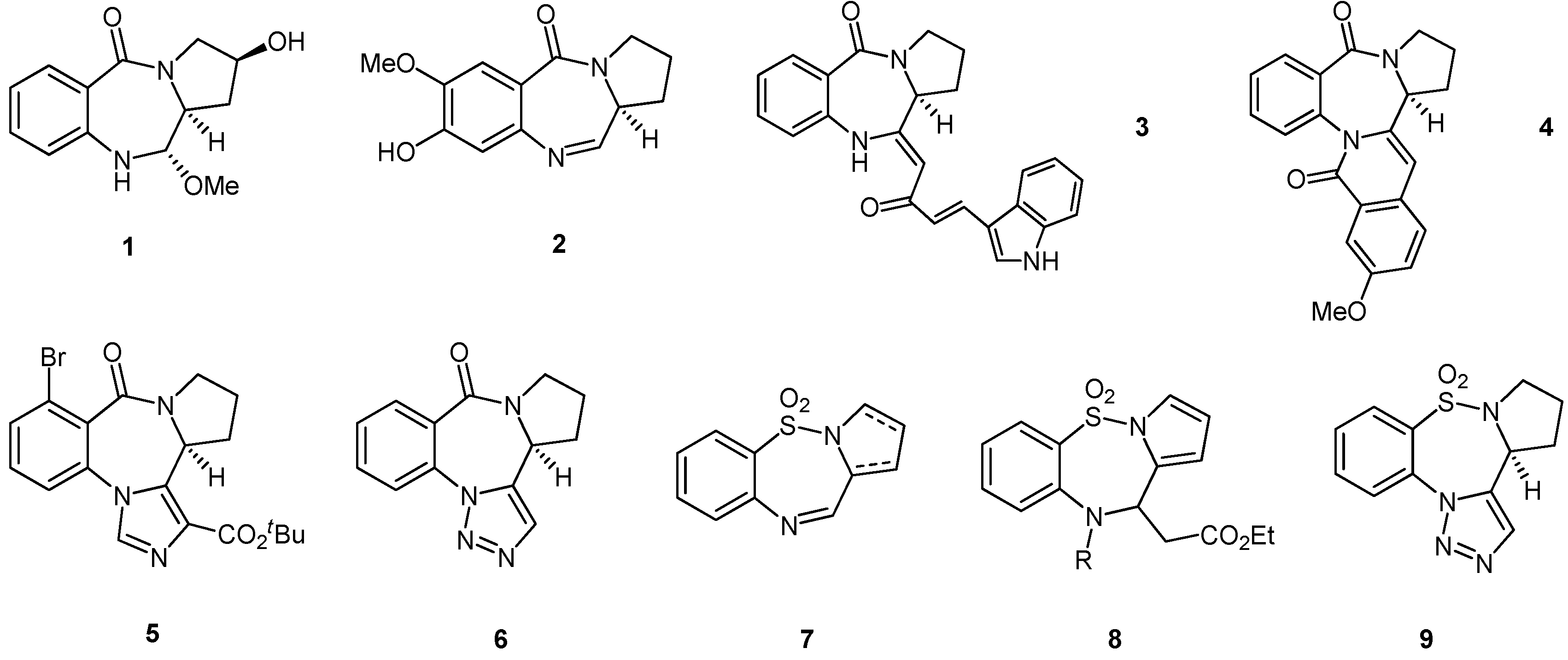
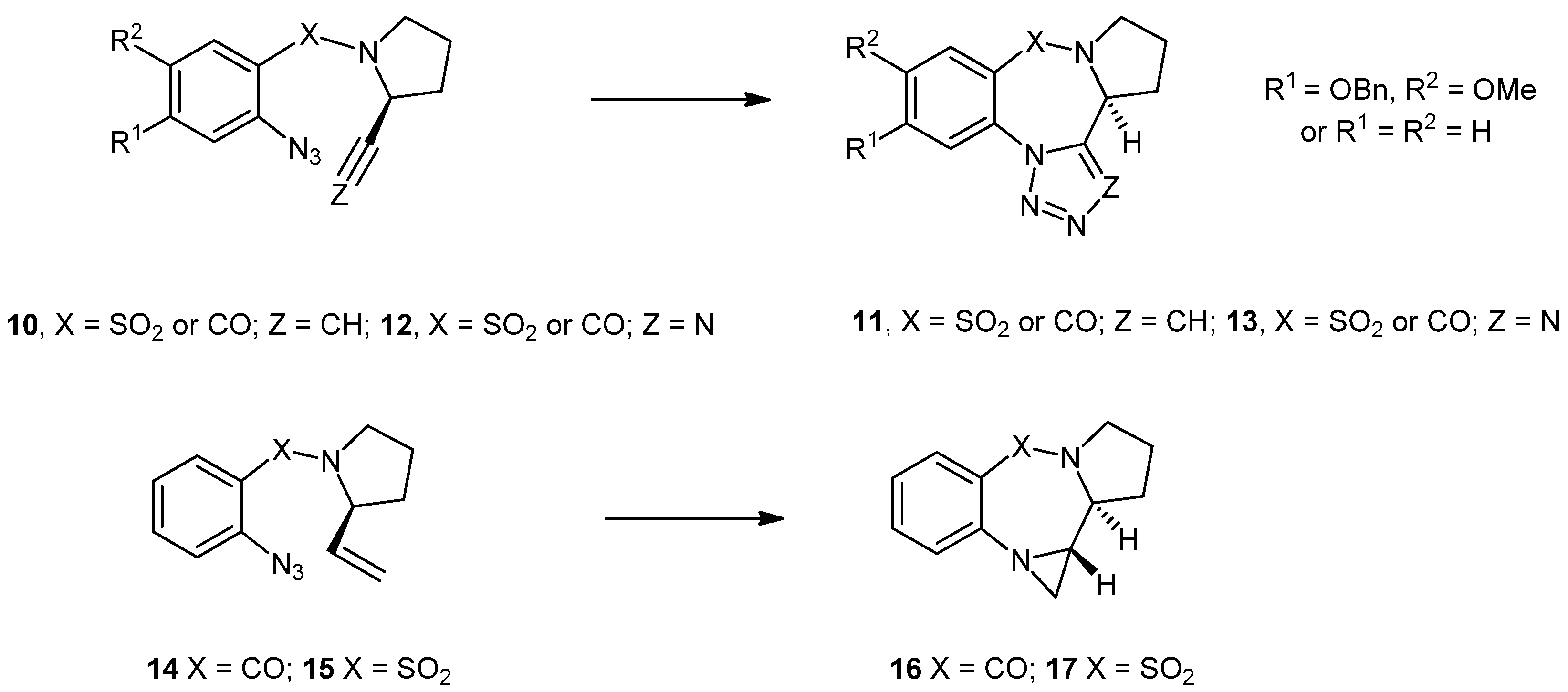
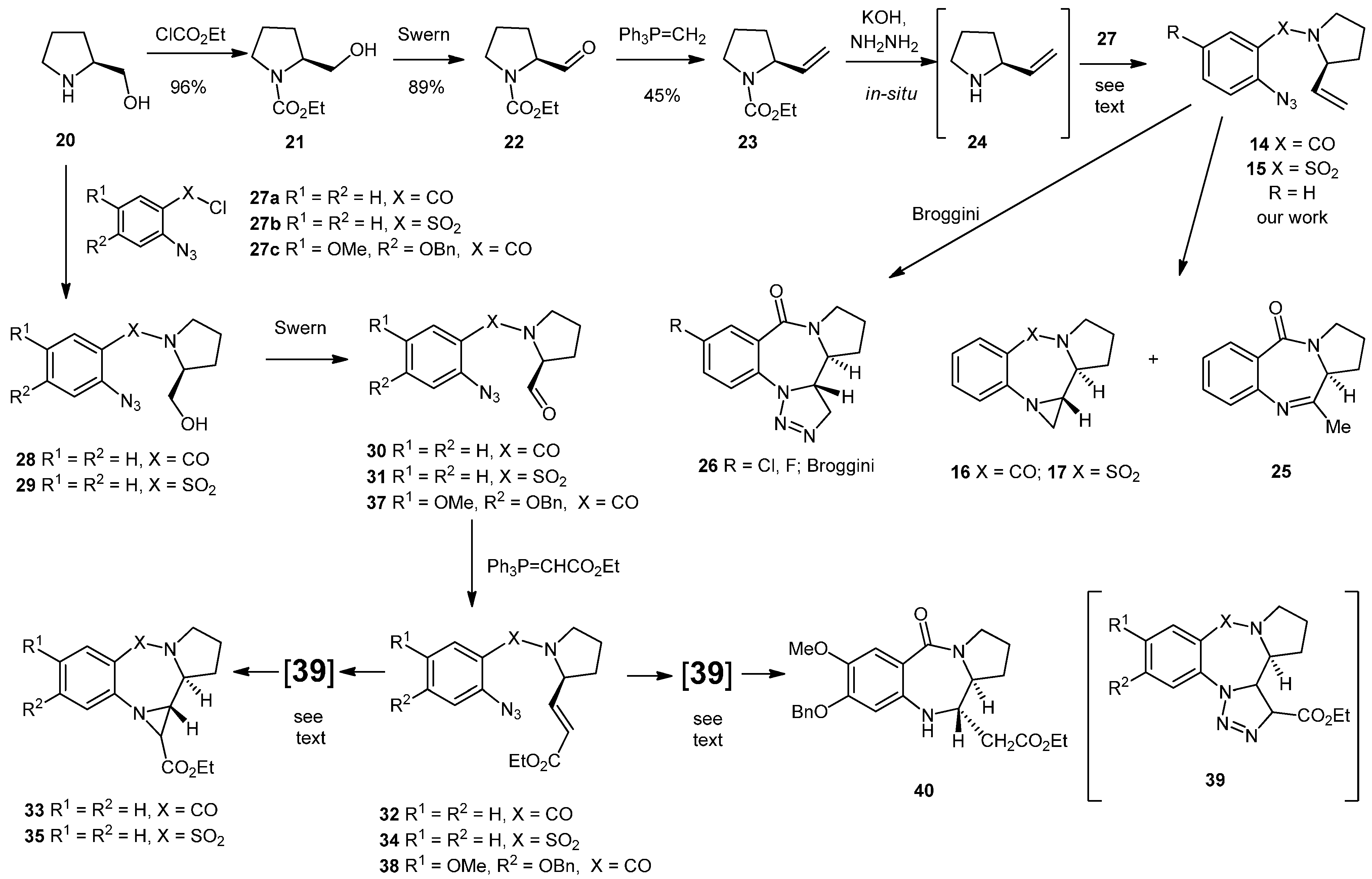
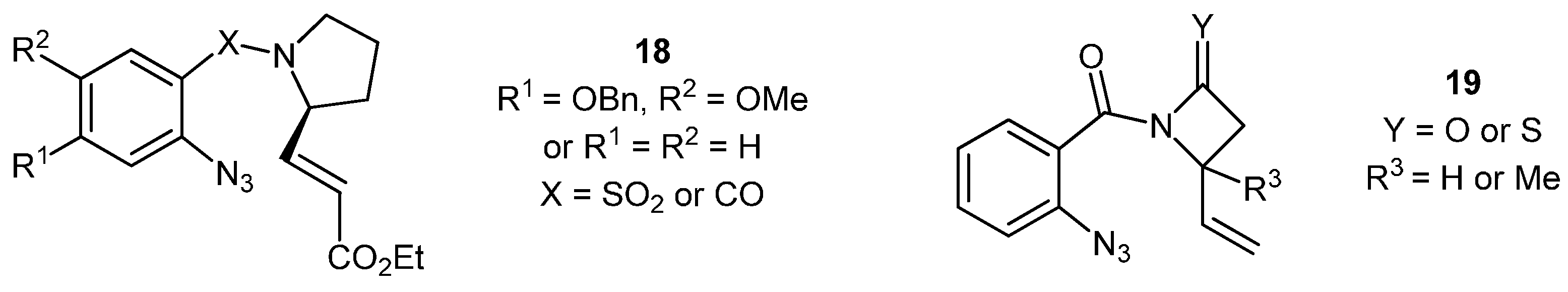3.2. Synthesis and Reactivity of the Pyrrolo-Based Systems
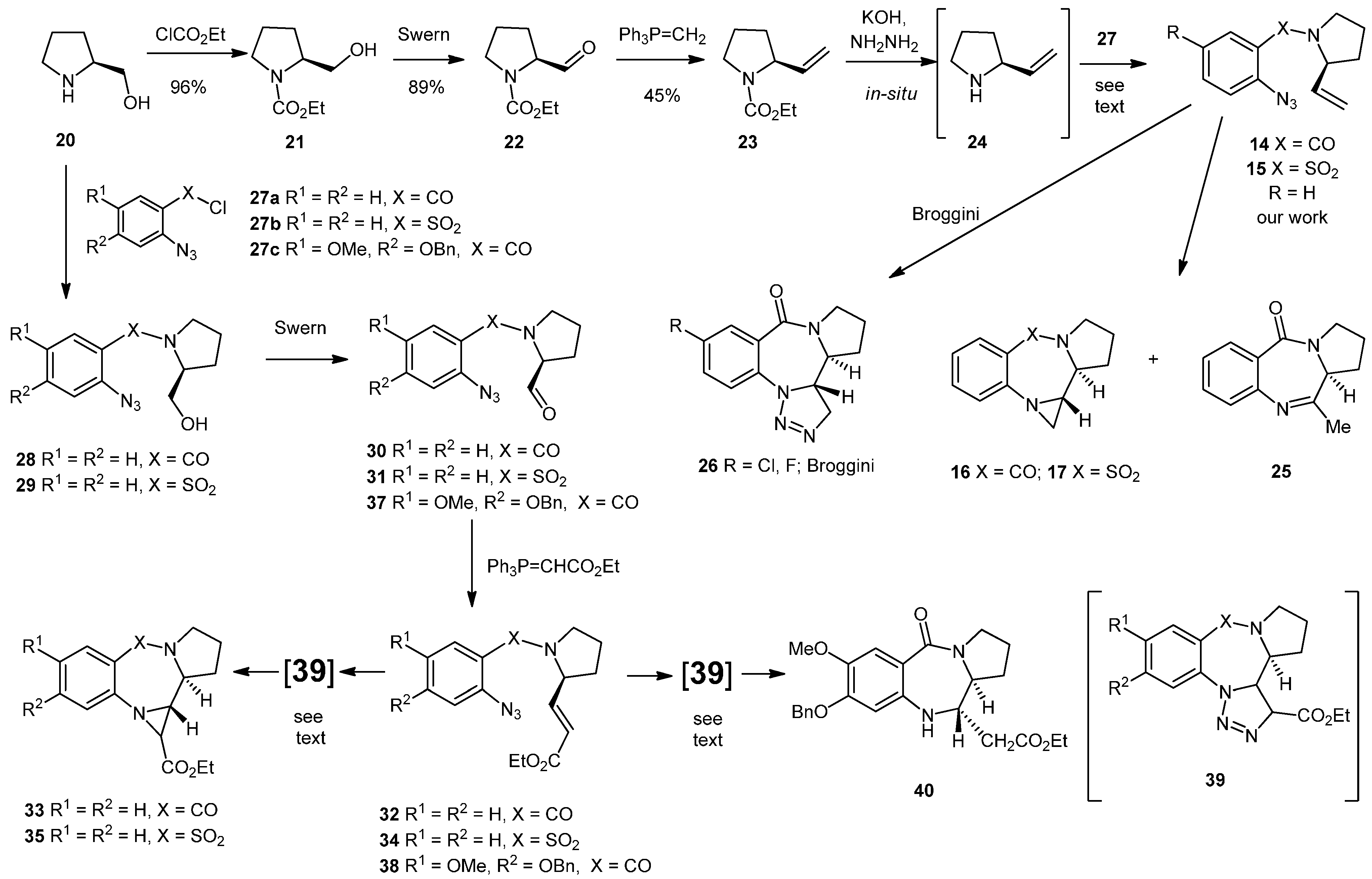
N-(Ethoxycarbonyl)-prolinol (21). To a stirring solution of S-prolinol (20, 1.00 g, 9.89 mmol) in 4 M NaOH (7 mL) was added ethyl chloroformate (1.13 mL, 1.29 g, 11.9 mmol) over 10 min at 0 °C. The reaction was stirred at 0 °C for 30 min followed by 30 min at ambient temperature. The reaction solution was neutralized with 2M HCl, the aqueous phase was separated and extracted with DCM (3 × 10 mL). The combined organic layers were dried (MgSO4), filtered and concentrated under reduced pressure to give the crude product 21 (1.64 g, 96%) as a yellow oil which was used directly in the next step. NMR: δH (400 MHz, CDCl3): 1.27 (3H, t, J = 7.1, COCH2CH3), 1.57 (1H, m, CHH), 1.74–1.91 (2H, m, CHH + CHH), 1.99–2.08 (1H, m, CHH), 3.31–3.78 (1H, m, NCHH), 3.49–3.54 (1H, m, NCHH), 3.59–3.69 (2H, m, CH2OH), 3.97–4.03 (1H, m, CH2OH), 4.14 (2H, q, J = 7.1, COCH2CH3), 4.61 (1H, dd, J = 7.6, 2.6, NCHCH2); δC (100 MHz, CDCl3): 14.7 (CH3), 24.1 (CH2), 28.6 (CH2), 47.3 (CH2), 60.6 (CH), 61.6 (CH2), 67.3 (CH2), 157.6 (q). IR: υmax (thin film cm−1): 770 (s), 906 (m), 1046 (s), 1106 (s), 1333 (s), 1379 (s), 1414 (s), 1667 (s), 2876 (w), 2975 (w), 3400–3500 (br). LRMS (ESI+): Found 196.1 [M+Na]+, C8H15N4NaO3 requires 196.1.
N-(Ethoxycarbonyl)-prolinal (22). A 2 M solution of (COCl)2 in DCM (4.72 mL, 9.43 mmol) was diluted with dry DCM (12 mL) and cooled to −78 °C under N2. DMSO (1.34 mL, 1.47 g, 18.9 mmol) in DCM (5 mL) was added followed by the alcohol 21 (1.36 g, 7.86 mmol) in DCM (5 mL), over 15 min. The whole was maintained at −78 °C for 30 min before the addition of Et3N (5.48 mL, 3.98 g, 39.3 mmol) dropwise over 10 min. The whole was allowed to reach room temperature over an hour before being quenched with a mixture of Et2O (12.5 mL) and H2O (12.5 mL). The organic layer was separated and the aqueous phase was extracted with DCM (3 × 10 mL). The combined organics were dried (MgSO4), filtered, concentrated under reduced pressure and purified by silica