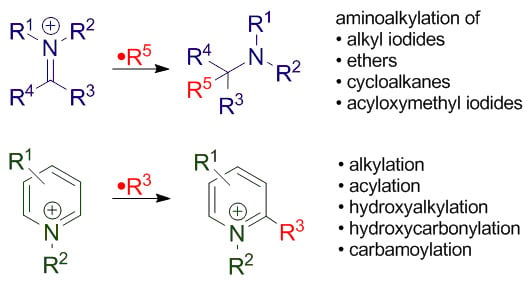
Radical Addition to Iminium Ions and Cationic Heterocycles


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
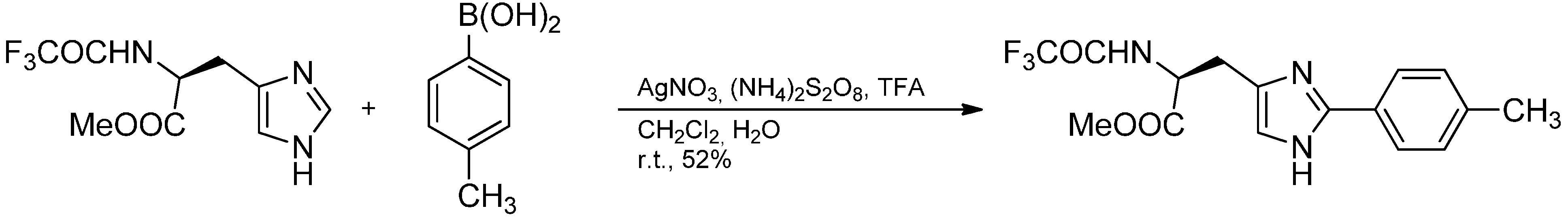{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
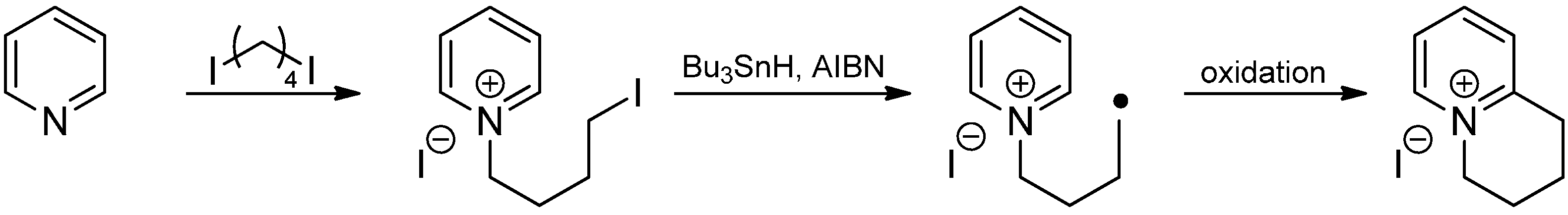{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
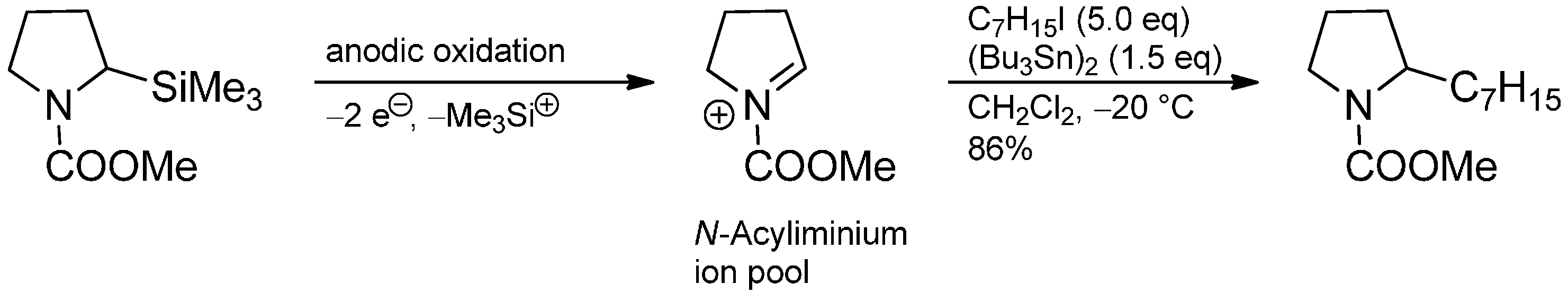{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
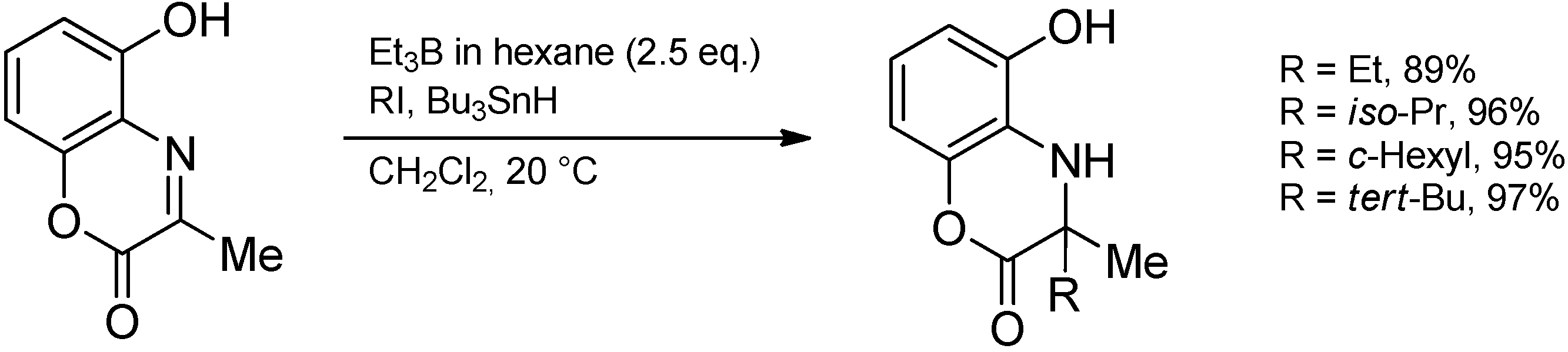{kind=link}
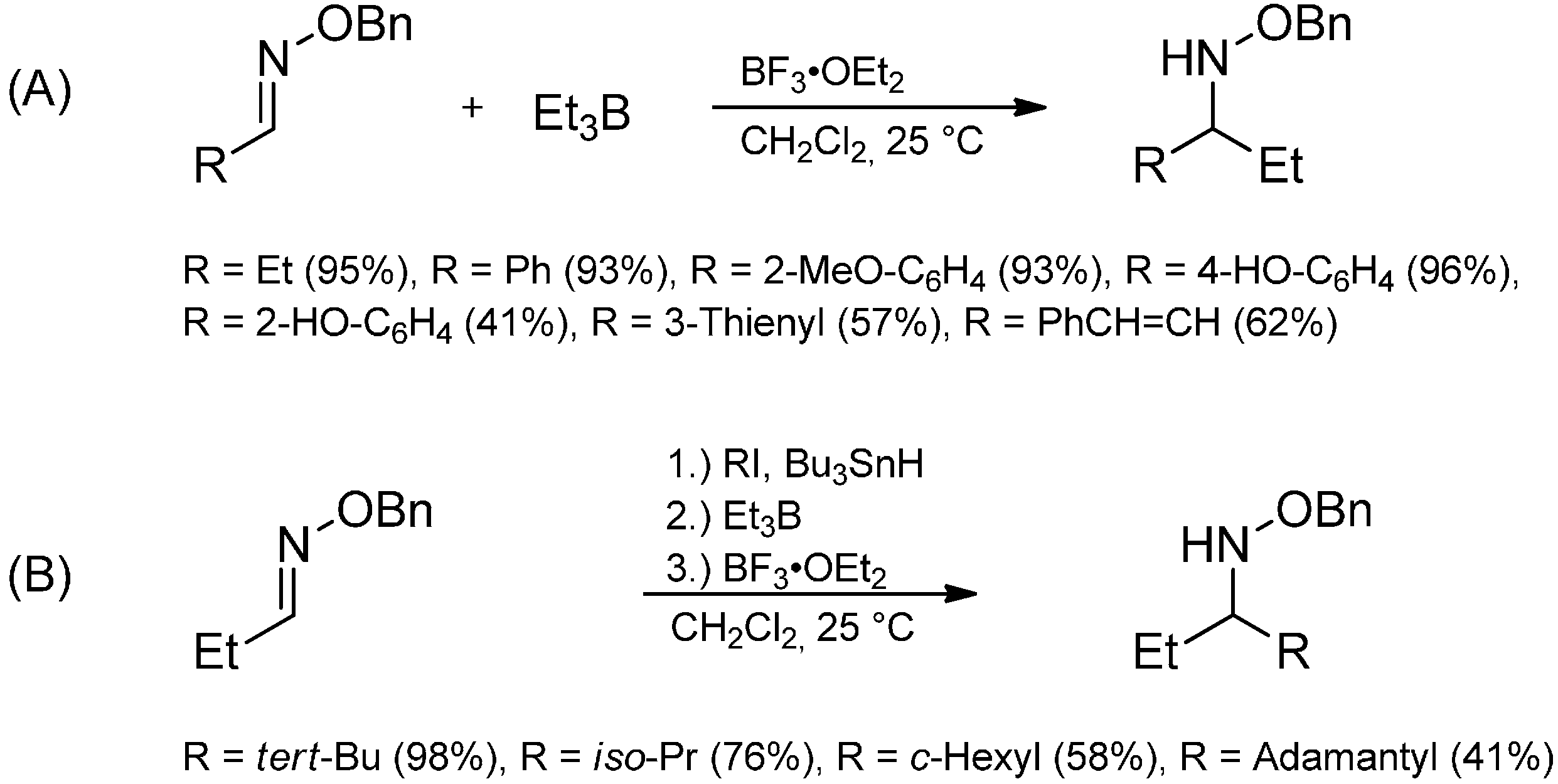{kind=link}
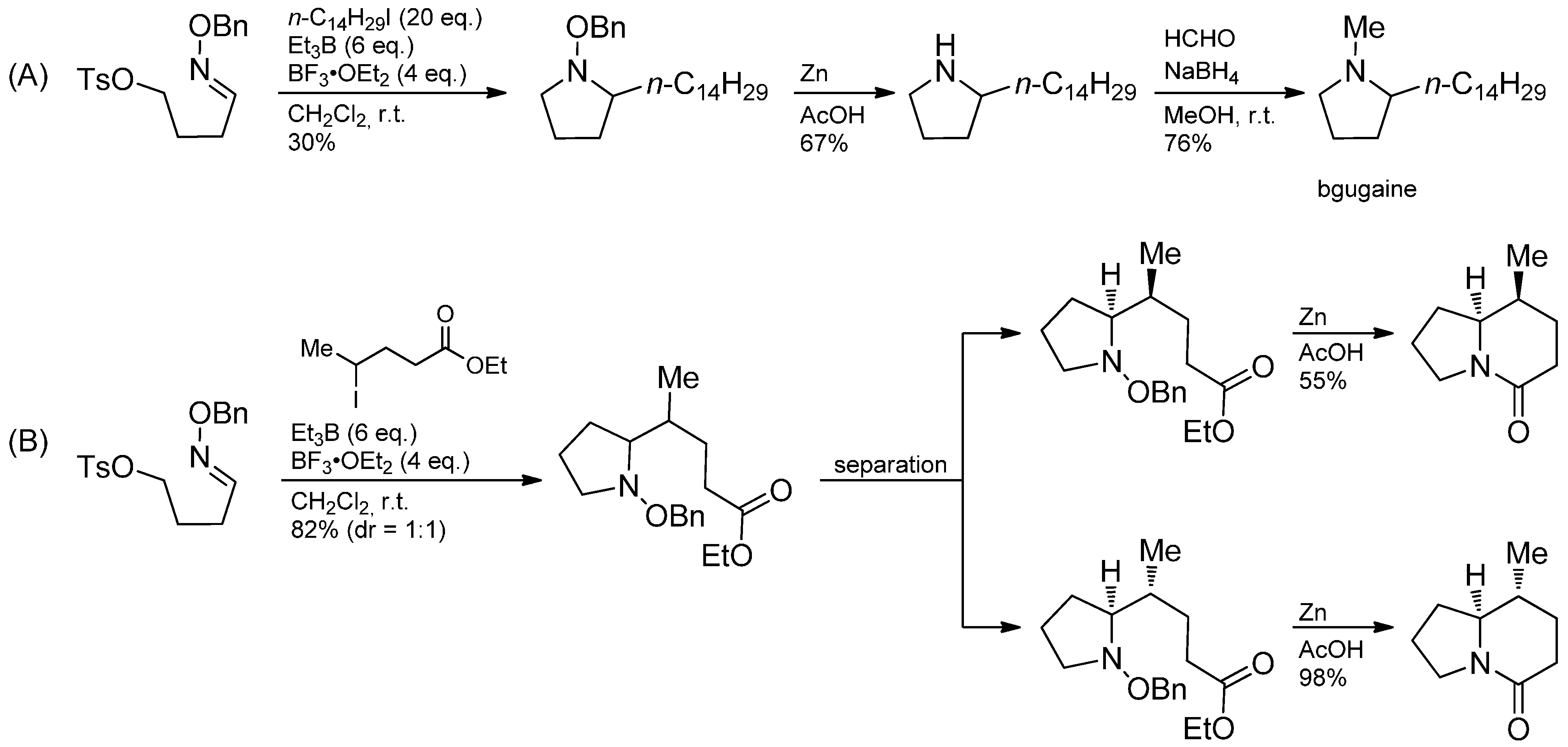{kind=link}
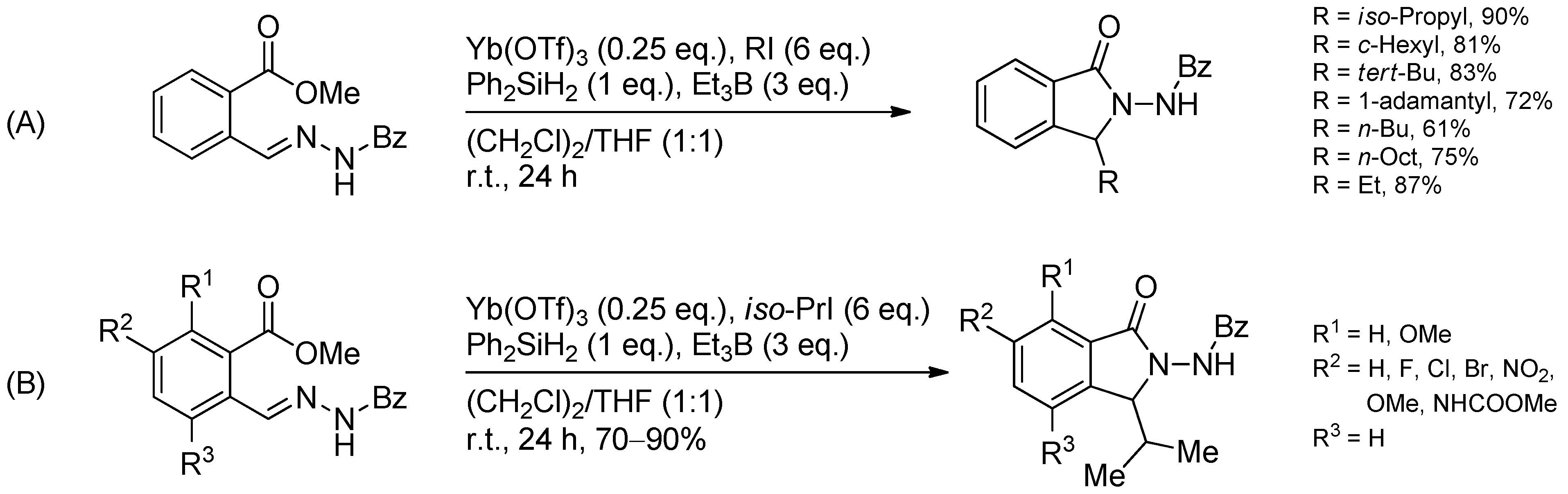{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
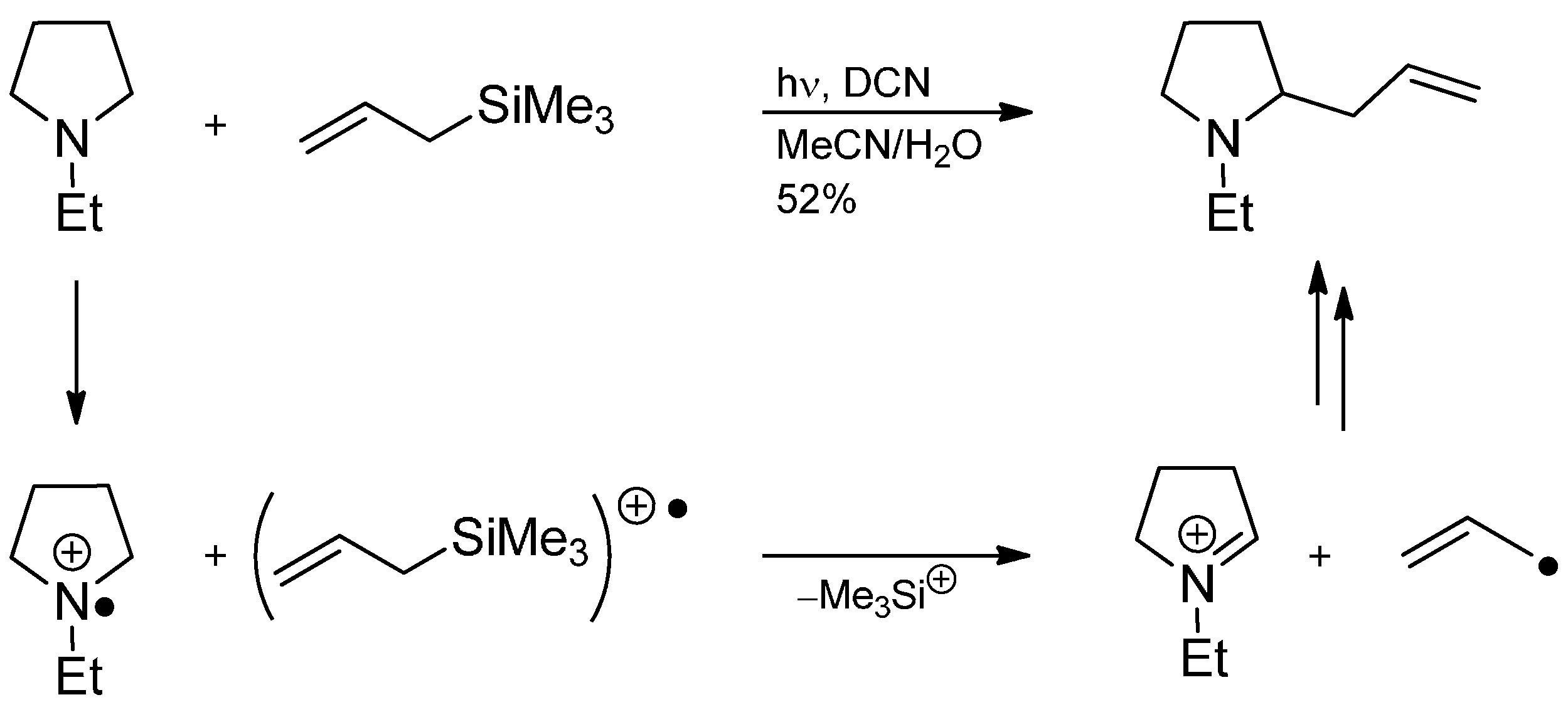{kind=link}
{kind=link}
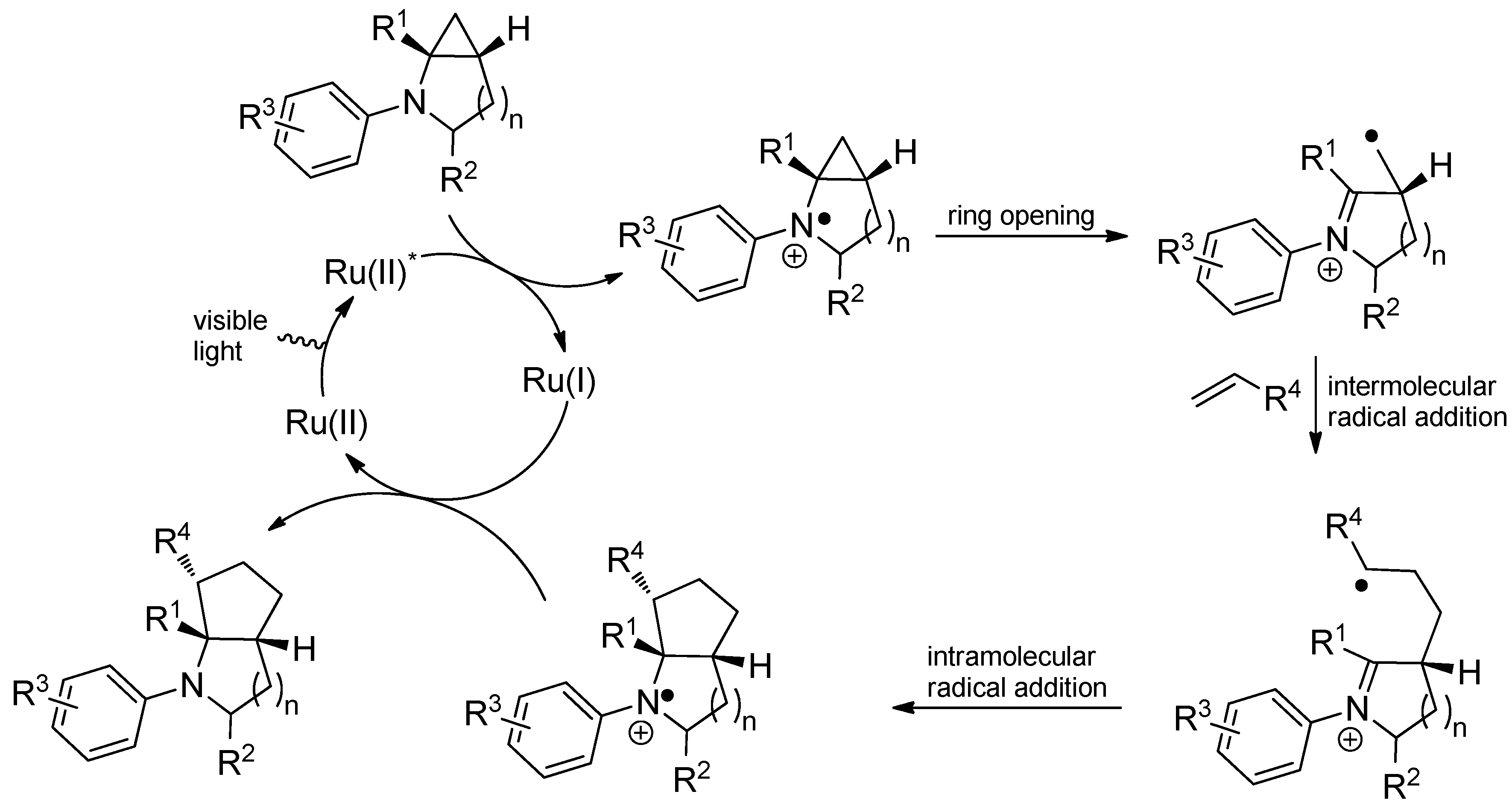{kind=link}
1. Introduction
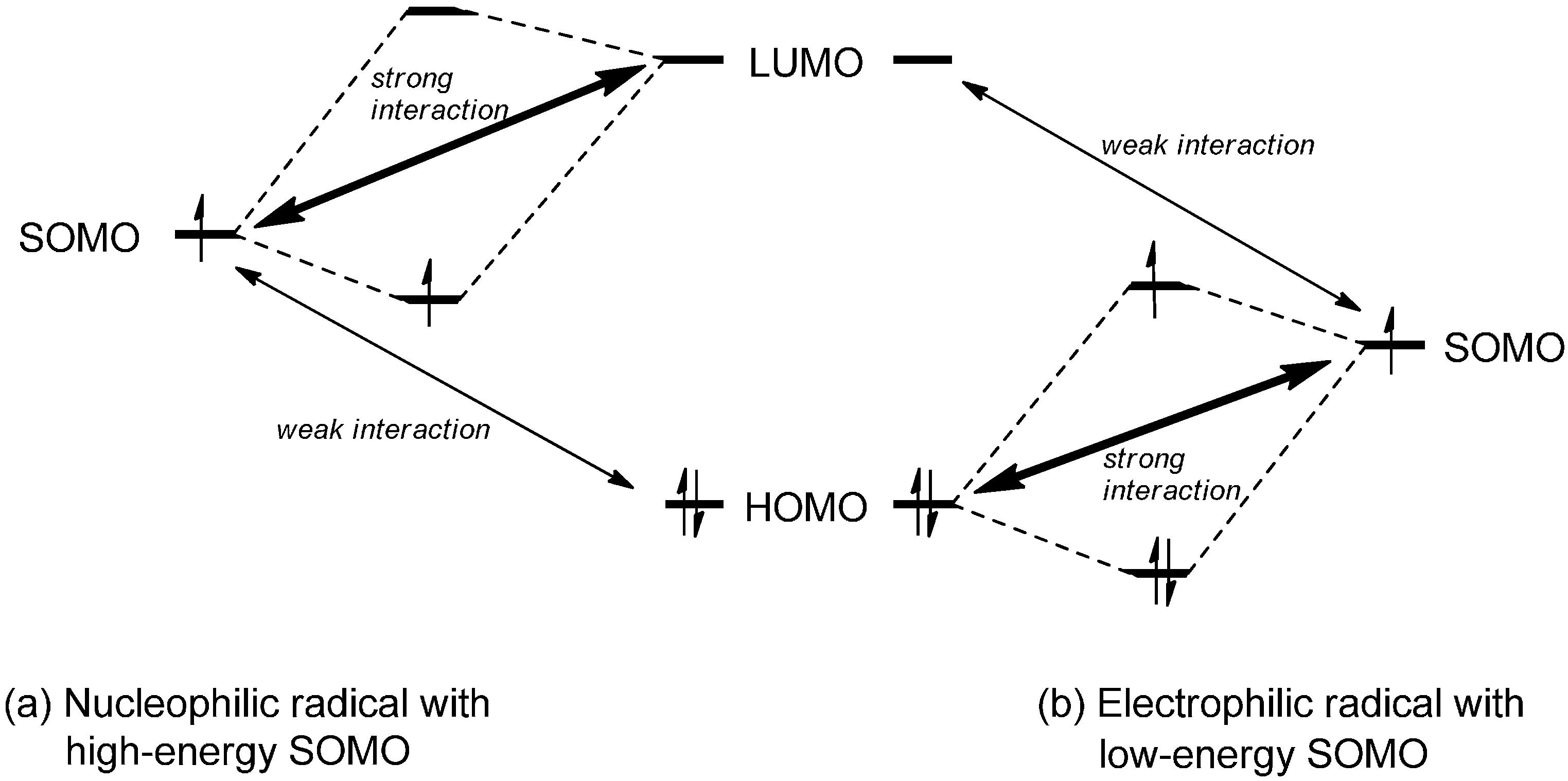
2. Addition of Carbon-Centered Radicals to Cationic Heterocycles
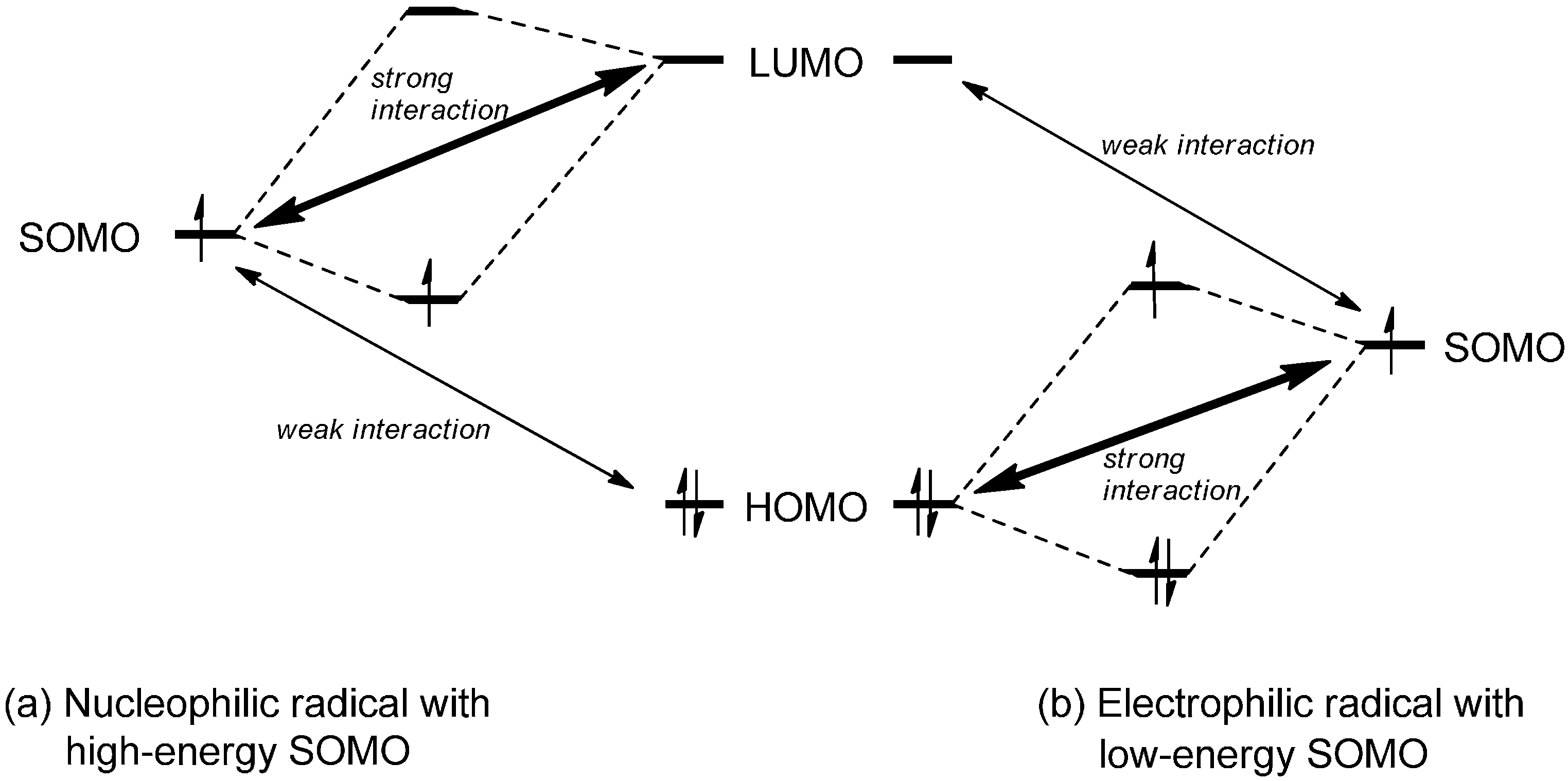
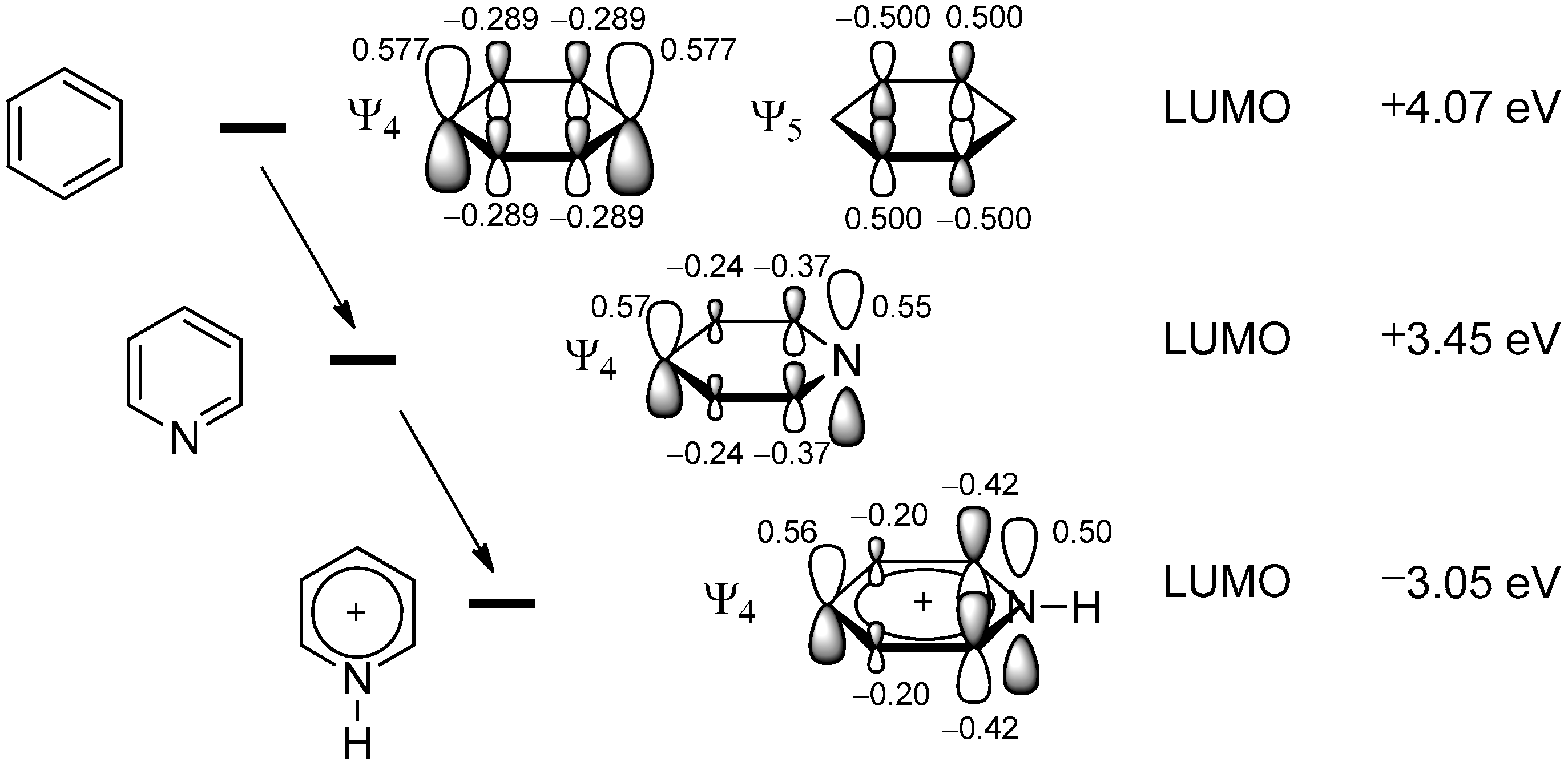
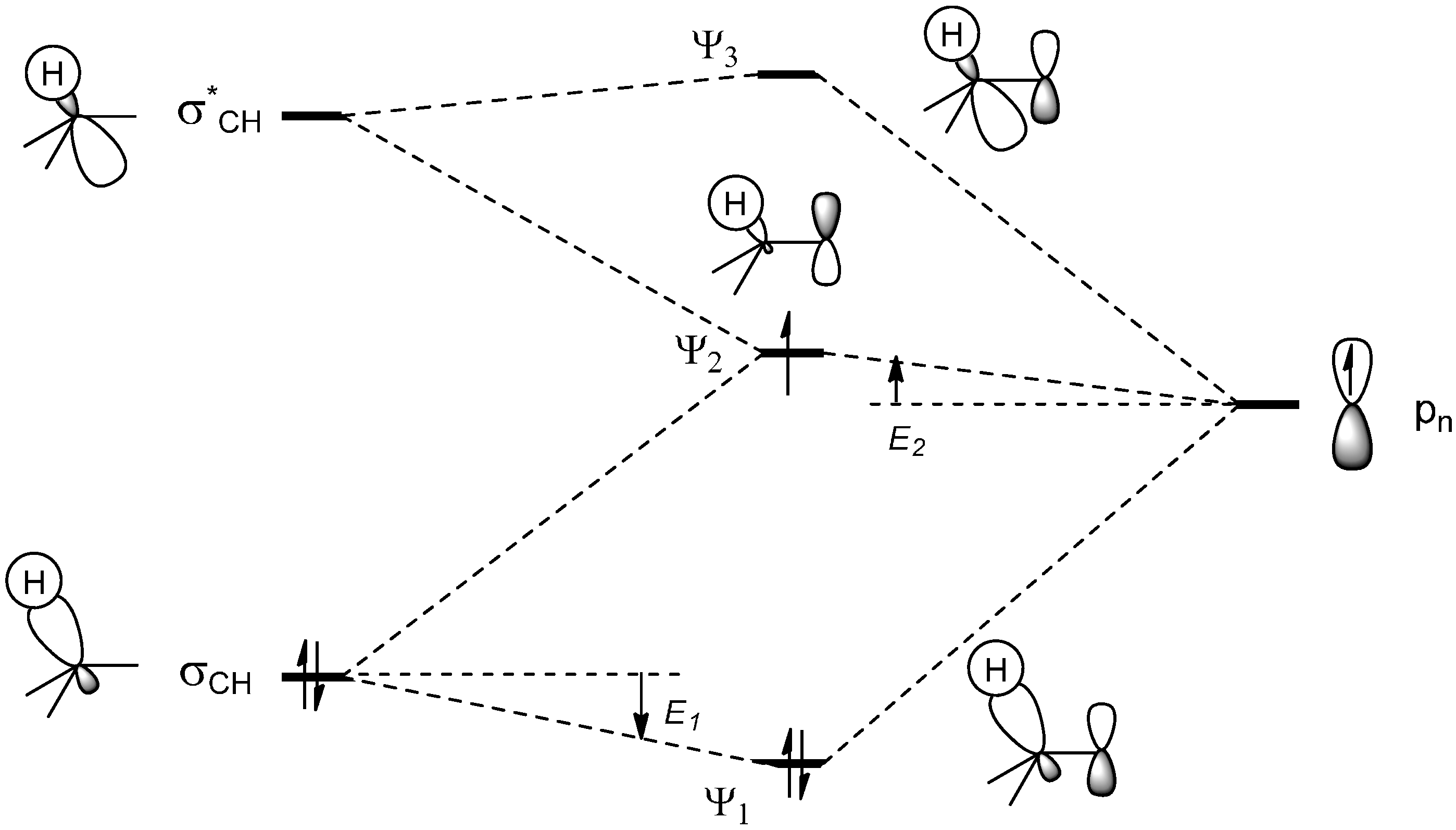
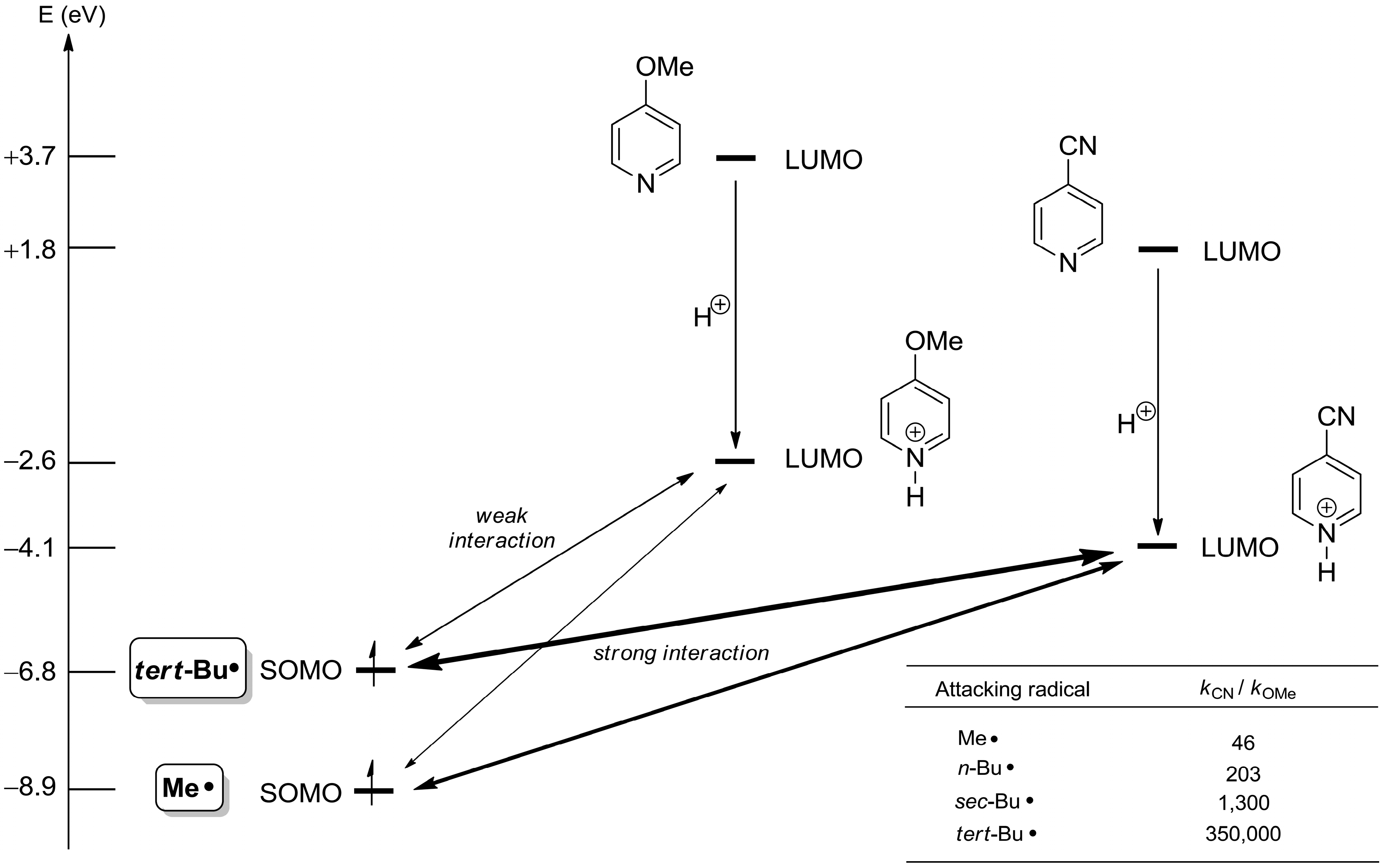
2.1. Theoretical Background, SOMO-HOMO/LUMO Interaction
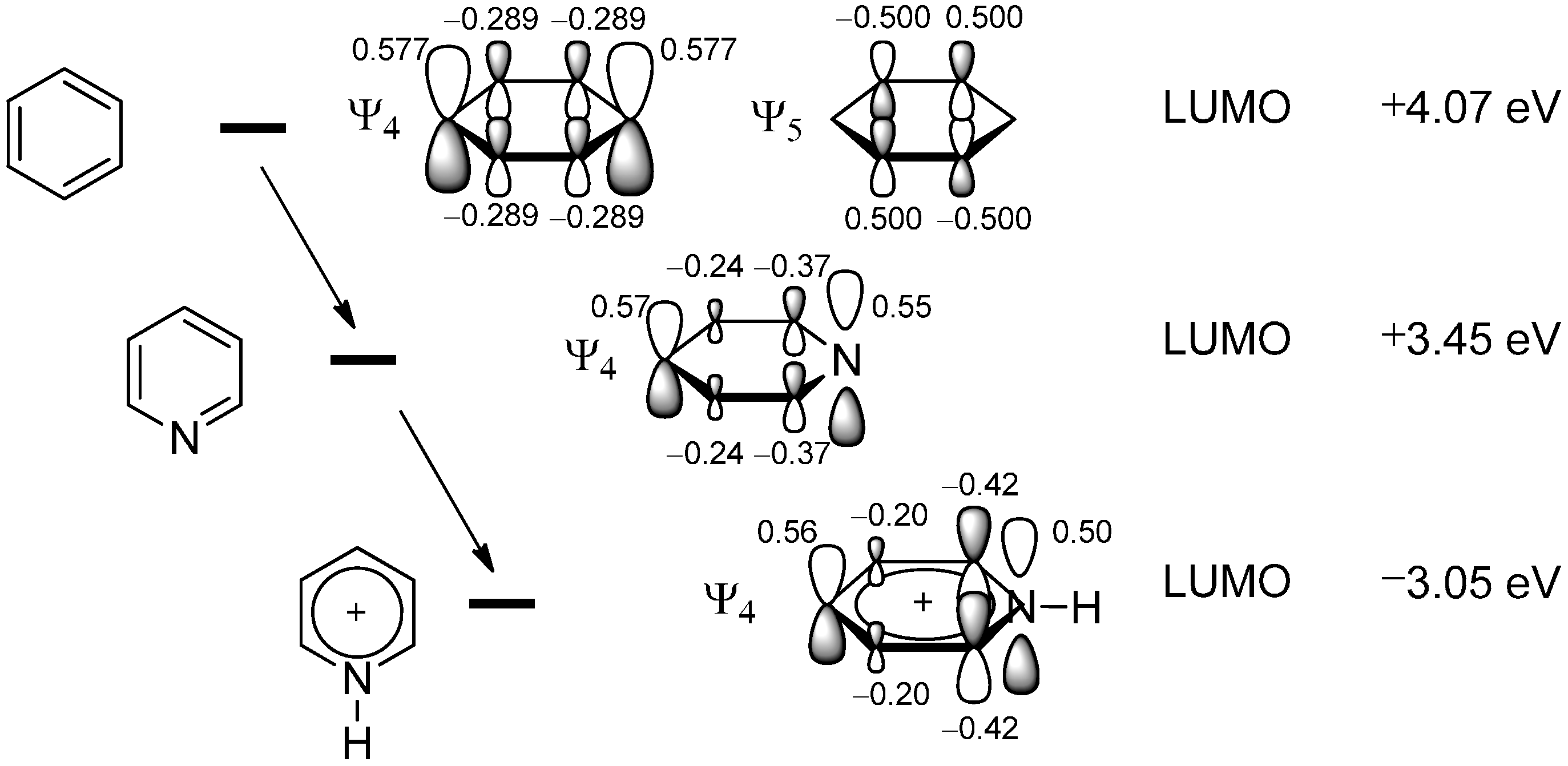

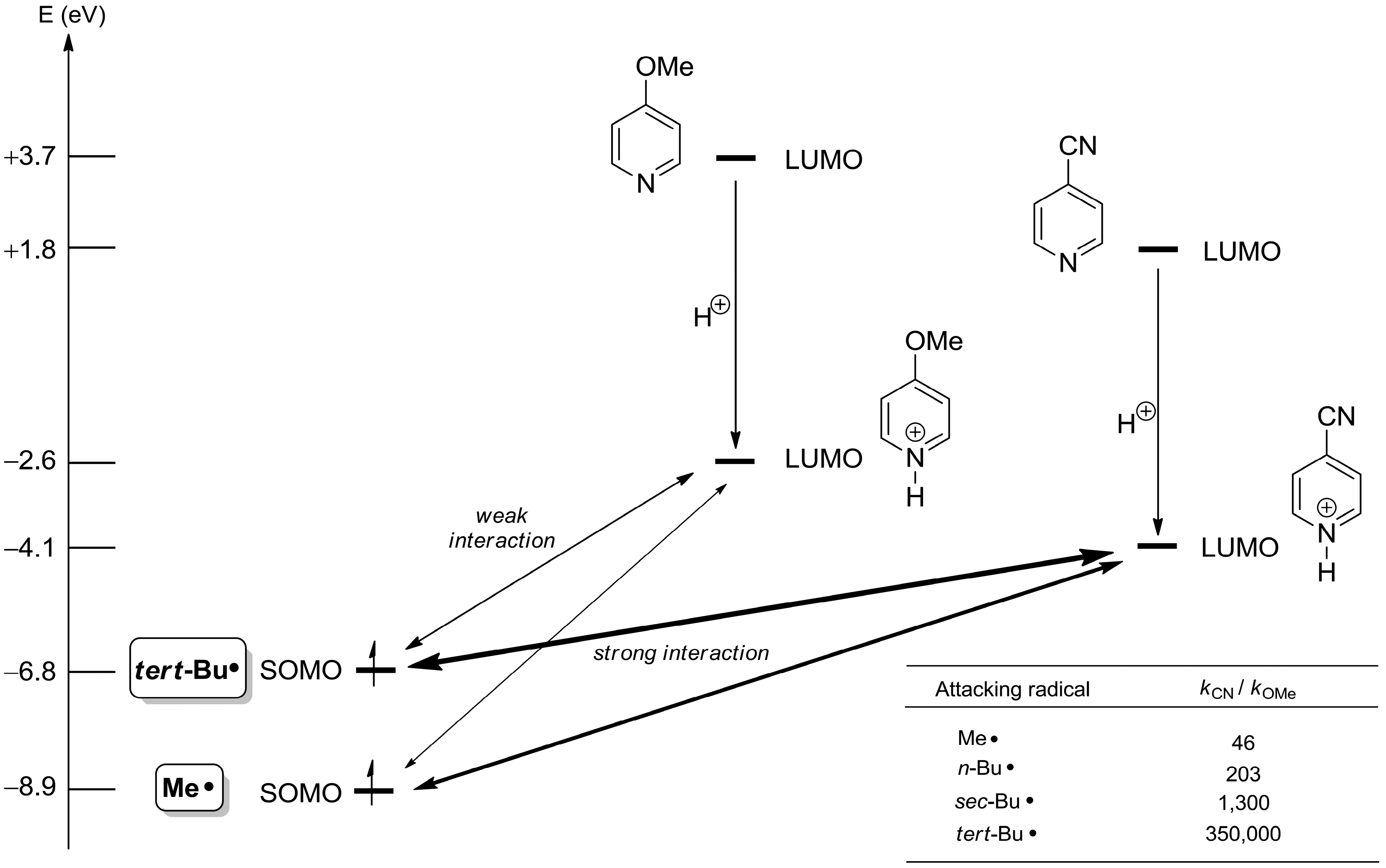
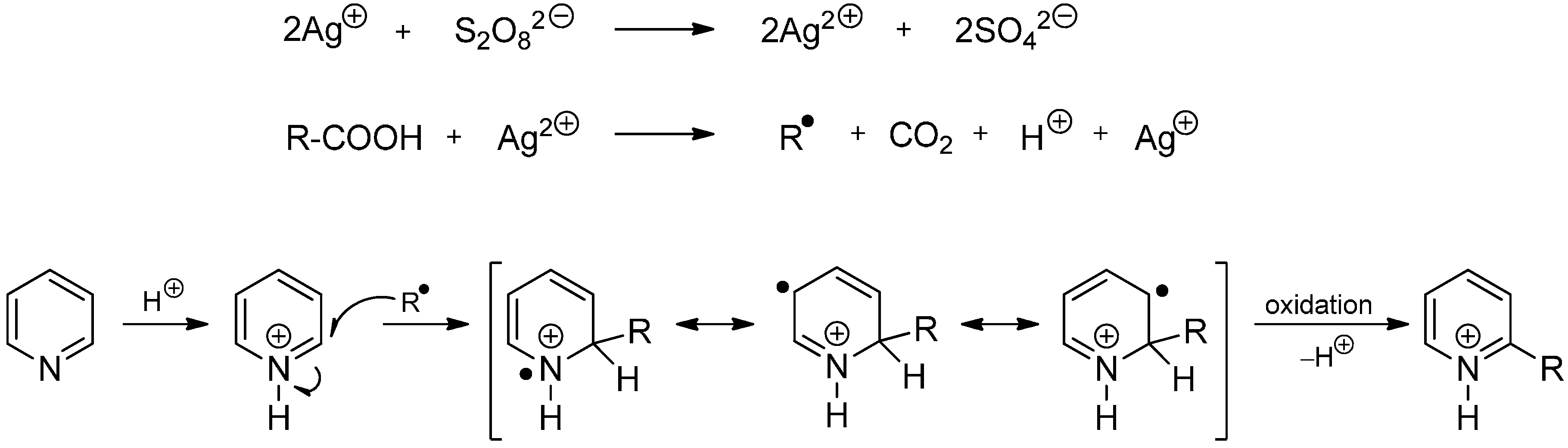
2.2. The Minisci Reaction
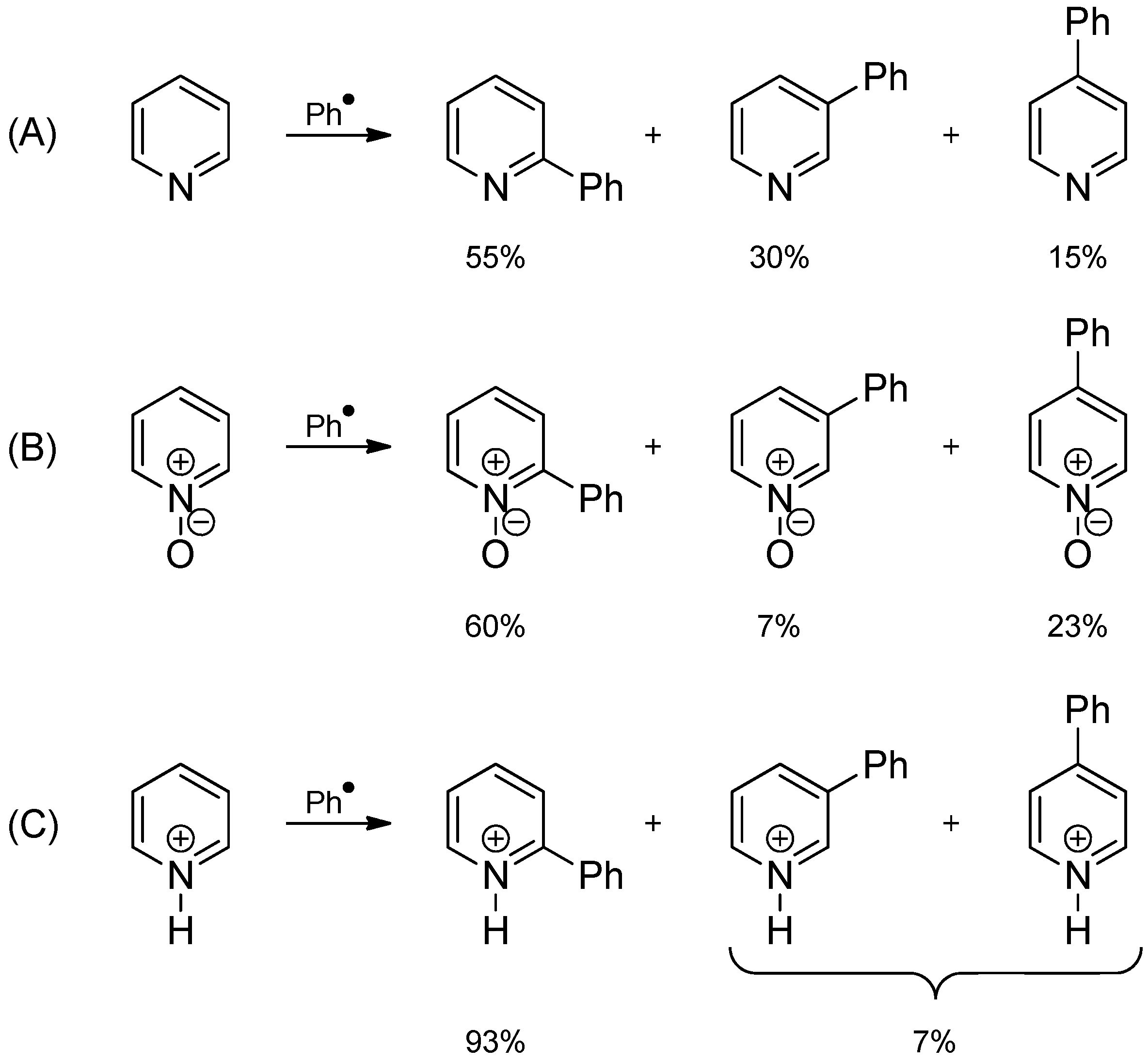
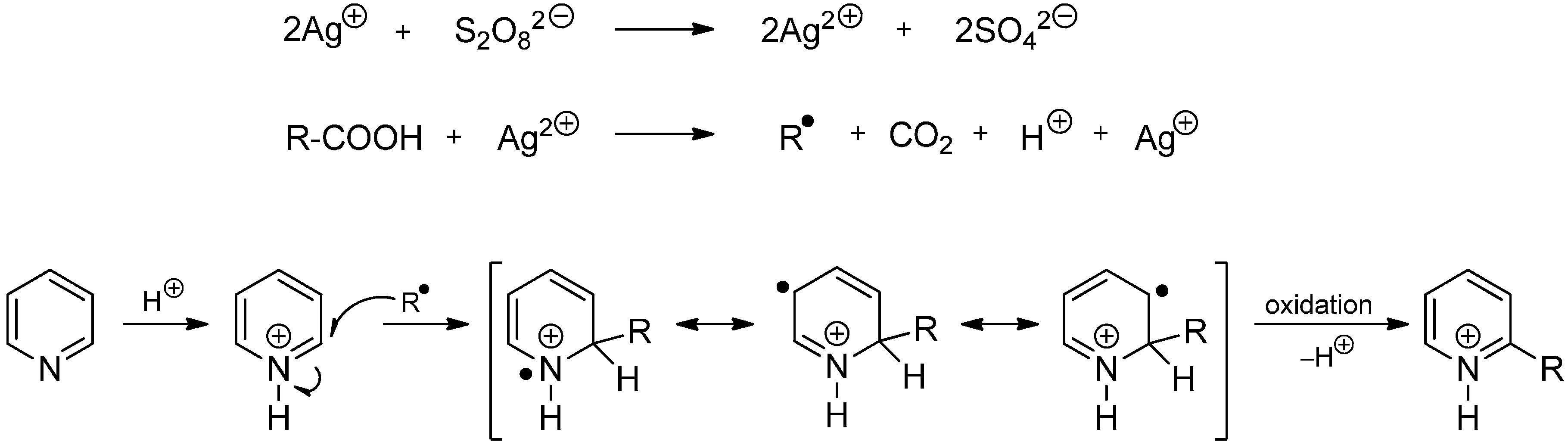

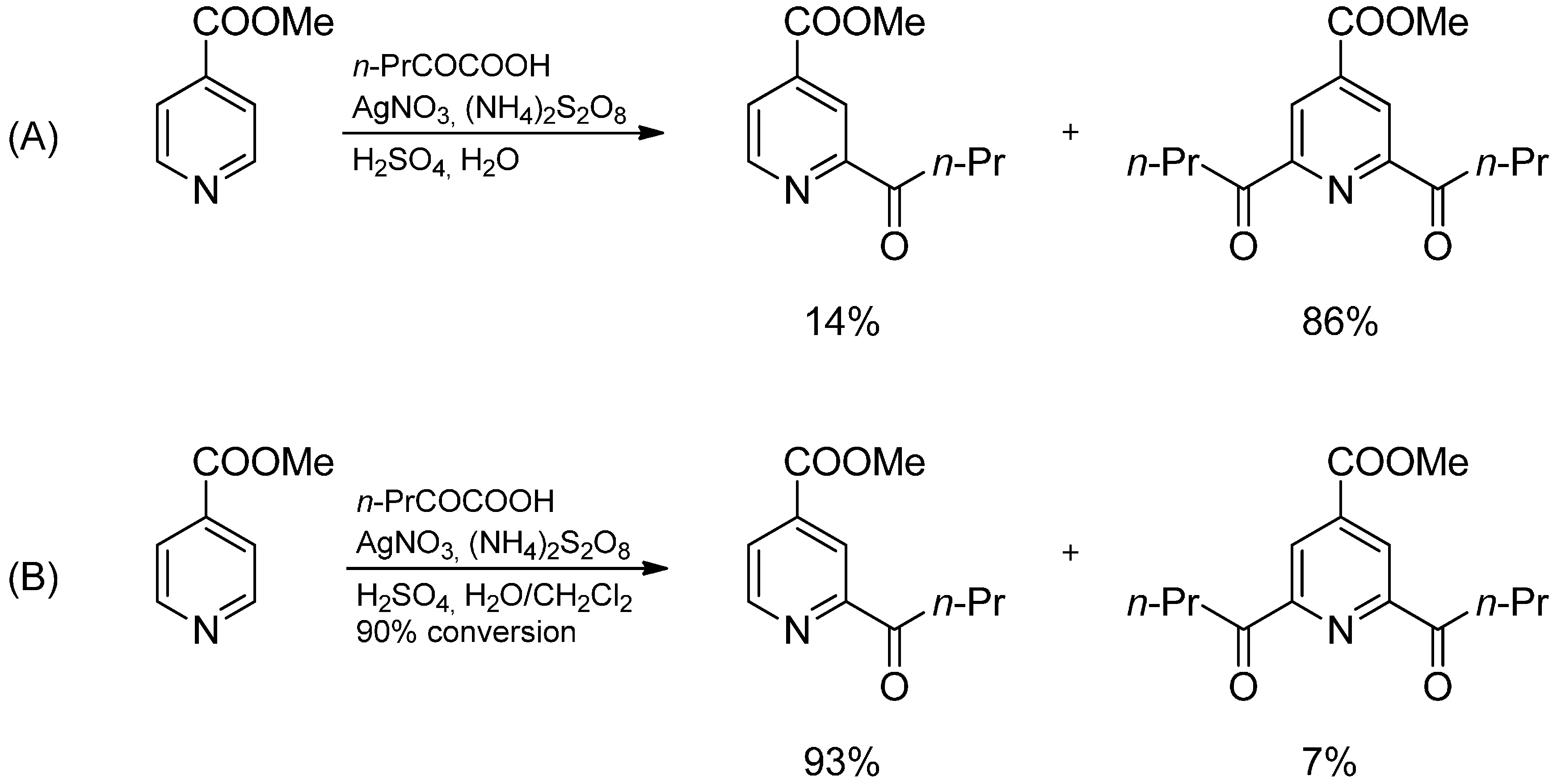
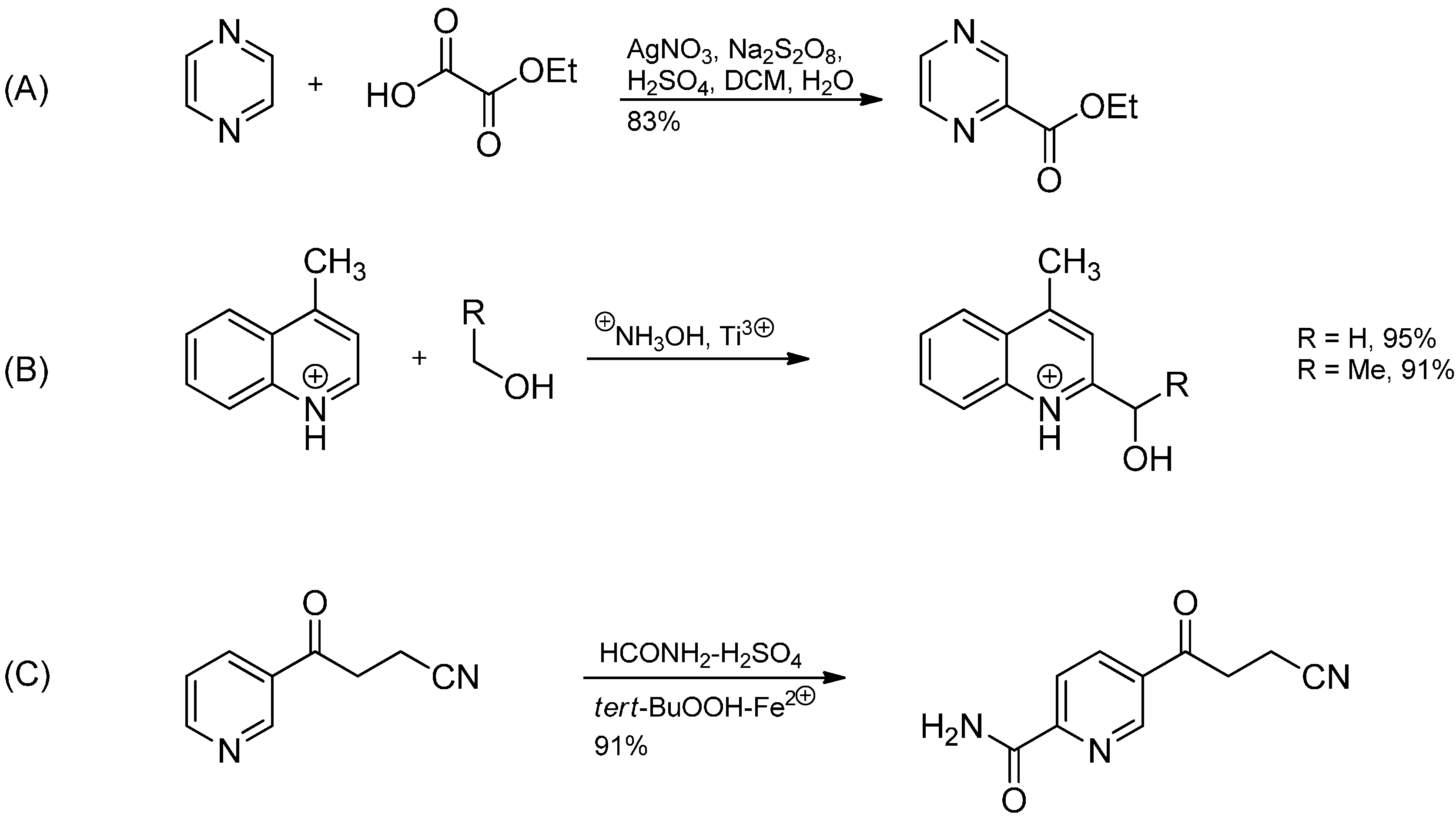
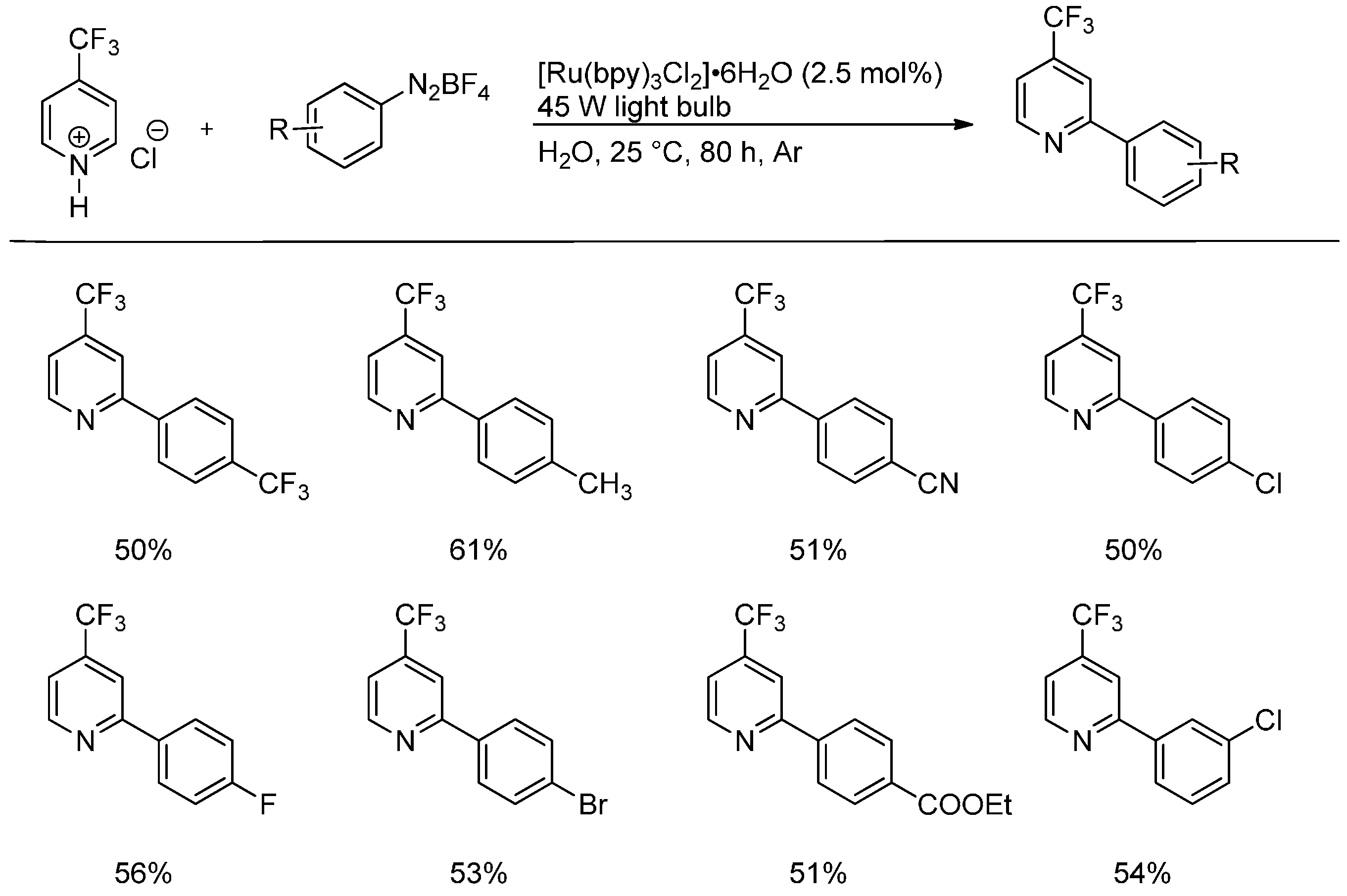

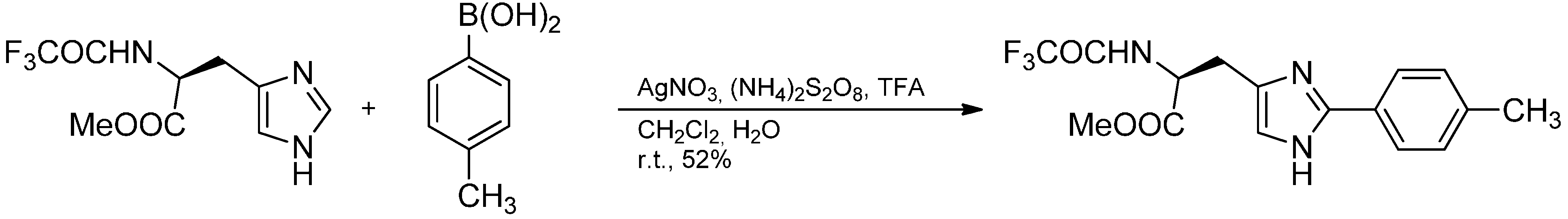
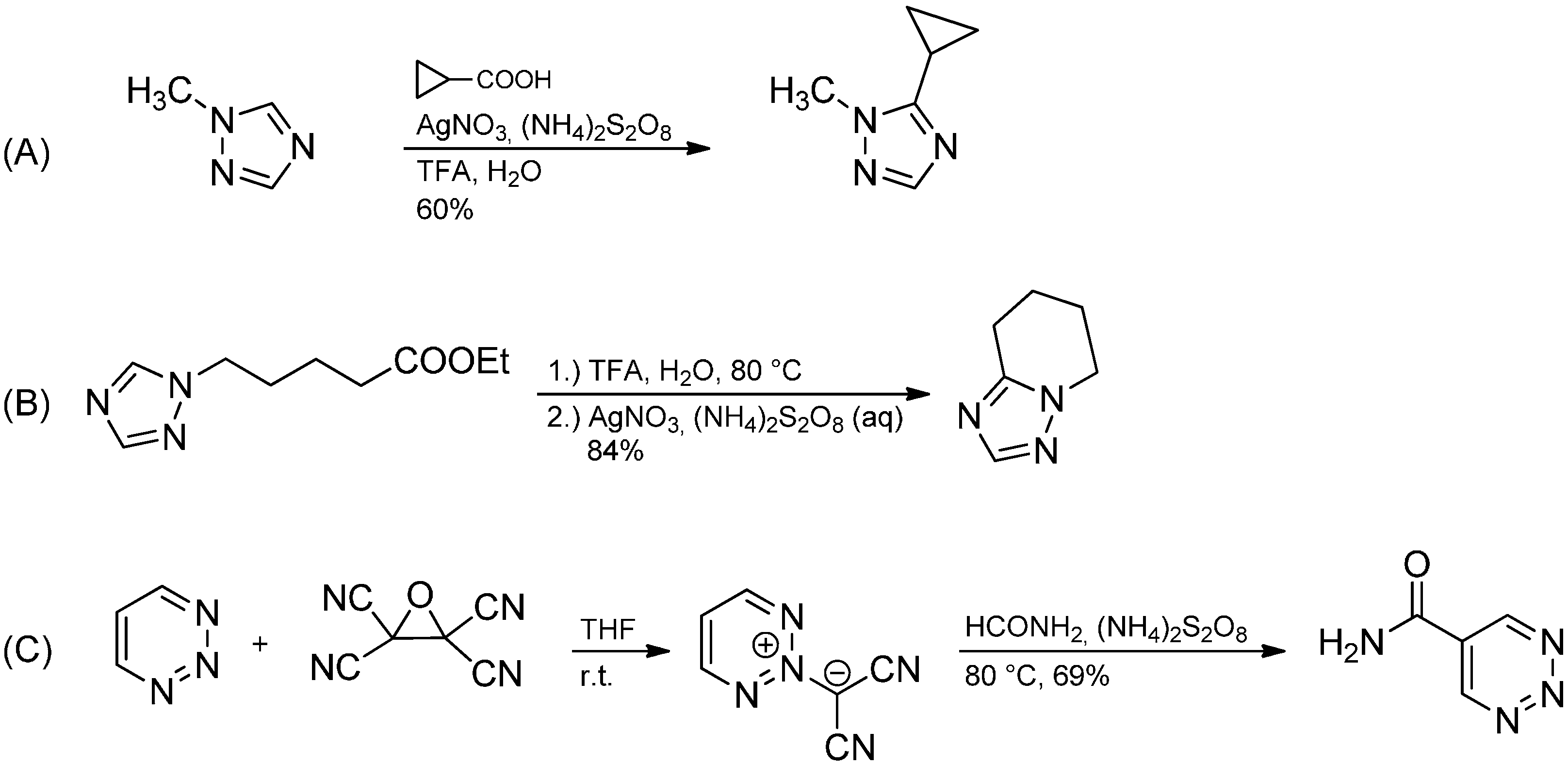
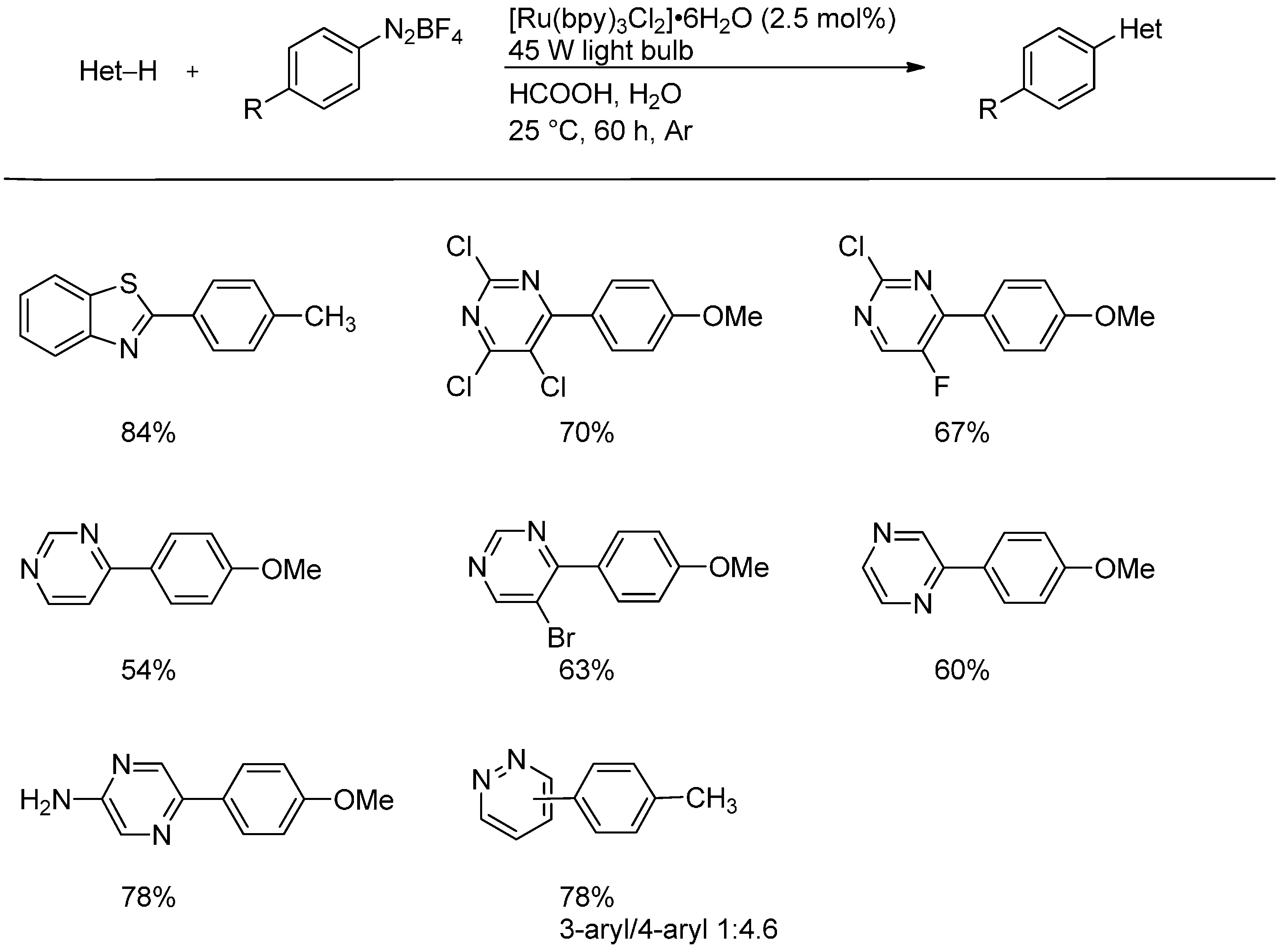
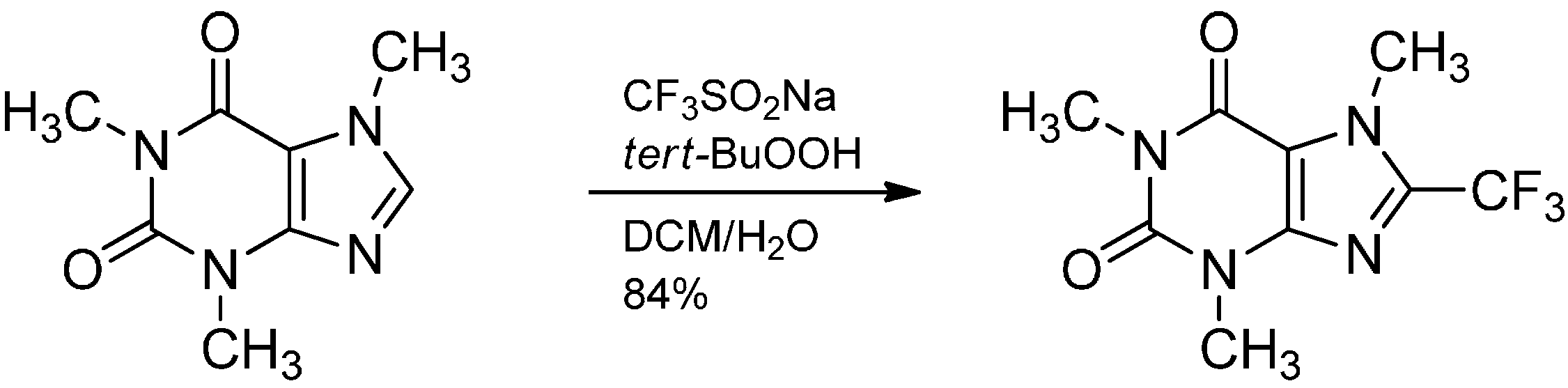

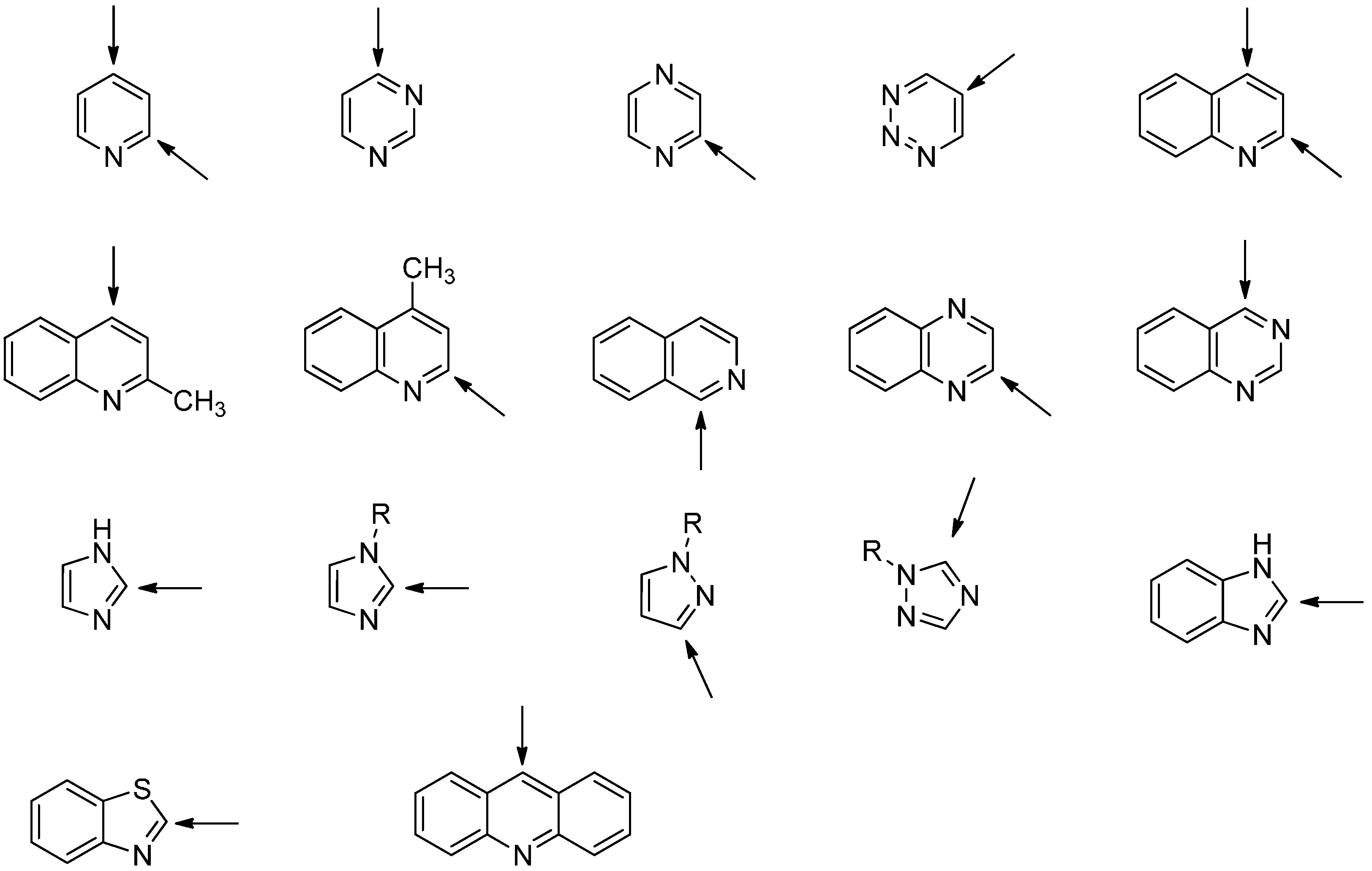
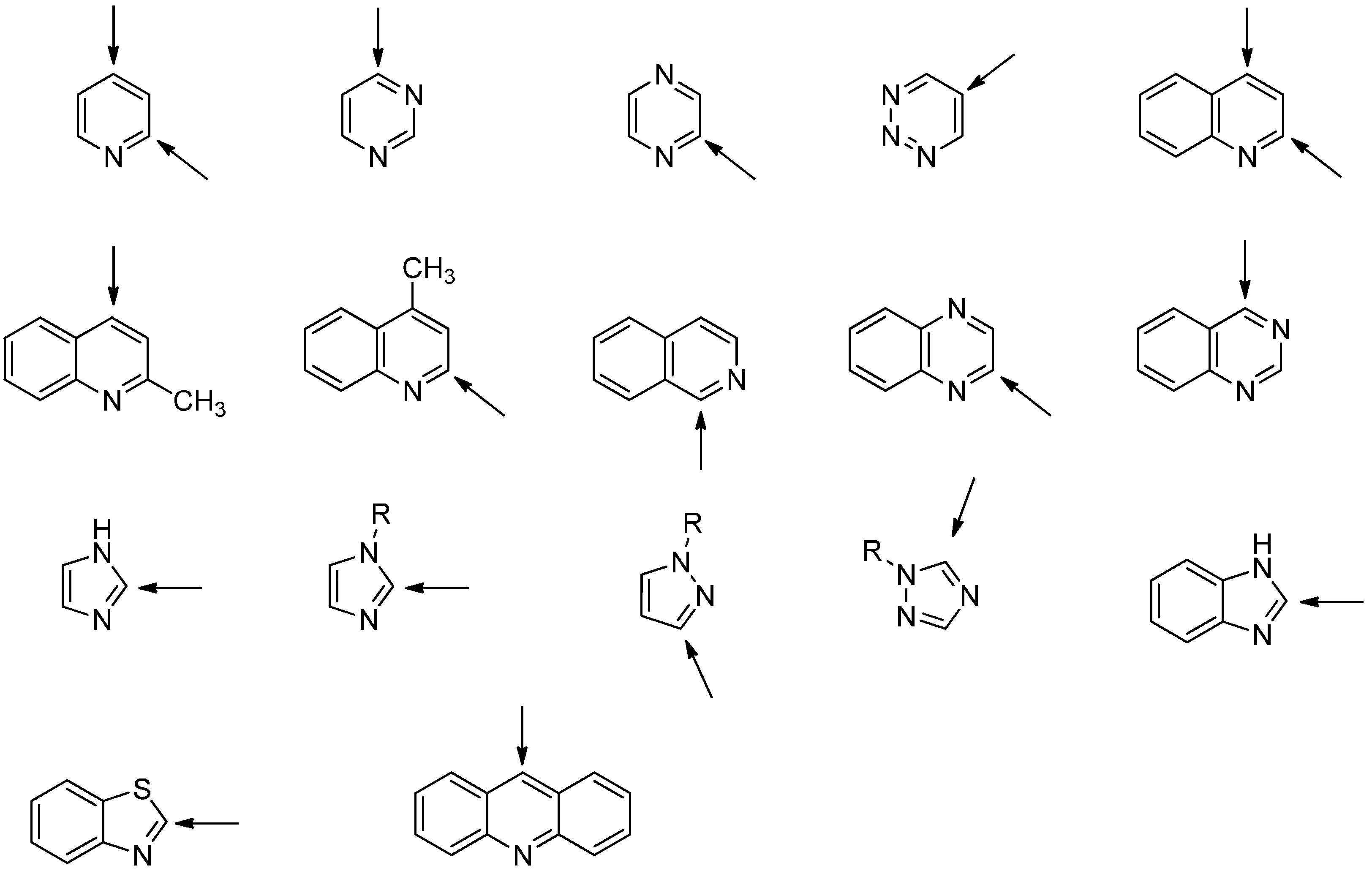
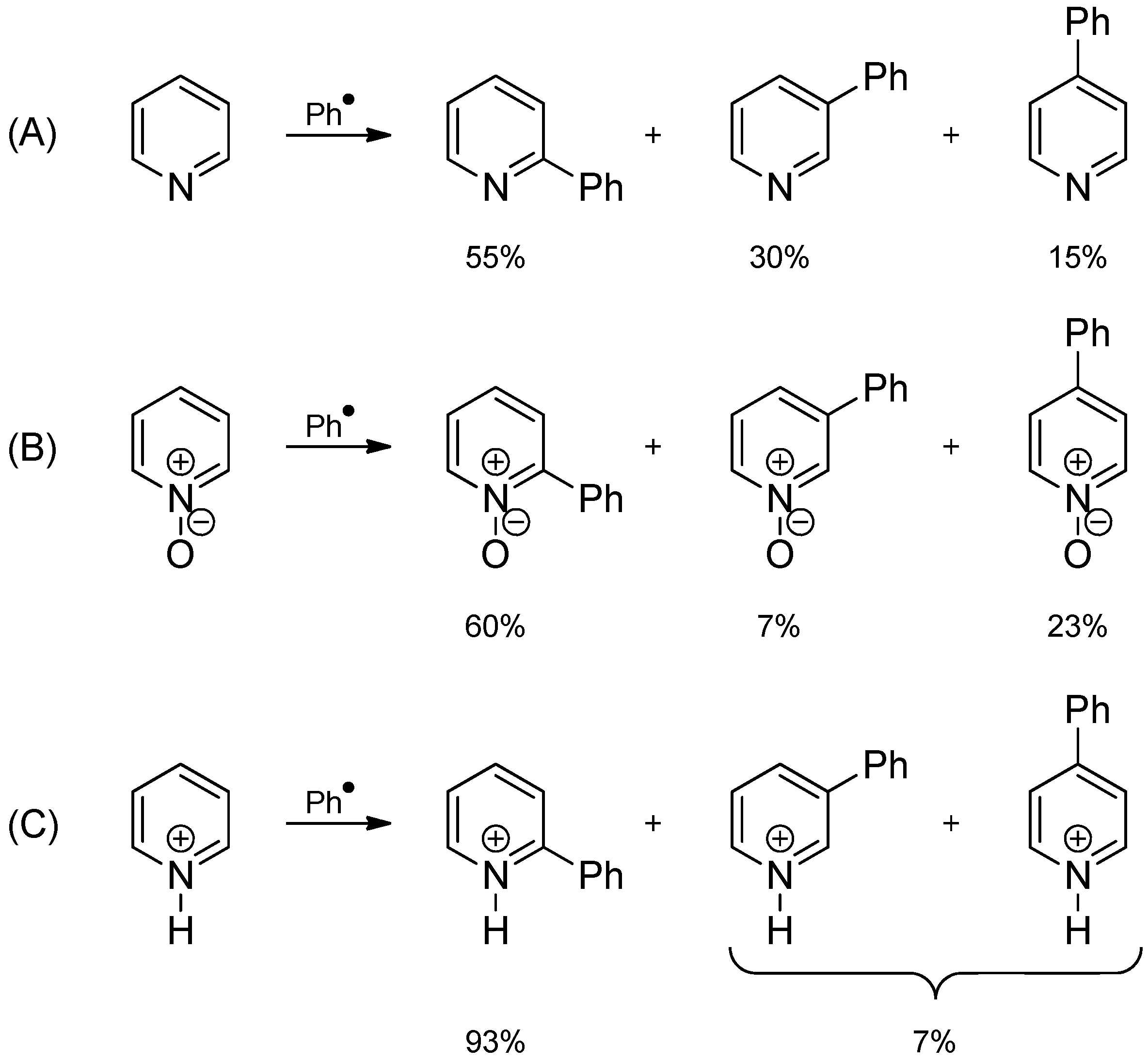
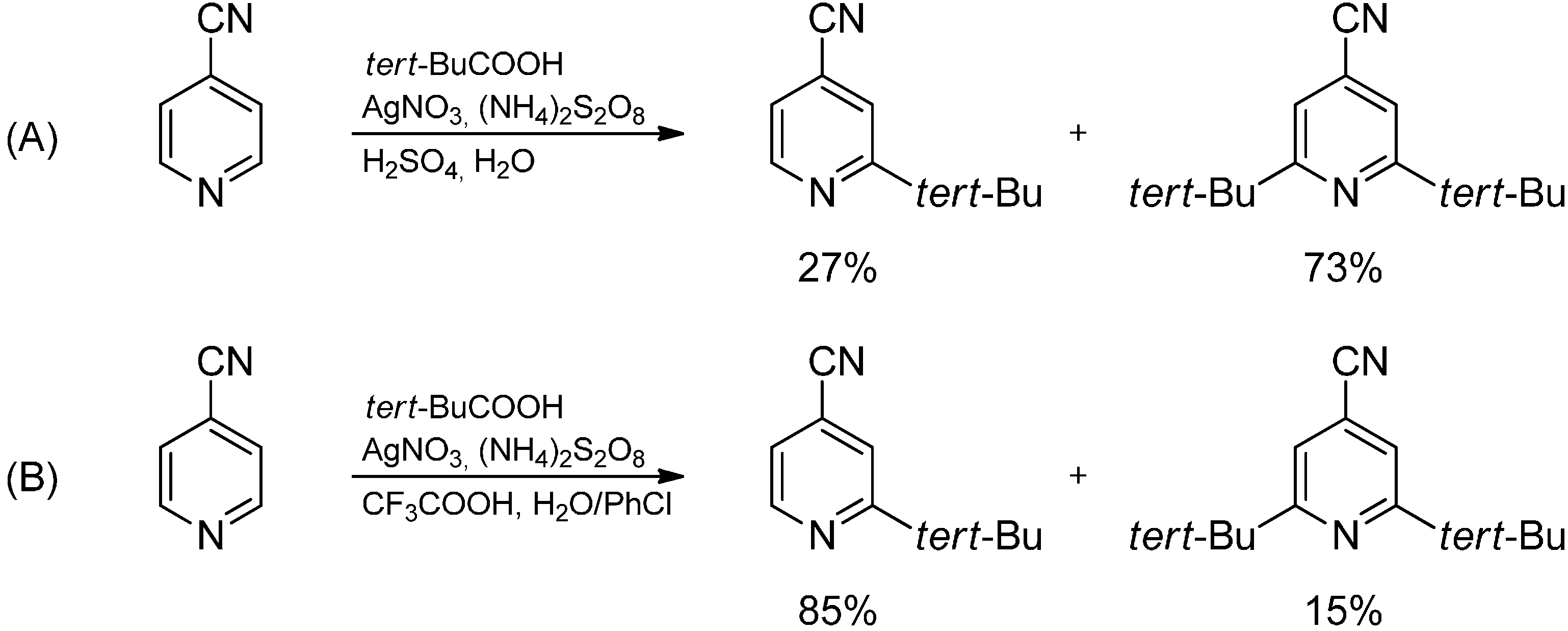
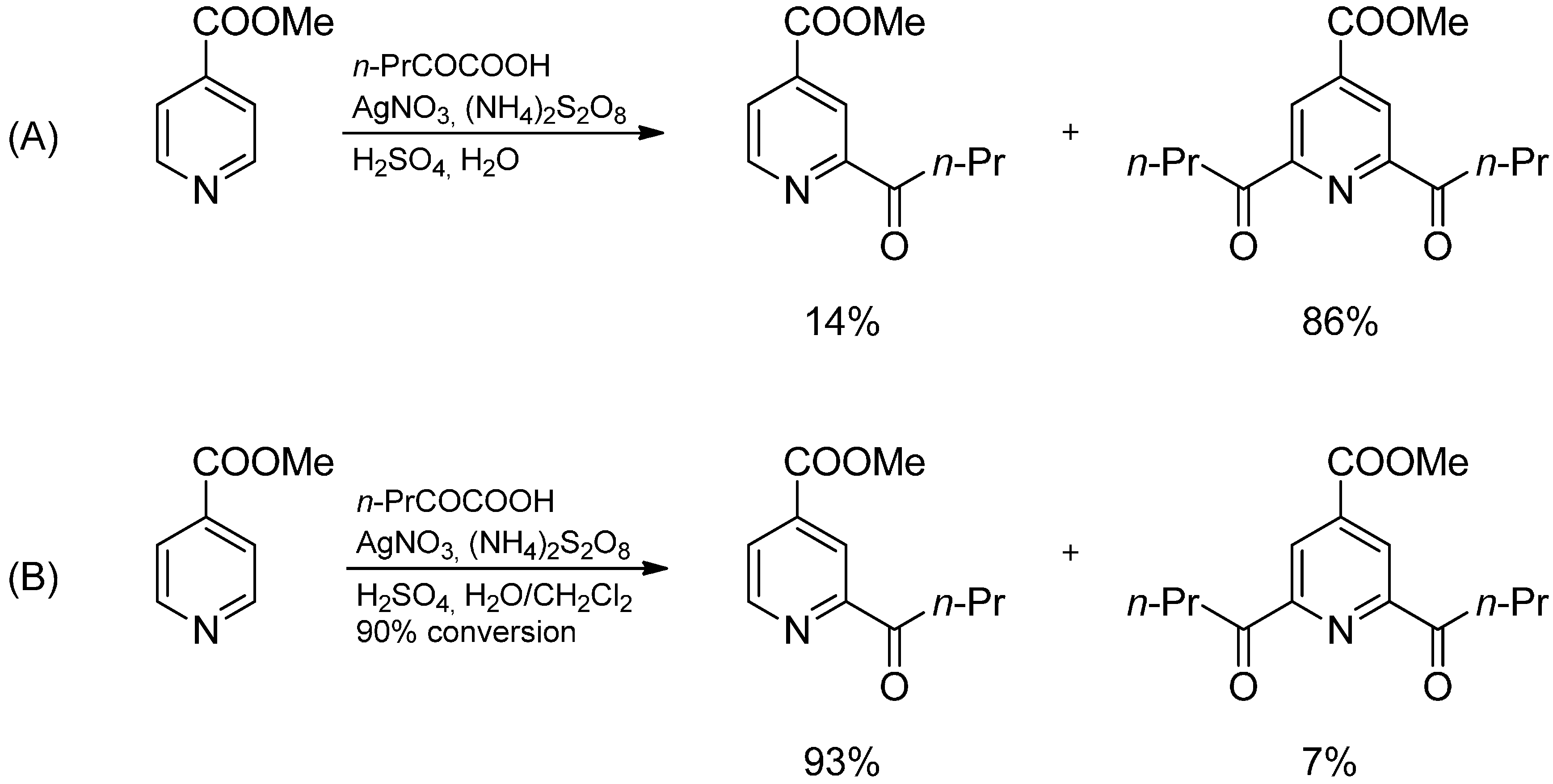
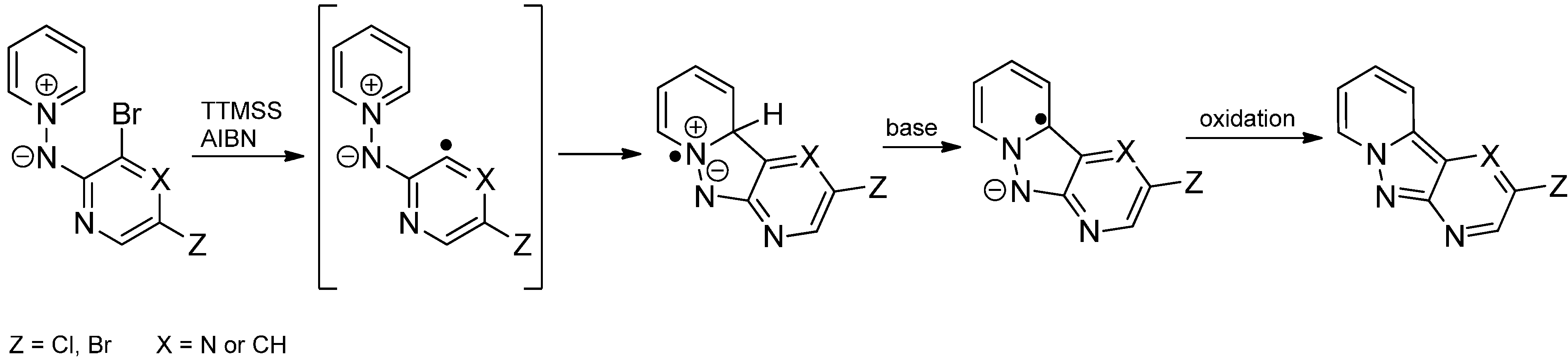
2.3. Addition to Quaternized Pyridines and Heterocycles
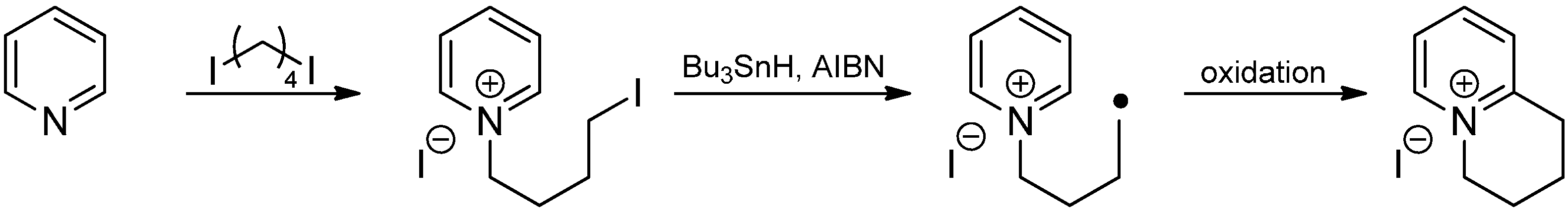
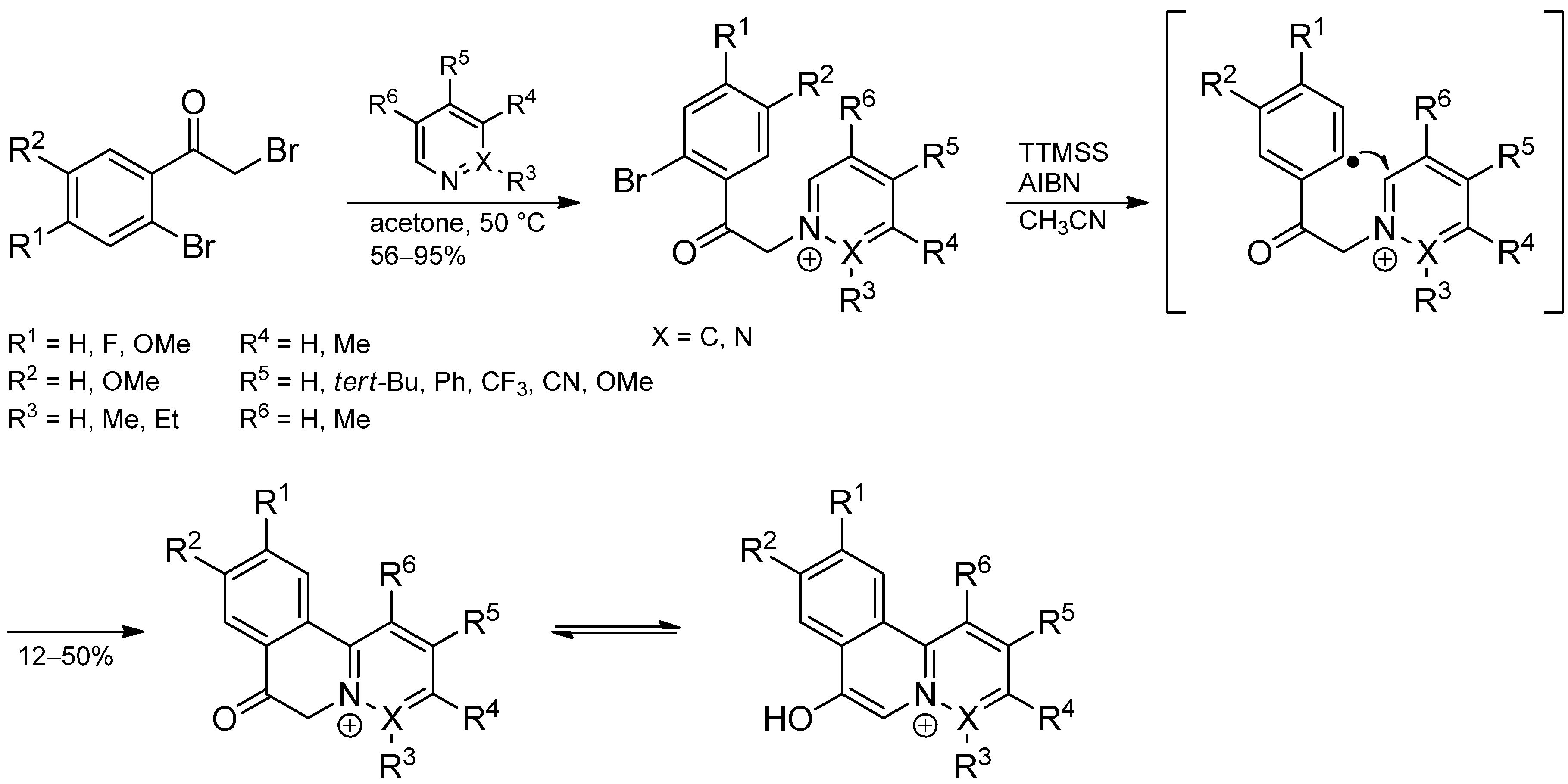
3. Addition to Aliphatic Iminium Ions
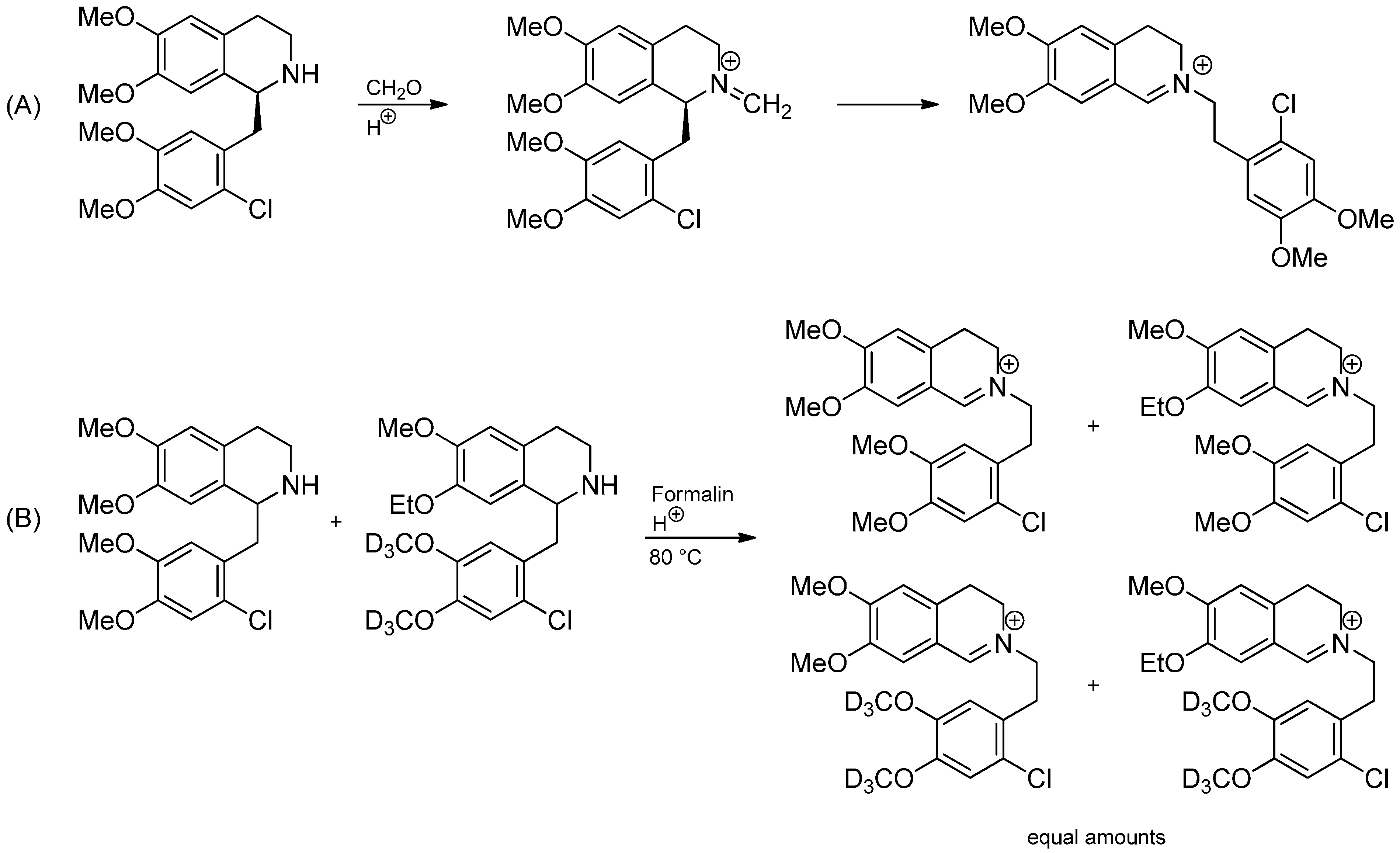
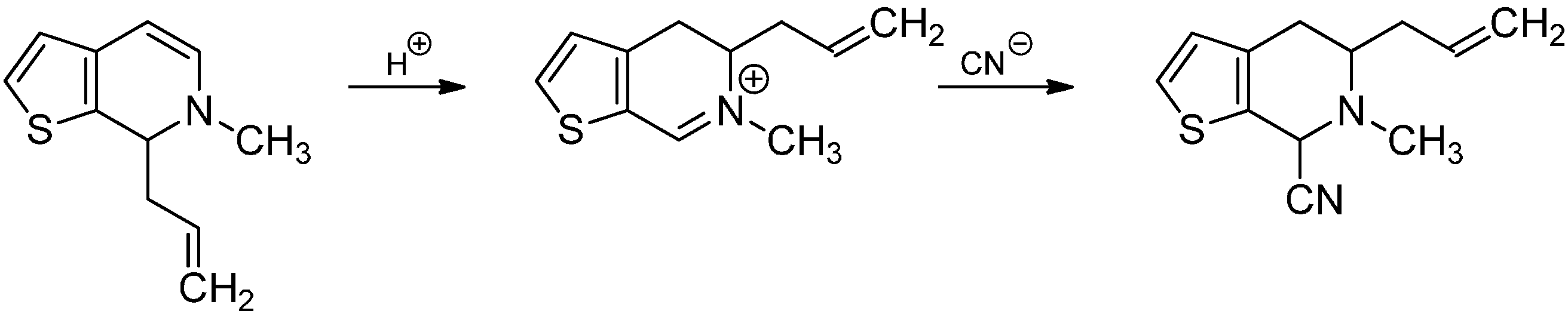
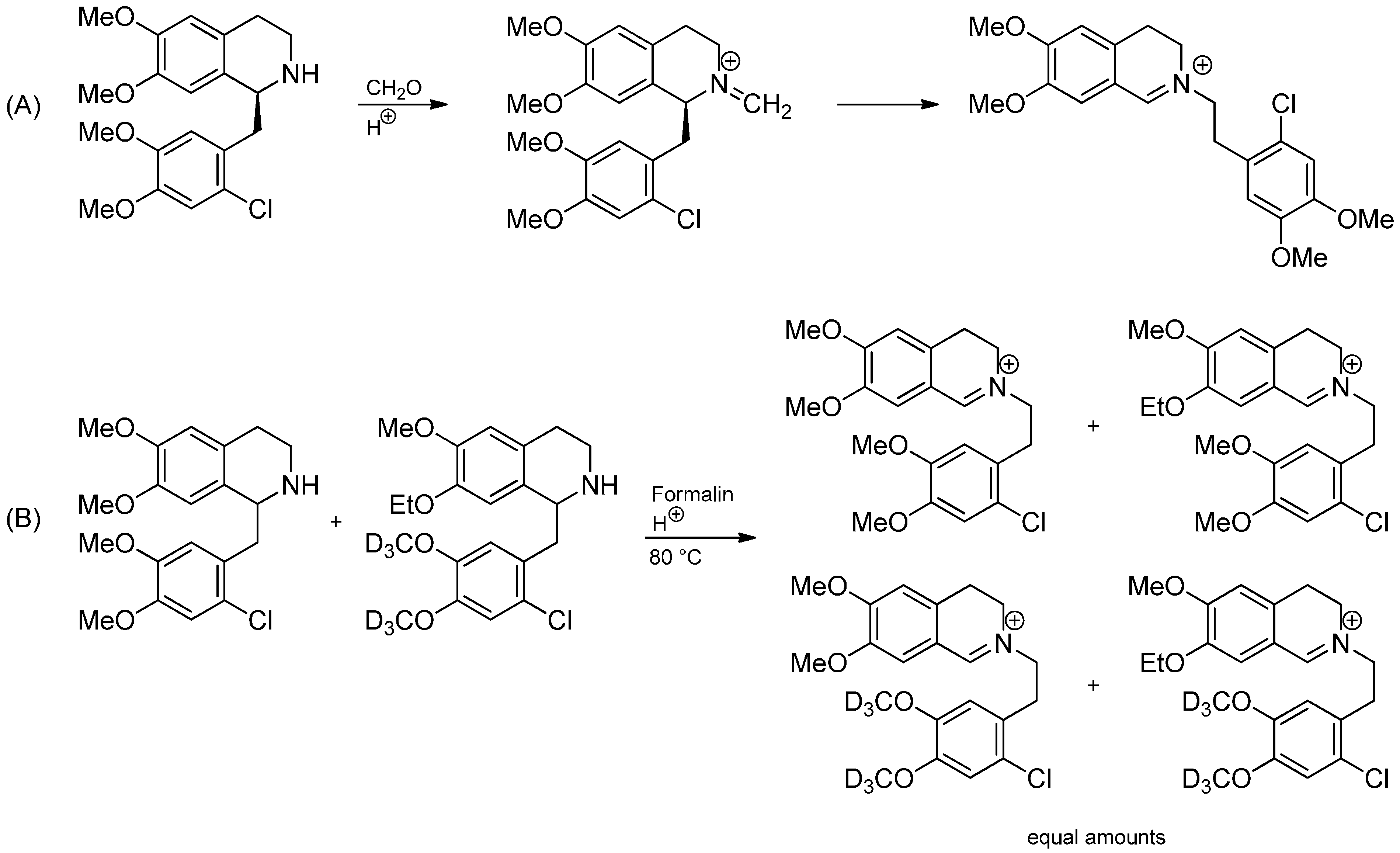
3.1. The Knabe Rearrangement
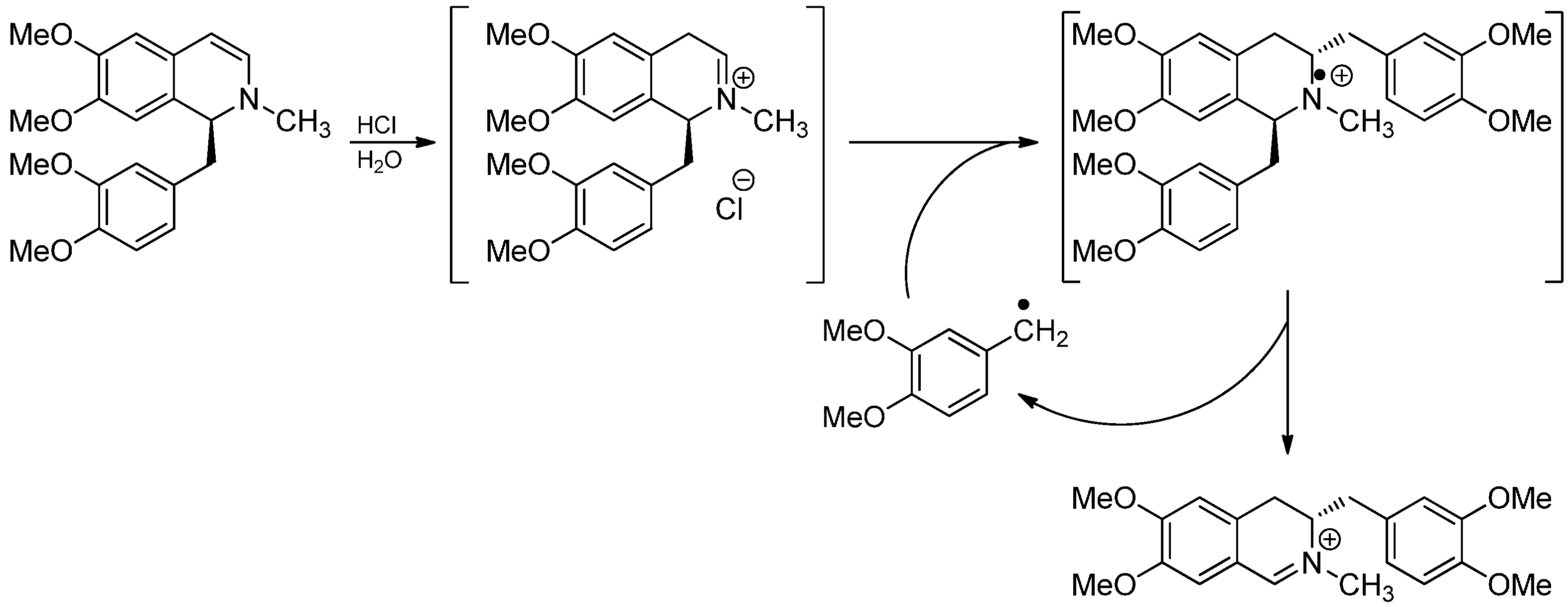
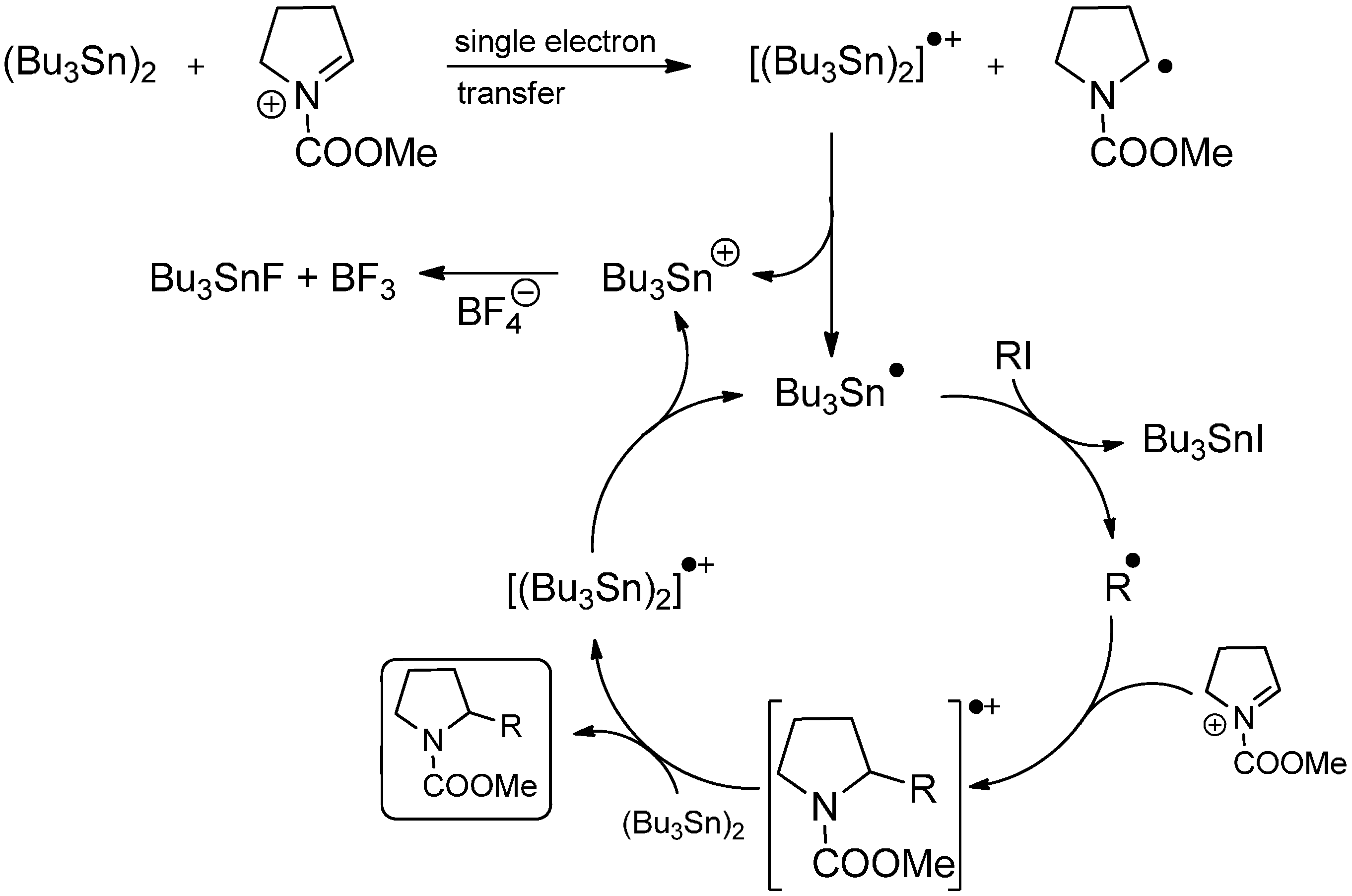
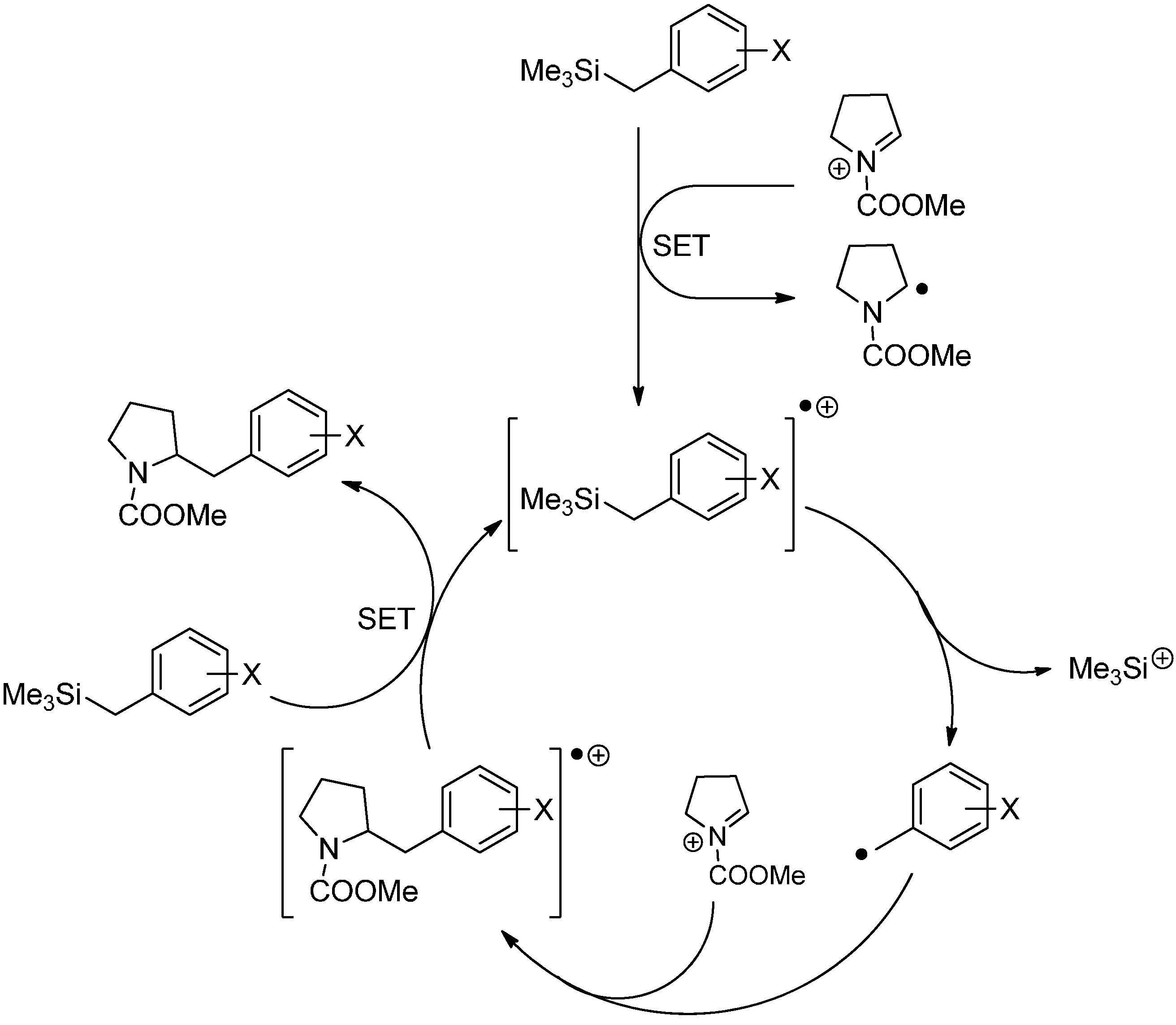
3.2. Radical Addition to “Cation Pool” Species
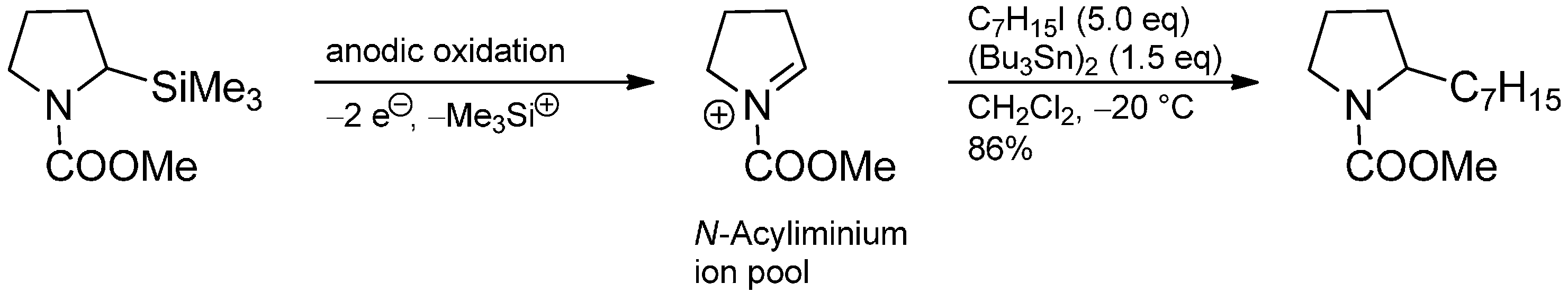
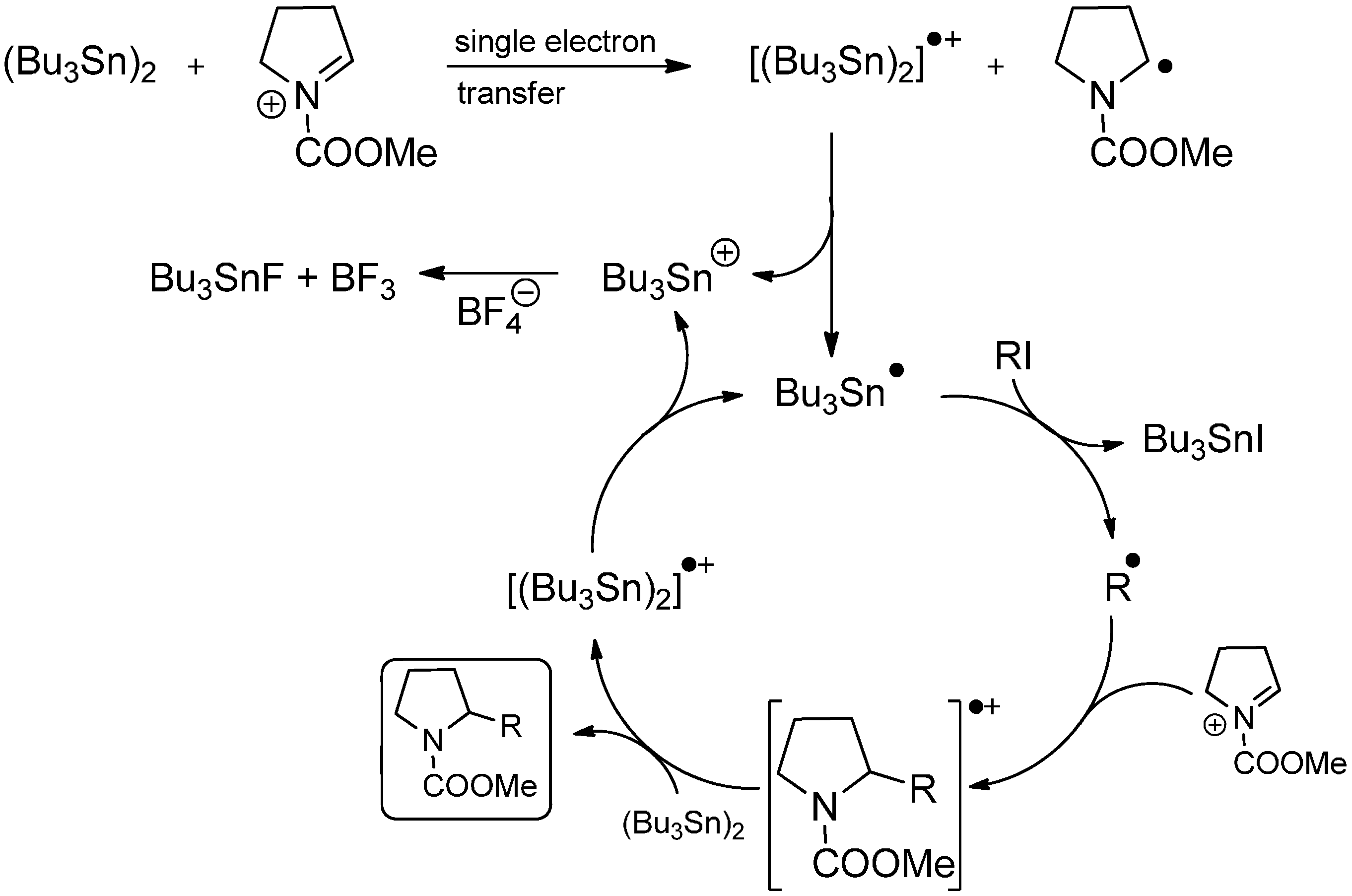
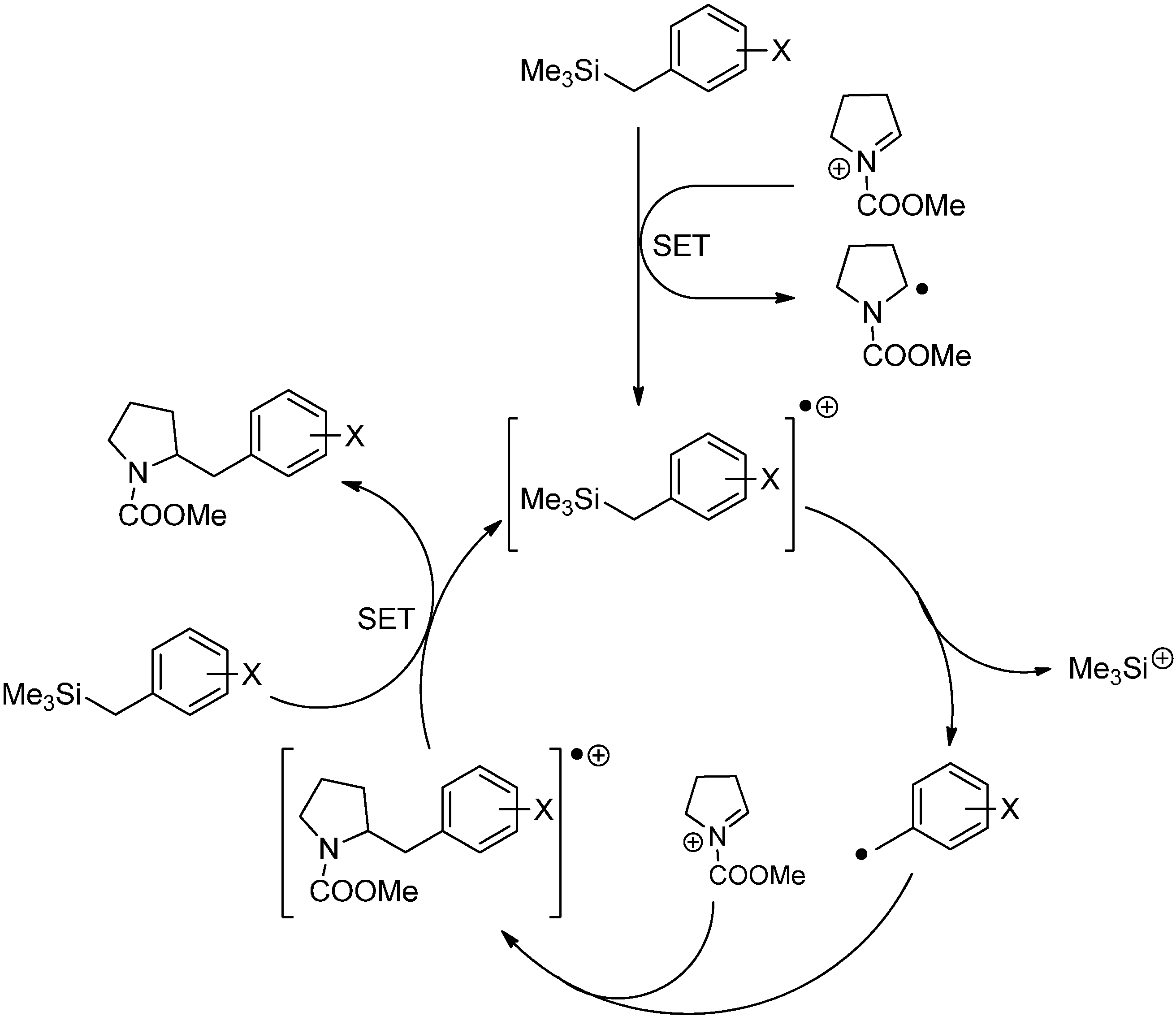
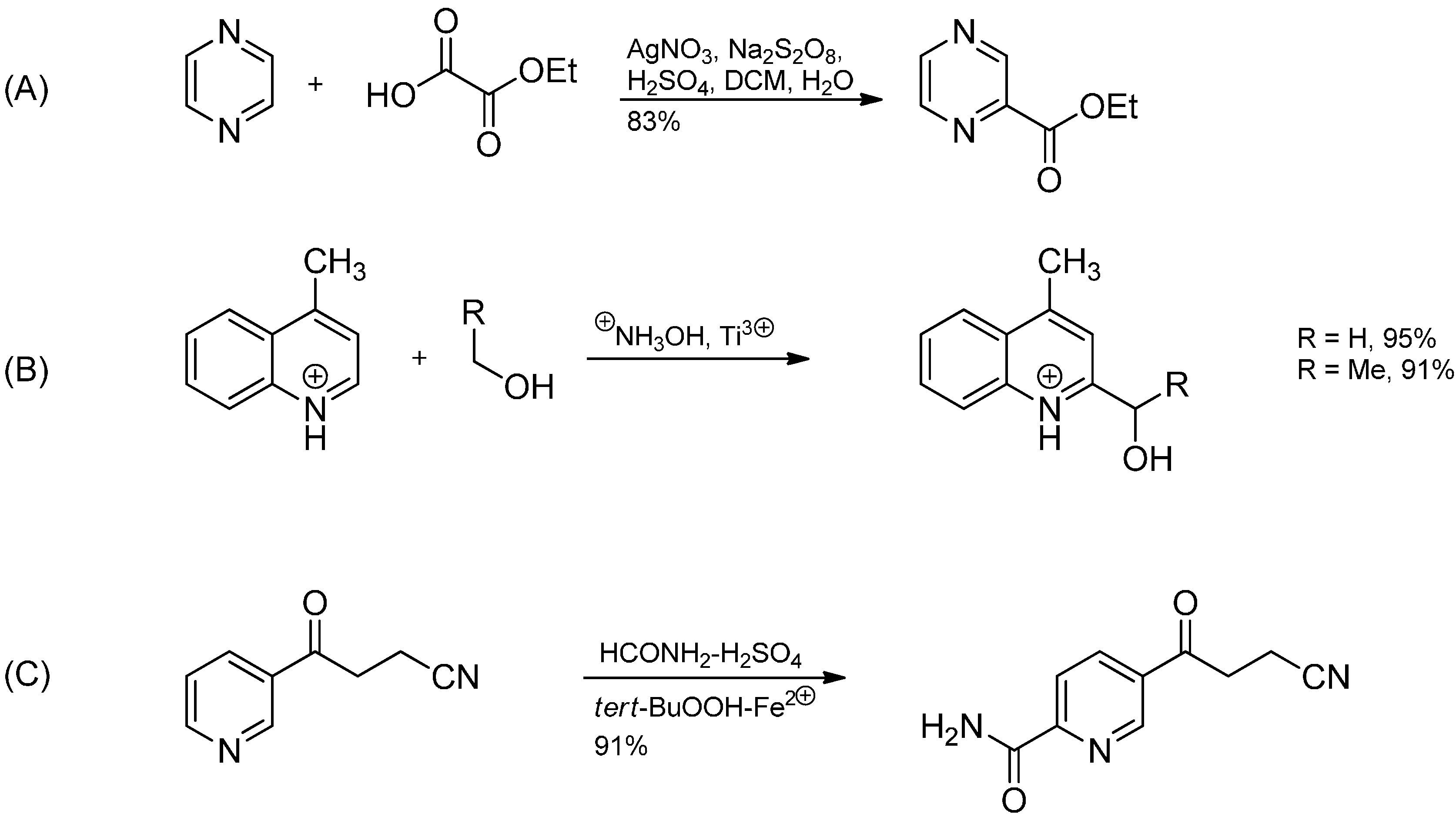
3.3. Addition to Activated Imines and Imine Derivatives
3.3.1. Porta Reaction
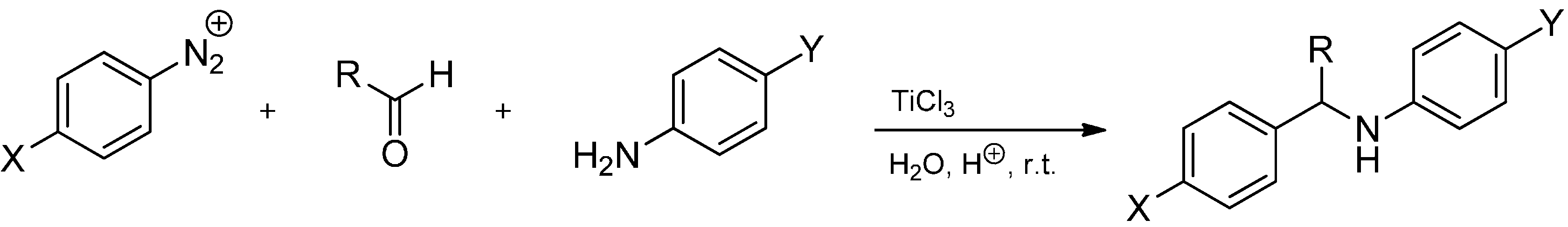

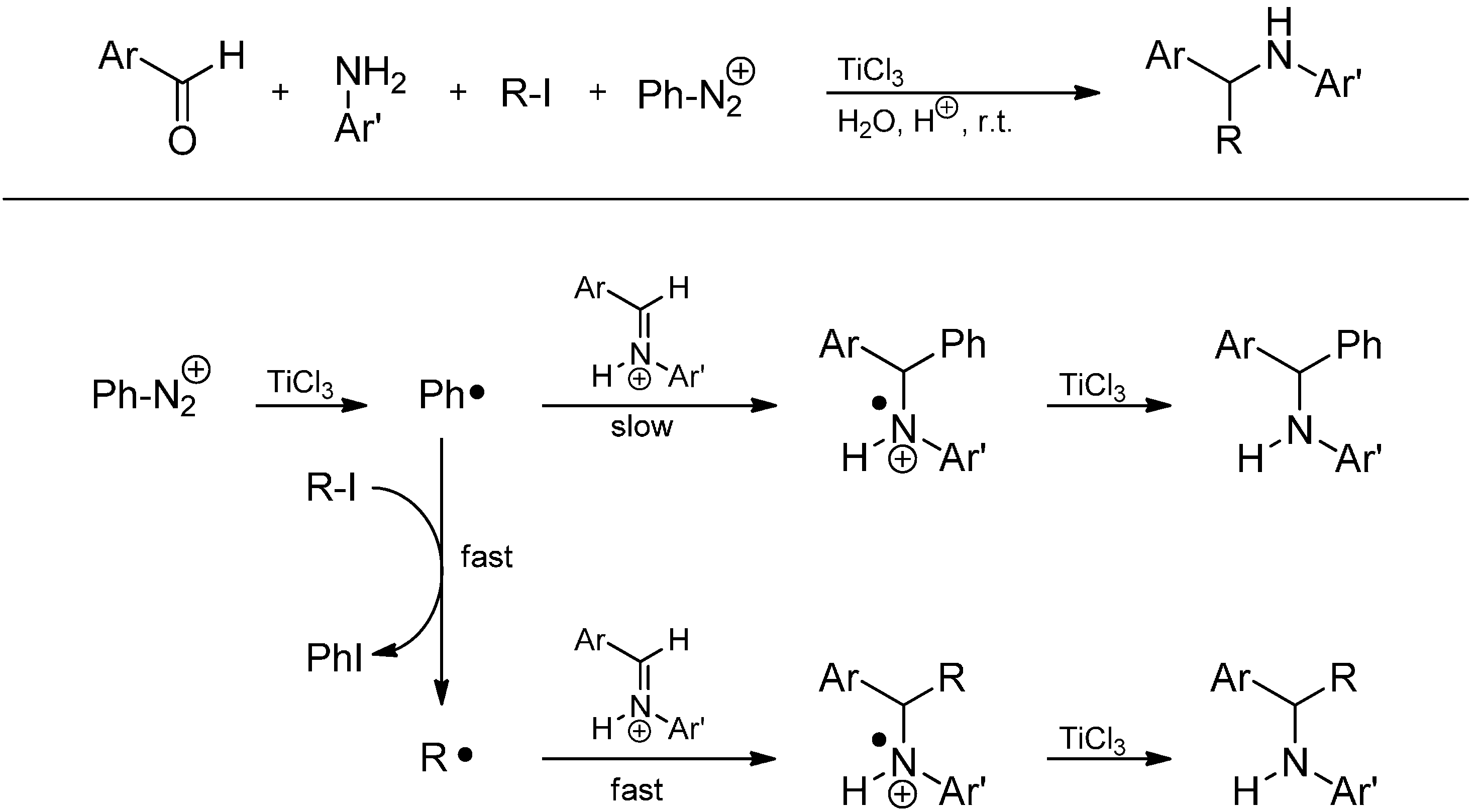
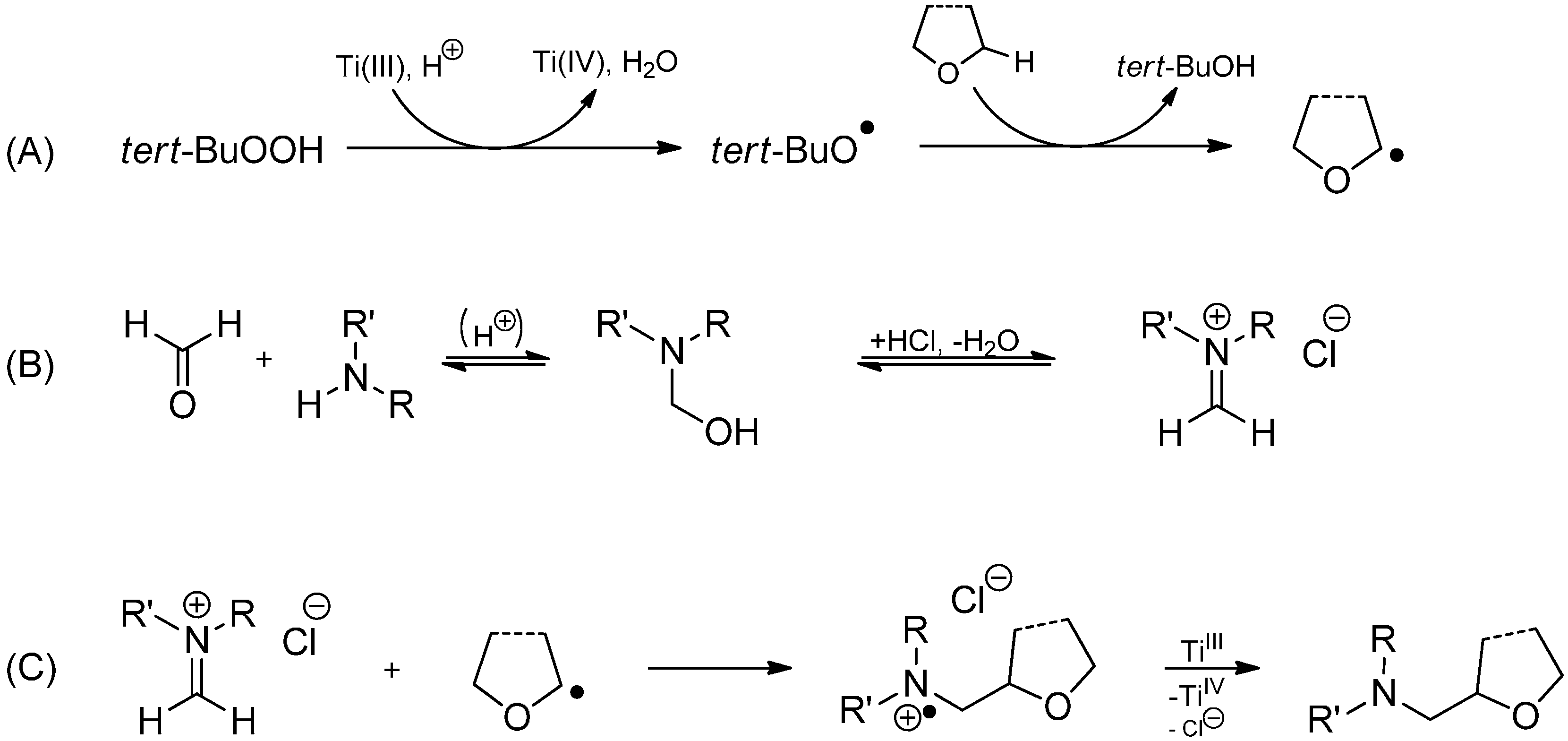
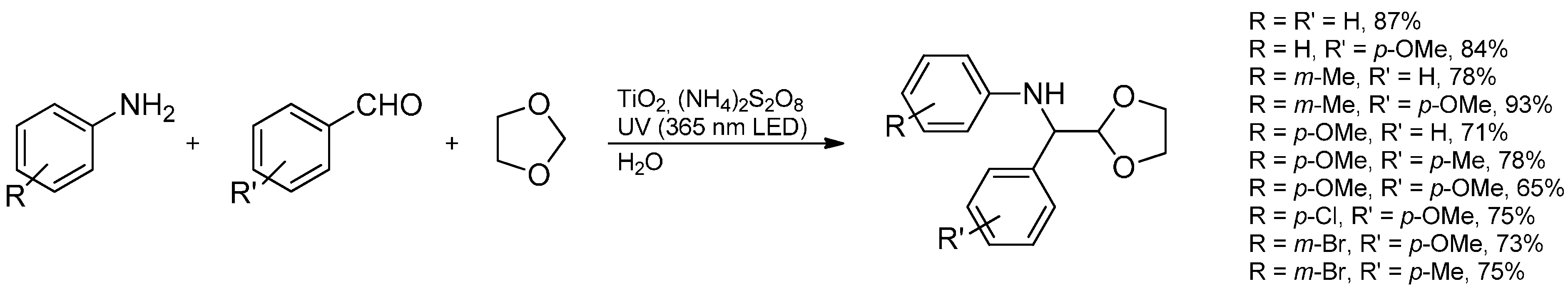
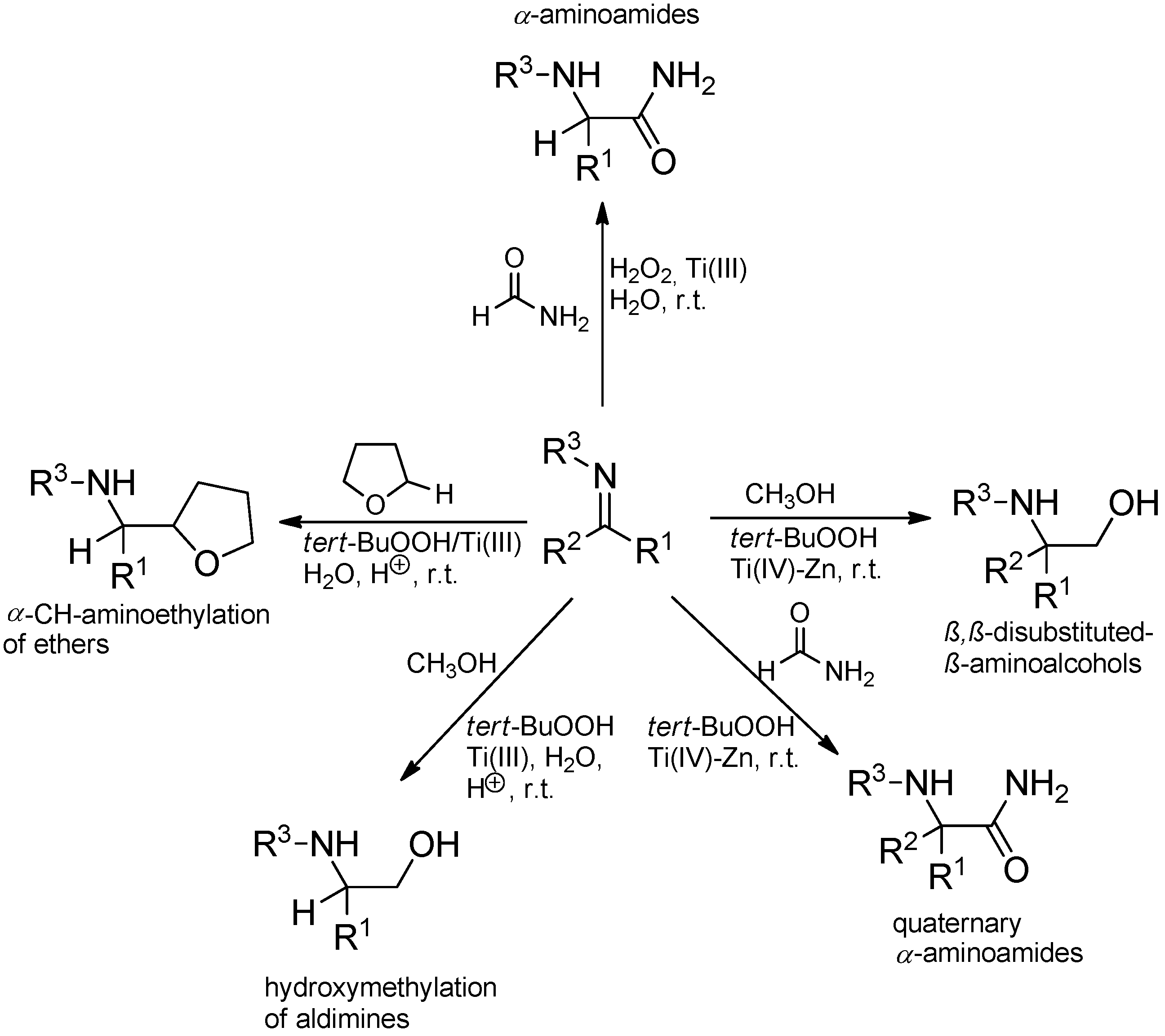
3.3.2. Intermolecular Radical Addition to Imines Initiated by Dimethylzinc-Air
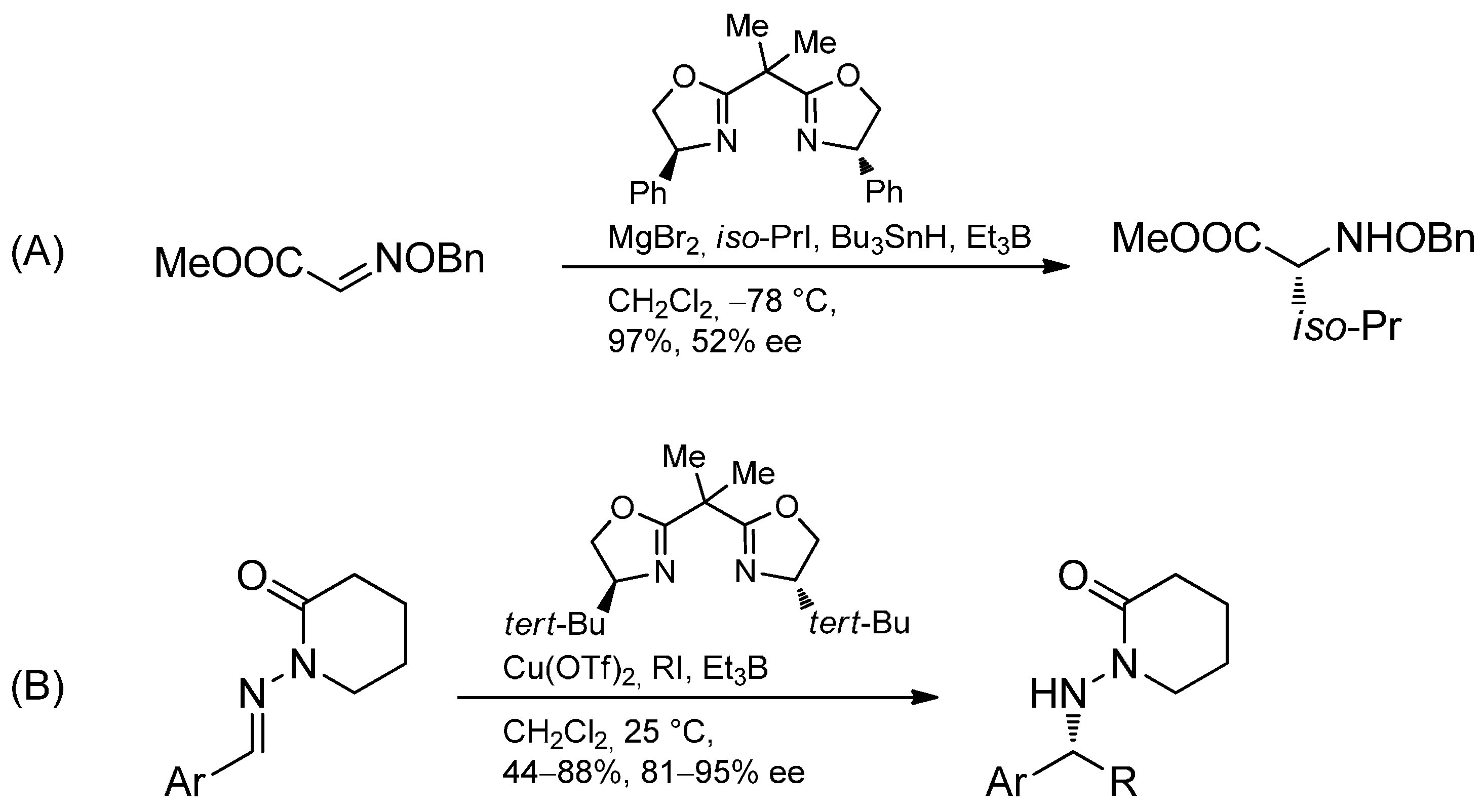
3.3.3. Intermolecular Radical Addition to Imines Initiated by Trialkylboranes
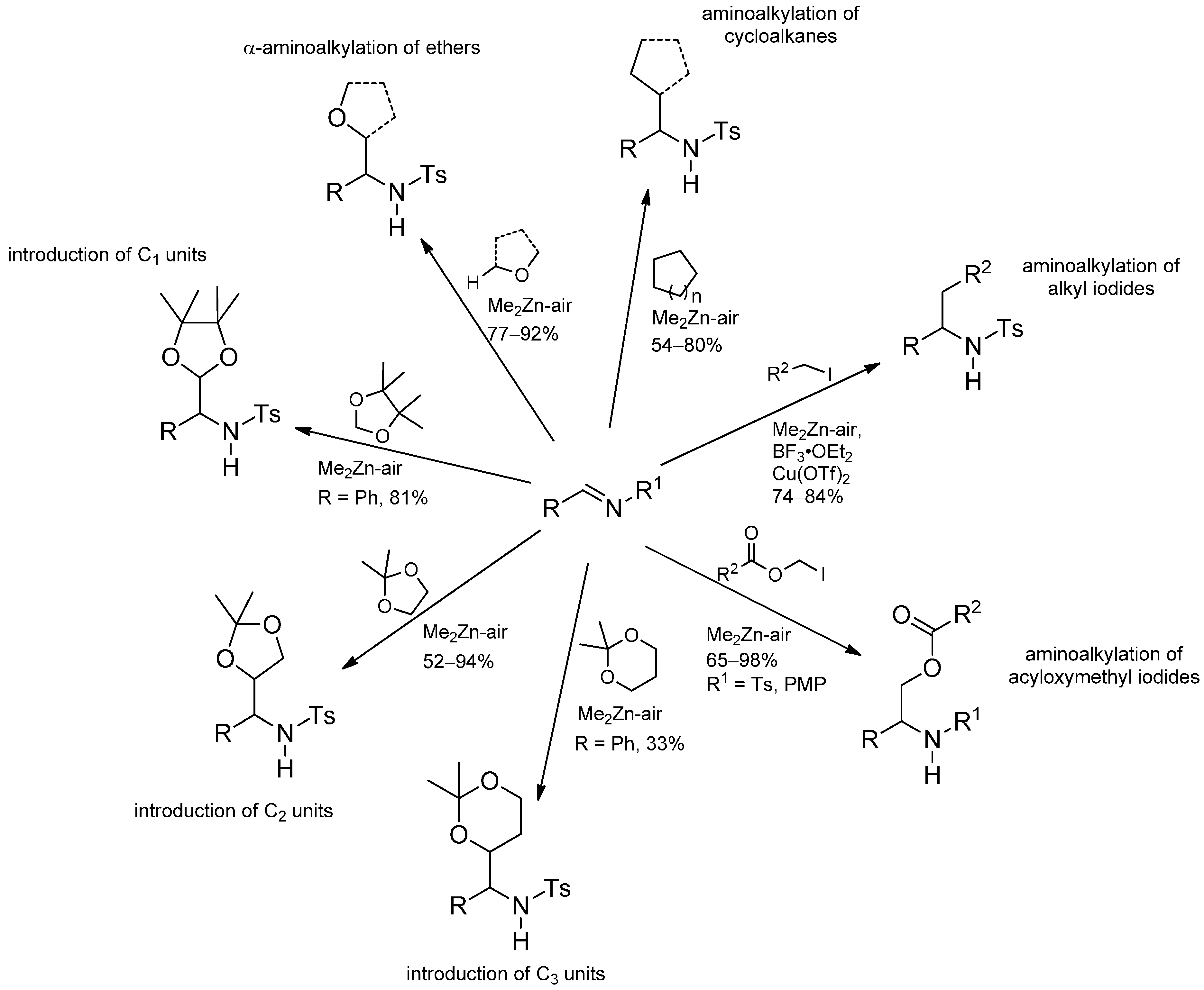
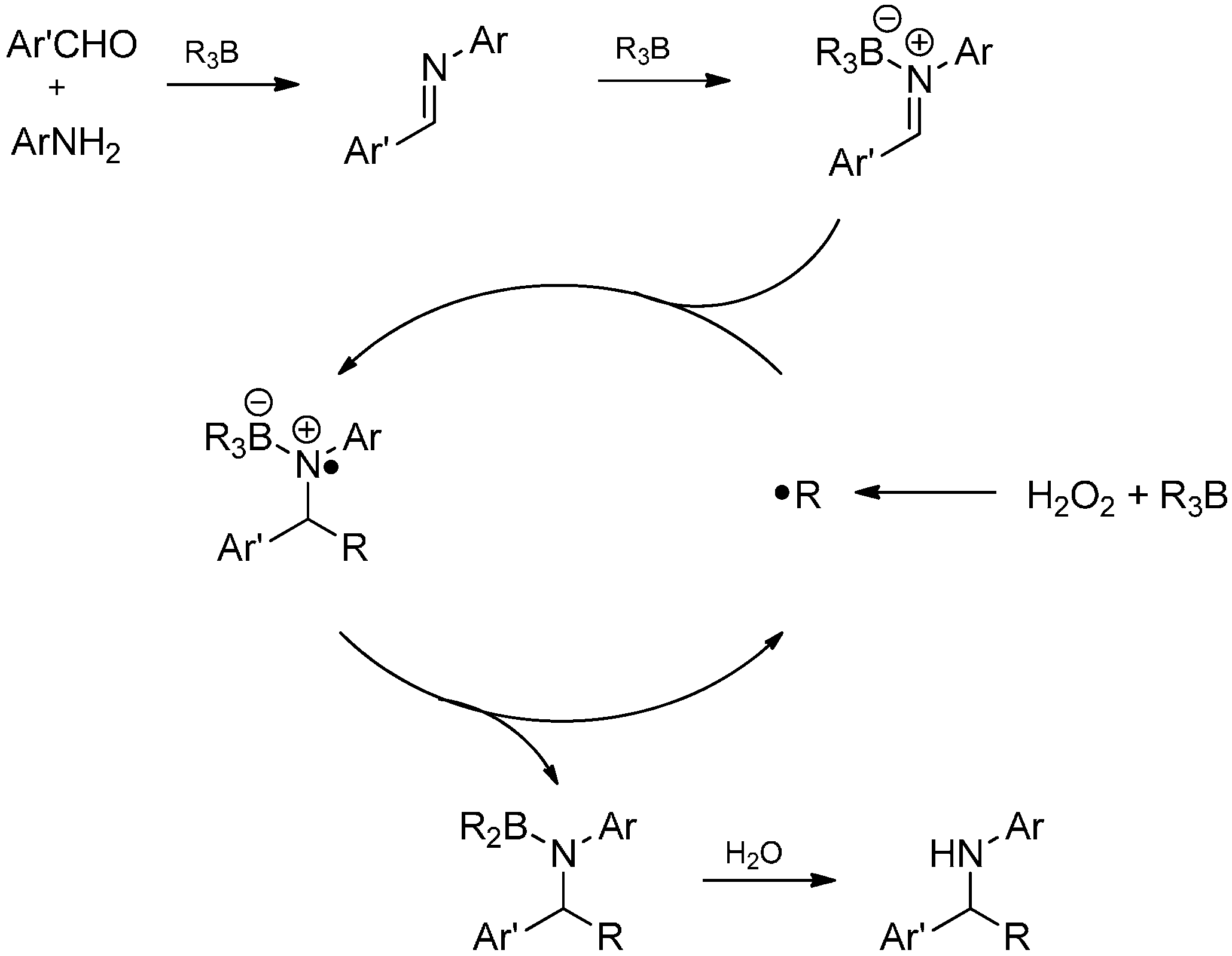
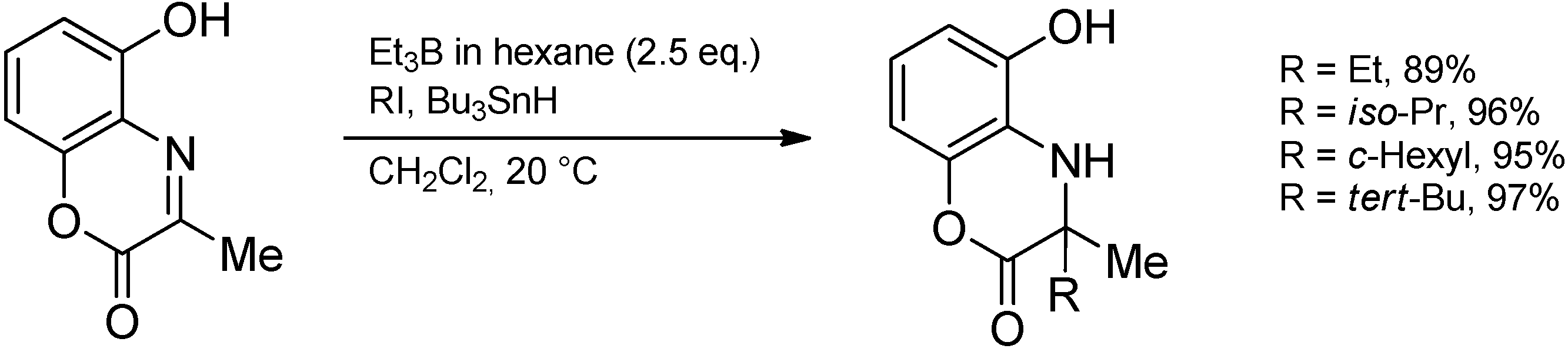
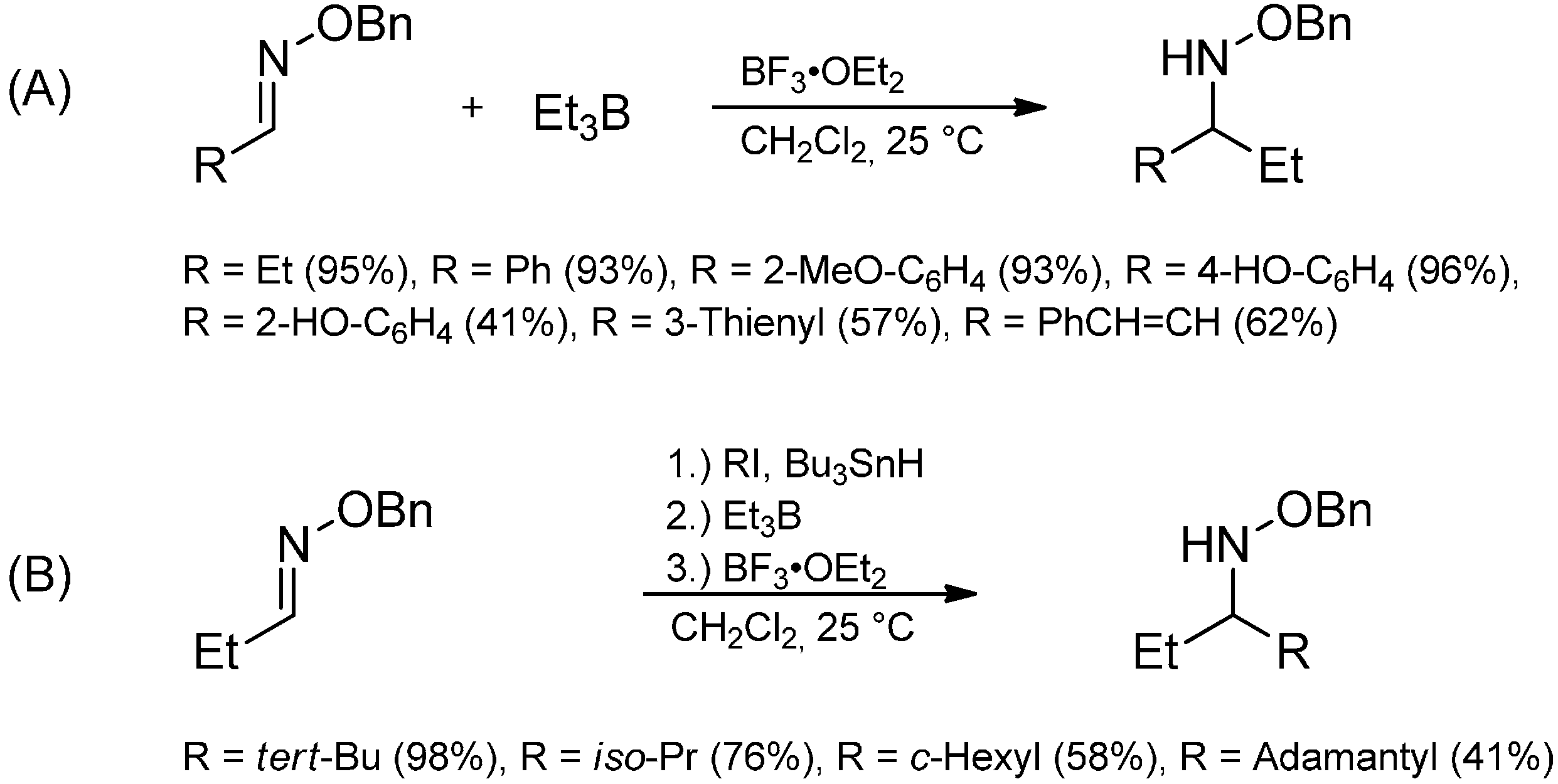
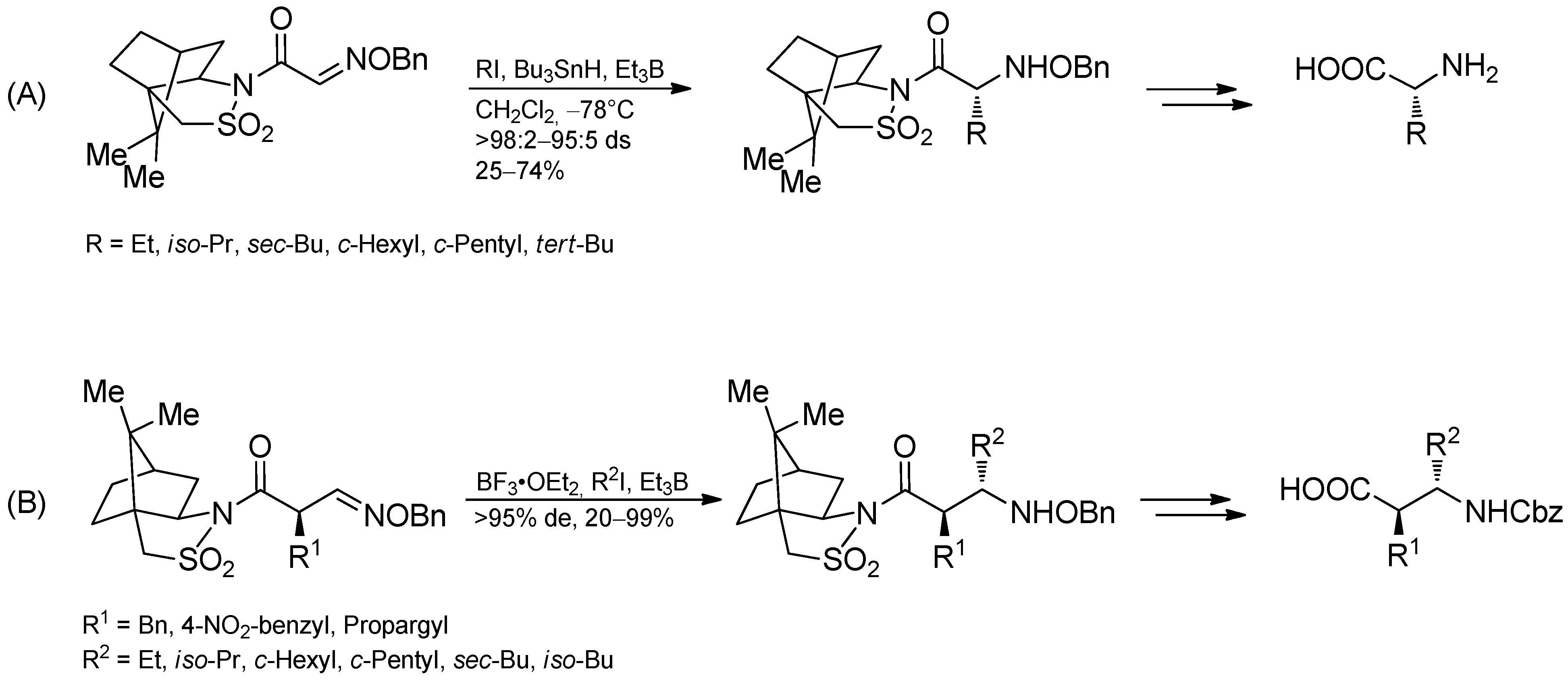
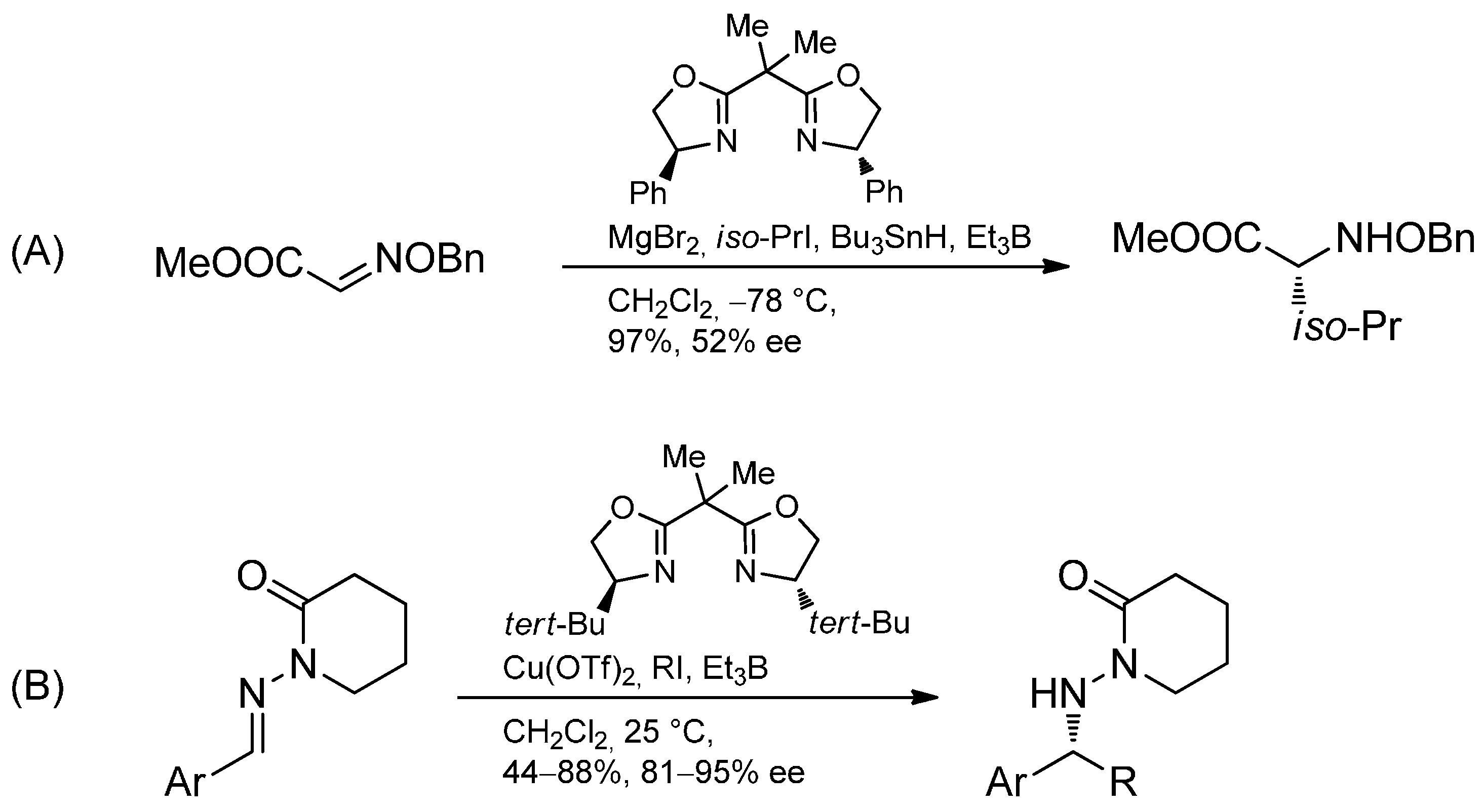
3.3.4. Intermolecular Radical Addition to Imine Derivatives
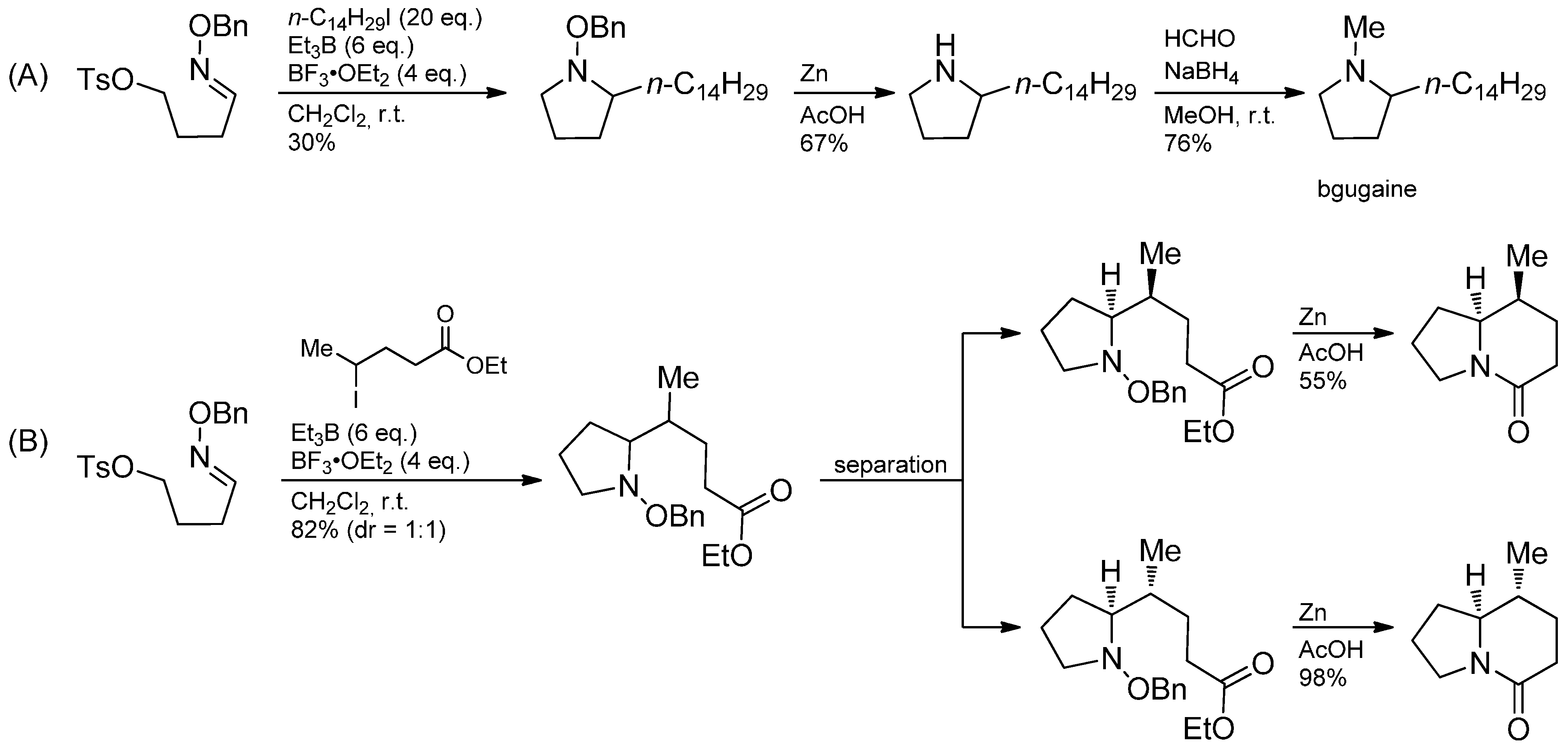
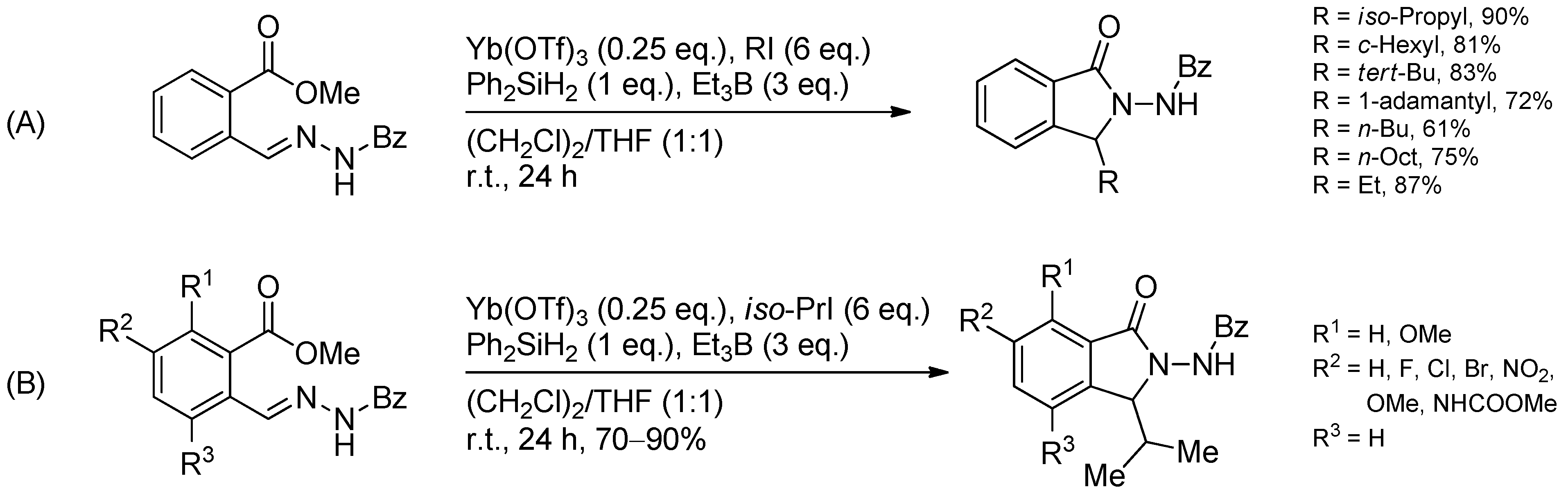
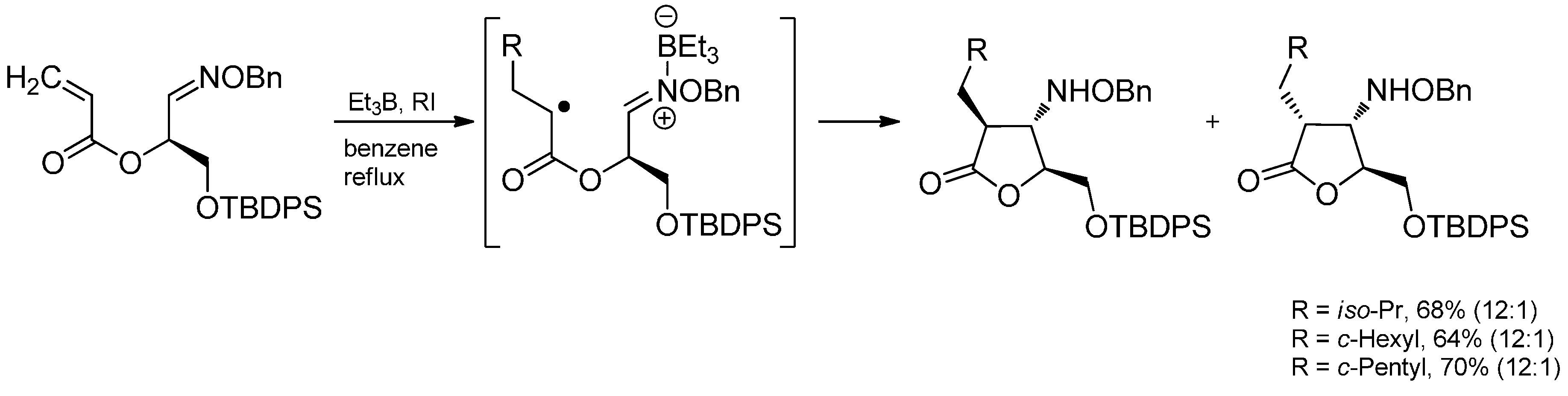
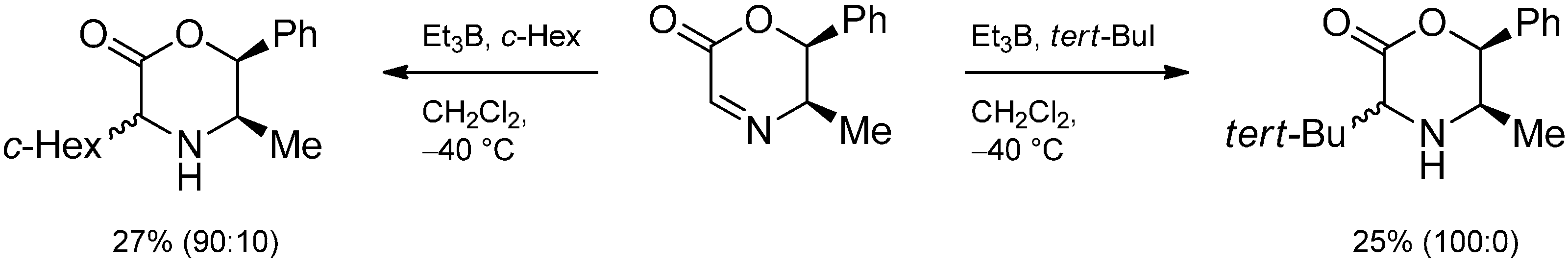

