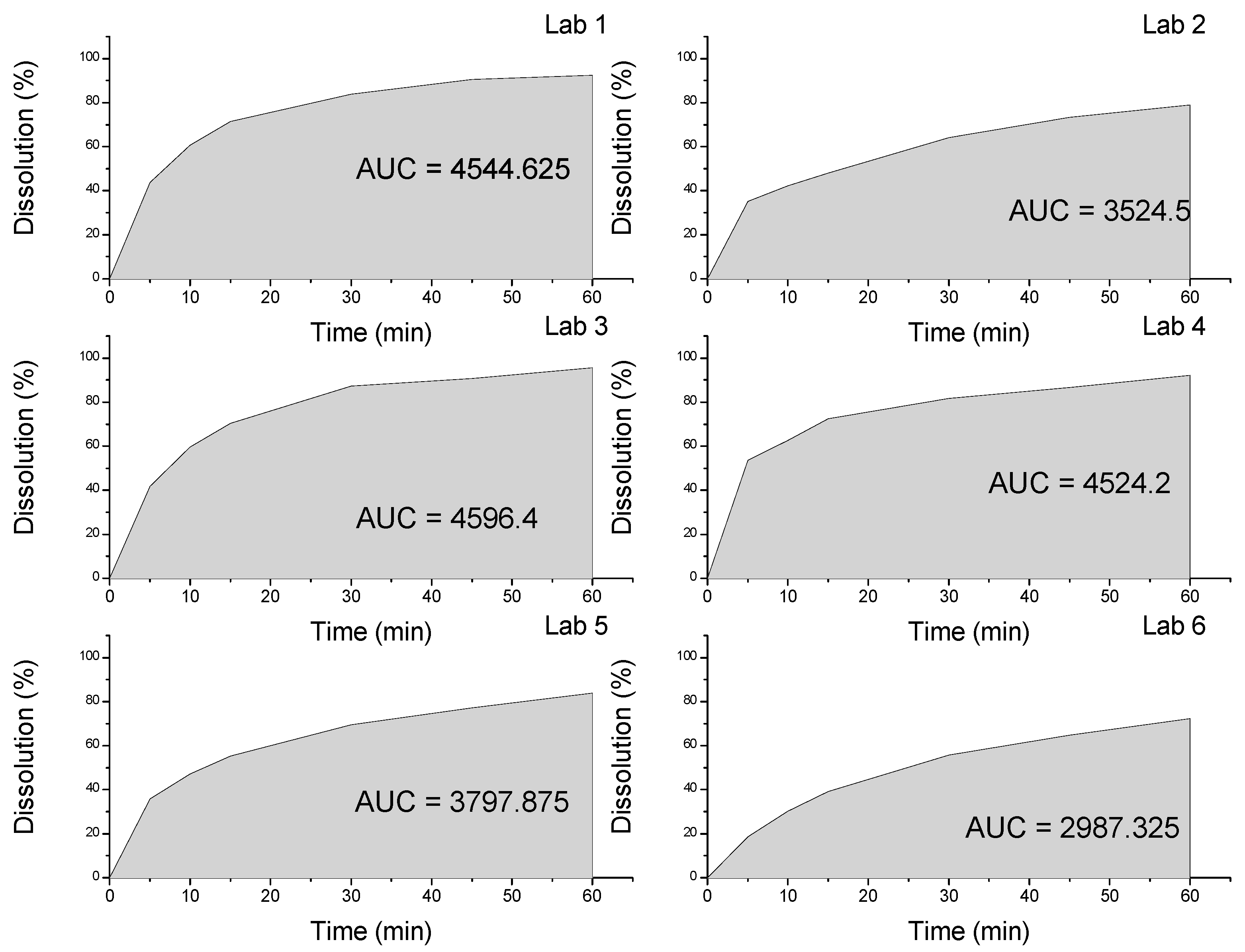
| Dissolution | 60% (Q) in 30 min | 83.89% | 64.22% | 87.28% | 81.77% | 33.27% | 55.83% |
| Assay | Minimum 93.0%LV and maximum 107.0%LV | 94.88% | 98.43% | 100.57% | 99.72% | 101.28% | 97.87% |
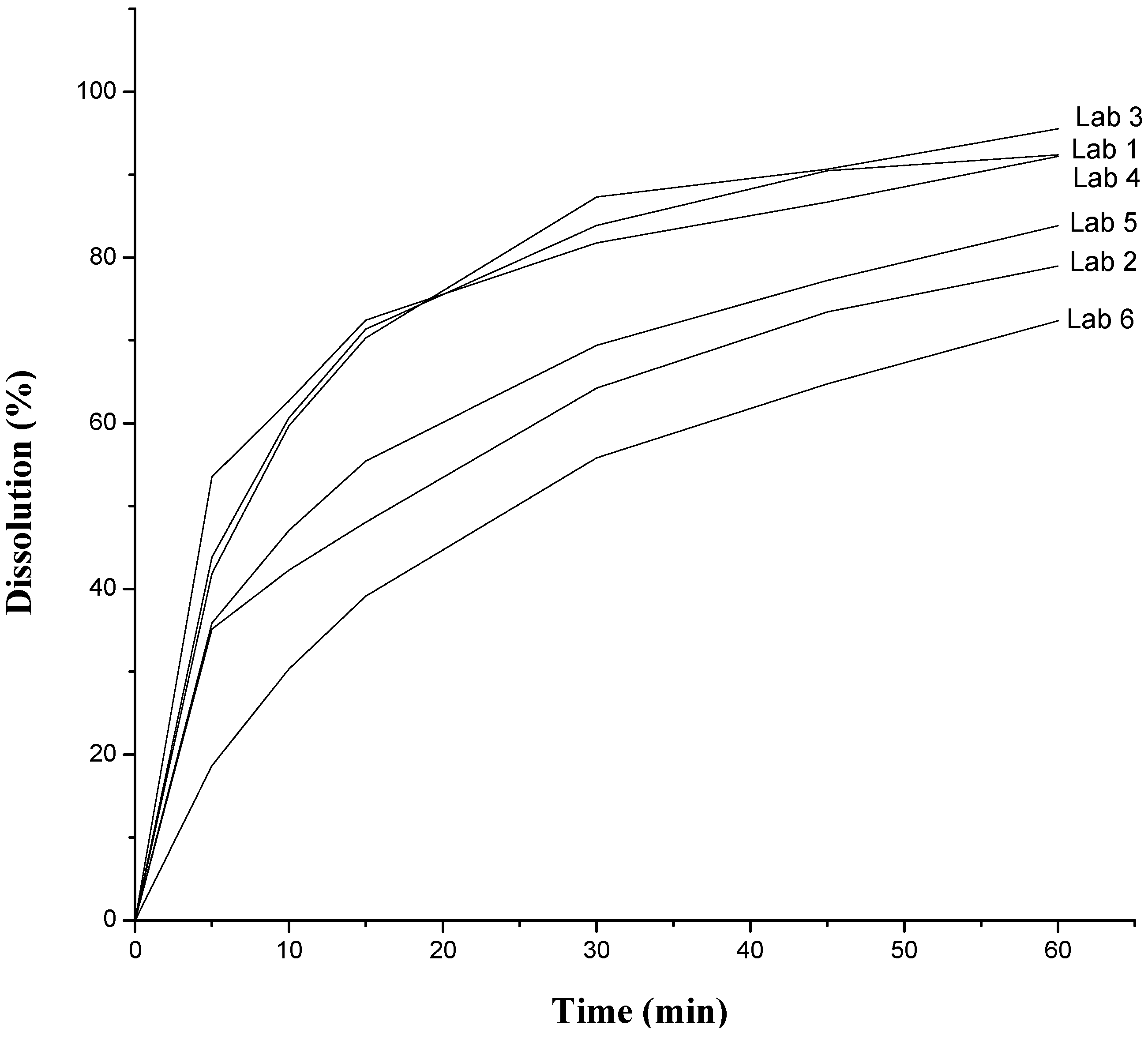
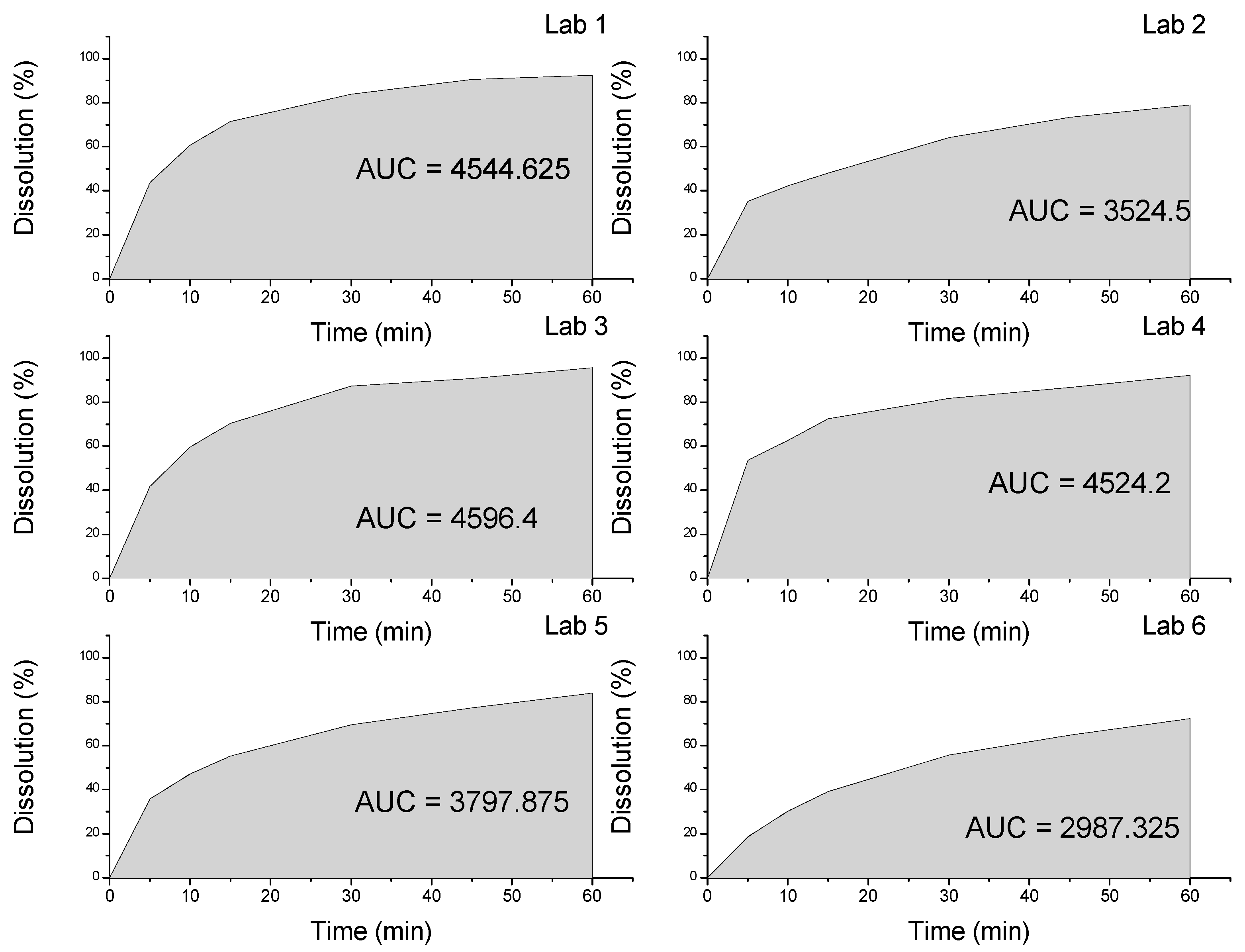
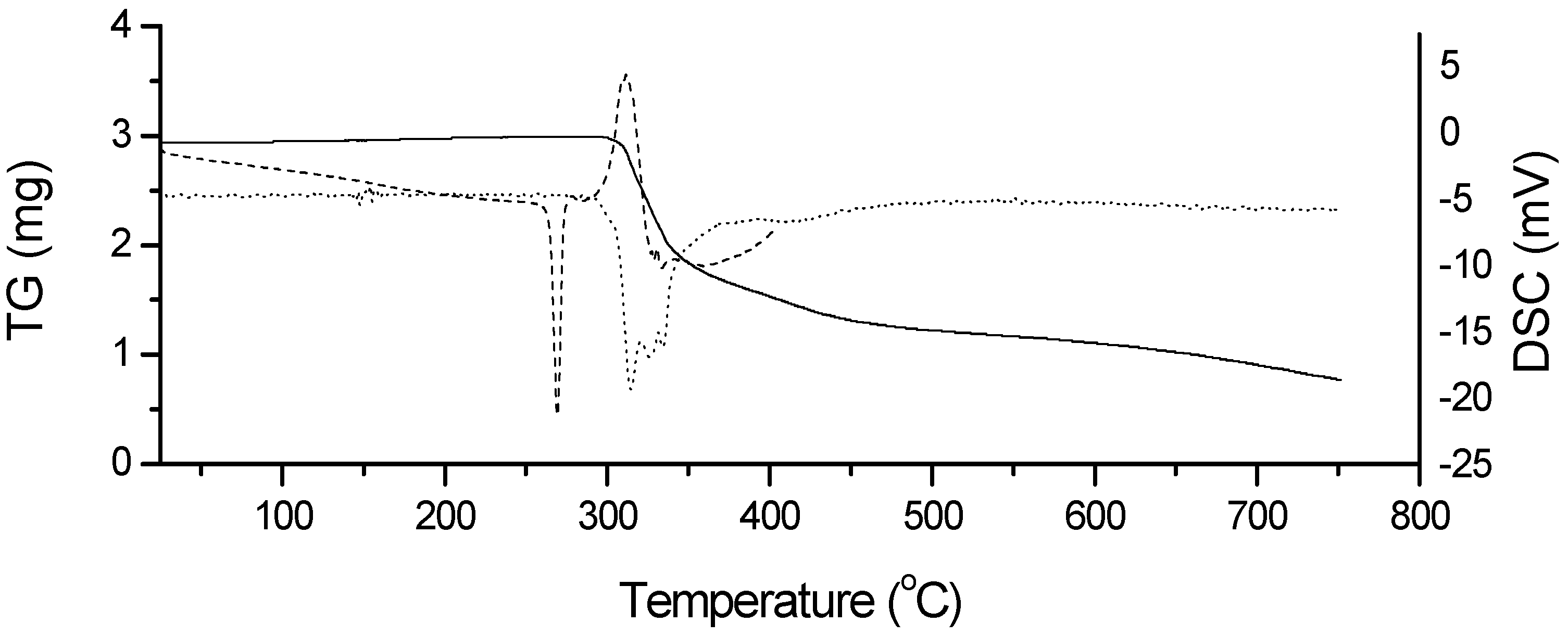
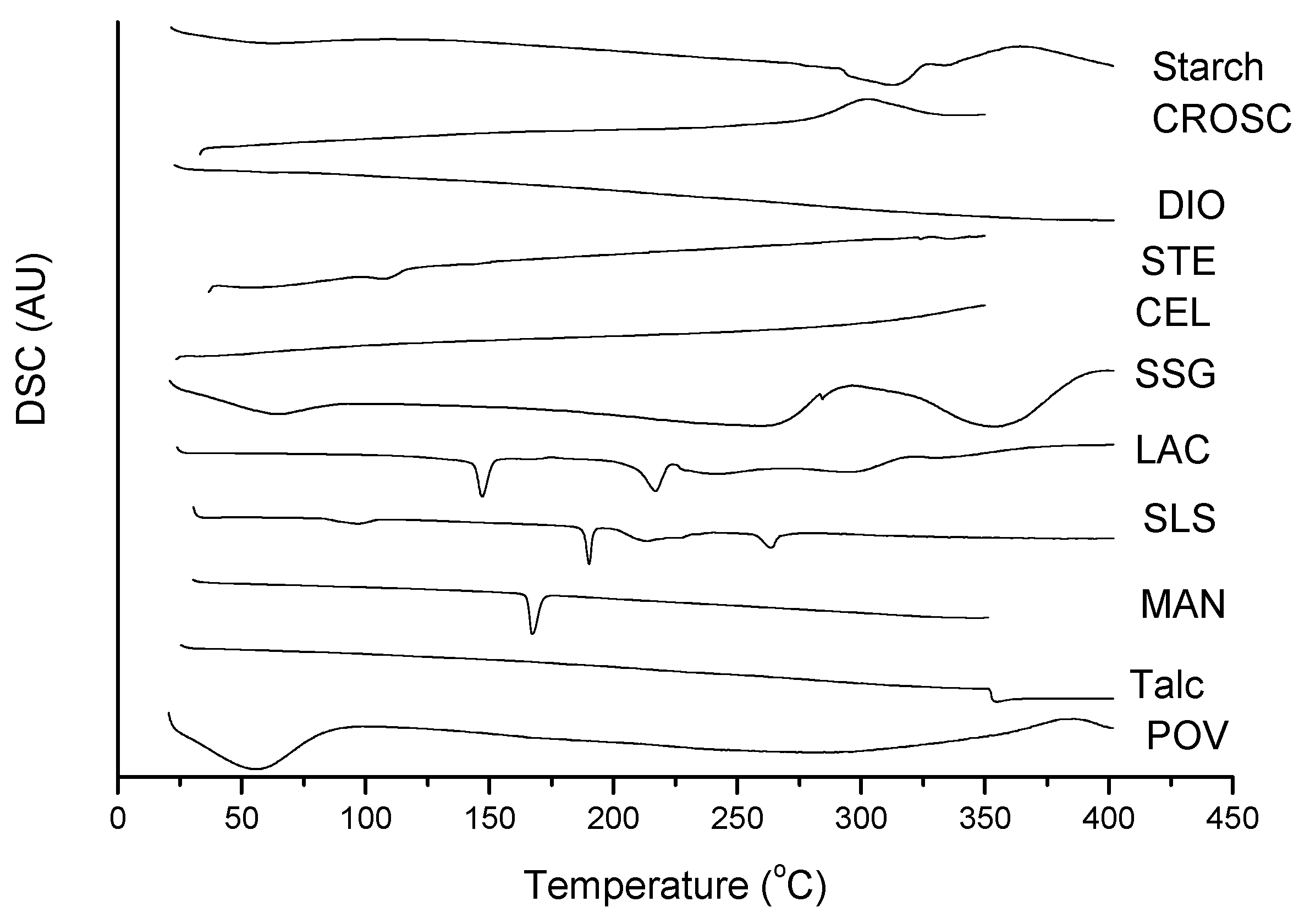
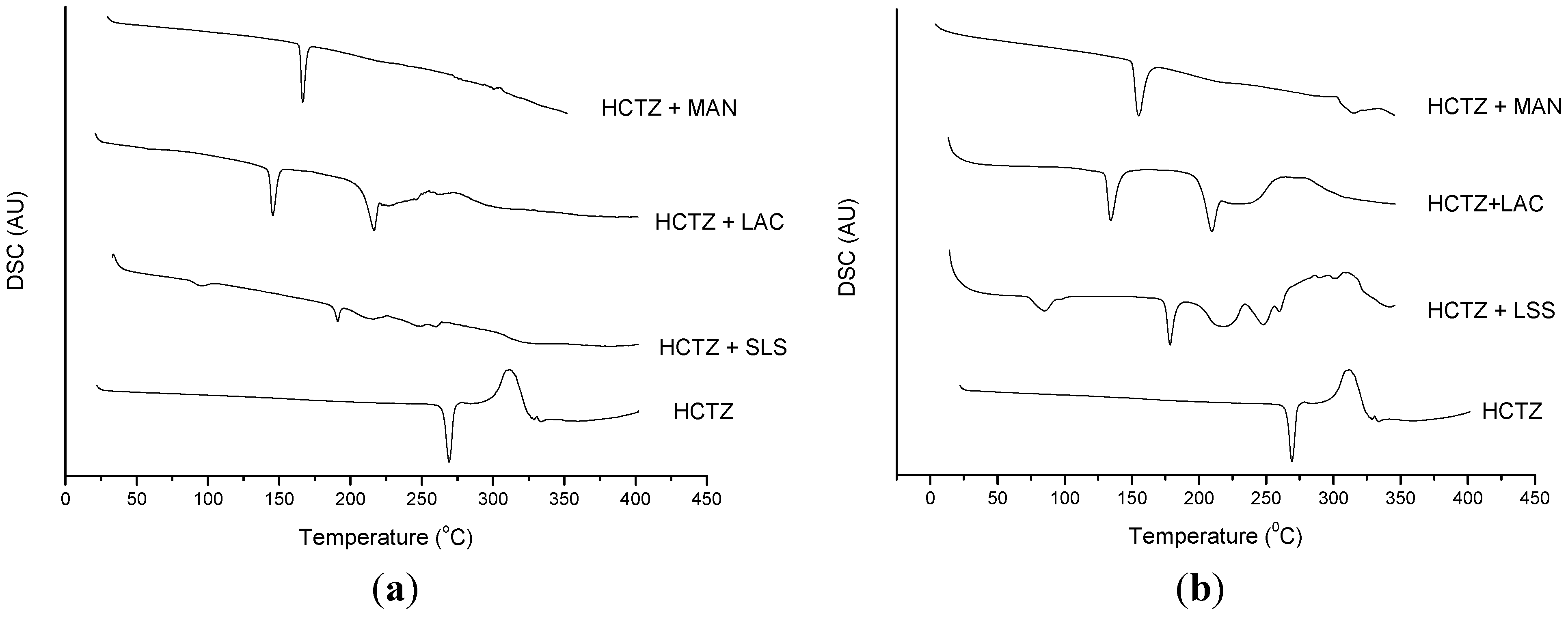
2.2. Thermal Characterization and Compatibility Study of Pharmaceutical Formulation