chromatography (20 g) (EtOAc/Hex; 2:3) to yield the aldehyde 22 as a mixture of rotamers in the form of a yellow oil (1.19 g, 89%). NMR: δH (400 MHz, CDCl3): 1.17 & 1.24 (3H, 2 × t, J = 7.1 & 7.1, COCH2CH3), 1.76–1.92 (2H, m, CH2), 1.93–2.09 (2H, m, CH2), 3.40–3.57 (2H, m, NCH2), 4.02–4.14 & 4.17–4.22 (3H, m, CO2CH2CH3 & NCHCHO), 9.48 & 9.56 (1H, 2 × d, J = 2.5, 1.7, CHO); δC (100 MHz, CDCl3): 14.5/14.7 (CH3), 23.8/24.5 (CH2), 26.6/27.8 (CH2), 46.6/47.1 (CH2), 61.5 (2 × CH2), 64.8/65.1 (CH), 154.7/155.6 (q), 200.2/200.3 (CHO). IR: υmax (thin film cm−1): 729 (s), 771 (s), 914 (m), 1021 (m), 1102 (s), 1172 (m), 1341 (s), 1380 (s), 1416 (s), 1466 (s), 1687 (s), 1733 (s), 2872 (m), 2980 (m). LRMS (ESI+): Found 194.1 [M+Na]+, C8H13NNaO3 requires 194.1.
N-2-Ethenyl-1-ethoxycarbonylpyrrolidine (23). n-BuLi in hexanes (1.6 M, 10.5 mL, 16.8 mmol) was added dropwise over 30 min to a stirring suspension of methyltriphenylphosphonium bromide (5.52 g, 15.4 mmol) in anhydrous THF (30 mL) at −78 °C under an inert atmosphere of nitrogen. The whole was allowed to reach −10 °C and kept at that temperature for 30 min before being cooled back to −78 °C. The aldehyde 22 (1.20 g, 7.02 mmol) in anhydrous THF (5 mL) was added dropwise over 10 min. The whole was allowed to warm to −10 °C, and maintained at that temperature for 2 h and then the mixture was allowed to reach room temperature overnight. The mixture was quenched with saturated aqueous NH4Cl (20 mL) and the aqueous layer was separated and extracted with EtOAc (3 × 10 mL). The combined organic layers were dried (MgSO4), filtered, concentrated under reduced pressure and purified by silica chromatography (50 g) (EtOAc/Hex; 3:2) yielding an air sensitive product as a mixture of rotamers in the form of a pale orange oil (536 mg, 45%). NMR: δH (400 MHz, CDCl3): 1.22–1.27 (3H, br, m, CH3), 1.71 (1H, bs, CH2CHHCH2), 1.81 (2H, br, m, NCHCH2), 1.97 (1H, m, br, CH2CHHCH2), 3.43 (2H, s, br, NCH2), 4.13 (2H, br, m, OCH2), 4.33 (1H, br, m, NCH), 5.04–5.12 (2H, br, m, CH=CH2), 5.72 (1H, br, m, CH=CH2); δC (100 MHz, CDCl3): 14.2/14.8 (CH3), 22.6/23.4 (CH2), 31.2/31.9 (CH2), 46.3/46.5 (CH2), 58.9/59.3 (CH), 60.4/60.8 (CH2), 113.8/114.1 (CH2), 138.2/138.5 (CH), 155.1/155.4 (q).