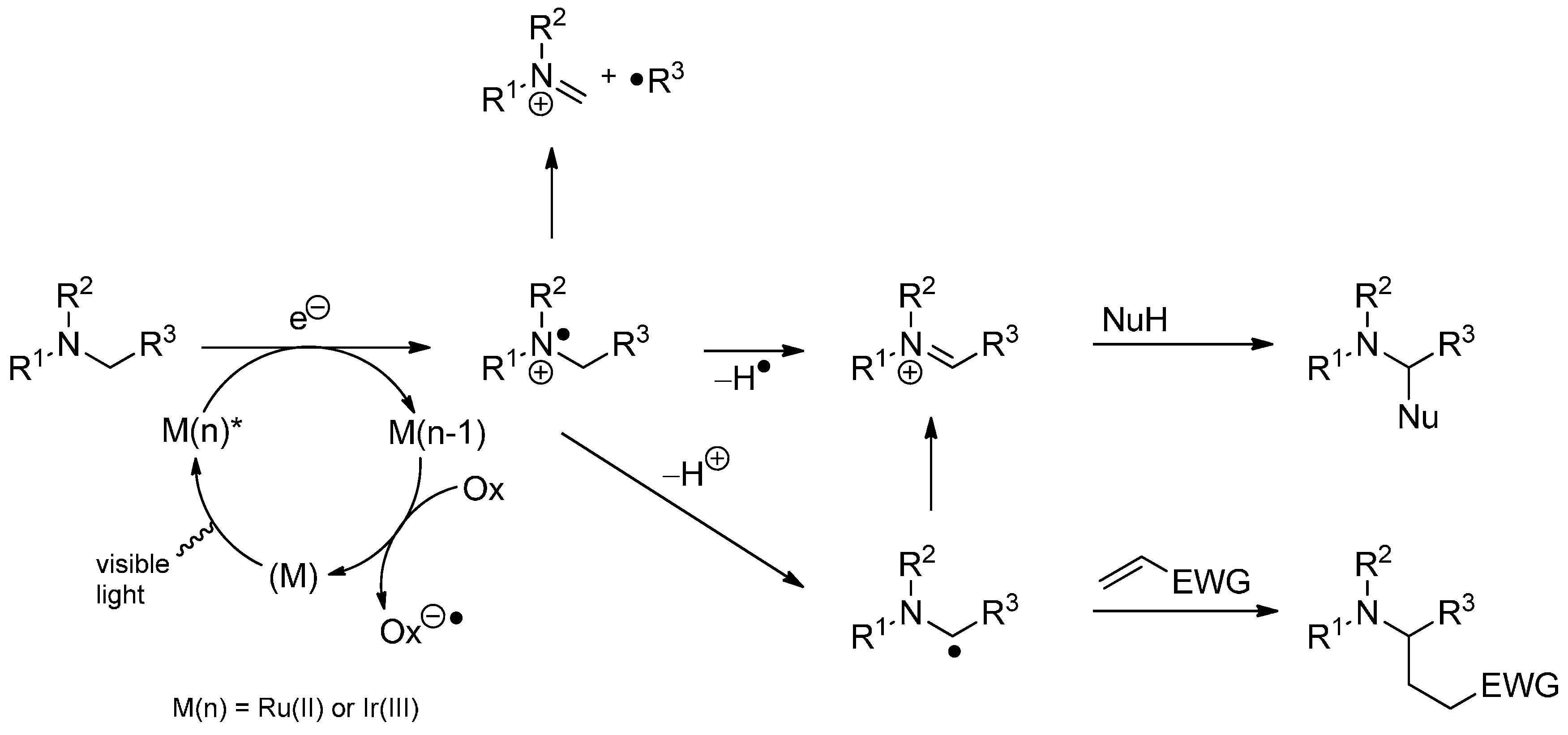
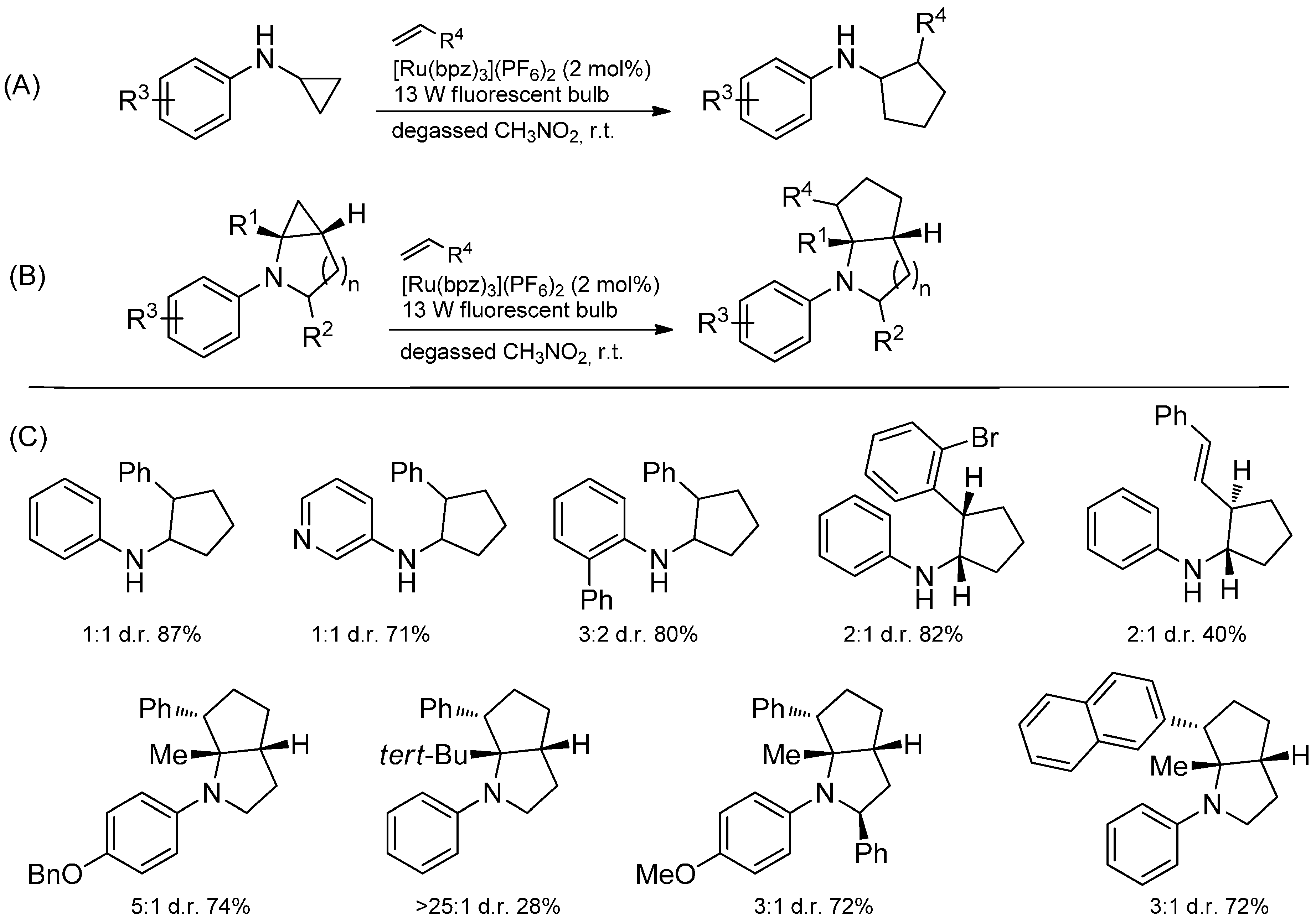
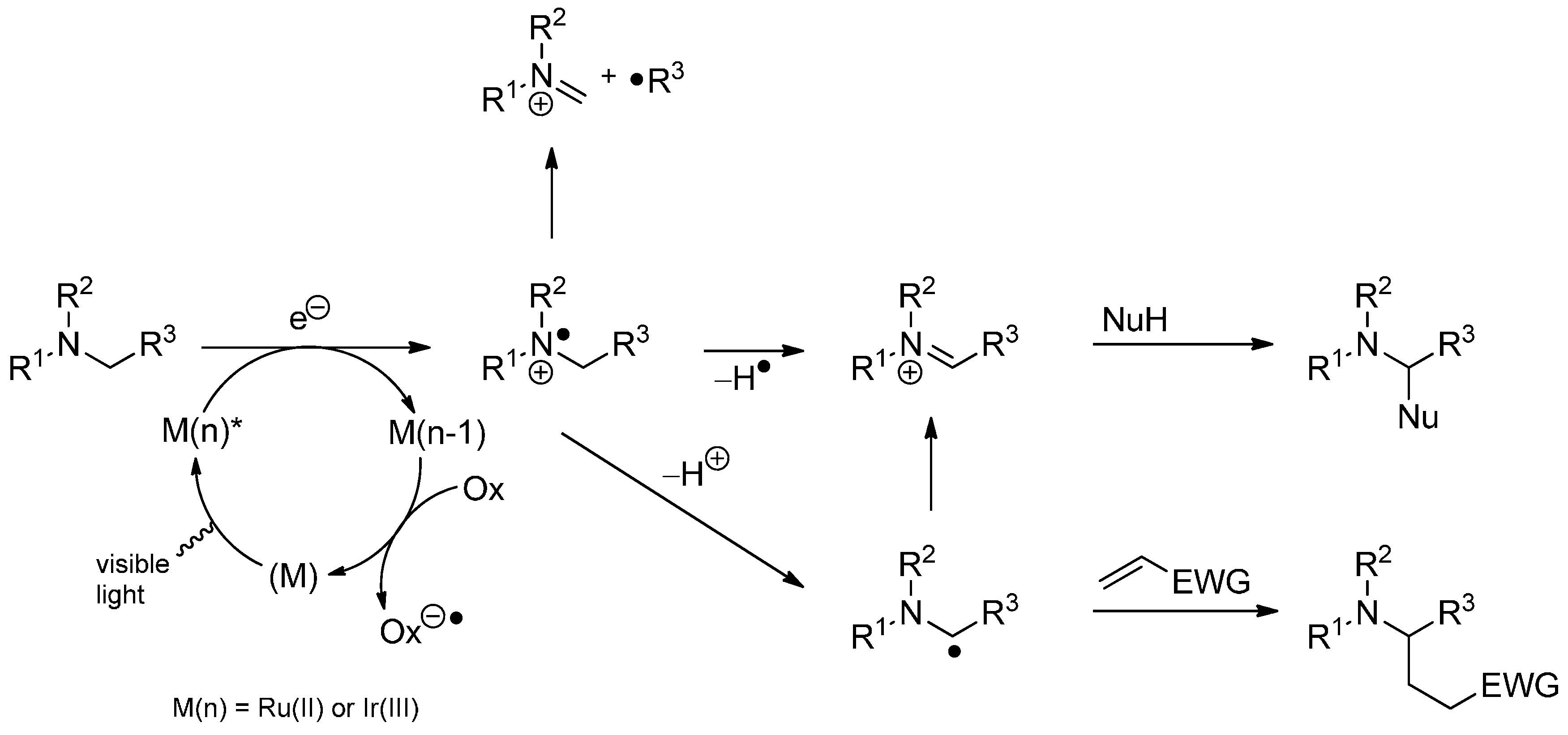
3.4. Iminium Ions in Photoredox Chemistry
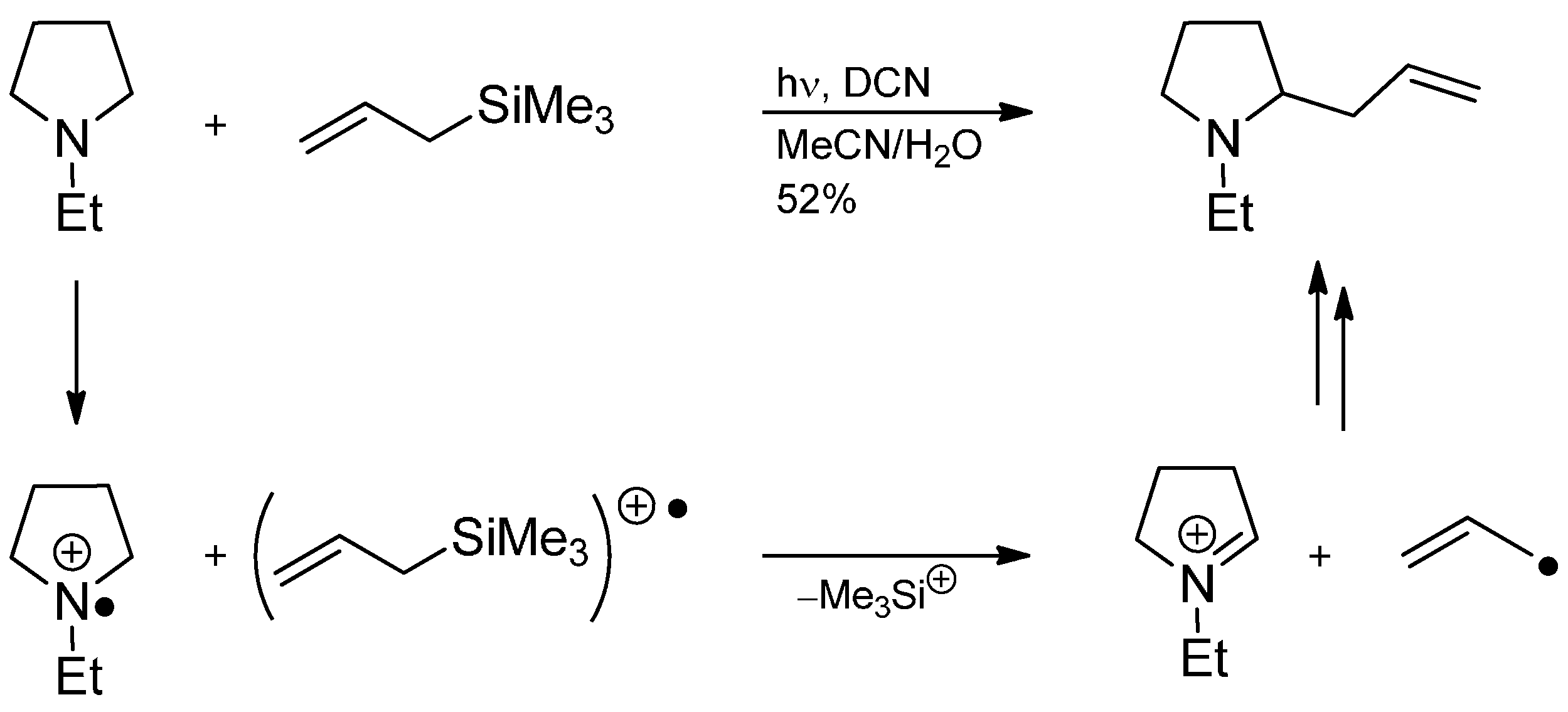
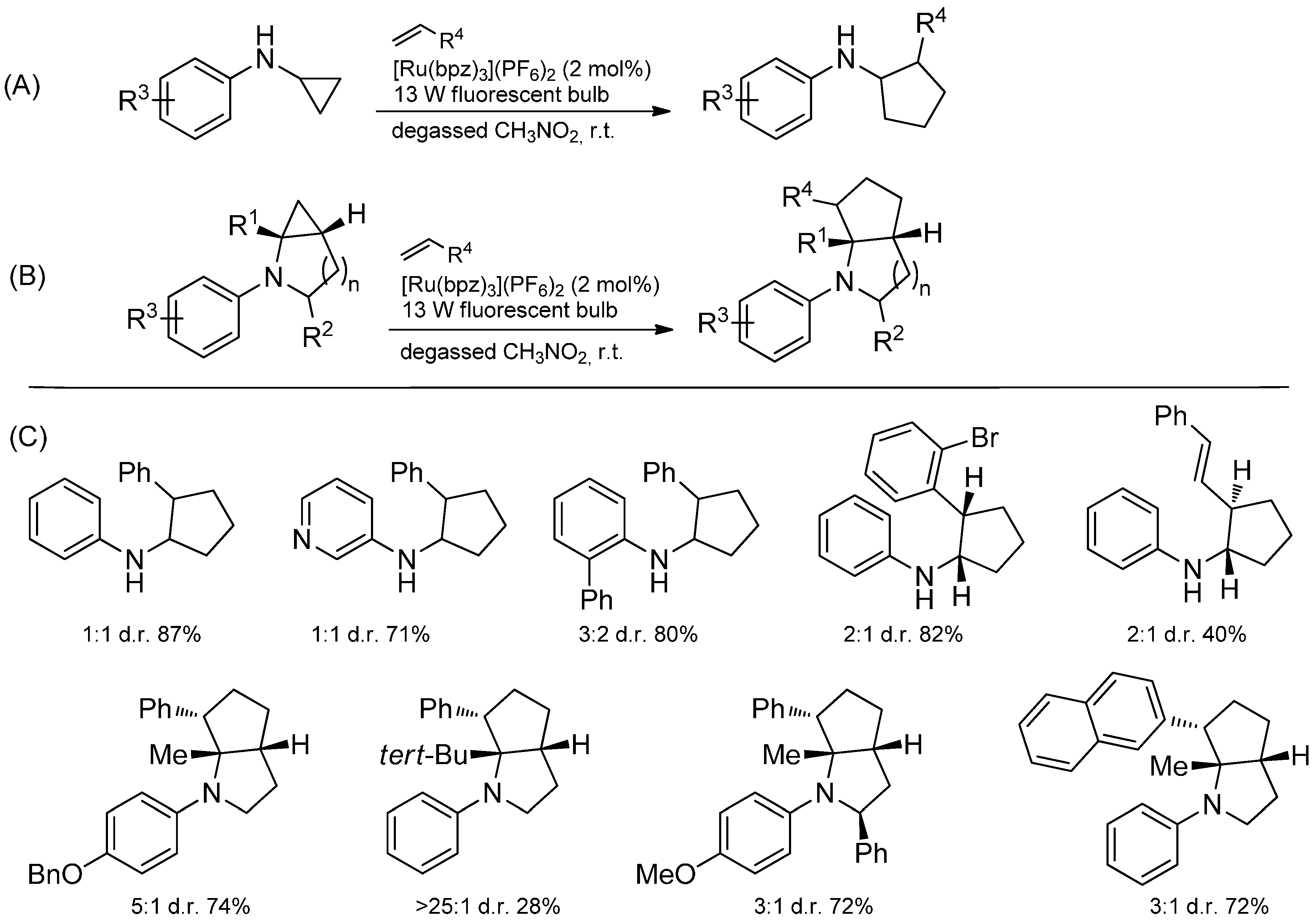
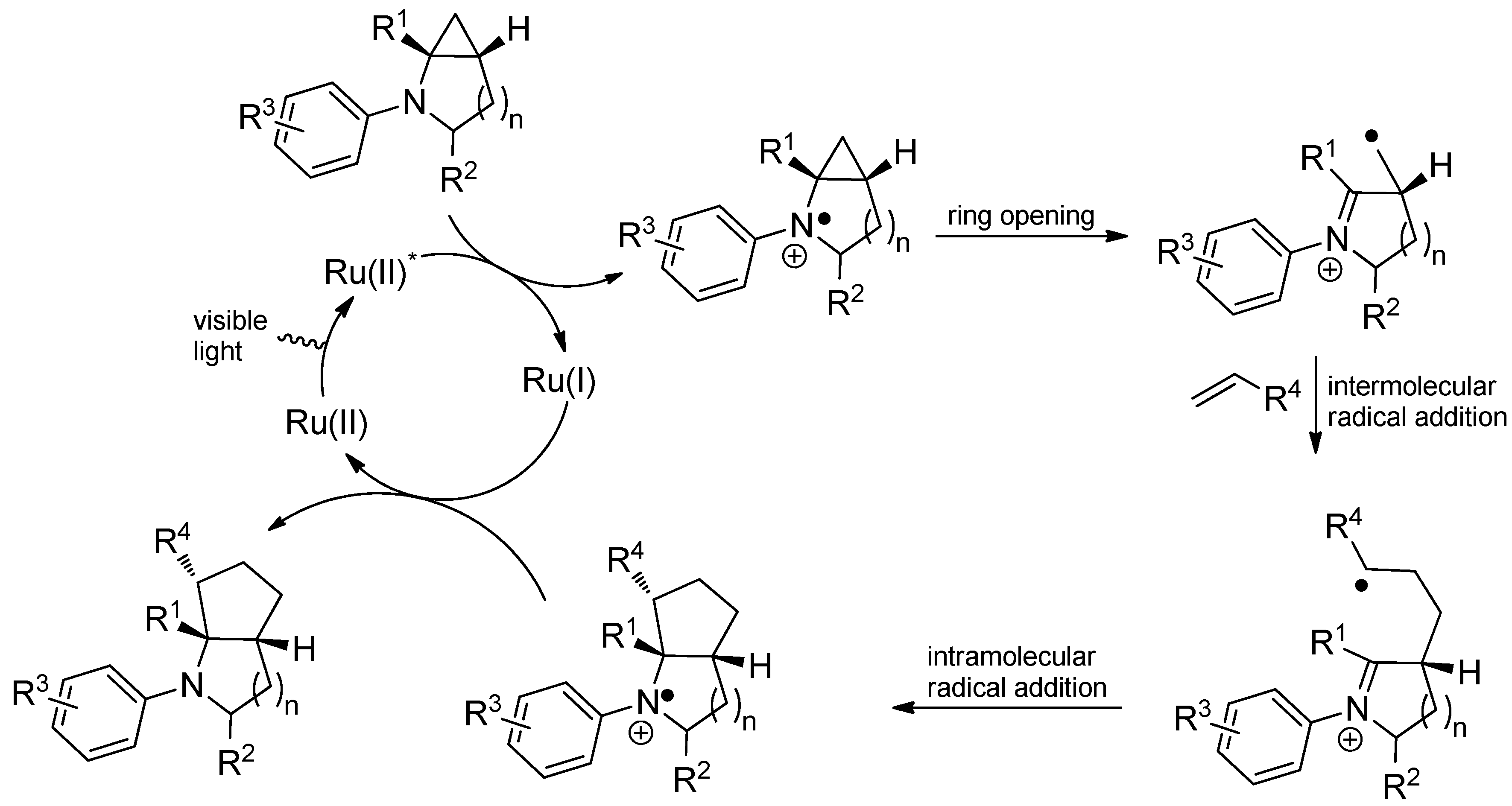
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Fleming, I. Molecular Orbitals and Organic Chemical Reactions, Reference Edition; Wiley: Chichester, UK, 2010. [Google Scholar]
- Minisci, F.; Bernardi, R.; Bertini, F.; Galli, R.; Perchinummo, M. Nucleophilic character of alkyl radicals—VI: A new convenient selective alkylation of heteroaromatic bases. Tetrahedron 1971, 27, 3575–3579. [Google Scholar] [CrossRef]
- Halland, N.; Jorgensen, K.A. Intermolecular addition of alkyl radicals to imines in the absence and in the presence of a Lewis acid. J. Chem. Soc. 2001, 1290–1295. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Wu, Y.D.; Lai, D.K.W.; Houk, K.N. Transition structures of hydride transfer-reactions of protonated pyridinium Ion with 1,4-dihydropyridine and protonated nicotinamide with 1,4-dihydronicotinamide. J. Am. Chem. Soc. 1995, 117, 4100–4108. [Google Scholar] [CrossRef]
- Foresman, J.B.; Frisch, A. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian, Inc.: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Augood, D.R.; Williams, G.H. Homolytic aromatic arylation. Chem. Rev. 1957, 57, 123–190. [Google Scholar] [CrossRef]
- Gomberg, M.; Bachmann, W.E. The synthesis of biaryl compounds by means of the diazo reaction. J. Am. Chem. Soc. 1924, 46, 2339–2343. [Google Scholar] [CrossRef]
- Duncton, M.A.J. Minisci reactions: Versatile CH-functionalizations for medicinal chemists. Med. Chem. Comm. 2011, 2, 1135–1161. [Google Scholar] [CrossRef]
- Lynch, B.M.; Chang, H.S. Free-radical phenylations of 1-methyldiazoles. Tetrahedron Lett. 1964, 5, 617–620. [Google Scholar] [CrossRef]
- Lynch, B.M.; Chang, H.S. Concentration-dependent orientations in free-radical phenylations of heteroaromatic compounds. Tetrahedron Lett. 1964, 5, 2965–2968. [Google Scholar] [CrossRef]
- Dou, H.J.M.; Lynch, B.M. Selective free-radical phenylations: Nitrogen-heteroaromatic compounds in acidic media. Tetrahedron Lett. 1965, 6, 897–901. [Google Scholar] [CrossRef]
- Dou, H.J.M. Méthylation radicalaire du thiazole et de ses derivés méthyles en milieu acétique. Bull. Soc. Chim. Fr. 1966, 5, 1678–1679. [Google Scholar]
- Dou, H.J.M.; Lynch, B.M. Phénylation radicalaire sélective en milieu acide-composés hétéroaromatiques azotes I. Methode et résultats expérimentaux. Bull. Soc. Chim. Fr. 1966, 12, 3815–3820. [Google Scholar]
- Dou, H.J.M.; Lynch, B.M. Phénylation radicalaire sélective en milieu acide-composés hétéroaromatiques azotes 2. Traitement théorique des résultats expérimentaux et discussion. Bull. Soc. Chim. Fr. 1966, 12, 3820–3823. [Google Scholar]
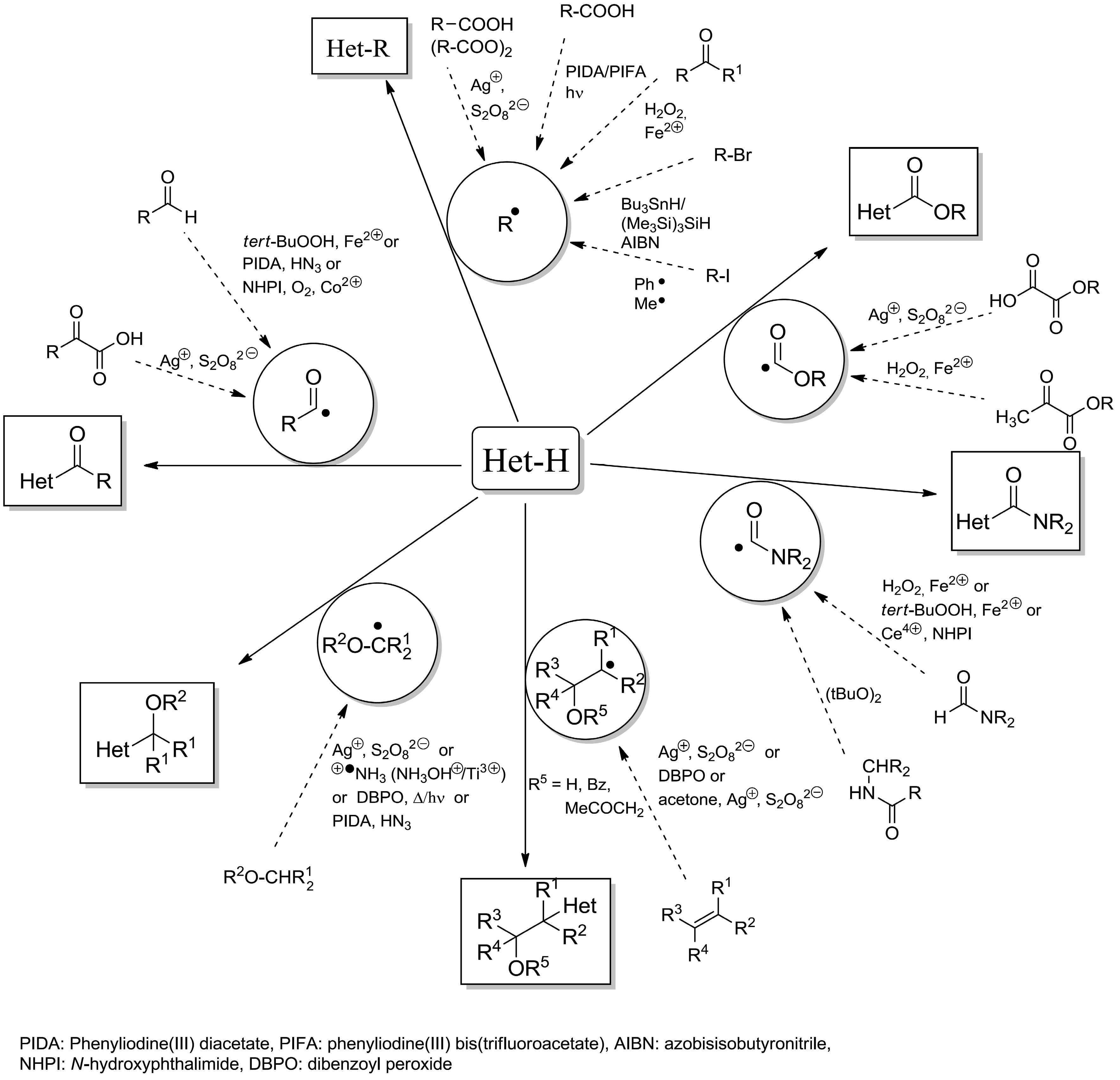
- Punta, C.; Minisci, F. Minisci reaction: A Friedel-Crafts type process with opposite reactivity and selectivity. Selective homolytic alkylation, acylation, carboxylation and carbamoylation of heterocyclic aromatic bases. Trends Heterocycl. Chem. 2008, 13, 1–68. [Google Scholar]
- Seiple, I.B.; Su, S.; Rodriguez, R.A.; Gianatassio, R.; Fujiwara, Y.; Sobel, A.L.; Baran, P.S. Direct C-H arylation of electron-deficient heterocycles with arylboronic acids. J. Am. Chem. Soc. 2010, 132, 13194–13196. [Google Scholar] [CrossRef]
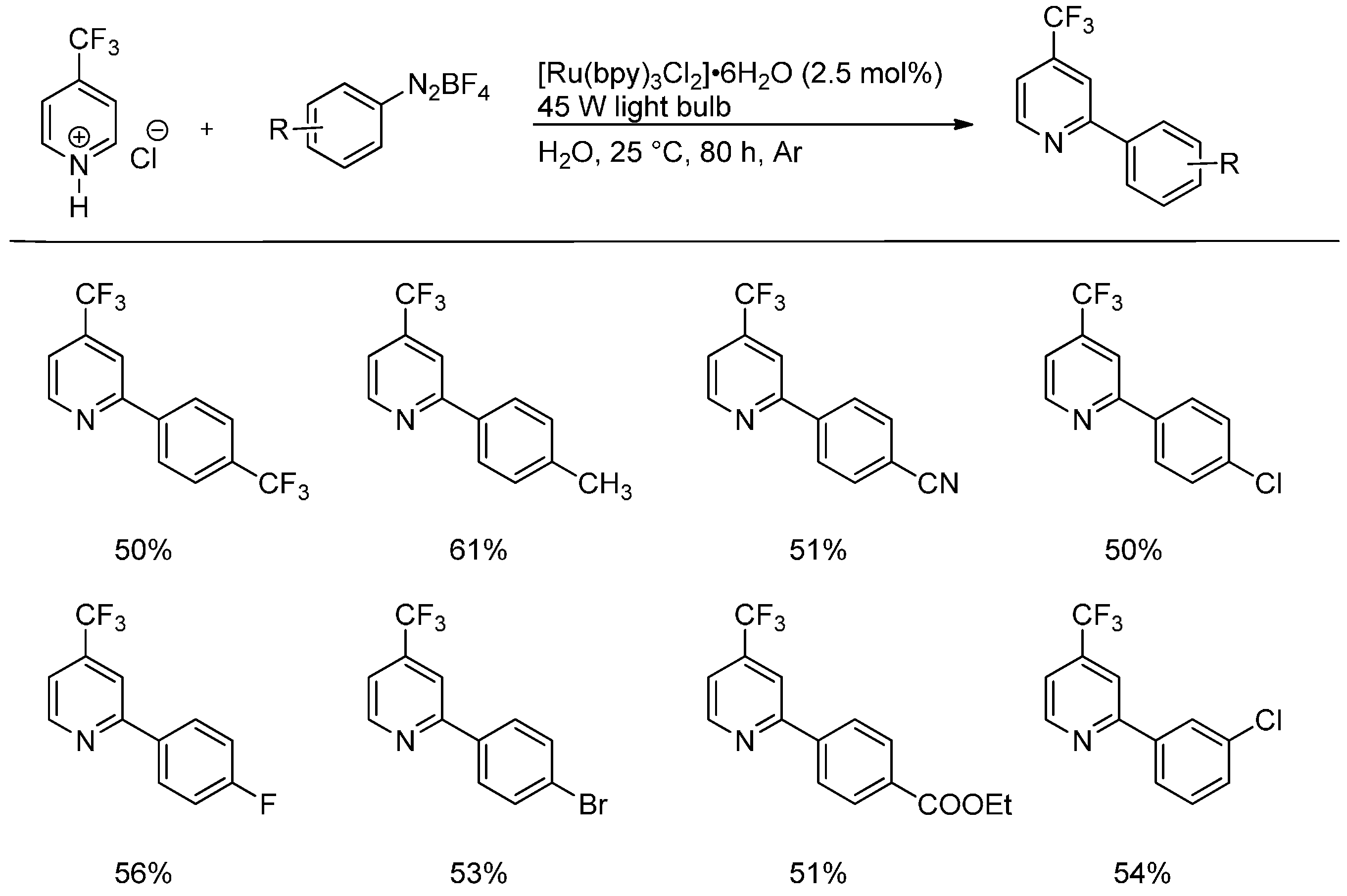
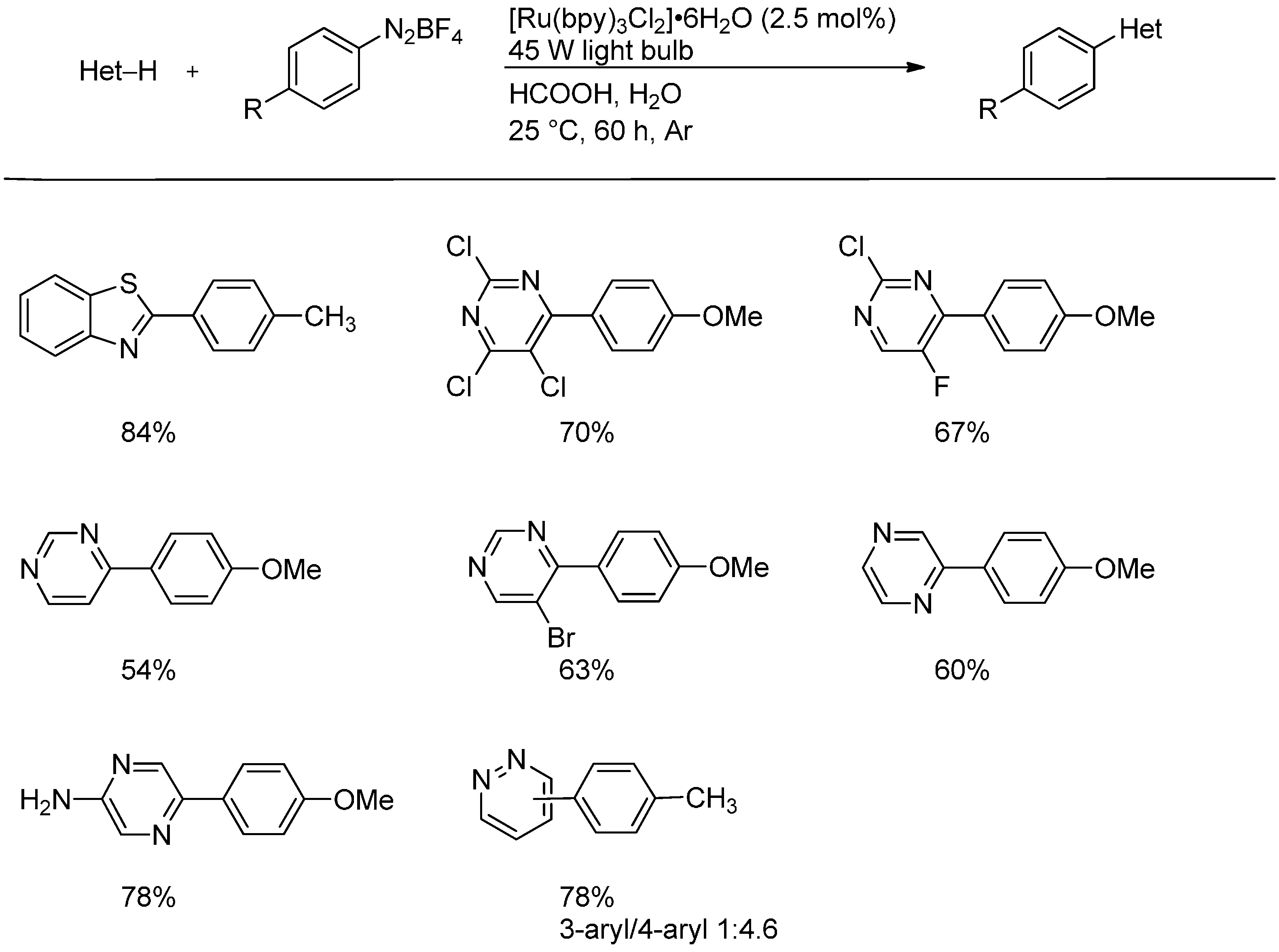
