Recombinant Expressed Vector pET32a (+) S Constructed by Ligation Independent Cloning

Abstract
:1. Introduction
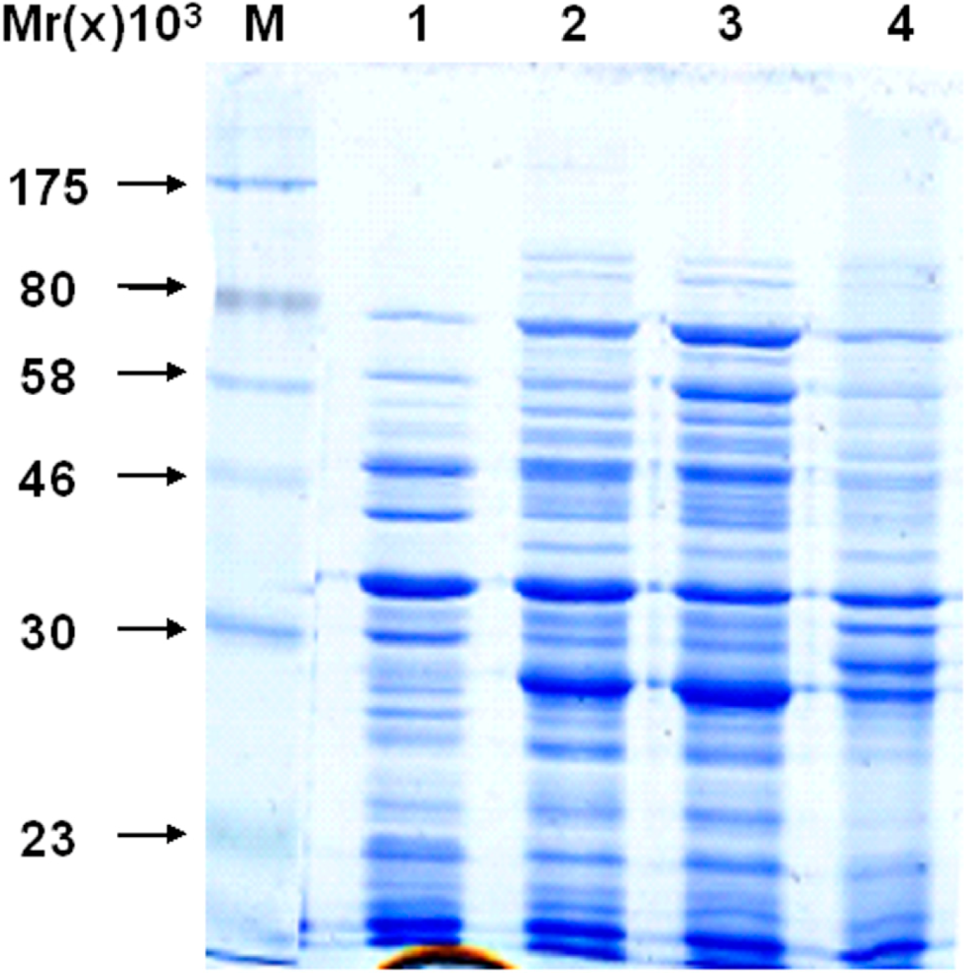
2. Results and Discussion
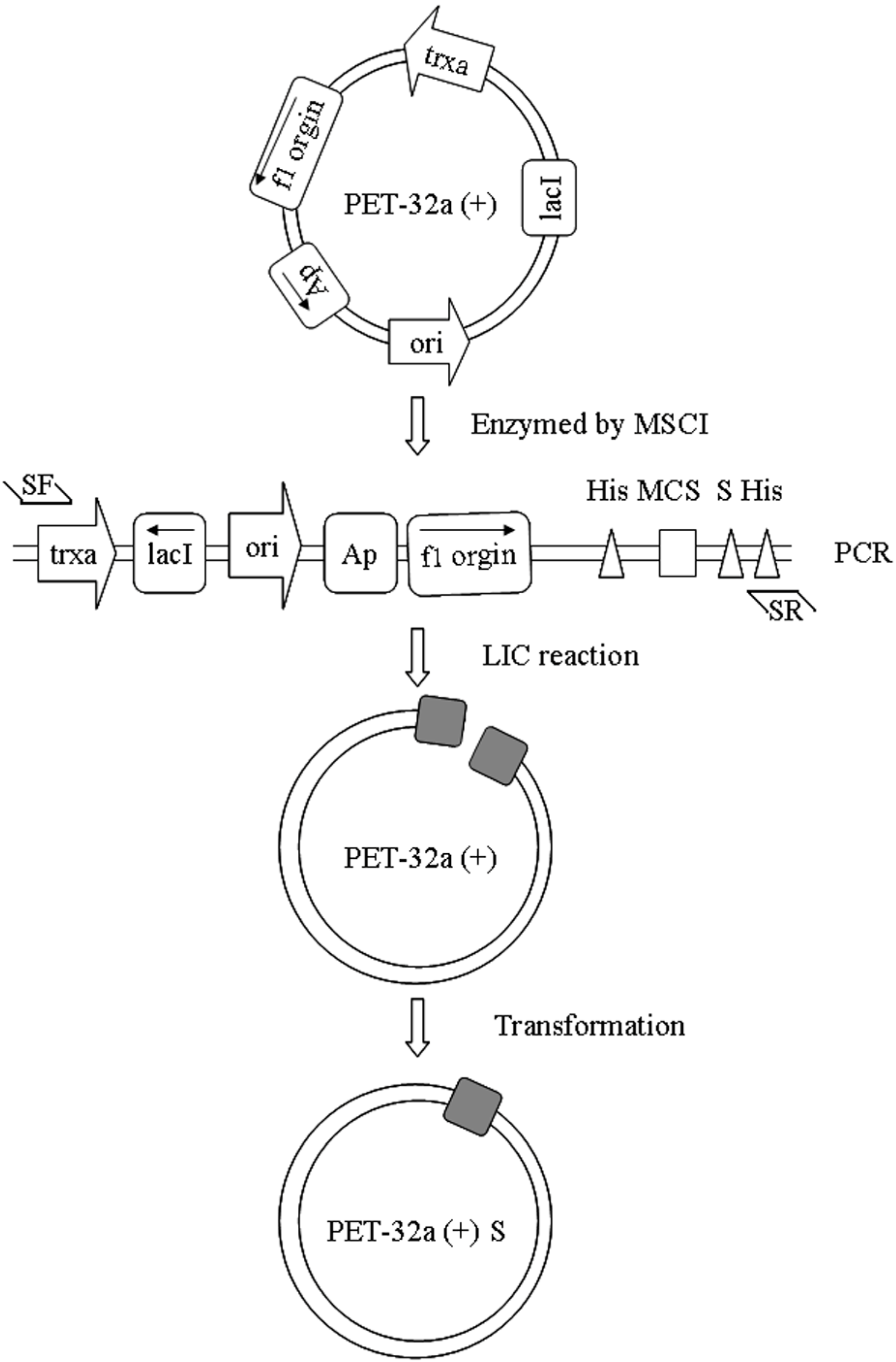
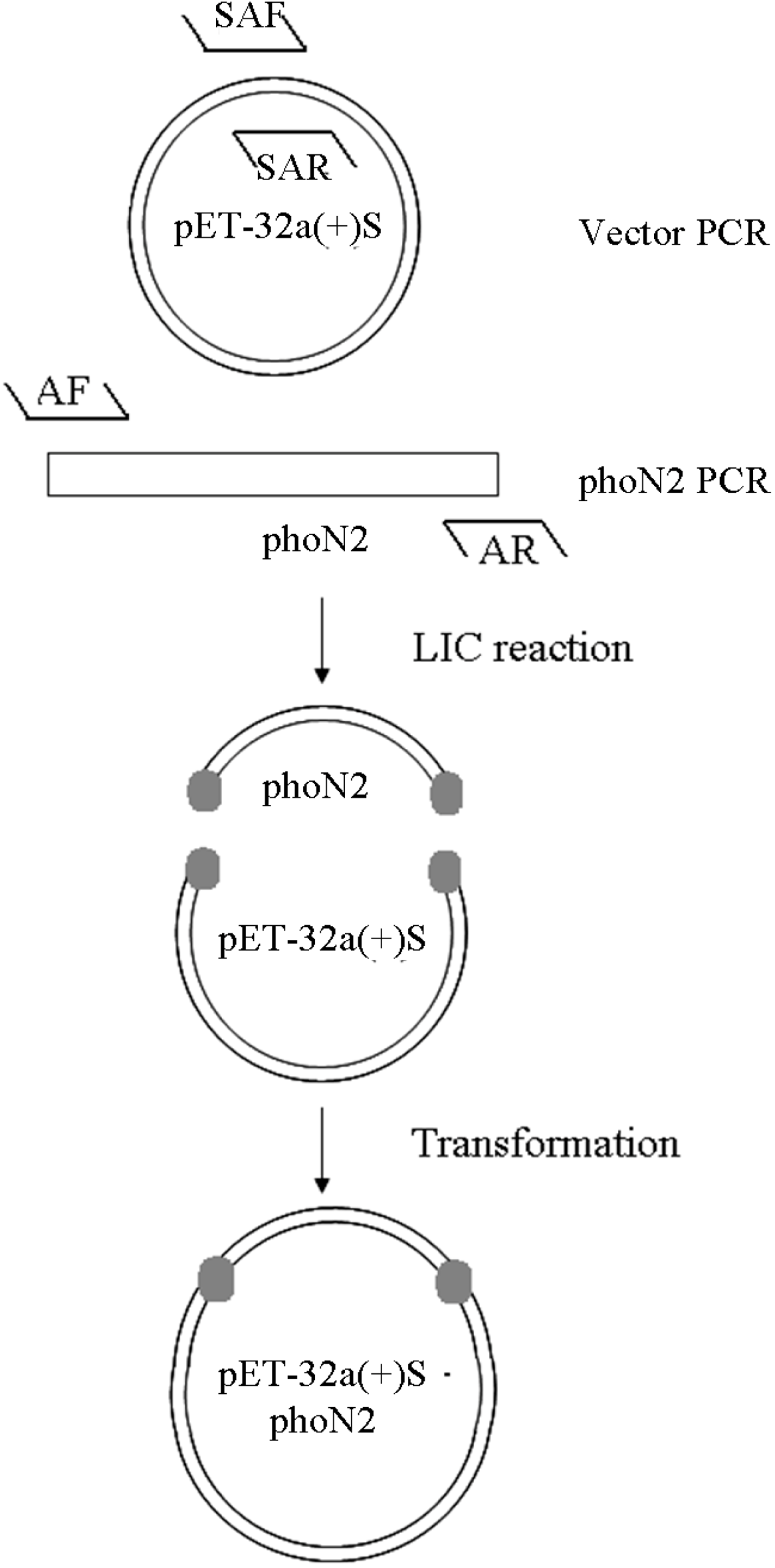