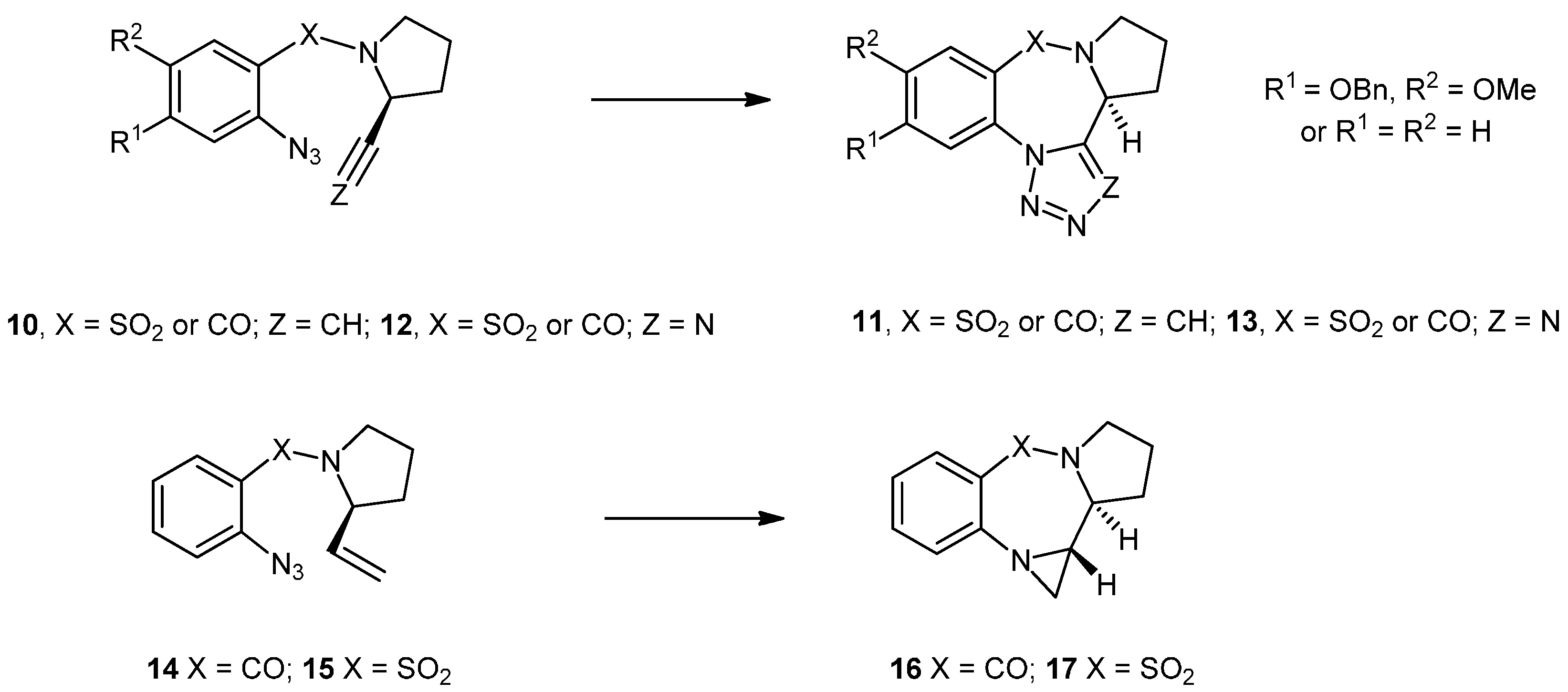
N-(2'-Azidobenzoyl)-2-ethenylpyrrolidine (14). To a vigorously stirred suspension of finely ground KOH (2.31 g, 41.2 mmol) in ethylene glycol (7.2 mL) was added hydrazine hydrate (0.25 mL, 7.92 mmol) and 2-ethenyl-1-ethoxycarbonylpyrrolidine (23) (0.2706 g, 1.58 mmol) and the whole was heated to reflux (195 °C) for 4 h. The reaction was allowed to reach ambient temperature before being diluted with a mixture of Et2O (4 mL) and H2O (4 mL). The thick syrup was extracted with Et2O (3 × 5 mL) and dried with finely ground NaOH. Et3N (0.33 mL, 2.37 mmol) was added to the ethereal solution containing the amine at 0 °C under an inert atmosphere and was stirred for 10 min before the freshly prepared acid chloride 27a (0.48 g, 2.30 mmol) in Et2O (5 mL) was added dropwise over 10 min and the whole was allowed to reach ambient temperature overnight. The reaction was diluted with water (20 mL), the ethereal layer was separated and the aqueous phase was extracted with ether (3 × 10 mL). The combined organics were dried (MgSO4), filtered, concentrated under reduced pressure and purified by silica chromatography (21 g) (EtOAc/Hex; 2:3) to yield the product as a mixture of rotamers (0.1230 g, 32%) in the form of a yellow oil. NMR: δH (400 MHz, CDCl3): 1.73–2.15 (4H, m, 2 × CH2), 3.17–3.23 & 3.30–3.36 (1H, m, CHHCH2), 3.64–3.71 & 3.74–3.82 (1H, m, CHHCH2), 4.11–4.15 & 4.84–4.87 (1H, br, m, CHCH2), 4.70 & 5.35 (1H, 2 × d, J = 16.9, 17.1, CHHCH), 4.89 & 5.20 (1H, 2 × d, J = 10.3 & 10.4, CHCHH), 4.89 & 5.20 (1H, 2 × d, J = 10.3 & 10.4, CHCHH), 5.56 & 5.90 (1H, 2 × ddd, J = 6.2, 10.4, 16.9 & 4.8, 10.4, 17.1, CHCH2) 7.10–7.24 (3H, m, ArH), 7.32–7.45 (1H, m, ArH); δC (100 MHz, CDCl3): 22.1/23.6 (CH2), 30.9/32.3 (CH2), 45.9/48.2 (CH2), 58.4/61.1 (CH), 114.5/114.9 (CH2), 118.4/118.5 (CH), 124.6/125.2 (CH), 127.9/128.3 (CH), 129.6/130.1 (q), 130.2/130.3 (CH), 136.1 (q), 136.9/137.6 (CH), 166.8/167.4 (q). IR: υmax (thin film cm−1): 750 (s), 932 (s), 1083 (m), 1149 (m), 1292 (s), 1415 (s), 1449 (s), 1479 (s), 1598 (s), 1631 (s), 2128 (s), 2878 (m), 2973 (m), 3078 (w). LRMS (ESI+): Found 265.1 [M+Na]+, 507.2 [2M+Na]+. HRMS (ESI+): Found 265.1064 [M+Na]+, C13H14N4NaO requires 265.1060.
Pyrrolobenzodiazepine (25) and aziridinopyrrolobenzodiazepine (16). The alkene 14 (78.0 mg, 0.32 mmol) was dissolved in CHCl3 (10 mL) and heated at reflux under an inert atmosphere of nitrogen for 16 h whilst being monitored by TLC (EtOAc/Hex; 3:2). The reaction mixture was concentrated under reduced pressure and purified by silica chromatography (20 g) (EtOAc/Hex; 3:2–4:1) to yield two inseparable close-running spots on TLC which were found to be the aziridine 16 and the methyl imine 25 in a 1:1 ratio (estimated by 1H-NMR, 43 mg, 55%). NMR: δH (400 MHz, CDCl3): 1.96–2.06 (2H, m, CH2), 2.00 (1H, d, J = 3.6, aziridine CH2), 2.09–2.22 (2H, m, 2 × CHH), 2.18–2.26 (3H, m, CH2 + CHH), 2.27 (3H, s, CH3), 2.29–2.37 (1H, m, CHH), 2.53 (1H, d, J = 4.3, aziridine CH2), 2.78 (1H, ddd, J = 9.5, 4.3, 3.6, aziridine CH), 3.34 (1H, ddd, J = 9.5, 2.9, 1.6, NCH [aziridine]), 3.45–3.52 (1H, m, NCH), 3.56–3.63 (1H, m, NCHH), 3.62–3.72 (2H, m, 2 × NCH), 3.74–3.84 (1H, m, NCH2), 3.81–3.95 (1H, m, NCH2), 7.01 (1H, dt, J 7.9, 0.7, ArH), 7.11 (1H, d, J 8.1, ArH), 7.14–7.18 (2H, m, 2 × ArH), 7.40 (1H, dt, J = 7.6, 1.6, ArH), 7.44–7.52 (1H, m, ArH), 7.74 (1H, d, J = 7.9, ArH), 7.93 (1H, dd, J = 7.9, 1.6, ArH); δC (100 MHz, CDCl3): 22.4 (CH3), 23.1 (CH2), 24.2 (CH2), 27.9 (CH2), 29.4 (CH2), 32.7 (CH2), 32.9 (CH2), 44.8 (CH), 46.1 (CH2), 55.6 (CH), 58.1 (CH), 122.0 (CH), 122.9 (CH), 125.5 (CH), 126.4 (CH), 126.8 (q),127.0 (q), 129.7 (CH),129.8 (CH), 131.2 (CH), 131.4 (CH), 145.6 (q), 145.7 (q), 150.3 (q), 165.5 (q), 169.9 (q). IR: υmax (thin film cm−1): 907 (s), 1244 (m), 1320 (m), 1403 (m), 1454 (s), 1614 (s), 1681 (m), 1743 (m), 2876 (w), 2924 (m), 2970 (w). LRMS (ESI+): Found 215.1 [M+H]+, 237.1 [M+Na]+, 451.2 [2M+Na]+, 665.3 [3M+Na]+. HRMS (ESI+): Found 215.1179 [M+H]+, C13H15N2O requires 215.1179.
N-(2'-Azidobenzenesulfonyl)-2-ethenylpyrrolidine (15). To a stirring suspension of KOH (4.280 g, 76.5 mmol) in ethylene glycol (15 mL), hydrazine hydrate (0.46 mL, 471 mg, 14.7 mmol) was added under nitrogen followed by the ester protected alkene 23 (497 mg, 2.94 mmol), and the whole was heated at reflux (195 °C) for 4.5 h. The reaction mixture was cooled to ambient temperature and was diluted with Et2O (9 mL) and water H2O (9 mL). The organic layer was separated and the aqueous layer was extracted with Et2O (3 × 5 mL) and dried over NaOH. Et3N (0.62 mL, 4.41 mmol) was added to the ethereal solution containing the amine at 0 °C and the mixture was stirred for 10 min under an inert atmosphere of nitrogen before addition of freshly prepared 2-azidobenzenesulfonyl chloride 27b [which was prepared by heating at reflux 2-azidobenzenesulfonic acid (900 mg, 4.41 mmol) with 2M oxalyl chloride in DCM (4.4 mL, 8.82 mmol) which was concentrated under reduced pressure and suspended in Et2O (10 mL)]. The whole was allowed to reach room temperature overnight. The reaction was diluted with water (30 mL), the ethereal layer was separated and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organics were dried (MgSO4), filtered, concentrated under reduced pressure and purified by silica chromatography (20 g) (EtOAc/Hex; 1:4) to yield the product as a yellow oil (163 mg, 20%). NMR: δH (400 MHz, CDCl3): 1.30–1.40 (1H, m, CHH), 1.83–1.92 (2H, m, CH2), 2.07–2.10 (1H, m, CHH), 2.92–2.99 (1H, m, CHH), 3.54–3.57 (1H, m, CHH), 3.93–3.98 (1H, m, NCH), 4.37–4.40 (1H, m, CH=CHH), 4.54 (1H, dd, J = 12.6, 17.8, CH=CHH), 5.30–5.37 (1H, m, CH=CH2), 7.16 (1H, dd, J = 8.0, 8.0, ArH), 7.45 (1H, dd, J = 8.4, 8.4, ArH), 7.83 (1H, d, J = 8.4, ArH), 7.91 (1H, d, J = 8.0, ArH). IR: υmax (thin film cm−1): 926 (m), 1026 (s), 1099 (m), 1157 (m), 1182 (s), 1296 (m), 1330 (m), 1445 (m), 1471 (s), 1518 (m), 1592 (s), 2137 (s), 2871 (m), 2977 (m). LRMS (ESI+): Found 301.1 [M+Na]+. HRMS (ESI+): Found 301.0722 [M+Na]+, C12H14N4O2S + Na+ requires 301.0730.