- Xue, D.; Jia, Z.-H.; Zhao, C.-J.; Zhang, Y.-Y.; Wang, C.; Xiao, J. Direct arylation of N-heteroarenes with aryldiazonium salts by photoredox catalysis in water. Chem. Eur. J. 2014, 20, 2960–2965. [Google Scholar] [CrossRef]
- Minisci, F.; Vismara, E.; Fontana, F. Recent developments of free-radical substitutions of heteroaromatic bases. Heterocycles 1989, 28, 489–519. [Google Scholar] [CrossRef]
- Minisci, F.; Fontana, F.; Vismara, E. Substitutions by nucleophilic free-radicals—A new general reaction of heteroaromatic bases. J. Heterocycl. Chem. 1990, 27, 79–96. [Google Scholar]
- Harrowven, D.C.; Sutton, B.J.; Coulton, S. Intramolecular radical additions to quinolines. Tetrahedron 2002, 58, 3387–3400. [Google Scholar] [CrossRef]
- Harrowven, D.C.; Sutton, B.J.; Coulton, S. Intramolecular radical additions to pyridines. Org. Biomol. Chem. 2003, 1, 4047–4057. [Google Scholar] [CrossRef]
- Harrowven, D.C.; Sutton, B.J. Radical additions to pyridines, quinolines and isoquinolines. Prog. Heterocycl. Chem. 2004, 16, 27–53. [Google Scholar]
- Bowman, W.R.; Storey, J.M.D. Synthesis using aromatic homolytic substitution-recent advances. Chem. Soc. Rev. 2007, 36, 1803–1822. [Google Scholar] [CrossRef]
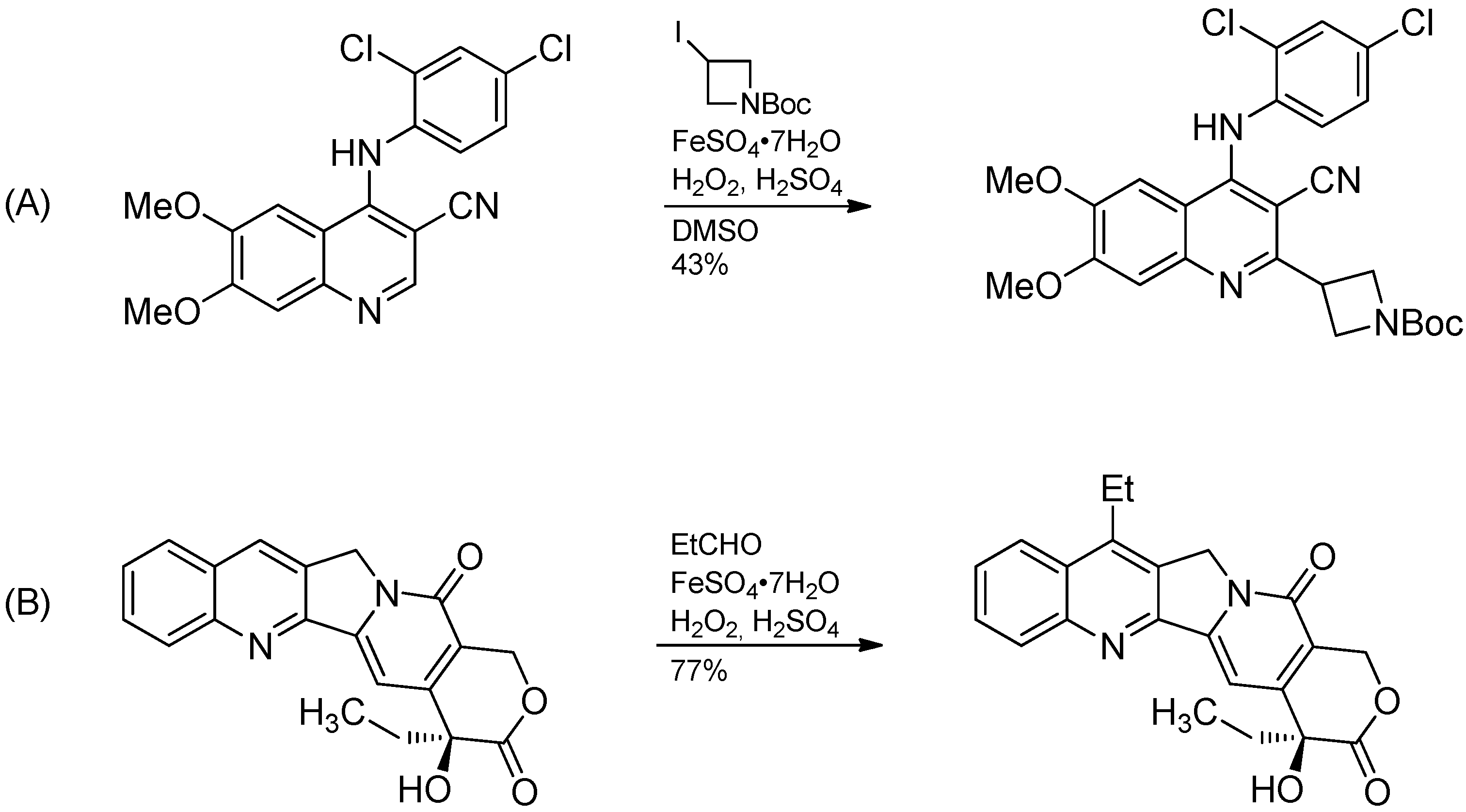
- O’Hara, F.; Blackmond, D.G.; Baran, P.S. Radical-based regioselective C-H functionalization of electron-deficient heteroarenes: Scope, tunability, and predictability. J. Am. Chem. Soc. 2013, 135, 12122–12134. [Google Scholar] [CrossRef]
- Begg, C.; Grimmett, M.; Yu-Man, L. The synthesis of 2-alkyl- and 2-acyl-imidazoles by substitution methods. Aust. J. Chem. 1973, 26, 415–420. [Google Scholar] [CrossRef]
- Jain, R.; Cohen, L.A.; El-Kadi, N.A.; King, M.M. Regiospecific alkylation of histidine and histamine at C-2. Tetrahedron 1997, 53, 2365–2370. [Google Scholar] [CrossRef]
- Mahindra, A.; Jain, R. Regiospecific direct C-H arylation at the 2-position of L-histidine using arylboronic acids. Synlett 2012, 1759–1764. [Google Scholar]
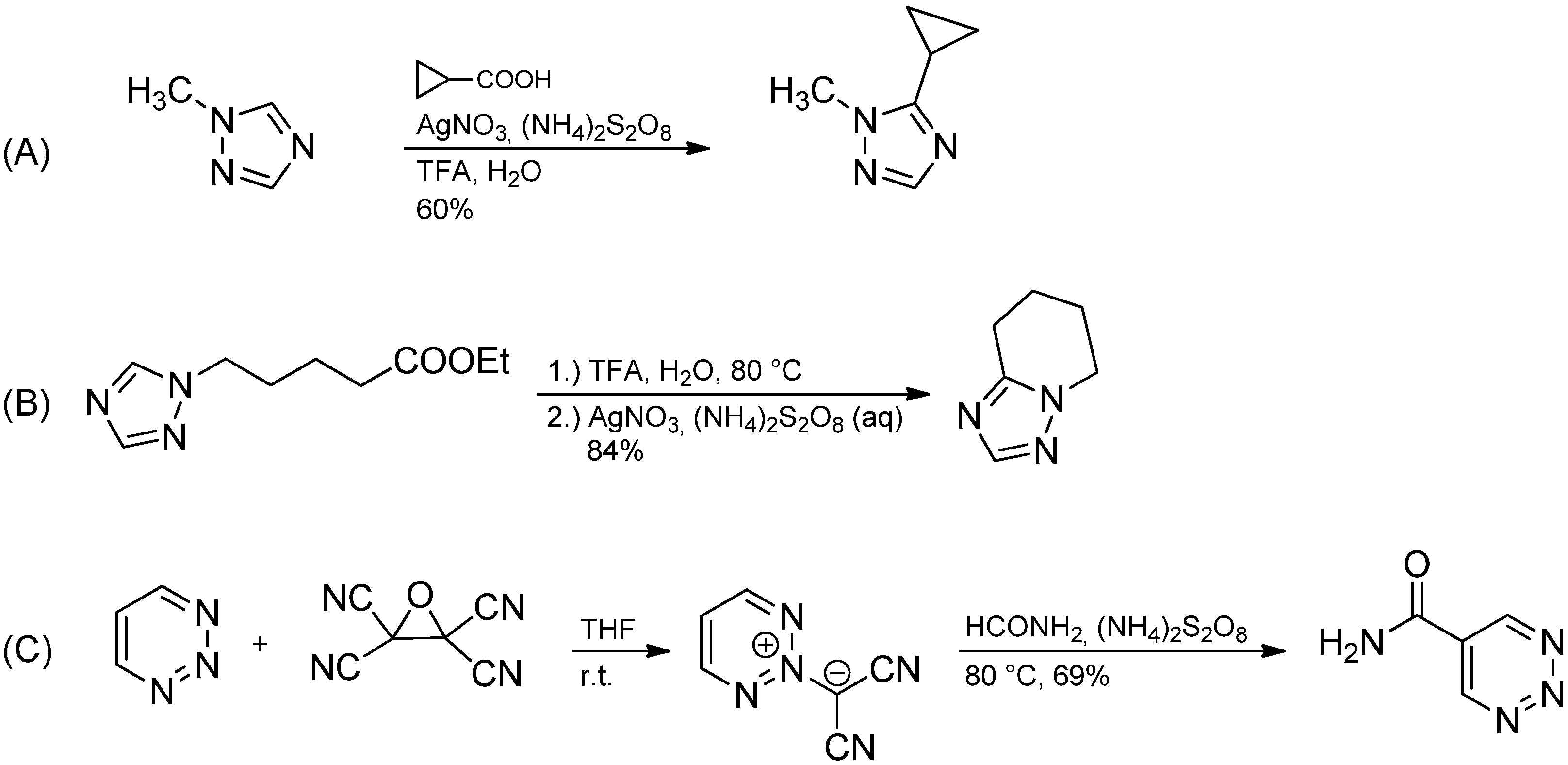
- Hansen, K.B.; Springfield, S.A.; Desmond, R.; Devine, P.N.; Grabowski, E.J.J.; Reider, P.J. Radical alkylation of N-alkyl 1,2,4-triazoles. Tetrahedron Lett. 2001, 42, 7353–7355. [Google Scholar] [CrossRef]
- Joule, J.A.; Mills, K. Heterocyclic Chemistry, 5th ed.; Wiley-Blackwell: Chichester, UK, 2013. [Google Scholar]
- Nagata, K.; Itoh, T.; Okada, M.; Takahashi, H.; Ohsawa, A. Radical carbamoylation of 1,2,3-triazinium 2-dicyanomethylides. Heterocycles 1991, 32, 855–857. [Google Scholar] [CrossRef]
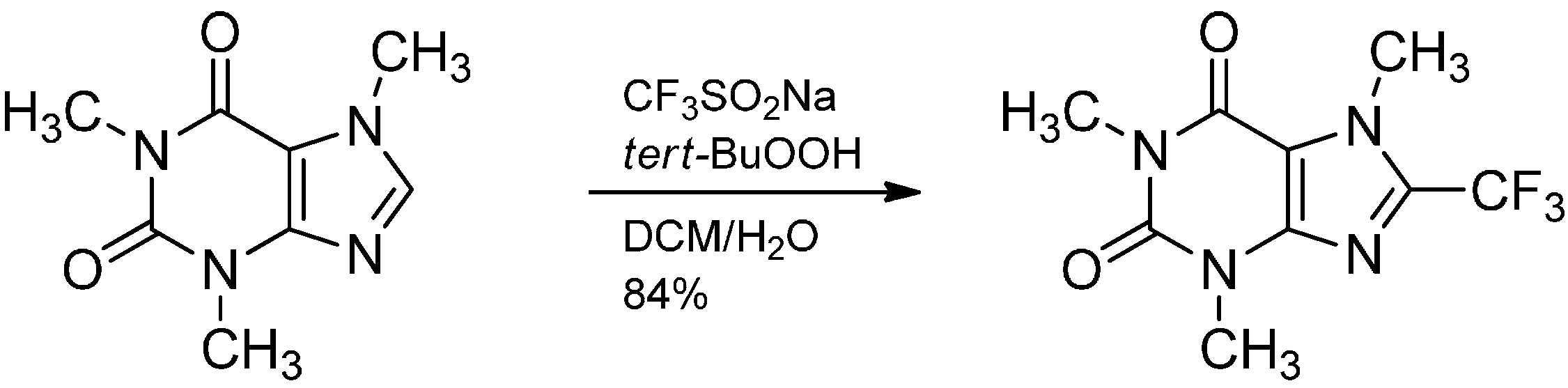
- Ji, Y.; Brueckl, T.; Baxter, R.D.; Fujiwara, Y.; Seiple, I.B.; Su, S.; Blackmond, D.G.; Baran, P.S. Innate C-H trifluoromethylation of heterocycles. Proc. Natl. Acad. Sci. USA 2011, 108, 14411–14415. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Dixon, J.A.; O’Hara, F.; Funder, E.D.; Dixon, D.D.; Rodriguez, R.A.; Baxter, R.D.; Herle, B.; Sach, N.; Collins, M.R.; et al. Practical and innate carbon-hydrogen functionalization of heterocycles. Nature 2012, 492, 95–99. [Google Scholar]
- Murphy, J.A.; Sherburn, M.S. Intramolecular addition of Free radicals to quaternised heterocyclic rings. Tetrahedron Lett. 1990, 31, 1625–1628. [Google Scholar] [CrossRef]
- Murphy, J.A.; Sherburn, M.S. Intramolecular free-radical substitution reactions of pyridinium rings: Efficient formation of [5,6]- and [6,7]-fused ring systems. Tetrahedron Lett. 1990, 31, 3495–3496. [Google Scholar] [CrossRef]
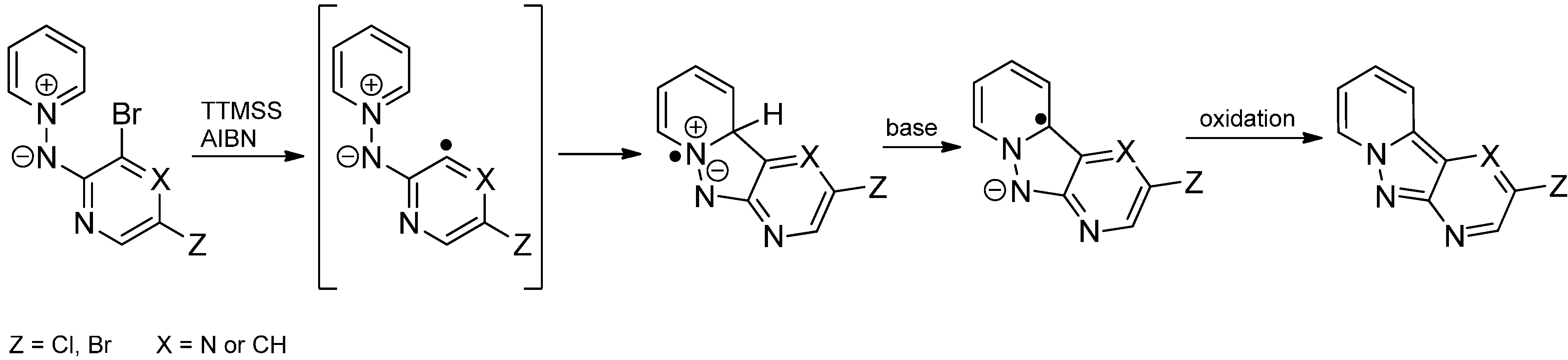