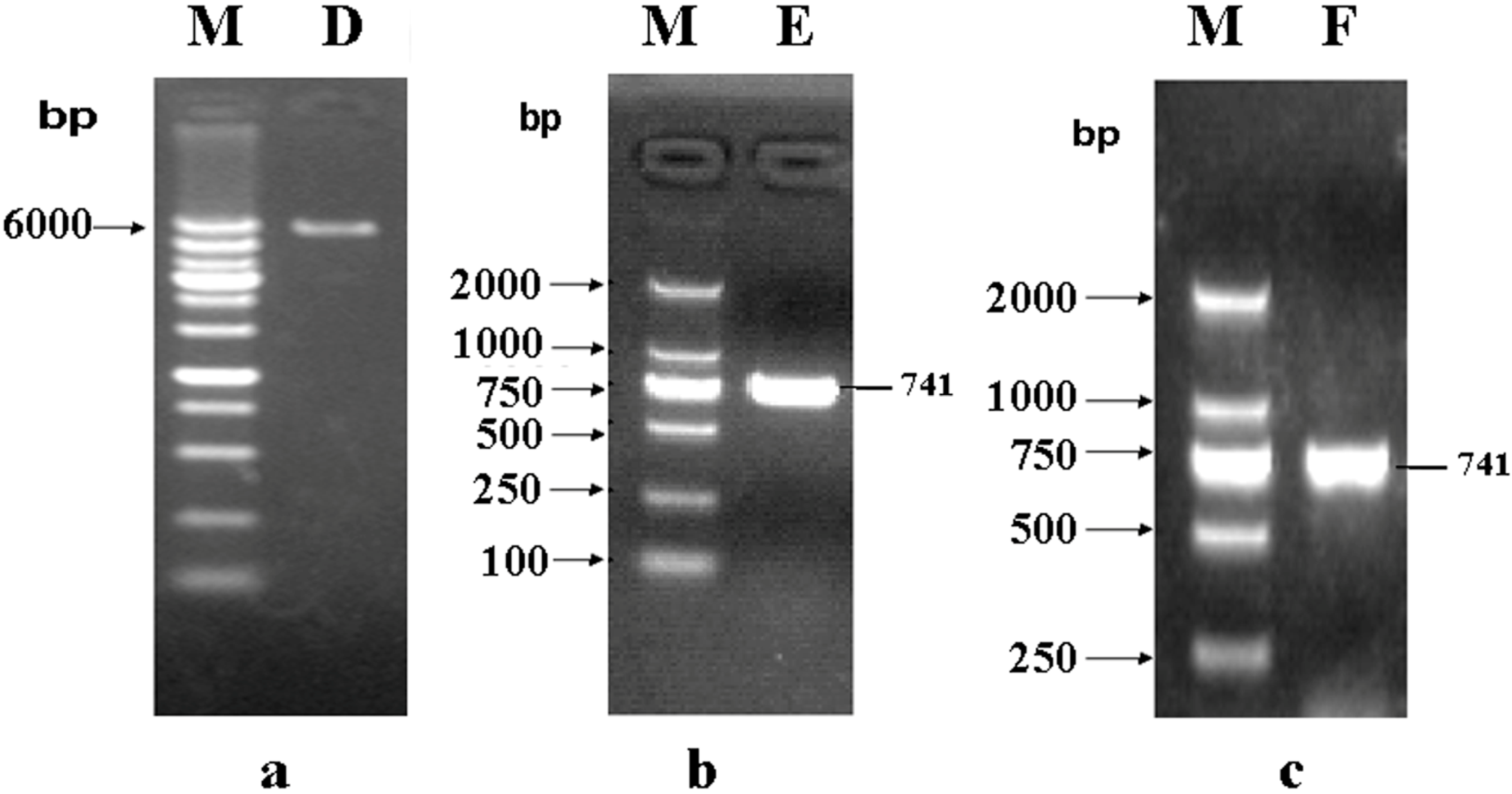
2.1. Construction of a pET32a (+) S Expression Vector
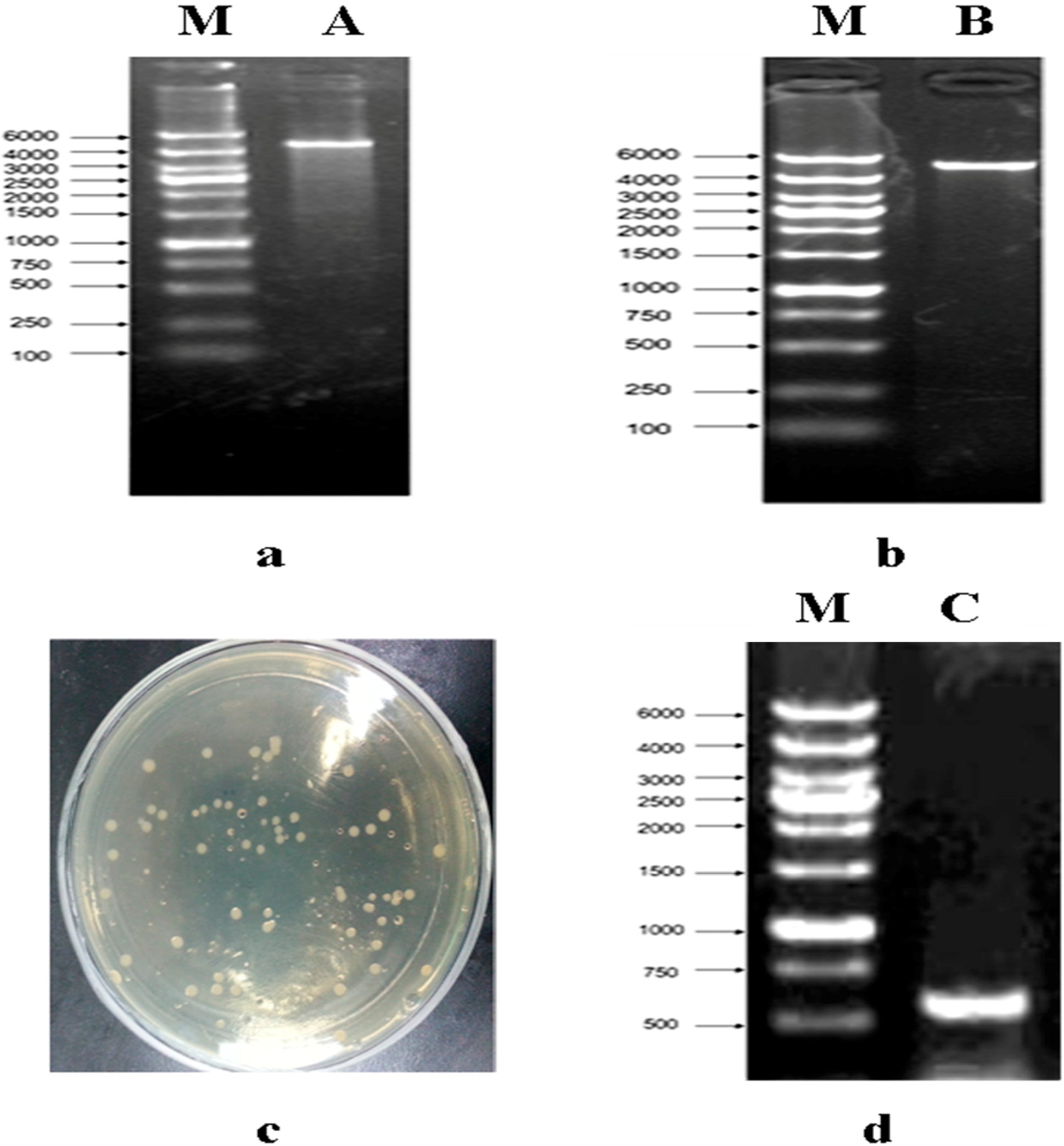

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAA GAC CCG TTT AGA GGC CCC AAG GGG TTA TGC TAG TTA TTG