Aziridinopyrrolobenzothiadiazepine (17). The N-(2'-azidobenzenesulfonyl)-2-ethenylpyrrolidine (15) (125 mg, 0.449 mmol) was dissolved in toluene (10 mL) and heated to reflux for 18 h. The reaction was allowed to reach room temperature before the solvent was removed and the crude was purified by silica chromatography (25 g) (EtOAc/Hex;2:3) to yield the aziridine product 17 as a tan colored oil (50 mg, 44%). NMR: δH (500 MHz, CDCl3): 1.81–2.07 (4H, m, 2 × CH2), 2.91–2.95 (1H, m, SO2NCHH), 3.32 (1H, dd, J = 9.9, 11.7, ArNCHH), 3.50 (1H, m, SO2NCHH), 3.57 (1H, dd, J = 3.9, 11.7, ArNCHH), 3.89 (1H, ddd, J = 2.9, 2.9, 7.9, SO2NCH), 4.84–4.87 (1H, m, ArNCH), 6.70 (1H, d, J = 8.1, ArH), 6.84 (1H, dd, J = 7.8, 7.8, ArH), 7.20 (1H, ddd, J = 7.8, 7.8, 1.3, ArH), 7.70 (1H, dd, J = 8.1, 1.3, ArH); δC (125 MHz, CDCl3): 24.5 (CH2), 25.8 (CH2), 43.4 (CH2), 49.2 (CH2), 59.3 (CH), 62.9 (CH), 118.6 (CH), 119.9 (CH), 125.7 (q), 129.2 (CH), 132.7 (CH), 144.6 (q). IR: υmax (thin film cm−1): 759 (s), 1009 (m), 1037 (m), 1081 (s), 1127 (s), 1274 (m), 1333 (s), 1482 (s), 1520 (m), 1591 (s), 2926 (m), 2970 (w), 3066 (m), 3362 (w). LRMS (ESI+): Found 273.1 [M+Na]+, 523.1 [2M+Na]+, C12H14N2NaO2S requires 273.1. HRMS (ESI+): Found 251.0845 [M+H]+, C12H14N2O2S + H+ requires 251.0849.
Synthesis of 1-[2'-Azido-4-(benzyloxy)-5-methoxybenzoyl]prolinal (37)
Step 1: Synthesis of 1-[2'-Azido-4-(benzyloxy)-5-methoxybenzoyl]prolinol
2'-Azido-4-(benzyloxy)-5-methoxybenzoic acid (236 mg, 0.789 mmol) was dissolved in dry toluene (5 mL) and SOCl2 (1.5 mL) was added. The solution was placed in a preheated oil bath at 85 °C for 3 h. The reaction was cooled to ambient temperature, concentrated in vacuo, dissolved in fresh DCM (3 × 10 mL) and concentrated in vacuo, to remove the excess SOCl2. K2CO3 (218 mg, 1.58 mmol) in water (2 mL) was added in one portion to a stirring solution of S-prolinol (0.162 mL, 168 mg, 1.66 mmol) in DCM (2 mL) and the whole was stirred for 10 min. The acid chloride in DCM (5 mL) was added drop-wise and the whole was stirred at ambient temperature overnight before the organic layer was separated. The aqueous phase was extracted with DCM (3 × 10 mL) and the combined organic layers were dried (MgSO4), filtered, concentrated and purified by silica chromatography (20 mg) (EtOAc/Hex; 3:1) to yield the product as an orange oil (262 mg, 87%). NMR: δH (400 MHz, CDCl3): 1.56–1.84 (3H, m, CH2 + CHH), 2.06–2.13 (1H, m, CHH), 3.20–3.30 (2H, m, CH2), 3.62–3.67 (1H, m, CHHOH), 3.73–3.77 (1H, m, CHHOH), 3.80 (3H, s, OMe), 4.23–4.29 (1H, m, CHCH2OH), 4.69 (1H, bs, CH2OH), 5.10 (2H, s, OCH2), 6.59 (1H, s, ArH), 6.76 (1H, s, ArH), 7.24–7.38 (5H, m, ArH); δC (100 MHz, CDCl3): 24.5 (CH2), 28.5 (CH2), 49.6 (CH2), 56.3 (CH3), 61.2 (CH), 66.6 (CH2), 71.2 (CH2), 104.2 (CH), 110.7 (CH), 121.4 (q), 127.3 (CH), 128.2 (CH), 128.7 (CH), 135.9 (q), 147.2 (q), 149.8 (q), 168.8 (q). IR: υmax (thin film cm−1): 742 (s), 1004 (m), 1048 (m), 1077 (m), 1178 (s), 1214 (s), 1242 (s), 1433 (s), 1511 (s), 1603 (s), 2109 (s), 2882 (w), 2937 (w), 3145–3593 (br). LRMS (ESI+): Found 383.2 [M+H]+, 765.3 [2M+H]+, 787.3 [2M+Na]+. HRMS (ESI+): Found 383.1708 [M+H]+, C20H22N4O4 + H+ requires 383.1714.
Step 2: Oxidation to 1-[2'-azido-4-(benzyloxy)-5-methoxybenzoyl]prolinal (37)
DMSO (0.14 mL, 154 mg, 1.97 mmol) in DCM (1 mL) and the alcohol from the previous step (250 mg, 0.654 mmol) in DCM (2 mL) were added dropwise, respectively, over 15 min, to a solution of 2 M oxalyl chloride (0.39 mL, 0.785 mmol) at −78 °C in DCM (1 mL). After 10 min, Et3N (0.24 mL, 174 mg, 1.72 mmol) was added dropwise to the mixture at −78 °C. The whole was allowed to reach room temperature over two h before being quenched with a mixture of Et2O (10 mL) and H2O (10 mL). The organic layer was separated and the aqueous layer was extracted with DCM (3 × 10 mL). The combined organics were dried (MgSO4), filtered, concentrated and purified by silica chromatography (20 g) (EtOAc/Hex; 3:1) to give the product as a mixture of rotamers in the form of an orange oil (161 mg, 65%). NMR: δH (400 MHz, CDCl3): 1.79–1.89 (2H, m, CH2), 1.97–2.04 (1H, m, CHH), 2.07–2.20 (1H, m, CHH), 3.28–3.34 & 3.35–3.41 (2H, m, NCH2), 3.76 & 3.81 (3H, 2 × s, OCH3), 4.12–4.17 & 4.52–4.57 (1H, m, NCHCHO), 5.08 & 5.12 (2H, 2 × s, OCH2), 6.53 & 6.61 (1H, 2 × s, ArH), 6.72 & 6.80 (1H, 2 × s, ArH), 7.24–7.39 (5H, m, ArH), 9.22 & 9.62 (1H, 2 × d, J = 1.4 & 1.8, CHO); δC (100 MHz, CDCl3): 24.9/26.4 (CH2), 46.8/48.6 (CH2), 56.3/56.4 (CH3), 60.4 (CH2), 64.8/66.5 (CH), 71.3/71.3 (CH2), 104.2/104.4 (CH), 111.2/111.5 (CH), 120.7/120.8 (q), 127.4 (CH), 128.3 (CH), 128.8 (CH), 135.9/136.0 (q), 147.3/147.3 (q), 150.0/150.1 (q), 167.2/167.4 (q), 198.0/199.4 (CH). IR: υmax (thin film cm−1): 1245 (s), 1430 (s), 1454 (s), 1512 (s), 1604 (s), 1622 (s), 1731 (m), 2110 (s), 2942 (m). LRMS (ESI+): Found 381.2 [M+H]+, 403.1 [M+Na]+, 783.3 [2M+Na]+. HRMS (ESI+): Found 403.1375 [M+Na]+, C20H20N4O4 + Na+ requires 403.1377.
(1-(2'-Azidobenzenesulfonyl)pyrrolidin-2-yl)methanol (29). 2-Azidobenzenesulfonic acid (2.95 g, 14.8 mmol) was heated at reflux in a 2M solution of (COCl)2 in dichloromethane (14.8 mL, 29.6 mmol) with a drop of DMF under an inert atmosphere of nitrogen for 5 h. The reaction was allowed to reach room temperature before the crude acid chloride was concentrated in vacuo and washed with dichloromethane (3 × 10 mL). K2CO3 (3.30 g, 23.7 mmol) in water (10 mL) was added in one portion to a stirring solution of L-prolinol (0.60 g, 5.9 mmol) in dichloromethane (15 mL). The crude sulfonyl chloride in dichloromethane (10 mL) was added slowly and the whole was stirred for 18 h at room temperature. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (3 × 10 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated to give the product as a pure orange oil (1.61 g, 96% from prolinol). NMR: δH (400 MHz, CDCl3): 1.67–1.78 (1H, m, CHH), 1.79–1.99 (3H, m, CHH + CH2), 2.79 (1H, bs, CH2OH), 3.38 (1H, dt, J = 6.3, 10.2, NCHH), 3.49–3.56 (1H, m, NCHH), 3.62 (1H, dd, J = 5.6, 11.5, CHHOH), 3.70 (1H, dd, J = 4.2, 11.5, CHHOH), 4.02–4.08 (1H, m, CHCH2OH), 7.27 (1H, ddd, J = 1.0, 7.8, 7.8, ArH), 7.32 (1H, dd, J = 1.0, 8.0, ArH), 7.62 (1H, ddd, J = 1.5, 7.8, 7.8, ArH), 8.02 (1H, dd, J = 1.5, 8.0, ArH); δC (100 MHz, CDCl3): 24.7 (CH2), 29.0 (CH2), 49.5 (CH2), 61.8 (CH), 65.5 (CH2), 119.9 (CH), 124.8 (CH), 129.0 (q), 132.6 (CH), 134.2 (CH), 138.2 (q). IR: υmax (thin film cm−1): 819, 870, 900, 928, 992, 1043, 1069, 1122, 1146, 1155, 1199, 1264, 1287, 1323, 1439, 1471, 1574, 1583, 1660, 2120, 2876, 2953, 3172–3693 (br). LRMS (ESI+): Found 305.1 [M+Na]+, 587.1 [2M+Na]+, C11H14N4O3S + Na+ requires 305.1. HRMS (ESI+): Found 305.0676 [M+Na]+, C11H14N4O3S + Na+ requires 305.0679.