- Nunez, A.; de Viedma, A.G.; Martinez-Barrasa, V.; Burgos, C.; Alvarez-Builla, J. N-Azinylpyridinium N-aminides: An approach to pyrazolopyridines via an intramolecular radical pathway. Synlett 2002, 1093–1096. [Google Scholar] [CrossRef]
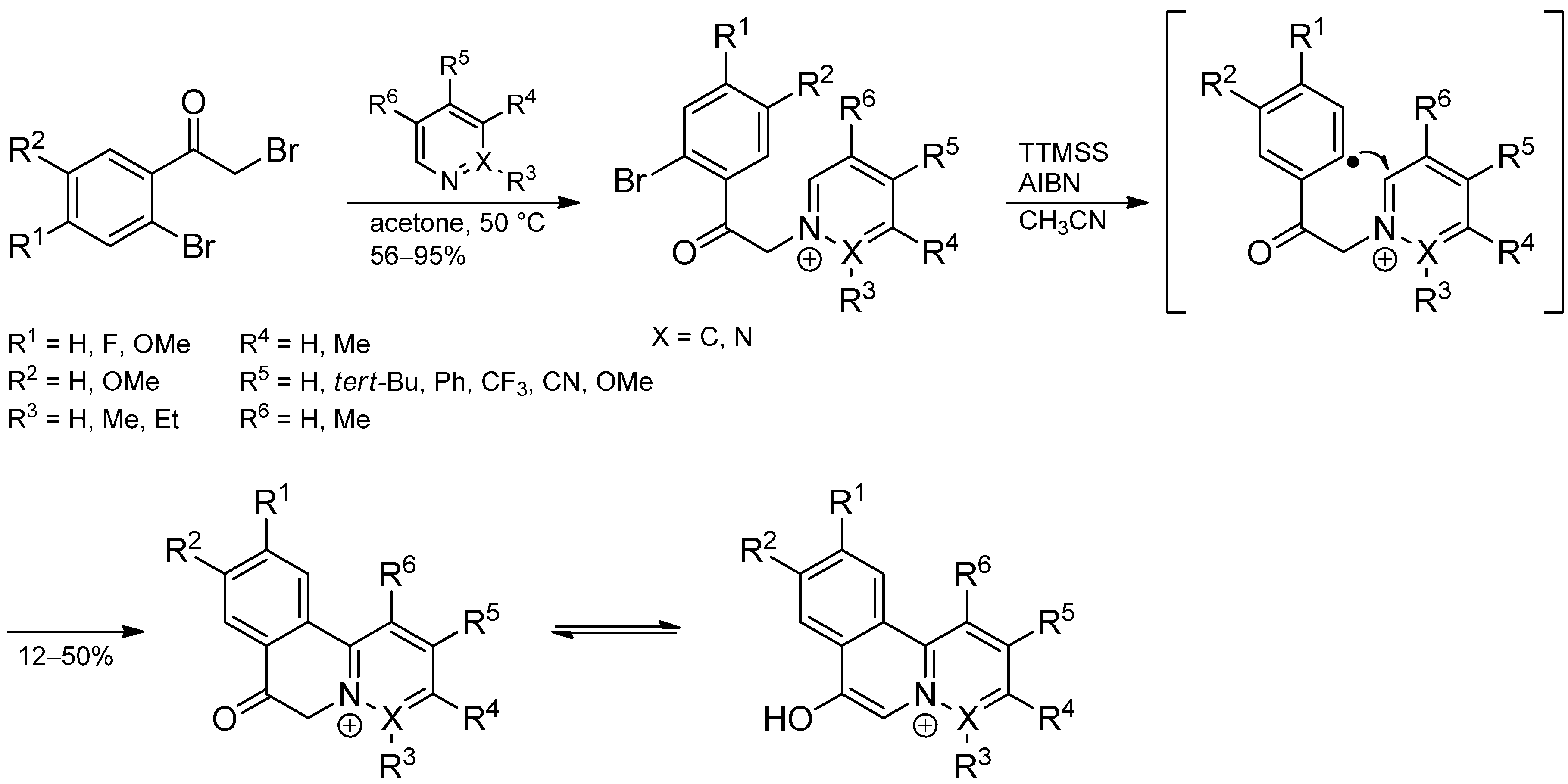
- Castillo, R.R.; Burgos, C.; Vaquero, J.J.; Alvarez-Builla, J. Radical intramolecular arylation of pyridinium salts: A straightforward entry to 7-hydroxypyrido[2,1-a]isoquinolinium salts. Eur. J. Org. Chem. 2011, 2011, 619–628. [Google Scholar] [CrossRef]
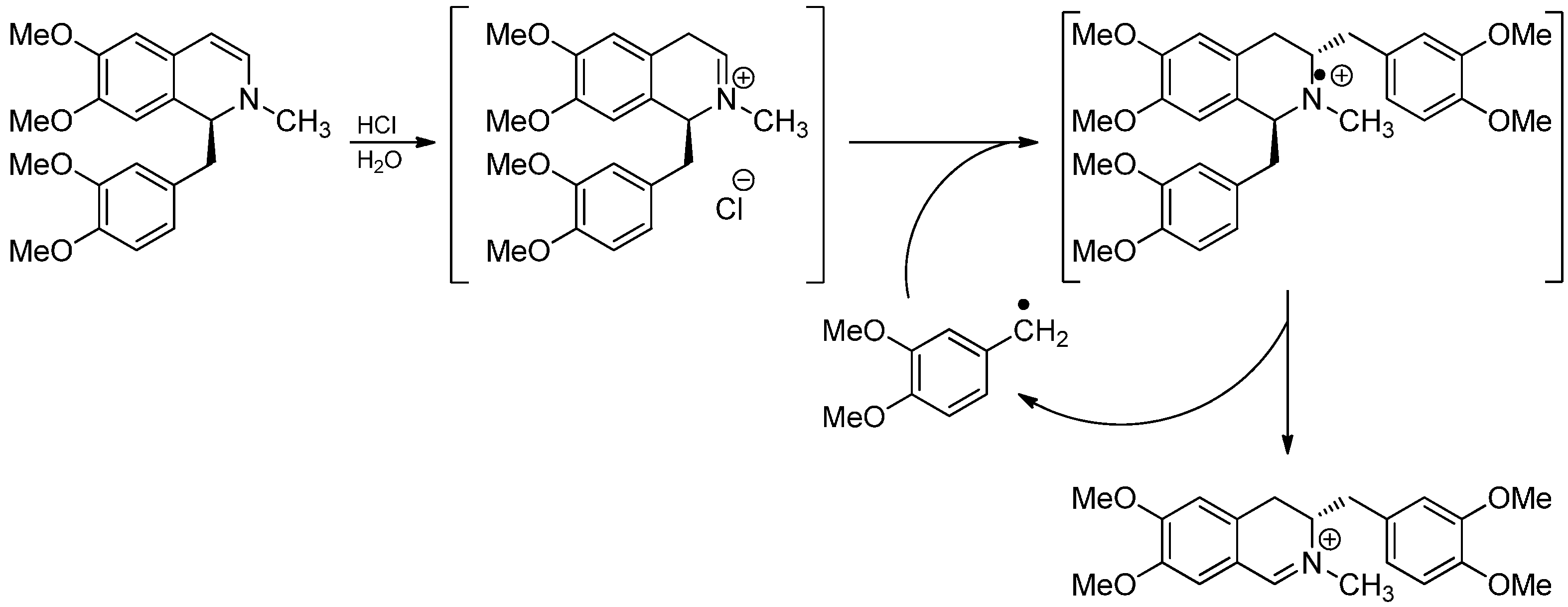
- Knabe, J.; Kubitz, J.; Ruppenthal, A.N. Umlagerung von N-Methyl-1.2-dihydropapaverin mit verdünnten Säuren. Angew. Chem. 1963, 75, 981. [Google Scholar]
- Knabe, J.; Kubitz, J. Über eine Umlagerung von N-Methyl-1,2-dihydropapaverin mit verdünnten Säuren. Arch. Pharm. Ber. 1964, 297, 129–140. [Google Scholar]
- Knabe, J.; Heckmann, R. Dihydroisoquinoline rearrangement 27. Proposal of a new mechanism. Arch. Pharm. 1980, 313, 1033–1042. [Google Scholar]
- Kinsman, R.G.; Dyke, S.F. Mechanism of the rearrangement of 1-benzyl-1,2-dihydroisoquinolines—Some criticisms answered. Tetrahedron 1979, 35, 857–860. [Google Scholar] [CrossRef]
- Powilleit, H. Untersuchungen zum Mechanismus der 1,2-Dihydroisochinolin-Umlagerung. Ph.D. Dissertation, University of Saarbrücken, Saarbrücken, Germany, 1969. [Google Scholar]
- Langhals, E.; Langhals, H.; Ruchardt, C. Evidence for a radical chain mechanism for the Knabe reaction of 1,2-dihydro-2-methylpapaverine. Chem. Ber. 1984, 117, 1436–1454. [Google Scholar]
- Knabe, J.; Grunewald, F.J. Rearrangement of dihydroisoquinolines 38. Behavior of 1,2-dihydroisoquinolines with bulky C-1-substituents towards acids. Arch. Pharm. 1987, 320, 492–499. [Google Scholar]
- Sainsbury, M.; Brown, D.W.; Dyke, S.F.; Kinsman, R.G.; Moon, B.J. 1,2-Dihydroisoquinolines—VIII: Rearrangement—II. Tetrahedron 1968, 24, 6695–6702. [Google Scholar] [CrossRef]
- Knabe, J.; Hanke, B. Dihydroisoquinoline Rearrangement 39. 6,7-Dimethoxy-2-methyl-1-(phenylpropargyl)-1,2-dihydroisoquinoline. Arch. Pharm. 1987, 320, 629–635. [Google Scholar]
- Knabe, J.; Holtje, H.D. Dihydroisoquinoline rearrangement 9: Vinylog principle in rearrangement of tertiary 1,2-dihydroisoquinolines. Tetrahedron Lett. 1969, 10, 2107–2108. [Google Scholar] [CrossRef]
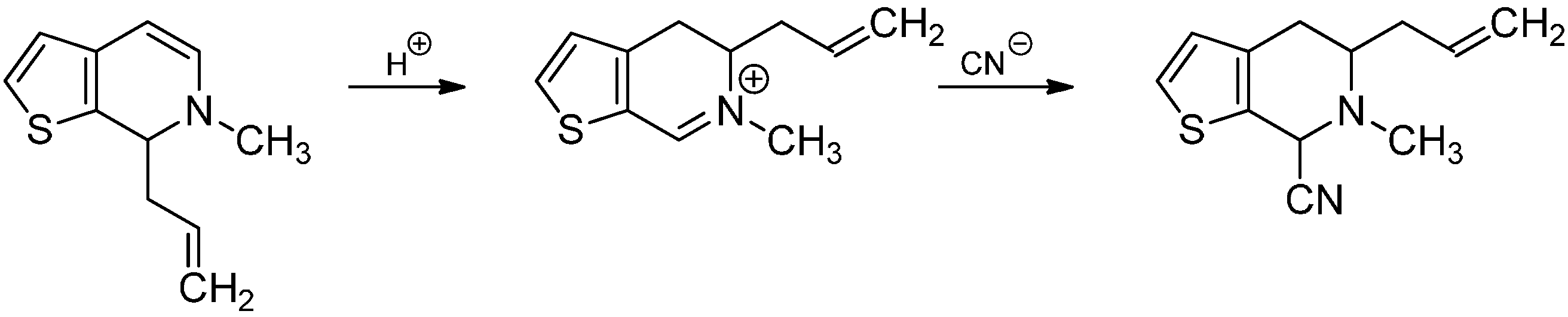
- Knabe, J.; Lorenz, J. Dihydroisoquinoline rearrangement 34. 7-Allyl-6-methyl-6,7-dihydrothieno[2,3-C]pyridine. Arch. Pharm. 1983, 316, 831–834. [Google Scholar]
- Blank, N.; Straub, B.F.; Opatz, T. 1,3-Benzyl migration in iminium ions: Evidence for a fast free-radical chain reaction. Eur. J. Org. Chem. 2011, 2011, 7355–7365. [Google Scholar] [CrossRef]
- Yoshida, J.; Suga, S.; Suzuki, S.; Kinomura, N.; Yamamoto, A.; Fujiwara, K. Direct oxidative carbon-carbon bond formation-using the “cation pool” method. 1. Generation of iminium cation pools and their reaction with carbon nucleophiles. J. Am. Chem. Soc. 1999, 121, 9546–9549. [Google Scholar]
- Yoshida, J.; Ashikari, Y.; Matsumoto, K.; Nokami, T. Recent developments in the “cation pool” method. J. Synth. Org. Chem. Jpn. 2013, 71, 1136–1144. [Google Scholar] [CrossRef]
- Maruyama, T.; Suga, S.; Yoshida, J.-I. Radical addition to “Cation Pool”. Reverse process of radical cation fragmentation. J. Am. Chem. Soc. 2005, 127, 7324–7325. [Google Scholar]
- Maruyama, T.; Suga, S.; Yoshida, J.I. Distannane mediated reaction of N-acyliminium ion pools with alkyl halides. A chain mechanism involving radical addition followed by electron transfer. Tetrahedron 2006, 62, 6519–6525. [Google Scholar]