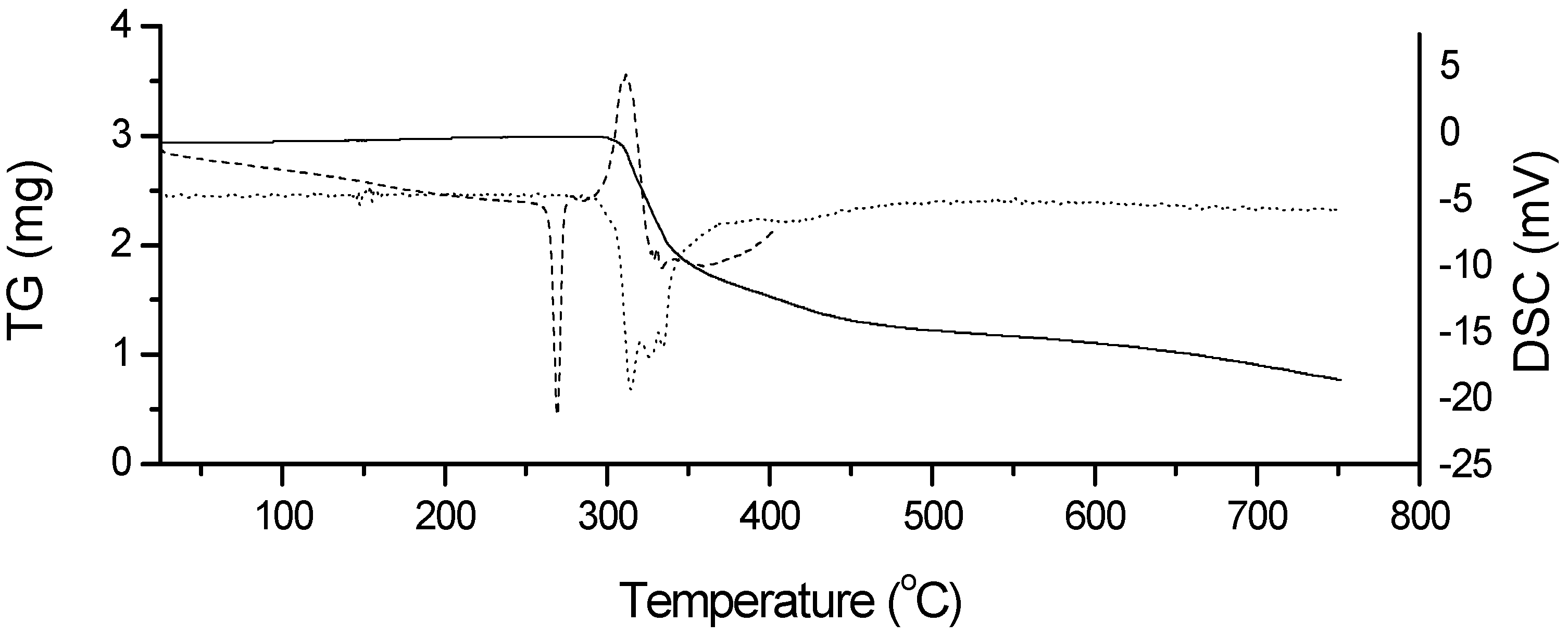
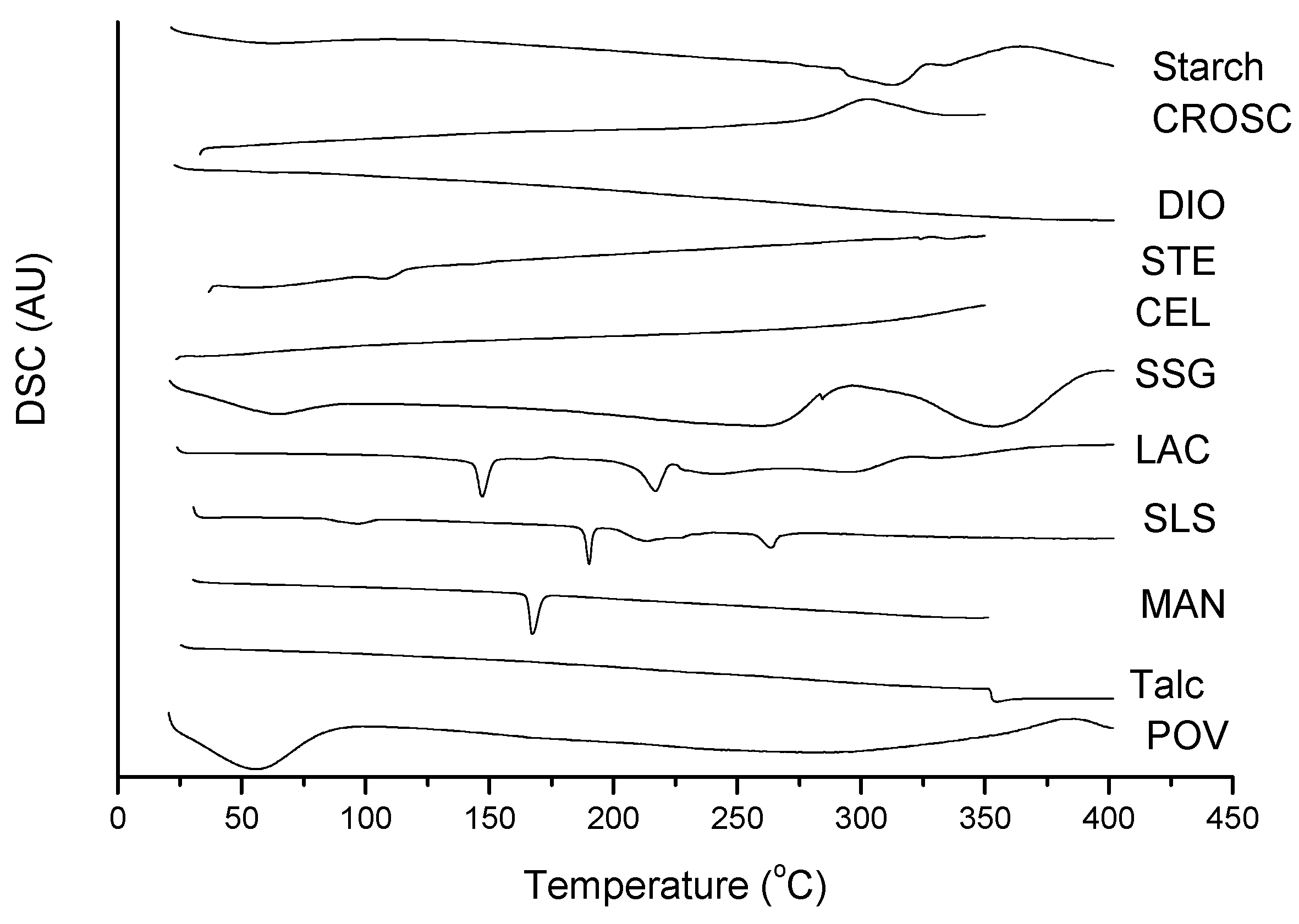
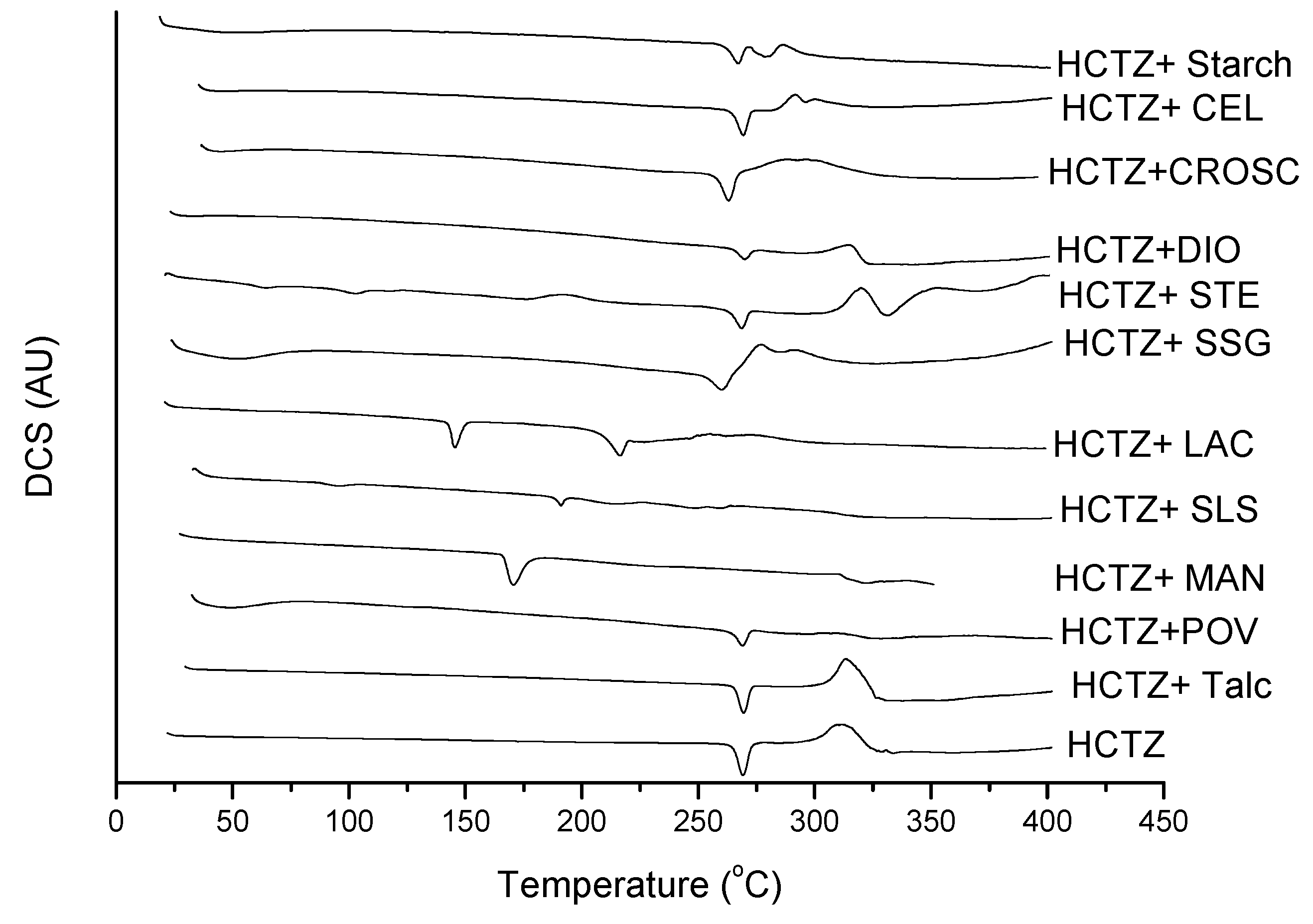

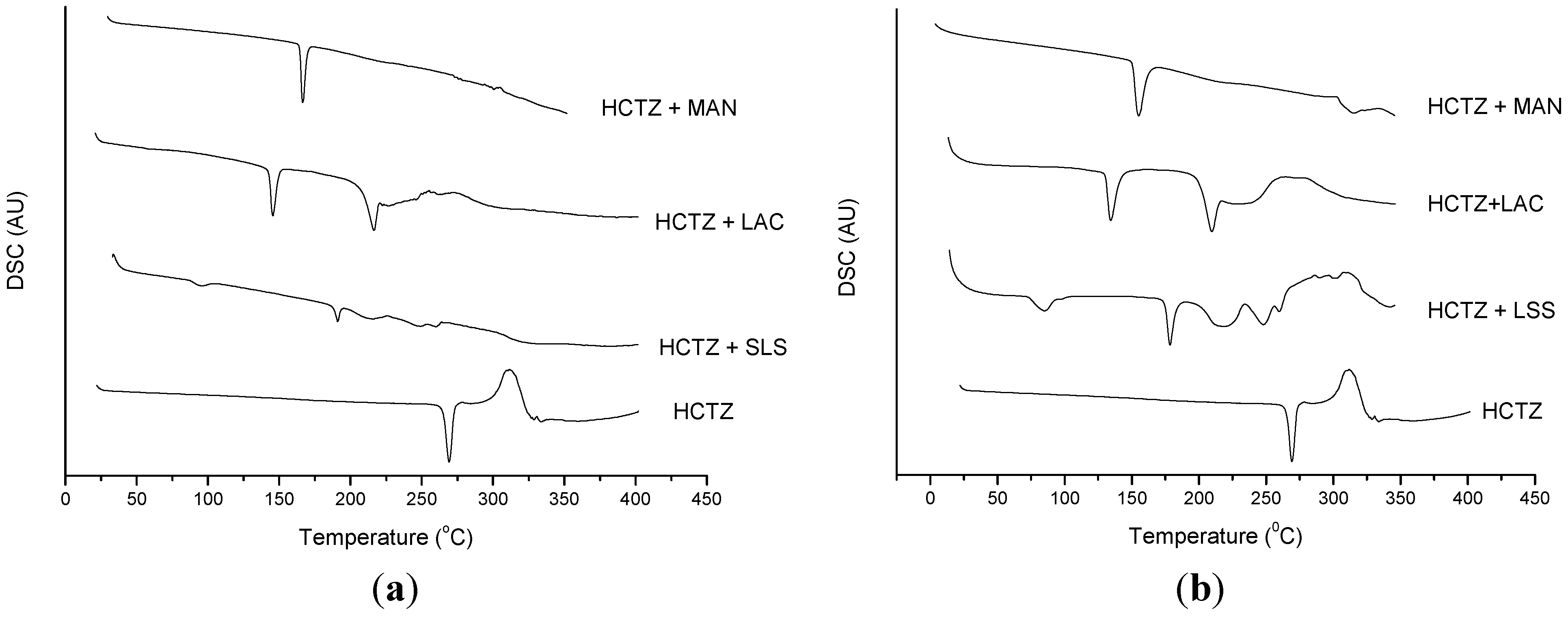
3. Experimental
3.1. Origin of Samples
| Laboratory | Pharmaceutical Formulation | Type Drug |
|---|---|---|
| Lab 1 | HCTZ, lactose monohydrate, starch, and magnesium stearate. | Reference |
| Lab 2 | HCTZ, sodium lauryl sulfate, croscarmellose sodium, crospovidone, sodium starch glycolate, lactose, microcrystalline cellulose, magnesium stearate, colloidal silicon dioxide. | Generic |