(1-(2′-Azidobenzenesulfonyl)pyrrolidin-2-yl)carbaldehyde (31). A 2M solution of oxalyl chloride in dichloromethane (1.88 mL, 3.75 mmol) was diluted with dichloromethane (10 mL) and cooled to −78 °C under nitrogen. DMSO (0.53 mL, 0.59 g, 7.5 mmol) in dichloromethane (10 mL) and the alcohol 29 (0.80 g, 2.83 mmol) in dichloromethane (5 mL) were each added over 10 min. The whole was maintained at −78 °C for 30 min before dropwise addition of Et3N (2.18 mL, 1.58 g, 15.6 mmol) after which the whole was allowed to reach room temperature. The reaction was quenched with a mixture of Et2O (10 mL) and H2O (10 mL). The organic layer was separated and the aqueous phase was extracted with dichloromethane (3 × 10 mL). The combined organic layers were dried (MgSO4), filtered, concentrated and purified on silica (20 g) (EtOAc/Hex; 2:3) to yield the product as a white solid (0.57 g, 72%) which rapidly decomposed to an orange oil and was used immediately in subsequent reactions. NMR: δH (400 MHz, CDCl3): 1.83–1.95 (2H, m, CH2), 1.98–2.09 (1H, m, CHH), 2.15–2.23 (1H, m, CHH), 3.40 (1H, dt, J = 9.7, 7.2, NCHH), 3.57 (1H, ddd, J = 5.5, 6.8, 9.7, CHH), 4.47 (1H, ddd, J = 1.9, 4.5, 8.5, CHCHO), 7.28 (1H, ddd, J = 7.8, 7.8, 1.0, ArH), 7.34 (1H, dd, J = 8.0, 1.0, ArH), 7.64 (1H, ddd, J = 7.8, 7.8, 1.6, ArH), 8.03 (1H, dd, J = 8.0, 1.6, ArH), 9.72 (1H, d, J = 1.6, CHO); δC (100 MHz, CDCl3): 25.0 (CH2), 27.7 (CH2), 48.8 (CH2), 67.1 (CH), 119.9 (CH), 124.9 (CH), 128.9 (q), 132.5 (CH), 134.4 (CH), 138.2 (q), 200.5 (CH). IR: υmax (thin film cm−1): 820, 865, 999, 1080, 1122, 1156, 1199, 1265, 1287, 1332, 1439, 1471, 1574, 1583, 1603, 1730, 2122, 2953. HRMS (ESI+): compound degraded.
N-(2'-Azidobenzoyl)-2-(carbethoxy-1''-ethenyl)-pyrrolidine (
32). The aldehyde
30 [
33,
34] (273 mg, 1.12 mmol) was dissolved in toluene (10 mL). (Carbethoxymethylene) triphenylphosphorane (390 mg, 1.12 mmol) was added in one portion and the whole was stirred at room temperature under an inert atmosphere of nitrogen for 12 h before being concentrated
in vacuo and purified by silica chromatography (40 g) (EtOAc/Hex; 3:2) to yield the product
32 in the form of a yellow oil as a mixture of rotamers (178 mg, 51%). NMR: δ
H (400 MHz, CDCl
3): 1.29 & 1.31 (3H, t,
J = 7.1, 2 × COCH
2C
H3), 1.80–1.99 & 2.01–2.43 (4H, m, CH
2), 3.21–3.28 & 3.78–3.86 (1H, m, C
HH), 3.34–3.40 & 3.69–3.75 (1H, m, CH
H), 4.13 & 4.21 (2H, q,
J = 7.1 & 7.1, COC
H2CH
3), 4.24–4.30 & 4.96–5.00 (1H, m, NC
HCH
2), 5.46 & 6.15 (1H, dd & d,
J = 15.6, 1.1 & 15.6, CHC
HCO
2Et), 6.59 & 6.94 (1H, dd & dd,
J = 15.6, 6.4 & 15.6, 4.9, C
HCHCO
2Et), 7.12 (1H, dd,
J = 7.5, 7.5, ArH), 7.14 (1H, d,
J = 7.3, ArH), 7.19–7.25 (1H, m, ArH), 7.40 & 7.45 (2H, 2 × m, ArH); δ
C (100 MHz, CDCl
3): 14.2/14.3 (CH
3), 22.3/23.8 (CH
2), 30.5/32.1 (CH
2), 46.1/48.2 (CH
2), 57.2 (CH), 60.4/60.6 (CH
2), 118.5 (CH), 121.2/121.3 (CH), 124.9/125.2 (CH), 127.9 (CH), 129.1/129.5 (q), 130.6 (CH), 133.7/136.2 (q), 146.5 (CH), 165.8/166.5 (q), 167.1/167.4 (q). IR: υ
max (thin film cm
−1): 753 (s), 1043 (m), 1093 (m), 1180 (s), 1301 (s), 1369 (m), 1414 (s), 1451 (s), 1475 (m), 1633 (s), 1716 (s), 2130 (s), 2238 (w), 2880 (m), 2979 (m). LRMS (ESI+): Found 315.1 [M+H]
+, 337.1 [M+Na]
+, 651.3 [2M+Na]
+. HRMS (ESI+): Found 315.1451 [M+H]
+, C
16H
18N
4O
3 + H
+ requires 315.1452.
Aziridinopyrrolobenzodiazepine (33). The carbethoxy alkene 32 (178 mg, 0.567 mmol) was heated at reflux under nitrogen in CHCl3 (10 mL) for 48 h before being concentrated and purified by silica chromatography (20 g) (EtOAc/Hex; 3:2) to yield the product 33 as a ~1:1 mixture of two diastereoisomers in the form of a yellow oil (47 mg, 30%). NMR: δH (400 MHz, CDCl3) [mixture of isomers]: 1.23 (3H, t, J = 7.1, COCH2CH3, isomer “a”), 1.29 (3H, t, J = 7.1, COCH2CH3, isomer “b”), 1.74–1.80 (2H, m, CH2),1.81–2.27 (4H, m, 2 × CH2), 2.77 (1H, d, J = 2.6, CHCO2Et, isomer “a”), 2.87 (1H, d, J = 2.7, CHCO2Et, isomer “b”), 3.08 (1H, dd, J = 9.6, 2.6, ArNCH, isomer “a”), 3.37 (1H, dd, J = 8.7, 2.7, ArNCH, isomer “b”), 3.57 (1H, ddd, J = 7.3, 7.3, 12.0, CHH), 3.66–3.73 (4H, m, 2 × CH2), 3.74–3.79 (1H, m, CONCH), 3.78–3.87 (1H, m, CHH), 4.20 (2H, quartet, J = 7.1, COCH2CH3, isomer “a”), 4.23 (2H, quartet, J = 7.1, COCH2CH3, isomer “b”), 4.38 (1H, bd, J = 9.6, CONCH), 6.69 (1H, d, J = 7.4, ArH), 6.94 (1H, ddd, J = 7.5, 7.5, 0.9, ArH), 7.01 (1H, ddd, J = 7.5, 7.5, 1.0, ArH), 7.07 (1H, d, J = 8.0, ArH), 7.19 (1H, ddd, J = 7.6, 7.6, 1.6, ArH), 7.28 (1H, ddd, J = 7.7, 7.8, 1.6, ArH), 7.70 (2H, dd, J = 7.8, 1.8, ArH); δC (100 MHz, CDCl3): 14.2 (CH3), 14.5 (CH3), 23.1 (CH2), 25.6 (CH2), 29.5 (2 × CH2), 42.6 (CH), 46.4 (CH2), 47.0 (CH), 50.4 (CH), 56.6 (CH), 57.2 (CH), 60.1 (CH), 61.4 (CH2), 62.0 (CH2), 68.0 (CH2), 121.3 (CH), 122.0 (CH), 123.1 (CH), 123.3 (CH), 125.3 (q), 126.6 (q), 130.7 (CH), 131.0 (CH), 132.0 (CH), 132.2 (CH), 142.9 (q), 148.1 (q), 166.0 (q), 166.3 (q), 167.9 (q), 168.8 (q). IR: υmax (thin film cm−1): 752 (m), 1024 (s), 1178 (s), 1217 (s), 1260 (s), 1372 (m), 1454 (m), 1503 (m), 1602 (s), 1622 (s), 1725 (s), 2871 (m), 2926 (m), 2977 (m). LRMS (ESI+): Found. 309.1 [M+Na]+. HRMS (ESI+): Found 309.1206 [M+Na]+, C16H18N2O3 + Na+ requires 309.1210.
Aziridinopyrrolobenzothiadiazepine (35). The aldehyde 31 (286 mg, 1.13 mmol) was dissolved in toluene (10 mL) and (carbethoxymethylene) triphenylphosphorane (500 mg, 1.43 mmol) was added in one portion and the whole was stirred at room temperature for 18 h. The reaction mixture was concentrated and purified by silica chromatography (40 g) (EtOAc/Hex; 3:2) to yield, as a single isomer, the aziridine 35 as a yellow oil (123 mg, 34%). NMR: δH (500 MHz, CDCl3): 1.41 (3H, t, J = 7.1, COCH2CH3), 1.75–1.80 (1H, m, CHH), 1.87–2.01 (1H, m, CHH), 2.09–2.16 (1H, m, CHH), 2.27–2.35 (1H, m, CHH), 3.19 (1H, ddd, J = 5.0, 9.6, 9.6, SO2NCHH), 3.67–3.73 (2H, m, SO2NCHH + CHCHCH), 4.08 (1H, ddd, J = 2.1, 7.4, 9.8, SO2NCH), 4.33–4.40 (2H, m, COCH2CH3), 4.94 (1H, d, J = 9.8, ArNCH), 7.43 (1H, ddd, J = 7.7, 7.7, 1.1, ArH), 7.55 (1H, dd, J = 8.0, 1.1, ArH), 7.60 (1H, ddd, J = 7.7, 7.7, 1.5, ArH), 8.04 (1H, dd, J = 8.0, 1.5, ArH); δC (125MHz, CDCl3): 14.1 (CH3), 22.7 (CH2), 28.9 (CH2), 46.4 (CH2), 60.7 (CH), 61.8 (CH), 63.0 (CH2), 85.4 (CH), 123.8 (CH), 126.6 (CH), 128.5 (CH), 131.6 (q), 133.6 (CH), 138.4 (q), 167.3 (q). IR: υmax (thin film cm−1): 1035 (m), 1066 (m), 1092 (s), 1135 (m), 1167 (s), 1205 (m), 1247 (m), 1271 (m), 1344 (s), 1469 (s), 1503 (m), 1589 (m), 1738 (s), 2981 (m). LRMS (ESI+): Found 345.1 [M+Na]+, 723.2 [2M+Na]+. HRMS (ESI+): Found 345.0875 [M+Na]+, C15H18N2O4S + Na+ requires 345.0879.
3-Benzyloxy-4-methoxy-11-ethyl-ethanoyl-[1,4]-pyrrolo[2,1-c] benzodiazepin-5-one (40). The aldehyde 37 (198 mg, 0.58 mmol) was dissolved in toluene (10 mL) and (carbethoxy-methylene)triphenylphosphorane (200 mg, 0.58 mmol) was added in one portion and the whole was stirred at room temperature for 18 h. The reaction was concentrated in vacuo and purified by silica chromatography (30 g) (EtOAc/Hex; 4:1) to yield the product 40 as a yellow oil (52 mg, 21%). NMR: δH (500 MHz, CDCl3): 1.32 (3H, t, J = 7.2, CO2CH2CH3), 1.74–1.80 (1H, m, CHH), 1.93–2.19 (3H, m, CHH + CH2), 2.31–2.24 (2H, m, CH2CO2Et), 3.43–3.47 (1H, m, NHCH), 3.62–3.66 (1H, m, NHCHCH), 3.68–3.73 (1H, m, NCHH), 3.76–3.80 (1H, m, NCHH), 3.89 (3H, s, OMe), 4.22 (2H, q, J = 7.2, CO2CH2CH3), 5.13 (1H, d, J = 12.3, PhCHHO), 5.18 (1H, d, J = 12.3, PhCHHO), 6.34 (1H, s, ArH), 7.31–7.50 (7H, m, 6 × ArH + NH); δC (125 MHz, CDCl3): 14.2 (CH3), 23.2 (CH2), 29.9 (CH2), 37.3 (CH2), 46.9 (CH2), 56.3 (CH3), 60.1 (CH), 61.0 (CH2), 62.7 (CH), 70.9 (CH2), 108.1 (CH), 113.1 (CH), 120.0 (q), 127.4 (CH), 128.0 (CH), 128.4 (CH), 136.5 (q), 137.8 (q), 145.0 (q), 150.9 (q), 168.3 (q), 171.9 (q). IR: υmax (thin film cm−1): 723 (m), 1025 (s), 1119 (s), 1178 (s), 1218 (s), 1260 (s), 1373 (m), 1432 (s), 1453 (m), 1503 (m), 1602 (s), 1623 (m), 1726 (m), 2860 (m), 2924 (s), 2953 (m). LRMS (ESI+): Found 447.2 [M+Na]+. HRMS (ESI+): Found 447.1895 [M+Na]+ requires C24H28N2O5 + Na+ requires 447.1890.