- Maillard, B.; Forrest, D.; Ingold, K.U. Kinetic applications of electron-paramagnetic resonance spectroscopy 27. Isomerization of cyclopropylcarbinyl to allylcarbinyl. J. Am. Chem. Soc. 1976, 98, 7024–7026. [Google Scholar] [CrossRef]
- Maruyama, T.; Mizuno, Y.; Shimizu, I.; Suga, S.; Yoshida, J.-I. Reactions of a N-acyliminium ion pool with benzylsilanes. Implication of a radical/cation/radical cation chain mechanism involving oxidative C-Si Bond Cleavage. J. Am. Chem. Soc. 2007, 129, 1902–1903. [Google Scholar]
- Suga, S.; Shimizu, I.; Ashikari, Y.; Mizuno, Y.; Maruyama, T.; Yoshida, J. Electro-initiated coupling reactions of N-acyliminium ion pools with arylthiomethylsilanes and aryloxymethylsilanes. Chem. Lett. 2008, 37, 1008–1009. [Google Scholar]
- Friestad, G.K. Addition of carbon-centered radicals to imines and related compounds. Tetrahedron 2001, 57, 5461–5496. [Google Scholar] [CrossRef]
- Miyabe, H.; Ueda, M.; Naito, T. Carbon-carbon bond construction based on radical addition to C=N bond. Synlett 2004, 7, 1140–1157. [Google Scholar]
- Pastori, N.; Gambarotti, C.; Punta, C. Recent developments in nucleophilic radical addition to imines: The key role of transition metals and the new Porta radical-type version of the Mannich and Strecker reactions. Mini-Rev. Org. Chem. 2009, 6, 184–195. [Google Scholar] [CrossRef]
- Miyabe, H.; Yoshioka, E.; Kohtani, S. Progress in intermolecular carbon radical addition to imine derivatives. Curr. Org. Chem. 2010, 14, 1254–1264. [Google Scholar] [CrossRef]
- Naito, T. Heteroatom radical addition-cyclization and its synthetic application. Heterocycles 1999, 50, 505–541. [Google Scholar] [CrossRef]
- Fallis, A.G.; Brinza, I.M. Free radical cyclizations involving nitrogen. Tetrahedron 1997, 53, 17543–17594. [Google Scholar] [CrossRef]
- Miyabe, H.; Ueda, M.; Nishimura, A.; Naito, T. Indium-mediated intermolecular alkyl radical addition to electron-deficient C=N bond and C=C bond in water. Org. Lett. 2002, 4, 131–134. [Google Scholar] [CrossRef]
- Yamada, K.; Fujihara, H.; Yamamoto, Y.; Miwa, Y.; Taga, T.; Tomioka, K. Radical addition of ethers to imines initiated by dimethylzinc. Org. Lett. 2002, 4, 3509–3511. [Google Scholar] [CrossRef]
- Clerici, A.; Porta, O. Arylative amination of aldehydes promoted by aqueous titanium trichloride. Tetrahedron Lett. 1990, 31, 2069–2072. [Google Scholar]
- Rossi, B.; Prosperini, S.; Pastori, N.; Clerici, A.; Punta, C. New advances in titanium-mediated free radical reactions. Molecules 2012, 17, 14700–14732. [Google Scholar] [CrossRef]
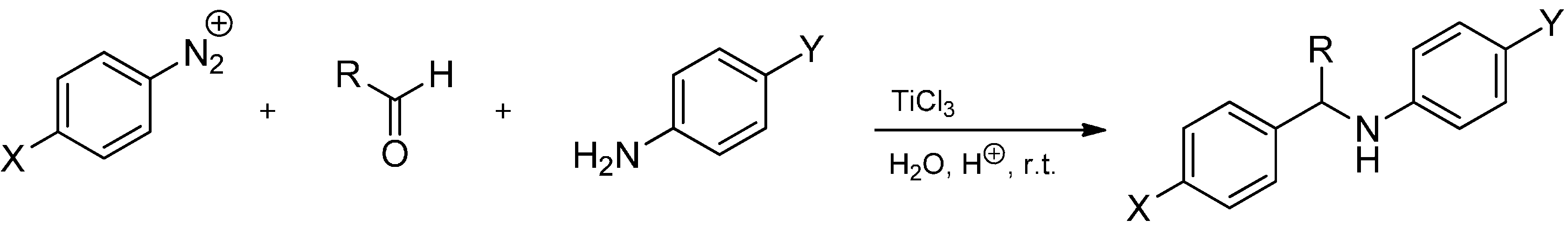
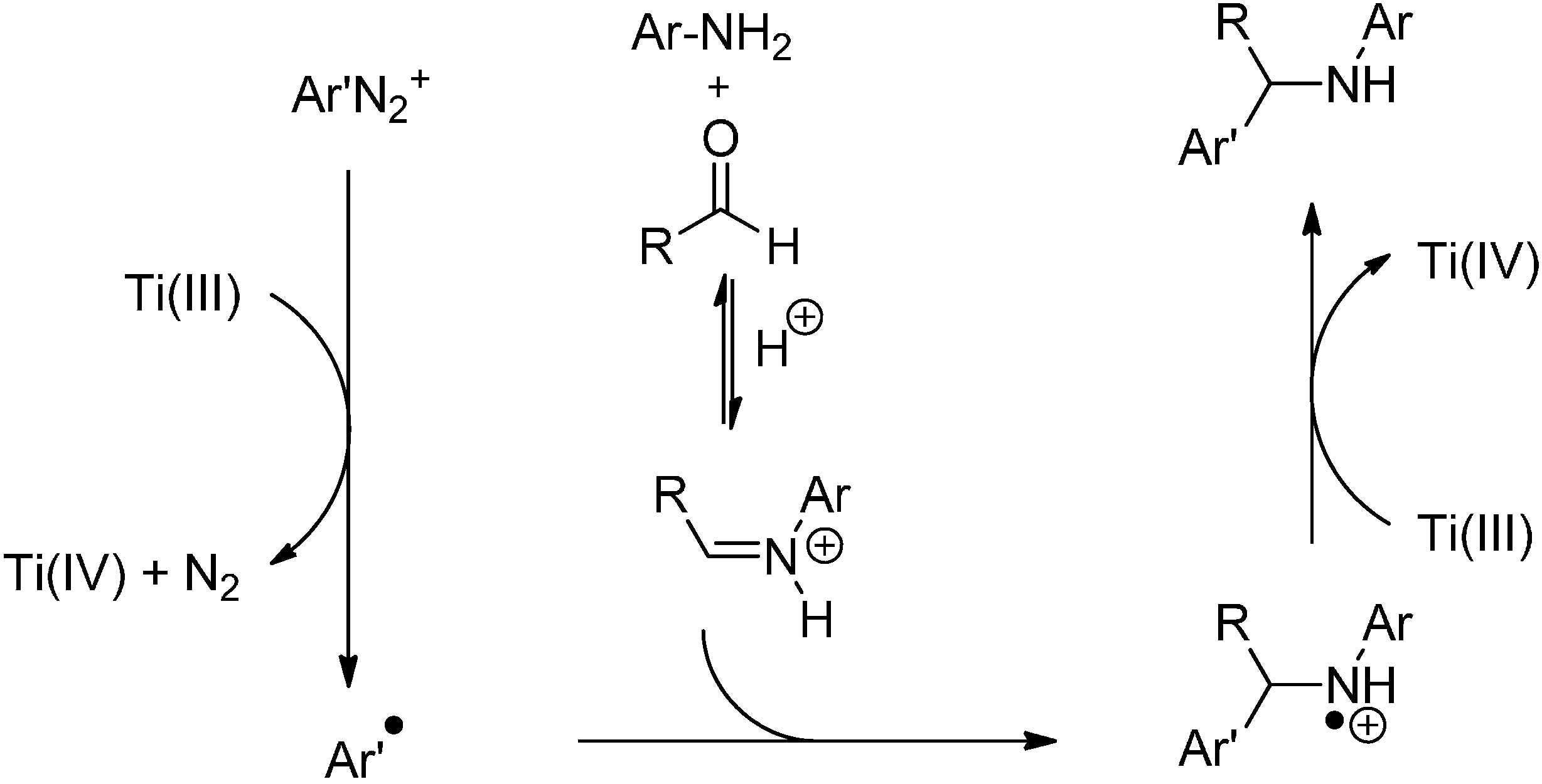
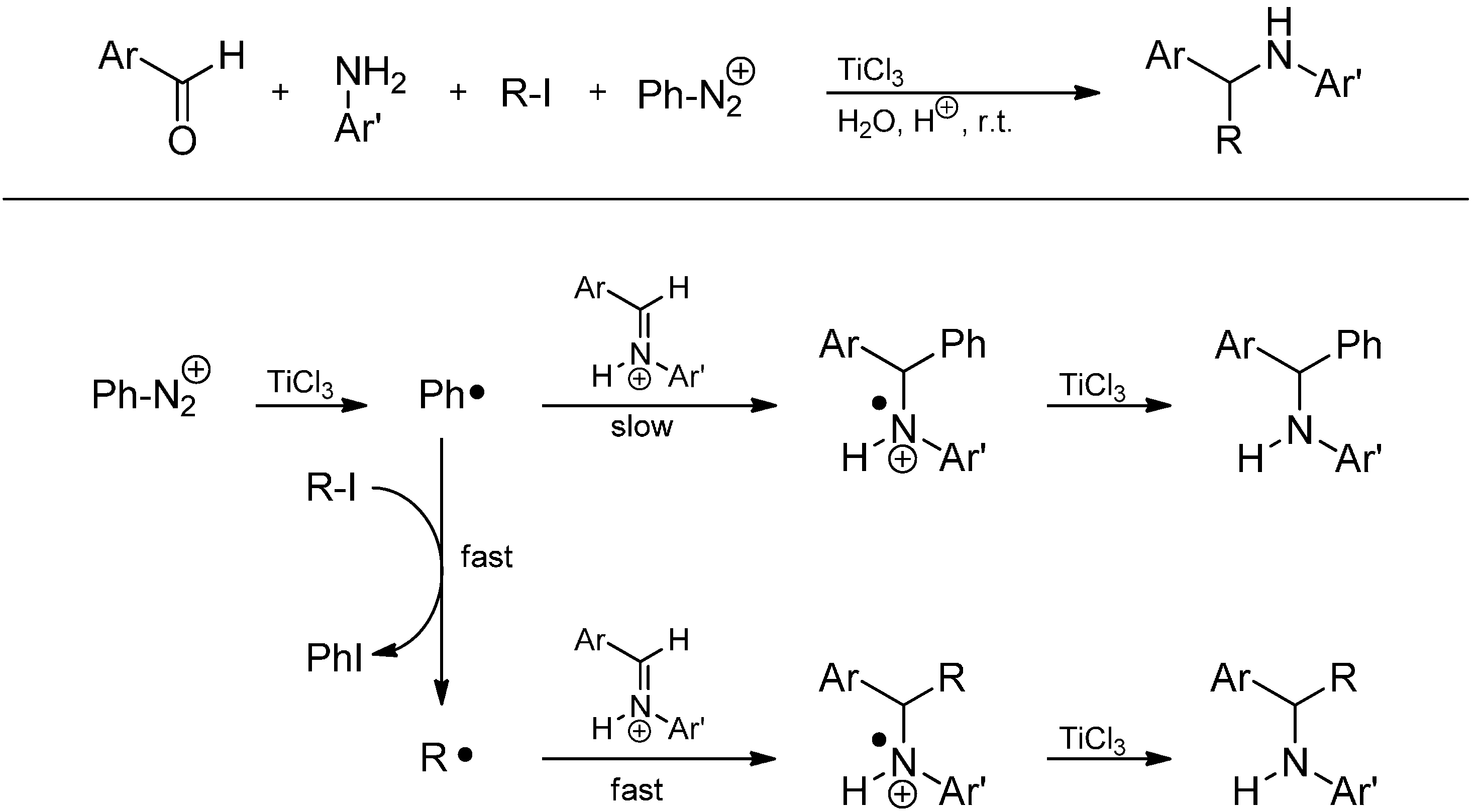
- Cannella, R.; Clerici, A.; Pastori, N.; Regolini, E.; Porta, O. One-pot four-component reaction: Aqueous TiCl3/PhN2+-mediated alkyl radical addition to imines generated in situ. Org. Lett. 2005, 7, 645–648. [Google Scholar]
- Clerici, A.; Cannella, R.; Panzeri, W.; Pastori, N.; Regolini, E.; Porta, O. TiCl3/PhN2+-mediated radical addition of ethers to aldimines generated in situ under aqueous conditions. Tetrahedron Lett. 2005, 46, 8351–8354. [Google Scholar] [CrossRef]
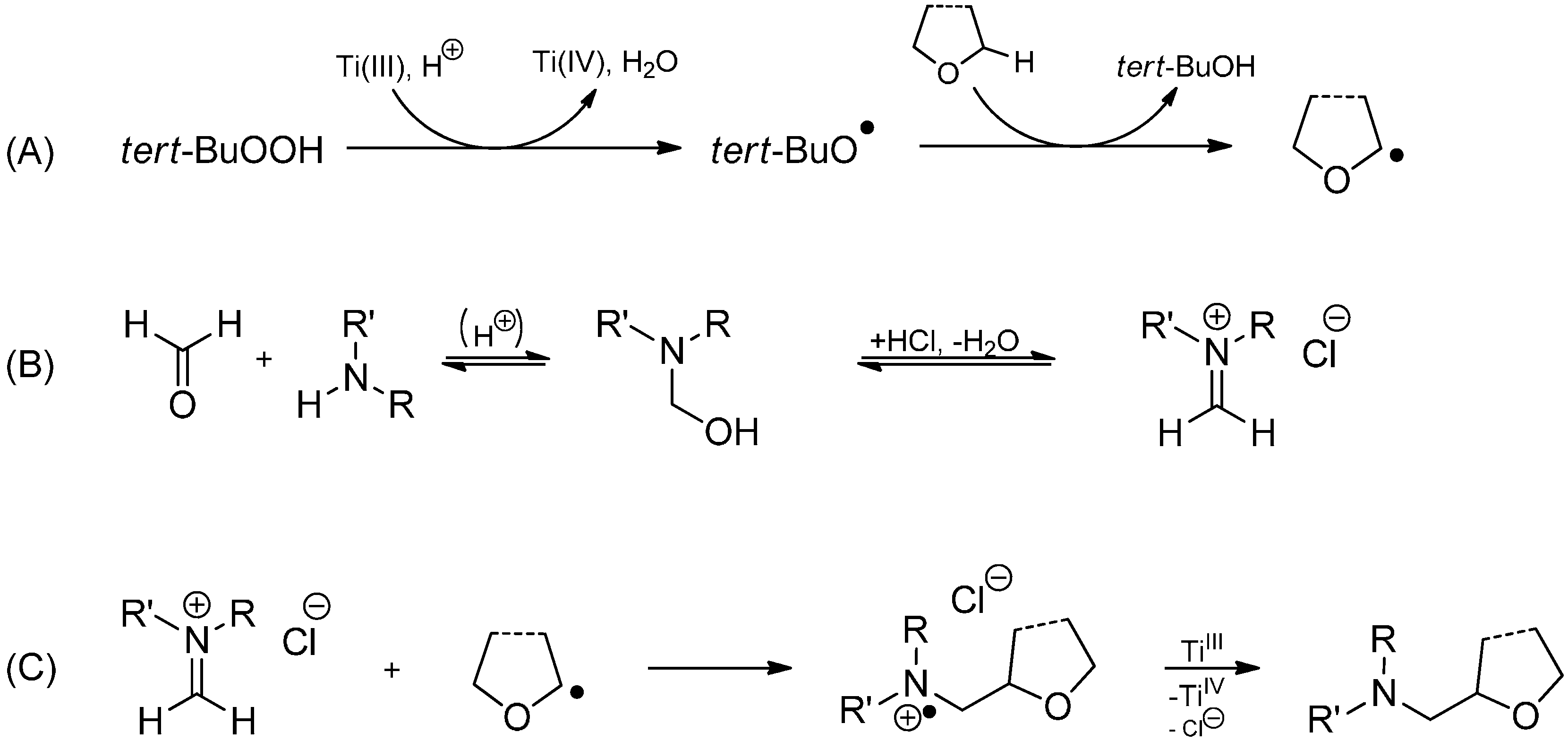
- Prosperini, S.; Pastori, N.; Ghilardi, A.; Clerici, A.; Punta, C. New domino radical synthesis of aminoalcohols promoted by TiCl4-Zn/t-BuOOH system: Selective hydroxyalkylation of amines in alcohol or in cyclic ether cosolvents. Org. Biomol. Chem. 2011, 9, 3759–3767. [Google Scholar] [CrossRef]