| Lab 3 | HCTZ, microcrystalline cellulose, colloidal silicon dioxide, lactose, magnesium stearate, sodium starch glycolate. | Similar |
| Lab 4 | HCTZ, starch, magnesium stearate, mannitol, talc, povidone (PVP) and ethanol. | Similar |
| Lab 5 | HCTZ, magnesium stearate, Aerosil® (colloidal silicon dioxide), talc, corn starch, sodium lauryl sulfate. | Compounded |
| Lab 6 | HCTZ, Aerosil® (colloidal silicon dioxide), microcrystalline cellulose and starch. | Compounded |
3.2. Quality Control Tests
3.2.1. Assay
3.2.2. Dissolution and Dissolution Profile
- Dissolution medium: 0.1 M hydrochloric acid, 900 mL.
- Apparatus 1 (basket): 100 rpm.
- Collection times in the dissolution profile: 5, 10, 15, 30, 45 and 60 min.
- Procedure: at the specified times, 15 mL aliquots of dissolution medium were taken, and filtered through filter paper. Then, 10 mL of the filtrate was pipetted into a 50 mL volumetric flask, and made up to the final volume with 0.1 M hydrochloric acid. The absorbance was measured at 272 nm using the same solvent for zero adjustment. The amount of C7H8ClN3O4S2 dissolved in the medium was calculated by comparing the readings obtained with the standard solution of hydrochlorothiazide at 0.01 mg·mL−1 prepared in the same solvent.
- Tolerance: No less than 60% (Q) of the labeled amount of C7H8ClN3O4S2 should dissolve within 30 min.
3.3. Thermal Characterization of Drugs and Compatibility Study of Pharmaceutical Formulation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gilman, A.G. Goodman & Gilman’s—The Pharmacological Basis of Therapeutics, 11th ed.; McGraw-Hill Interamericana: Rio de Janeiro, Brazil, 2006; p. 1821. [Google Scholar]