3.3. Synthesis and Reactivity of the Azetidino-Based Systems
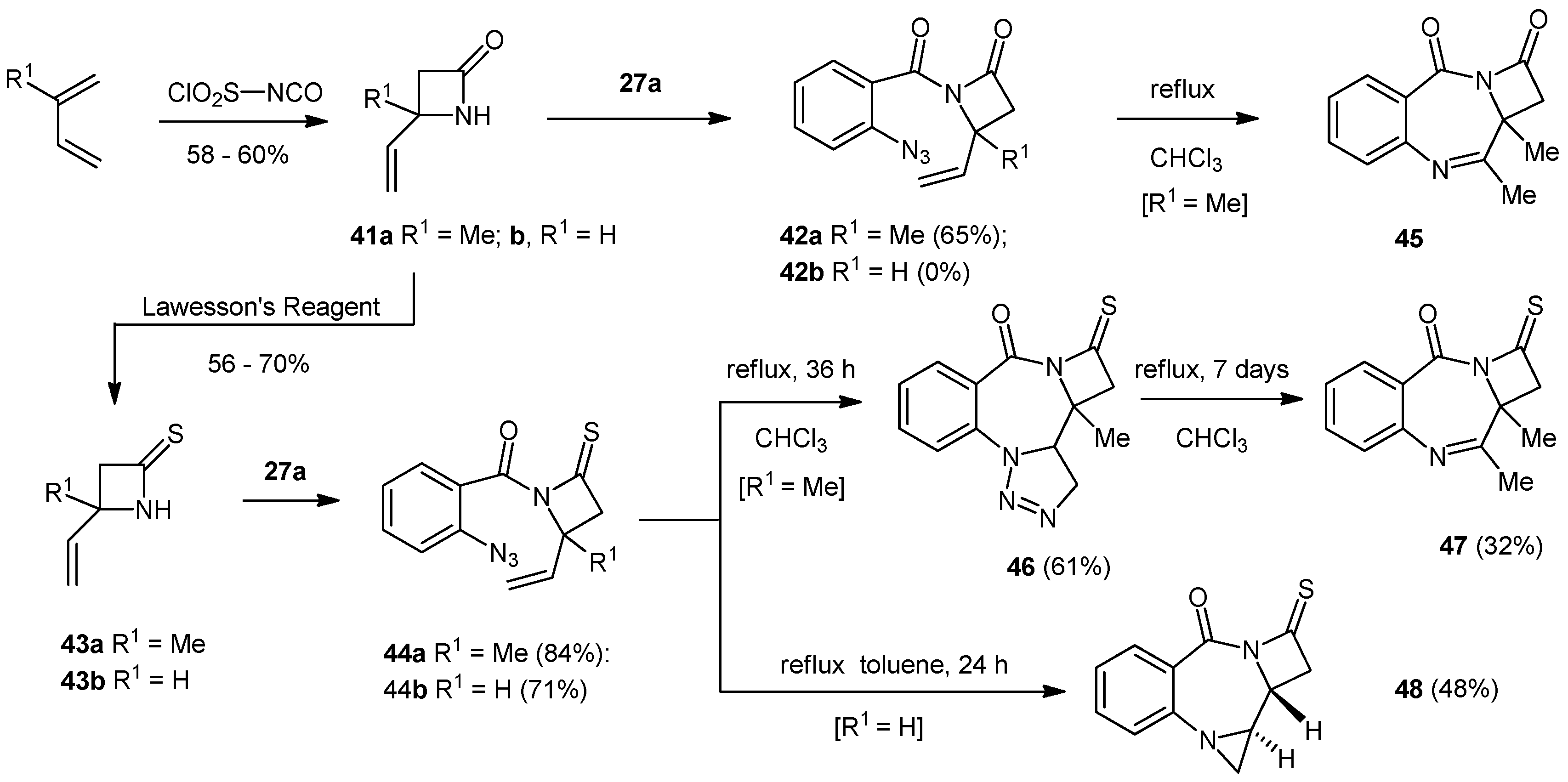
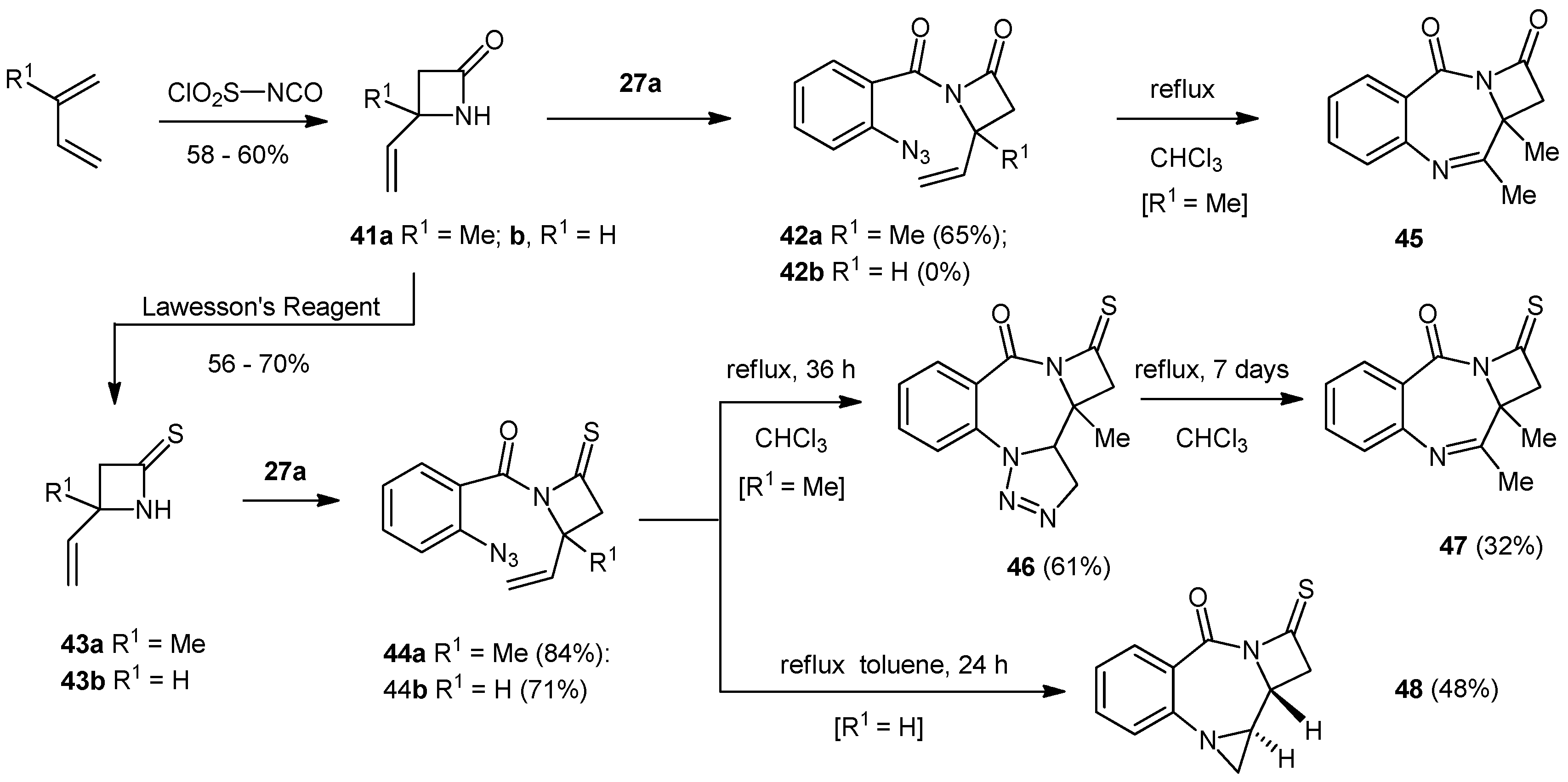
4-Methyl-4-ethenyl-1-azetidin-2-one (41a). To a stirred solution of isoprene (2.33 g, 3.43 mL, 34.41 mmol) in dry diethyl ether (15 mL) at –78 °C was added a solution of chlorosulfonyl isocyanate (4.88 g, 3.01 mL, 34.01 mmol) in dry ether (10 mL) dropwise over one hour. The reaction mixture was allowed to warm to −10) dropwise over one hour. The reaction mixture was allowed to warm to −10 °C and then the reaction flask was transferred to an ice-salt bath and stirred for 30 min. The cooled solution was added dropwise to a vigorously stirred solution of water (50 mL), sodium carbonate (9.00 g), sodium sulfite (6.01 g) and ice (30 g) over 10 min. The mixture was stirred at −10 °C for 1 h and then allowed to warm to room temperature and extracted with diethyl ether (6 × 20 mL). The combined organic extracts were dried (MgSO4) and the solvent removed under reduced pressure to give the product as a pale yellow oil (2.20 g, 58%). NMR: δH (400 MHz, CDCl3): 1.50 (3H, s, Me), 2.79 (2H, s, CH2), 5.10 (1H, dd, J = 10.6, 0.7, CH=CHH), 5.22 (1H, dd, J = 17.2, 0.7, CH=CHH), 6.02 (1H, dd, J = 10.6, 17.2, CH=CH2), 6.83 (1H, br, NH); δC (100 MHz, CDCl3): 24.8 (CH2), 50.7 (CH3), 54.5 (q), 113.8 (CH), 141.1 (CH2), 167.6 (q). IR: υmax (thin film cm−1): 923 (m), 1153 (m), 1186 (m), 1226 (m), 1274 (w), 1304 (m), 1372 (m), 1412 (m), 1643 (m), 1720 (s), 2970 (w), 3235 (m). LRMS (ESI+): Found 134.1 [M+Na]+, 291.2 [2M+3Na]+.
4-Methyl-4-ethenyl-1-azetidin-2-thione (43a). To the azetidin-2-one (0.55 g, 4.97 mmol) in dry THF (15 mL) was added Lawesson’s reagent (1.00 g, 2.48 mmol) and the whole was stirred at room temperature for an hour before being heated to reflux for two h. The reaction was cooled to ambient temperature before being concentrated under reduced pressure and purified by silica chromatography (75 g) (EtOAc/petroleum ether; 1:3) to give the product as a yellow oil (0.35 g, 56%). The reaction was higher yielding (up to 70%) on a larger scale (2 g of lactam), but less convenient to purify (stench). NMR: δH (400 MHz, CDCl3): 1.60 (3H, s, Me), 2.98 (2H, s, CSCH2), 5.21 (1H, d, J = 10.6, CH=CHH), 5.28 (1H, d, J = 17.2, CH=CHH), 6.05 (1H, dd, J = 10.6, 17.2 CH=CH2), 8.78 (1H, bs, NH); δC (100 MHz, CDCl3): 23.8 (CH3), 54.6 (CH2), 63.7 (q), 115.1 (CH2), 139.0 (CH), 202.2 (q). IR: υmax (thin film cm−1): 924 (s), 989 (m), 1016 (m), 1081 (s), 1217 (m), 1288 (m), 1374 (m), 1404 (s), 1466 (s), 2971 (w), 3147 (m).