- Clerici, A.; Cannella, R.; Pastori, N.; Panzeri, W.; Porta, O. A free radical Mannich type reaction: Selective α-CH aminomethylation of ethers by Ti(III)/t-BuOOH system under aqueous acidic conditions. Tetrahedron 2006, 62, 5986–5994. [Google Scholar] [CrossRef]
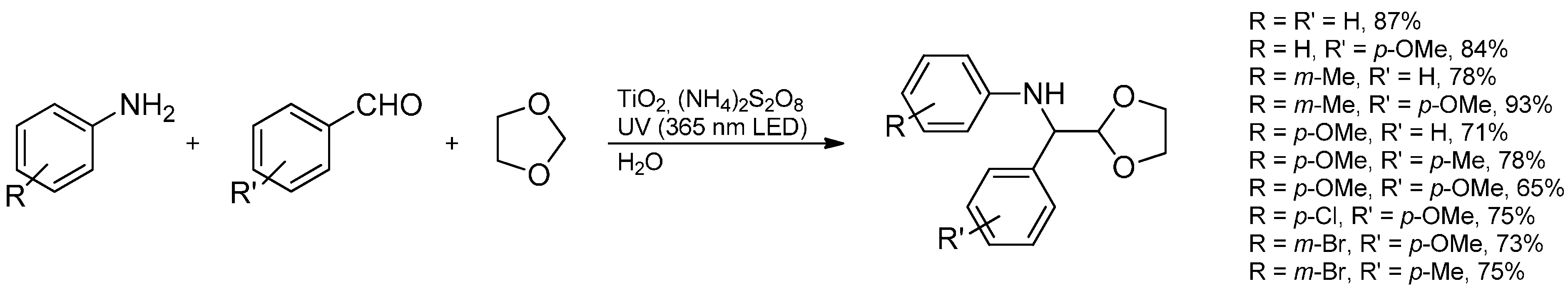
- Zhang, L.; Deng, Y.Q.; Shi, F. Light promoted aqueous phase amine synthesis via three-component coupling reactions. Tetrahedron Lett. 2013, 54, 5217–5219. [Google Scholar] [CrossRef]
- Caronna, T.; Gambarotti, C.; Palmisano, L.; Punta, C.; Recupero, F. Sunlight induced functionalisation of some heterocyclic bases in the presence of polycrystalline TiO2. Chem. Commun. 2003, 18, 2350–2351. [Google Scholar] [CrossRef]
- Gambarotti, C.; Punta, C.; Recupero, F.; Caronna, T.; Palmisano, L. TiO2 in organic photosynthesis: Sunlight induced functionalization of heterocyclic bases. Curr. Org. Chem. 2010, 14, 1153–1169. [Google Scholar] [CrossRef]
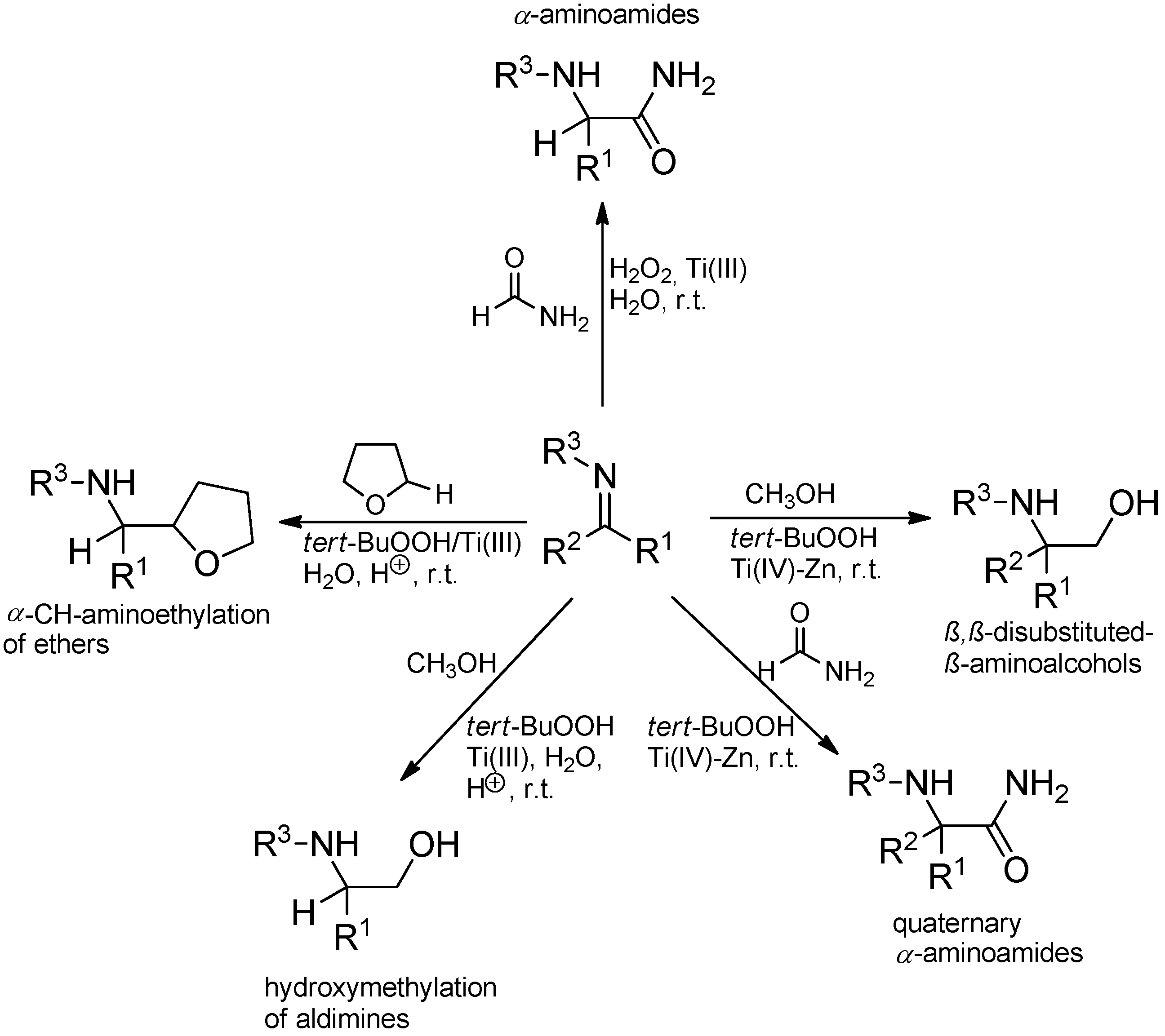
- Clerici, A.; Ghilardi, A.; Pastori, N.; Punta, C.; Porta, O. A new one-pot, four-component synthesis of 1,2-amino alcohols: TiCl3/t-BuOOH-mediated radical hydroxymethylation of imines. Org. Lett. 2008, 10, 5063–5066. [Google Scholar] [CrossRef]
- Spaccini, R.; Ghilardi, A.; Pastori, N.; Clerici, A.; Punta, C.; Porta, O. Efficient radical domino approach to β-aminoalcohols from arylamines and alcohols triggered by Ti(III)/t-BuOOH. Tetrahedron 2010, 66, 2044–2052. [Google Scholar]
- Pastori, N.; Greco, C.; Clerici, A.; Punta, C.; Porta, O. Free-radical addition to ketimines generated in situ. New one-pot synthesis of quaternary α-aminoamides promoted by a H2O2/TiCl4-Zn/HCONH2 System. Org. Lett. 2010, 12, 3898–3901. [Google Scholar]
- Rossi, B.; Pastori, N.; Clerici, A.; Punta, C. Free-radical hydroxymethylation of ketimines generated in situ: A one-pot multicomponent synthesis of β,β-disubstituted-β-aminoalcohols. Tetrahedron 2012, 68, 10151–10156. [Google Scholar] [CrossRef]
- Cannella, R.; Clerici, A.; Panzeri, W.; Pastori, N.; Punta, C.; Porta, O. Free-radical version of the strecker synthesis of α-aminoamides promoted by aqueous H2O2/TiCl3/HCONH2 system. J. Am. Chem. Soc. 2006, 128, 5358–5359. [Google Scholar]
- Bertrand, M.P.; Feray, L.; Nouguier, R.; Perfetti, P. Diethylzinc mediated radical additions to glyoxylate imines. Synlett 1999, 7, 1148–1150. [Google Scholar] [CrossRef]
- Bertrand, M.P.; Feray, L.; Nouguier, R.; Perfetti, P. Diethylzinc: A chain-transfer agent in intermolecular radical additions. A parallel with triethylborane. J. Org. Chem. 1999, 64, 9189–9193. [Google Scholar]
- Yamada, K.; Yamamoto, Y.; Tomioka, K. Initiator-dependent chemoselective addition of THF radical to aldehyde and aldimine and its application to a three-component reaction. Org. Lett. 2003, 5, 1797–1799. [Google Scholar]
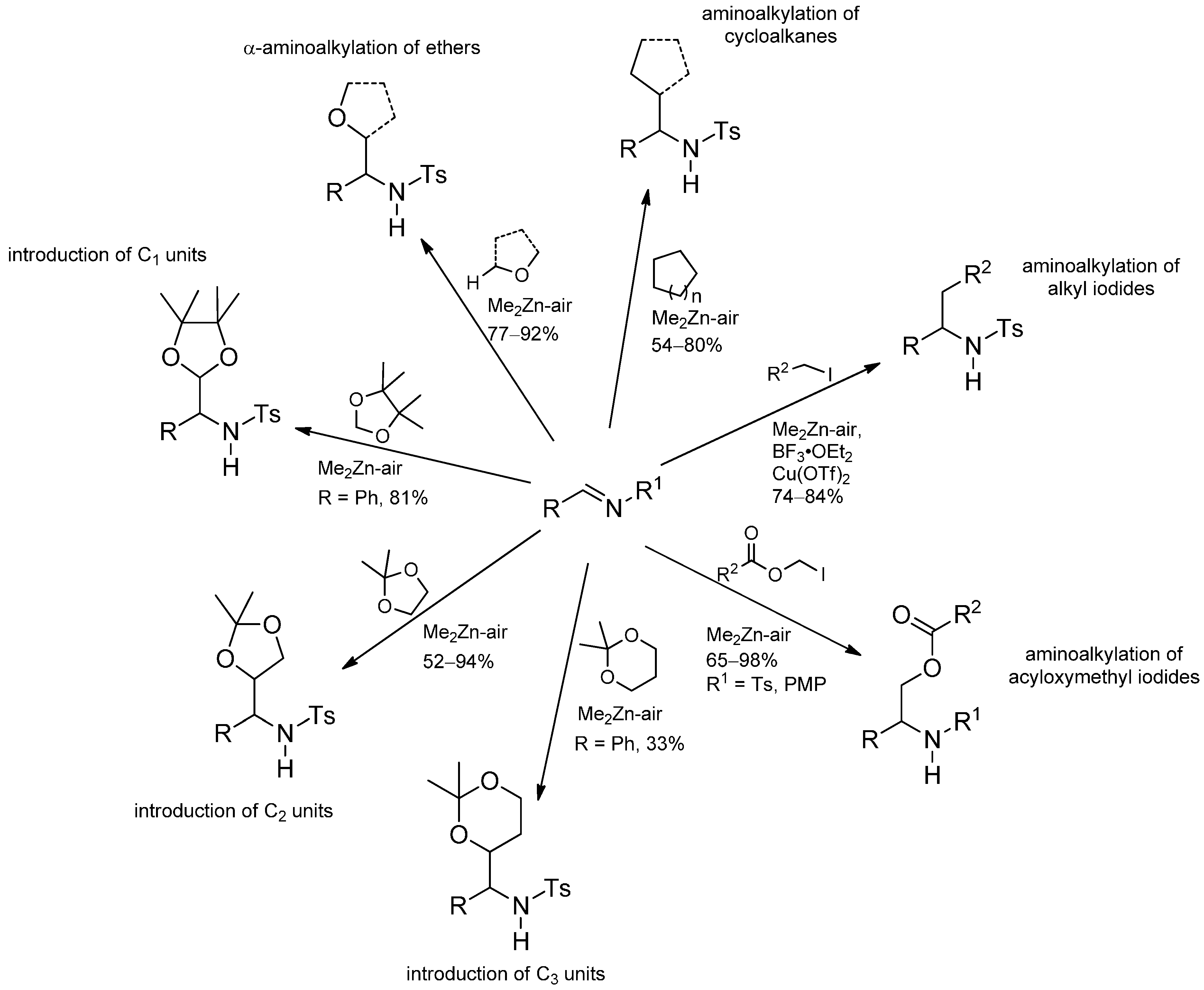
- Yamamoto, Y.; Maekawa, M.; Akindele, T.; Yamada, K.; Tomioka, K. Dimethylzinc-initiated radical reaction of cyclic ethers with arylamines, alkoxyamines, and dialkylhydrazines. Tetrahedron 2005, 61, 379–384. [Google Scholar] [CrossRef]
- Yamada, K.; Umeki, H.; Maekawa, M.; Yamamoto, Y.; Akindele, T.; Nakano, M.; Tomioka, K. Conjugate addition reaction of THF-2-yl radical with α,β-unsaturated N-tosyl imines using a dimethylzinc-air initiator. Tetrahedron 2008, 64, 7258–7265. [Google Scholar]
- Yamada, K.; Yamamoto, Y.; Maekawa, M.; Chen, J.B.; Tomioka, K. Direct aminoalkylation of cycloalkanes through dimethylzinc-initiated radical process. Tetrahedron Lett. 2004, 45, 6595–6597. [Google Scholar] [CrossRef]