- National Agency of Sanitary Vigilance. Resolution—RDC No. 49 of 23 November 2010. In Brazilian Pharmacopoeia, 5th ed.; National Agency of Sanitary Vigilance: Brasilia, Brazil, 2010. [Google Scholar]
- Amidon, G.L.; Kasim, N.A.; Whitehouse, M.; Ramachandran, C.; Bermejo, M.; Lennernas, H.; Hussain, A.S.; Junginger, H.E.; Stavchansky, S.A.; Midha, K.K.; et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 2004, 1, 85–96. [Google Scholar] [CrossRef]
- Aulton, M.E. Delineation of Dosage Forms, 2nd ed.; ARTMED: Porto Alegre, Brazil, 2006. [Google Scholar]
- Mahle, F.; Goelser, F.; Adriano, J.; Felippe, M.; Vier, N.; Carli, R.B.G.; Rosa, T.; Couto, A.G.; Lucinda-Silva, R.M. Evaluation of the dissolution profile of hydrochlorothiazide tablets marketed in Brazil. J. Basic Appl. Pharm. Sci. 2007, 28, 265–271. [Google Scholar]
- Brazil. National Agency of Sanitary Vigilance. Drugs Reference. Available online: http://www.anvisa.gov.br/medicamentos/referencia/lmr_a.pdf (accessed on 1 February 2014).