4-Ethenyl-2-azetidinone (41b). To 1,3-butadiene (10 mL) condensed into anhydrous ether (40 mL) at −10 °C, was added a solution of chlorosulfonyl isocyanate (3.0 mL, 34.5 mmol) in anhydrous ether (10 mL) over one hour, under an atmosphere of nitrogen. The temperature was maintained at −10 °C for a further period of 3 h and warmed slowly to room temperature overnight, to produce a clear, yellow solution. The solution was added to an ice cold mixture of water (70 mL), ice (30 g), NaHCO3 (9.0 g) and Na2SO3 (6.0 g) and stirred for one hour at −10 °C. The reaction was allowed to warm to room temperature before being extracted with ether (6 × 20 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered and evaporated to dryness under reduced pressure to yield 4-ethenyl-2-azetidinone (2.01 g, 60% yield) as a clear yellow oil. NMR: δH (400 MHz, CDCl3): 6.49 (1H, s, N−H), 5.92 (1H, ddd, J = 17.2, 10.2, 3.2, =CH), 5.32 (1H, d, J = 17.2, =CH2), 5.20 (1H, d, J = 10.2, =CH2), 4.13 (1H, m, NCH), 3.25 (1H, m, ring-CH2), 2.70 (1H, d, J = 17.2 ring-CH2); δC (100 MHz, CDCl3): 167.82 (C=O), 137.43 (CH), 116.93 (CH2), 49.40 (CH2), 48.93 (CH). IR: υmax (thin film cm−1): 3274.6 (m, broad), 1755.4 (s), 1448.2 (m), 1413.8 (m), 1380.1 (m), 1254.7 (w).
4-Ethenylazetidin-2-thione (43b). To the lactam (1.00 g, 10.3 mmol) dissolved in anhydrous THF (15 mL), was added Lawesson’s reagent (2.08 g, 5.15 mmol) in one portion, with stirring. The reaction was stirred at ambient temperature, under an inert atmosphere of dry nitrogen for one hour, before being heated at reflux for an additional hour. The reaction was monitored for completion by tlc and was subsequently cooled to room temperature, to give a crude product as a clear orange liquid. The sample was concentrated by rotary evaporation and purified using silica column chromatography (eluent: petroleum ether-ethyl acetate 4:1), to yield 4-ethenyl-azetidin-2-thione (0.67 g, 58%) as a clear, yellow oil. NMR: δH (400 MHz, CDCl3): 8.68 (1H, s, N−H), 5.96 (1H, ddd, J = 17.1, 10.3, 3.0, =CH), 5.35 (1H, d, J = 17.1, =CH2), 5.26 (1H, d, J = 10.3, =CH2), 4.62 (1H, m, NCH), 3.30 (1H, m, ring-CH2), 2.84 (1H, m, ring-CH2); δC (100 MHz, CDCl3): 203.65 (C=S), 135.14 (CH), 118.34 (CH2), 58.01 (CH), 48.34 (CH2). IR: υmax (thin film cm−1): 3174.1 (m, broad), 1482.6 (s), 1426.0 (m), 1407.2 (w), 1237.1 (m).
1-(2′-Azidobenzoyl)-4-methyl-4-vinylazetidin-2-one (42a). 2-Azidobenzoic acid (508 mg, 3.02 mmol) was heated to reflux in SOCl2 (4 mL) for 3 h under nitrogen. The excess SOCl2 was removed in vacuo and the crude acid chloride was dissolved in DCM (3 × 5 mL) which was removed in vacuo to yield the crude 2-azidobenzoylchloride. The β-lactam 41 (457 mg, 4.12 mmol) in DCM (25 mL) and DMAP (100 mg) were chilled to −10 °C. The crude acid chloride in DCM (5 mL) was added dropwise to the solution over 10 min. The whole was maintained at −10 °C for 30 min before the addition of Et3N (0.89 mL, 646 mg, 6.40 mmol) and the whole was allowed to reach room temperature overnight. The reaction was concentrated under reduced pressure and purified by silica chromatography (70 g) (EtOAc/Hex; 1:4) to give the product 42a as a mixture of rotamers in the form of a dark yellow oil (498 mg, 65%). NMR: δH (400 MHz, CDCl3) rotamer 1: 1.87 (3H, s, Me), 2.98 (1H, d, J = 16.2, COCHH), 3.08 (1H, d, J = 16.2, COCHH), 5.35 (1H, d, J = 10.7, CHH=CH), 5.45 (1H, d, J = 17.3, CHH=CH), 6.24 (1H, dd, J = 10.7, 17.3, CH=CH2), 7.22 (1H, dd, J = 7.6, 7.6, ArH), 7.23 (1H, d, J = 8.3, ArH), 7.42 (1H, dd, J = 7.6, 1.4, ArH), 7.52 (1H, ddd, J = 8.3, 8.3, 1.4, ArH). δH (400 MHz, CDCl3) rotamer 2: 1.51 (3H, s, Me), 2.68 (1H, d, J = 15.8, COCHH), 2.82 (1H, d, J = 15.8, COCHH), 5.20 (1H, d, J = 10.6, CHHCH), 5.32 (1H, d, J = 17.2, CHHCH), 5.91 (1H, dd, J = 10.6, 17.2, CHCH2), 7.21 (1H, ddd, J = 7.6, 7.6, 0.9, ArH), 7.24 (1H, d, J = 7.6, ArH), 7.51 (1H, ddd, J = 7.8, 7.8, 1.5, ArH), 7.67 (1H, dd, J = 7.8, 1.5, ArH); δC (100 MHz, CDCl3) rotamer 1: 21.7 (CH3), 49.2 (CH2), 58.9 (q), 114.9 (CH2), 117.5 (CH), 123.7 (CH), 125.8 (q), 128.1 (CH), 131.2 (CH), 137.1 (q), 137.2 (CH), 162.4 (q), 162.5 (q); δC (100 MHz, CDCl3) rotamer 2: 27.6 (CH3), 39.5 (CH2), 58.6 (q), 114.3 (CH2), 119.9 (CH), 123.5 (q), 124.7 (CH), 130.8 (CH), 132.1 (CH), 138.9 (q), 139.9 (CH), 151.5 (q), 165.3 (q). IR: υmax (thin film cm−1): 1084 (m), 1216 (s), 1329 (s), 1391 (s), 1451 (m), 1478 (m), 1519 (s), 1604 (m), 1656 (m), 1682 (m), 1801 (s), 2131 (s), 2853 (m), 2925 (m). LRMS (ESI+): 279.1 [M+Na]+, 535.2 [2M+Na]+. C13H12N4NaO2 requires 279.1. HRMS (ESI+): 279.0862 [M+Na]+, C13H12N4NaO2 requires 279.0852.
8,9-Dimethylazetidino[2,1-a][1,4]benzodiazepin-2,11-dione (45). The azetidinone 42a (250 mg, 0.97 mmol) was heated to reflux in CHCl3 (10 mL) under an atmosphere of dry nitrogen and monitored by NMR every 24 h. After 72 h, the reaction mixture was concentrated under reduced pressure and purified by silica chromatography (22 g) using graduated elution (EtOAc/Hex; 1:4–3:1) to give the imine 45 as a yellow oil (84 mg, 38%). NMR: δH (500 MHz, CDCl3): 2.00 (3H, s, Me), 2.49 (1H, s, Me), 3.42 (1H, d, J = 16.0, COCHH), 3.77 (1H, d, J = 16.0, COCHH), 7.52 (1H, ddd, J = 7.7, 7.7, 1.0, ArH), 7.70 (1H, dd, J = 8.0, 1.0, ArH), 7.78 (1H, ddd, J = 7.7, 7.7, 1.6, ArH), 8.32 (1H, dd, J = 8.0, 1.6, ArH); δC (125MHz, CDCl3): 20.1 (CH3), 26.2 (CH3), 43.6 (CH2), 73.3 (q), 123.7 (q), 126.5 (CH), 126.7 (CH), 127.4 (CH), 134.3 (CH), 149.7 (q), 155.2 (q), 158.1(q), 204.2 (q). IR: υmax (thin film cm−1): 702 (s), 730 (s), 1264 (s), 1421 (m), 1463 (m), 1609 (m), 1657 (m), 1686 (m), 1721 (s), 1799 (m), 2928 (w). LRMS (ESI+): Found 229.1 [M+H]+, 251.1 [M+Na]+, 479.2 [2M+Na]+. HRMS (ESI+): Found 229.0978 [M+H]+, 479.1703 [2M+Na], C13H13N2O2 requires 229.0972 and C26H24N4NaO4 requires 479.1690.
1-(2′-Azidobenzoyl)-4-methyl-4-vinyl-1-azetidin-2-thione (44a). 2-Azidobenzoic acid (214 mg, 1.3 mmol) was heated to reflux under nitrogen in SOCl2 (4 mL) for 4 h. The excess thionyl chloride was removed in vacuo and the product was dissolved in DCM (3 × 5 mL) and concentrated in vacuo to give the crude acid chloride. The thiolactam (250 mg, 1.97 mmol) in DCM (25 mL) and DMAP (100 mg) was chilled to −10 °C. The crude acid chloride in DCM (5 mL) was added dropwise over 10 min and the whole was maintained at −10 °C for 30 min before dropwise addition of Et3N (0.41 mL, 299 mg, 2.95 mmol). The whole was allowed to reach room temperature overnight and the reaction was concentrated and purified by silica chromatography (22 g) (EtOAc/Hex; 1:4) to give the product 44a as an orange oil (296 mg, 84%). NMR: δH (400 MHz, CDCl3): 1.92 (3H, s, Me), 2.93 (1H, d, J = 16.9, CSCHH), 3.04 (1H, d, J = 16.9, CSCHH), 5.38 (1H, d, J = 10.8, CHH=CH), 5.45 (1H, d, J = 17.3, CHH=CH), 6.31 (1H, dd, J = 17.3, 10.8 CHH=CH), 7.20 (1H, dd, J = 8.2, 0.8, ArH), 7.24 (1H, ddd, J = 7.6, 7.6, 0.8, ArH), 7.36 (1H, dd, J = 7.6, 1.5, ArH), 7.54 (1H, ddd, J = 8.2, 8.2, 1.5, ArH); δC (100 MHz, CDCl3): 22.3 (CH3), 54.5 (CH2), 67.5 (q), 116.4 (CH2), 118.5 (CH), 125.1 (CH), 126.6 (q), 128.9 (CH), 132.1 (CH), 137.7 (CH), 138.3 (q), 164.2 (q), 201.7 (q). IR: υmax (thin film cm−1): 909 (s), 931 (m), 1215 (s), 1322 (s), 1358 (m), 1448 (w), 1478 (m), 1674 (s), 2131 (s). LRMS (ESI+): Found 295.1 [M+Na]+, 567.1 [2M+Na]+. HRMS (ESI+): Found 295.0623 [M+Na]+, C13H12N4NaOS requires 295.0624.