- Yamada, K.; Yamamoto, Y.; Maekawa, M.; Akindele, T.; Umeki, H.; Tomioka, K. Tin-free intermolecular addition of primary alkyls to imines via the dimethylzinc-air radical process. Org. Lett. 2006, 8, 87–89. [Google Scholar] [CrossRef]
- Yamada, K.; Nakano, M.; Maekawa, M.; Akindele, T.; Tomioka, K. Tin-free radical addition of acyloxymethyl to imines. Org. Lett. 2008, 10, 3805–3808. [Google Scholar] [CrossRef]
- Yamada, K.I.; Yamamoto, Y.; Maekawa, M.; Tomioka, K. Introduction of functionalized C1, C2, and C3 units to imines through the dimethylzinc—Air-initiated radical addition. J. Org. Chem. 2004, 69, 1531–1534. [Google Scholar] [CrossRef]
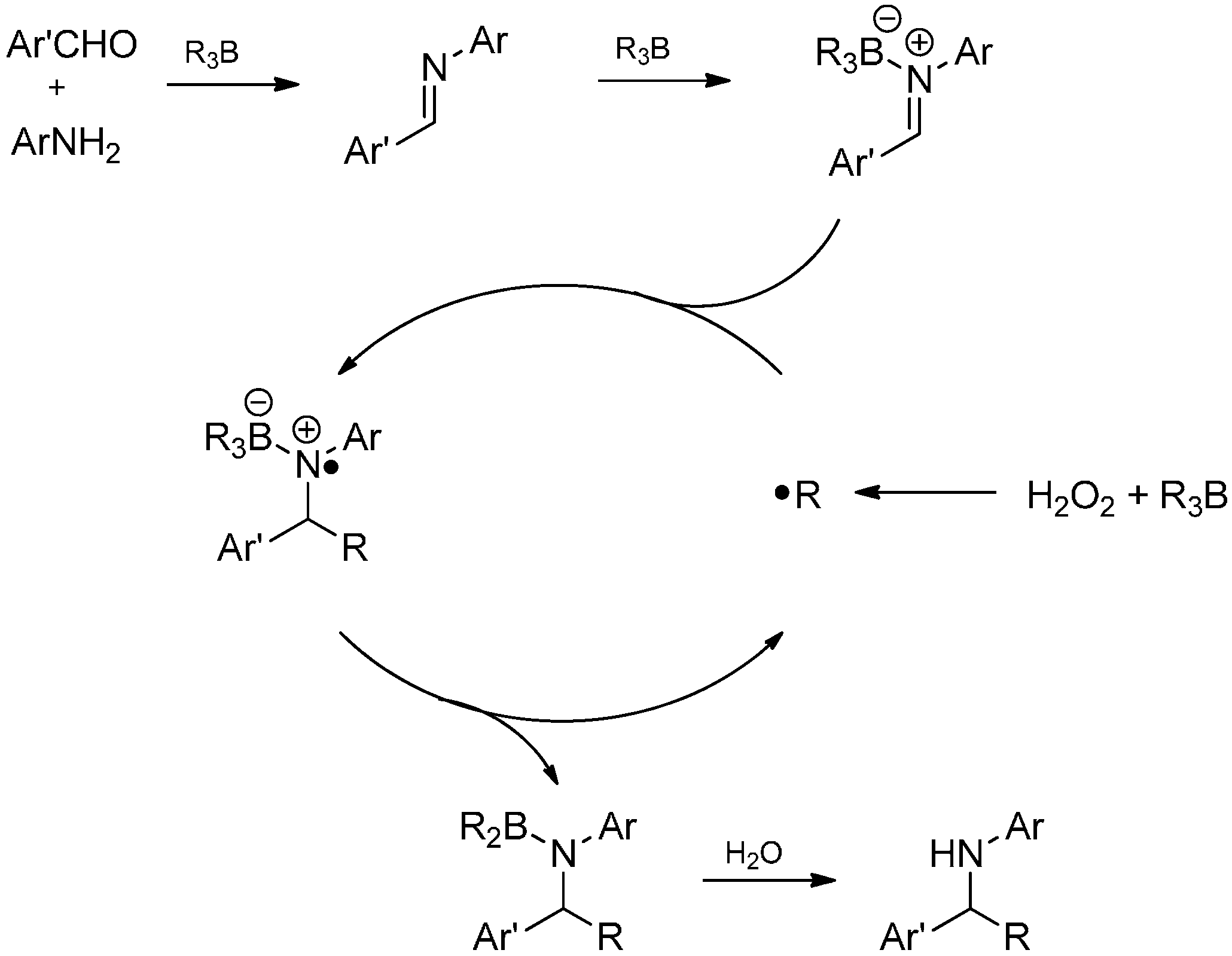
- Valpuesta, M.; Munoz, C.; Diaz, A.; Torres, G.; Suau, R. Multicomponent C-alkylation reactions of aromatic aldimines with trialkylboranes reagents. Eur. J. Org. Chem. 2010, 1934–1942. [Google Scholar] [CrossRef]
- Miyabe, H.; Yamaoka, Y.; Takemoto, Y. Reactive ketimino radical acceptors: Intermolecular alkyl radical addition to imines with a phenolic hydroxyl group. J. Org. Chem. 2006, 71, 2099–2106. [Google Scholar] [CrossRef]
- Miyabe, H.; Shibata, R.; Ushiro, C.; Naito, T. Carbon-carbon bond formation via intermolecular carbon radical addition to aldoxime ethers. Tetrahedron Lett. 1998, 39, 631–634. [Google Scholar] [CrossRef]
- Miyabe, H.; Shibata, R.; Sangawa, M.; Ushiro, C.; Naito, T. Intermolecular alkyl radical addition to the carbon-nitrogen double bond of oxime ethers and hydrazones. Tetrahedron 1998, 54, 11431–11444. [Google Scholar] [CrossRef]
- Miyabe, H.; Yamakawa, K.; Yoshioka, N.; Naito, T. Alkylative amination of aldehydes via carbon-carbon bond formation based on radical addition to carbon-nitrogen double bond. Tetrahedron 1999, 55, 11209–11218. [Google Scholar] [CrossRef]
- Song, Y.C.; Okamoto, S.; Sato, F. A concise asymmetric synthesis of 5,8-disubstituted indolizidine alkaloids. Total synthesis of (−)-indolizidine 209B. Tetrahedron Lett. 2002, 43, 8635–8637. [Google Scholar] [CrossRef]
- Aube, J.; Rafferty, P.S.; Milligan, G.L. Application of the intramolecular Schmidt reaction to the asymmetric-synthesis of (−)-indolizidine-209b from pulegone. Heterocycles 1993, 35, 1141–1147. [Google Scholar] [CrossRef]
- Satake, A.; Shimizu, I. A chiral synthesis of (8r,8as)-hexahydro-8-methyl-5(1h)-indolizinone. Tetrahedron: Asymmetry 1993, 4, 1405–1408. [Google Scholar] [CrossRef]
- Miyata, O.; Takahashi, S.; Tamura, A.; Ueda, M.; Naito, T. Novel synthesis of substituted pyrrolidines and piperidines via radical addition—Ionic cyclization reaction of oxime ethers. Tetrahedron 2008, 64, 1270–1284. [Google Scholar] [CrossRef]
- Zhang, L.; Kim, J.B.; Jang, D.O. Lewis acid-catalyzed cascade radical addition/cyclization for the synthesis of 3-substituted isoindolin-1-one derivatives. Tetrahedron Lett. 2014, 55, 2654–2658. [Google Scholar] [CrossRef]
- Miyabe, H. Inter- and intramolecular carbon-carbon bond-forming radical reactions. Synlett 2012, 23, 1709–1724. [Google Scholar] [CrossRef]
- Bertrand, M.P.; Feray, L.; Nouguier, R.; Stella, L. 1,3-stereoinduction in radical additions to glyoxylate imines. Synlett 1998, 7, 780–782. [Google Scholar] [CrossRef]
- Kim, B.H.; Curran, D.P. Asymmetric thermal-reactions with oppolzer camphor sultam. Tetrahedron 1993, 49, 293–318. [Google Scholar] [CrossRef]
- Miyabe, H.; Fujii, K.; Naito, T. Highly diastereoselective radical addition to oxime ethers: Asymmetric synthesis of beta-amino acids. Org. Lett. 1999, 1, 569–572. [Google Scholar] [CrossRef]