- National Agency of Sanitary Vigilance. Resolution—RDC No. 67, Official Gazette, Executive: Brasilia, Brazil, 2007; Section 1, Good Practices for Handling of Magistral Preparations and Workshop for Human Use in Pharmacies. 62.
- National Agency of Sanitary Vigilance. Resolution—RDC nº 31, Official Gazette, Executive: Brasilia, Brazil, 2010; Section 1, Carrying out of Pharmaceutical Equivalence and Comparative Dissolution Profile. 36.
- National Agency of Sanitary Vigilance. Resolution—RDC nº 17, Official Gazette, Executive: Brasilia, Brazil, 2007; Technical Regulation of Similar Drugs, 2007.
- National Agency of Sanitary Vigilance. Resolution—RDC nº 16, Official Gazette, Executive: Brasilia, Brazil, 2007; Technical Regulation of Generic Drugs, 2007.
- Gasparotto, F.S. Factors Related to the Synthesis of Raw Materials that may Alter the Bioavailability of Generic Drugs. Available online: http://www.lume.ufrgs.br/bitstream/handle/10183/5395/000469676.pdf?sequence=1 (accessed on 1 February 2014).
- ICH. Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances. Available online: http://www.ich.org/products/guidelines/quality/quality-single/article/specifications-test-procedures-and-acceptance-criteria-for-new-drug-substances-and-new-drug-produc.html (accessed on 1 February 2014).
- Guidance for Industry: ANDAs: Pharmaceutical Solid Polymorphism: Chemistry, Manufacturing, and Controls Information. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072866.pdf (accessed on 1 February 2014).
- National Agency of Sanitary Vigilance. Resolution RE No. 1, Official Gazette, Executive: Brasilia, Brazil, 2005; Publication of the Guide to Realization of Stability Studies, 2005.
- ICH. Stability Testing of New Drug Substances and Products. Available online: http://www.ich.org/products/guidelines/quality/quality-single/article/stability-testing-of-new-drug-substances-and-products.html (accessed on 1 February 2014).
- Oliveira, M.A.; Yoshida, M.I.; Gomes, E.C.L. Thermal analysis applied to drugs and pharmaceutical formulations in pharmaceutical industry. Quím. Nova 2011, 37, 1224–1230. [Google Scholar] [CrossRef]
- Oliveira, M.A.; Gomes, E.C.L.; Mussel, W.N.; Soares, C.D.V.; Pianetti, G.A.; Yoshida, M.I. Análise térmica aplicada à caracterização de sinvastatina em formulações farmacêuticas. Quím. Nova 2010, 33, 1653–1657. [Google Scholar] [CrossRef]
- Yoshida, M.I.; Oliveira, M.A.; Lacerda, C.D.; Valotto, R.S.; Bonella, A.F. Thermal characterization of atorvastatin and compatibility studies of pharmaceutical formulations. Braz. J. Thermal Anal. 2012, 1, 73–78. [Google Scholar]
- Yoshida, M.I.; Oliveira, M.A.; Gomes, E.C.L.; Mussel, W.N.; Castro, W.V.; Soares, C.D.V. Thermal characterization of lovastatin in pharmaceutical formulations. J. Thermal Anal. Calor. 2011, 106, 657–664. [Google Scholar] [CrossRef]
- Yoshida, M.I.; Gomes, E.C.L.; Soares, C.D.V.; Cunha, A.F.; Oliveira, M.A. Thermal analysis applied to verapamil hydrochloride characterization in pharmaceutical formulations. Molecules 2010, 15, 2439–2452. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.S.; Bharate, S.B.; Bajaj, A.N. Interactions and incompatibilities of pharmaceutical excipients with active pharmaceutical ingredients: A comprehensive review. J. Excip. Food Chem. 2010, 1, 3–26. [Google Scholar]
- Thermalcal 15 dez. 2009. Calorimetry: The Universal Detector. Available online: http://thermalcal.com (accessed on 1 February 2014).
- Anderson, N.H.; Bauer, M.; Boussac, N.; Khan-Malek, R.; Munden, P.; Sardaro, M. An evaluation of fit factors and dissolution efficiency for the comparison of in vitro dissolution profiles. J. Pharm. Biomed. Anal. 1998, 17, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.A.; Lacerda, C.D.; Bonella, A.F. Developing methods to compare tablet formulations of atorvastatin. Braz. J. Pharm. Sci. 2012, 48, 1–10. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Oliveira, M.A.; Yoshida, M.I.; Silva, D.C.G.M.d. Quality Evaluation of Pharmaceutical Formulations Containing Hydrochlorothiazide. Molecules 2014, 19, 16824-16836. https://doi.org/10.3390/molecules191016824
De Oliveira MA, Yoshida MI, Silva DCGMd. Quality Evaluation of Pharmaceutical Formulations Containing Hydrochlorothiazide. Molecules. 2014; 19(10):16824-16836. https://doi.org/10.3390/molecules191016824
Chicago/Turabian StyleDe Oliveira, Marcelo Antonio, Maria Irene Yoshida, and Daphne Carina Gonçalves Monteiro da Silva. 2014. "Quality Evaluation of Pharmaceutical Formulations Containing Hydrochlorothiazide" Molecules 19, no. 10: 16824-16836. https://doi.org/10.3390/molecules191016824