12-Methyl-1,2,3-triazolino[1,5-a]azetidino[1,4-c][1,4]benzodiazepin-2-on-14-thione (46). The 1-(2'-azidobenzoyl)-azetidin-2-thione 44a (295 mg, 1.08 mmol) was heated at reflux in CHCl3 (10 mL) under nitrogen for 36 h before being concentrated under reduced pressure and purified by silica chromatography (22 g) (EtOAc/Hex; 1:1) to give the triazolino product 46 as a yellow oil (180 mg, 61%). NMR: δH (500 MHz, CDCl3): 1.22 (3H, s, Me), 2.89 (1H, d, J = 16.8, SCCHH), 2.94 (1H, d, J = 16.8, SCCHH), 4.27 (1H, dd, J = 6.1, 12.2, CH3CHN), 4.37 (1H, dd, J = 6.1, 17.7, N3CHH), 4.71 (1H, dd, J = 12.2, 17.7, N3CHH), 7.14 (1H, ddd, J = 7.1, 7.1, 1.0, ArH), 7.52 (1H, ddd, J = 7.2, 7.2, 1.6, ArH), 8.04 (1H, dd, J = 8.4, 1.0, ArH), 8.2 (1H, dd, J = 8.4, 1.6, ArH); δC (125 MHz, CDCl3): 16.8 (CH3), 50.5 (CH2), 59.6 (CH), 65.0 (q), 70.4 (CH2), 115.9 (q), 118.6 (CH), 123.4 (CH), 134.2 (CH), 134.5 (CH), 138.3 (q), 161.8 (q), 198.1 (q). IR: υmax (thin film cm−1): 745 (s), 1103 (m), 1162 (m), 1211 (m), 1251 (m), 1322 (s), 1461 (s), 1483 (s), 1607 (s), 1651 (s), 1678 (s), 1720 (m), 2921 (w), 3000 (w). LRMS (ESI+): Found 295.1 [M+Na]+, 567.1 [2M+Na]+. HRMS (ESI+): Found 295.0622 [M+Na]+, C13H12N4NaOS requires 295.0624.
11-Thioxo-8,9-dimethyl-azetidino[2,1-c][1,4]benzodiazepin-2-one (47). A sample of the triazolo compound 46 from above (87 mg, 0.32 mmol) was heated at reflux in CHCl3 (10 mL) under nitrogen for a week before being concentrated under reduced pressure and purified by silica chromatography (15 g) (EtOAc/Hex; 3:2) to give the methyl imine product as a yellow oil (25 mg, 32%). NMR: δH (400 MHz, CDCl3): 1.98 (3H, s, Me), 2.47 (3H, s, Me), 3.40 (1H, d, J = 16.0, CSCHH), 3.77 (1H, d, J = 16.0, CSCHH), 7.50 (1H ddd, J = 7.7, 7.7, 1.0, ArH), 7.68 (1H, d, J = 8.1, ArH), 7.77 (1H, ddd, J = 7.7, 7.7, 1.4, ArH), 8.30 (1H, dd, J = 8.1, 1.4, ArH); δC (100 MHz, CDCl3): 20.1 (CH3), 26.3 (CH3), 43.6 (CH2), 73.3 (q), 123.6 (q), 126.6 (CH), 126.7 (CH), 127.3 (CH), 134.3 (CH), 149.6 (q), 155.2 (q), 158.0 (q), 204.4 (q). IR: υmax (thin film cm−1): 647 (s), 770 (s), 1103 (m), 1115 (m), 1132 (m), 1300 (m), 1323 (s), 1346 (s), 1460 (s), 1606 (s), 1651 (s), 1676 (s), 2928 (w), 2975 (w). LRMS (ESI+): Found 267.1 [M+Na]+. HRMS (ESI+): Found 267.0551 [M+Na]+, C13H12N2NaOS requires 267.0563.
1-(2′-Azidobenzoyl)-4-ethenyl-1-azetidin-2-thione (44b). To a solution of 4-ethenyl-1-azetidin-2-thione (43b, 0.16 g, 1.42 mmol) and dimethylaminopyridine (0.1 g, 0.82 mmol) in anhydrous dichloromethane (20 mL), was added, with stirring and under an atmosphere of dry nitrogen, 2-azidobenzoylchloride (0.28 g, 1.56 mmol) in anhydrous dichloromethane (10 mL) [prepared as described previously], dropwise over 20 min at −10 °C. The reaction was stirred for 30 min before triethylamine (0.29 mL, 2.83 mmol) was added dropwise over 10 min at −10 °C. The reaction mixture was warmed to room temperature and left to stir at ambient temperature for 24 h, before being concentrated by rotary evaporation under reduced pressure and purified by flash silica column chromatography (eluent: petroleum ether-ethyl acetate, 4:1) to yield 1-(o-azidobenzoyl)-4-ethenyl-1-azetidin-2-thione (0.26 g, 71%) as a yellow solid, melting point: 79–82 °C. NMR: δH (500 MHz, CDCl3): 2.84 (1H, dd, J = 17.1, 3.1, CHH). 3.24 (1H, dd, J = 17.1, 5.9, CHH), 5.15 (1H, m, CH), 5.38 (1H, d, J = 10.4, CHH=CH), 5.49 (1H, d, J = 17.2, CHH=CH), 6.09 (1H, ddd, J = 17.2, 10.4, 7.0, =CH), 7.21 (2H, m, 2 × ArH), 7.39 (1H, dd, J = 7.6, 1.2, ArH), 7.53 (1H, ddd, J = 7.8, 7.8, 1.5, ArH); δC (125 MHz, CDCl3): 47.10 (CH2), 59.42 (CH), 118.52 (CH), 119.46 (CH2), 124.97 (CH), 126.06 (q), 129.32 (CH), 132.40 (CH), 133.59 (CH), 163.97 (C=O), 201.89 (C=S). IR: υmax (thin film cm−1): 2927.9 (s), 2853.4 (s), 2128.3 (s), 1687.0 (s), 1462.9 (m), 1375.5 (m), 1303.6 (m). HRMS (ESI+): calc. for C12H10N4OS + H+ = 259.0648, measured = 259.0648.
(Aziridino[1,2-a]azetidino[2,1-c])[1,4]benzodiazepine-7-one-9-thione (48). A solution of 1-(2'-azidobenzoyl)-4-ethenyl-1-azetidin-2-thione (44b, 0.074 g, 2.87 mmol) dissolved in anhydrous toluene (6 mL) was heated at reflux under an atmosphere of dry nitrogen for 24 h at which point TLC confirmed that the reaction had gone to completion. The sample was cooled to room temperature, concentrated under reduced pressure, and purified by silica column chromatography (eluent: petroleum ether:ethyl acetate, 1:2) to yield the title compound 48 (0.032 g, 48% yield) as a yellow oil. NMR: δH (500 MHz, CDCl3): 2.26 (1H, d, J = 3.5, NCHH), 2.81 (1H, d, J = 4.4, NCHH), 3.10 (1H, dd, J = 16.8, 2.8, CSCHH), 3.17 (1H, ddd, J = 8.9, 4.4, 3.5, ArNCH), 3.31 (1H, dd, J = 16.8, 5.7, CSCHH), 4.20 (1H, ddd, J = 8.9, 5.7, 2.8, CONCH), 7.10 (1H, ddd, J = 7.6, 7.6, 0.8, ArH), 7.17 (1H, d, J = 8.1, ArH), 7.45 (1H, ddd, J = 7.7, 7.7, 1.6, ArH), 7.80 (1H, dd, J = 8.1, 1.6, ArH); δC (125 MHz, CDCl3): 35.13 (CH2), 42.00 (CH), 45.14 (CH2), 58.86 (CH), 122.90 (CH), 123.47 (CH), 132.20 (CH), 133.76 (CH), 150.04 (q), 163.89 (C=O), 199.55 (C=S). IR: υmax (thin film cm−1): 2873.3 (w), 1698.2 (s), 1653.5 (s), 1601.9 (m), 1483.4 (m), 1355.8 (m), 1345.6 (s), 1195.4 (m). HRMS (ESI+): calc. for C12H10N2OS + H+ = 231.0587, measured = 231.0588.

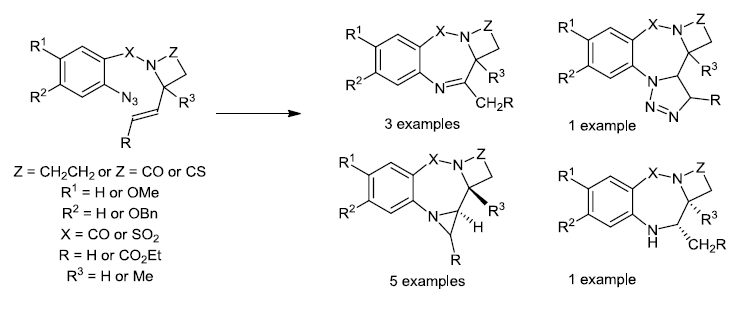
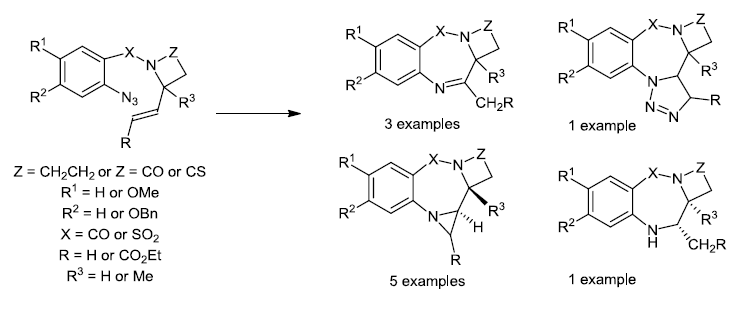{kind=link}
{kind=link}
{kind=link}
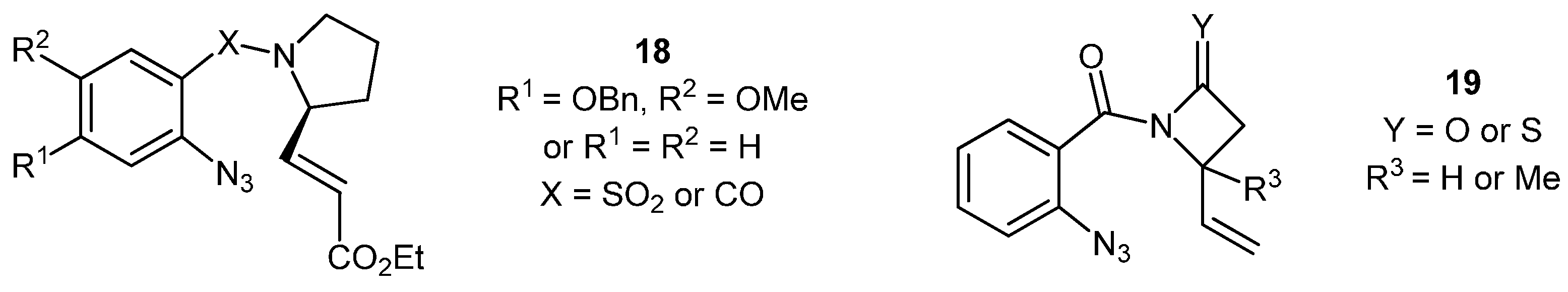{kind=link}
{kind=link}
{kind=link}