- Miyabe, H.; Fujii, K.; Naito, T. Radical addition to oxime ethers for asymmetric synthesis of beta-amino acid derivatives. Org. Biomol. Chem. 2003, 1, 381–390. [Google Scholar] [CrossRef]
- Friestad, G.K.; Qin, J. Highly stereoselective intermolecular radical addition to aldehyde hydrazones from a chiral 3-amino-2-oxazolidinone. J. Am. Chem. Soc. 2000, 122, 8329–8330. [Google Scholar] [CrossRef]
- Friestad, G.K.; Qin, J. Intermolecular alkyl radical addition to chiral N-acylhydrazones mediated by manganese carbonyl. J. Am. Chem. Soc. 2001, 123, 9922–9923. [Google Scholar] [CrossRef]
- Friestad, G.K.; Marie, J.C.; Deveau, A.M. Stereoselective Mn-mediated coupling of functionalized iodides and hydrazones: A synthetic entry to the tubulysin gamma-amino acids. Org. Lett. 2004, 6, 3249–3252. [Google Scholar] [CrossRef]
- Friestad, G.K.; Draghici, C.; Soukri, M.; Qin, J. Radical addition approach to asymmetric amine synthesis: Design, implementation, and comparison of chiral N-acylhydrazones. J. Org. Chem. 2005, 70, 6330–6338. [Google Scholar] [CrossRef]
- Friestad, G.K.; Marie, J.C.; Suh, Y.S.; Qin, J. Mn-mediated coupling of alkyl iodides and chiral N-acylhydrazones: Optimization, scope, and evidence for a radical mechanism. J. Org. Chem. 2006, 71, 7016–7027. [Google Scholar] [CrossRef]
- Korapala, C.S.; Qin, J.; Friestad, G.K. Quinine synthesis studies: A radical-ionic annulation via Mn-mediated addition to chiral N-acylhydrazones. Org. Lett. 2007, 9, 4243–4246. [Google Scholar] [CrossRef]
- Friestad, G.K.; Ji, A. Mn-mediated coupling of alkyl iodides and ketimines: A radical addition route to α,α-disubstituted α-aminoesters. Org. Lett. 2008, 10, 2311–2313. [Google Scholar] [CrossRef]
- Friestad, G.K.; Banerjee, K. Synthesis of γ-amino esters via Mn-mediated radical addition to chiral γ-Hydrazonoesters. Org. Lett. 2009, 11, 1095–1098. [Google Scholar] [CrossRef]
- Miyabe, H.; Ushiro, C.; Ueda, M.; Yamakawa, K.; Naito, T. Asymmetric synthesis of alpha-amino acids based on carbon radical addition to glyoxylic oxime ether. J. Org. Chem. 2000, 65, 176–185. [Google Scholar] [CrossRef]
- Friestad, G.K.; Shen, Y.H.; Ruggles, E.L. Enantioselective radical addition to N-acyl hydrazones mediated by chiral Lewis acids. Angew. Chem. Int. Ed. 2003, 42, 5061–5063. [Google Scholar] [CrossRef]
- Hu, J.; Wang, J.; Nguyen, T.H.; Zheng, N. The chemistry of amine radical cations produced by visible light photoredox catalysis. Beilstein J. Org. Chem. 2013, 9, 1977–2001. [Google Scholar] [CrossRef]
- Albini, A.; Mella, M.; Freccero, M. A new method in radical chemistry—Generation of radicals by photoinduced electron-transfer and fragmentation of the radical-cation. Tetrahedron 1994, 50, 575–607. [Google Scholar] [CrossRef]
- Pandey, G.; Rani, K.S.; Lakshmaiah, G. Direct carbon-carbon bond formation strategy at α-position of tertiary-amines by photoinduced electron-transfer (PET) processes. Tetrahedron Lett. 1992, 33, 5107–5110. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tauber, J.; Imbri, D.; Opatz, T. Radical Addition to Iminium Ions and Cationic Heterocycles. Molecules 2014, 19, 16190-16222. https://doi.org/10.3390/molecules191016190
Tauber J, Imbri D, Opatz T. Radical Addition to Iminium Ions and Cationic Heterocycles. Molecules. 2014; 19(10):16190-16222. https://doi.org/10.3390/molecules191016190
Chicago/Turabian StyleTauber, Johannes, Dennis Imbri, and Till Opatz. 2014. "Radical Addition to Iminium Ions and Cationic Heterocycles" Molecules 19, no. 10: 16190-16222. https://doi.org/10.3390/molecules191016190