CTC AGC GGT GGC AGC AGC CAA CTC AGC TTC CTT TCG GGC TTT GTT AGC AGC CGG ATC TCA GTG GTG GTG GTG GTG GTG CTC GAG TGC GGC CGC AAG CTT GAC GAC GGA GCT CGA ATT CGG ATC CGA TAT CAG CCA TGG CCT TGT CGT CGT CGT CGG TAC CCA GAT CTG GGC TGT CCA TGT GCT GGC GTT CGA ATT TAG CAG CAG CGG TTT CTT TAT GTA TAT CTC CTT CTT AAA GTT AAA CAA AAT TAT TTC TAG AGG GGA ATT GTT ATC CGC TCA CAA TTC CCC TAT AGT GAG TCG TAT TAA TTT CGC GGG ATC GAG ATC GAT CTC GAT CCT CTA CGC CGG ACG CAT CGT GGC CGG CAT CAC CGG CGC CAC AGG TGC GGT TGC TGG CGC CTAA TAT CGC CGA CAT CAC CGA TGG GGA AGA TCG GGC TCG CCA CTT CGG GCT CAT GAG CGC TTG TTT CGG CGT GGG TAT GGT GGC AGG CCC CGT GGC CGG GGG ACT GTT GGG CGC CAT CTC CTT GCA TGC ACC ATT CCT TGC GGC GGC GGT GCT CAA CGG CCT CAA CCT ACT ACT # |
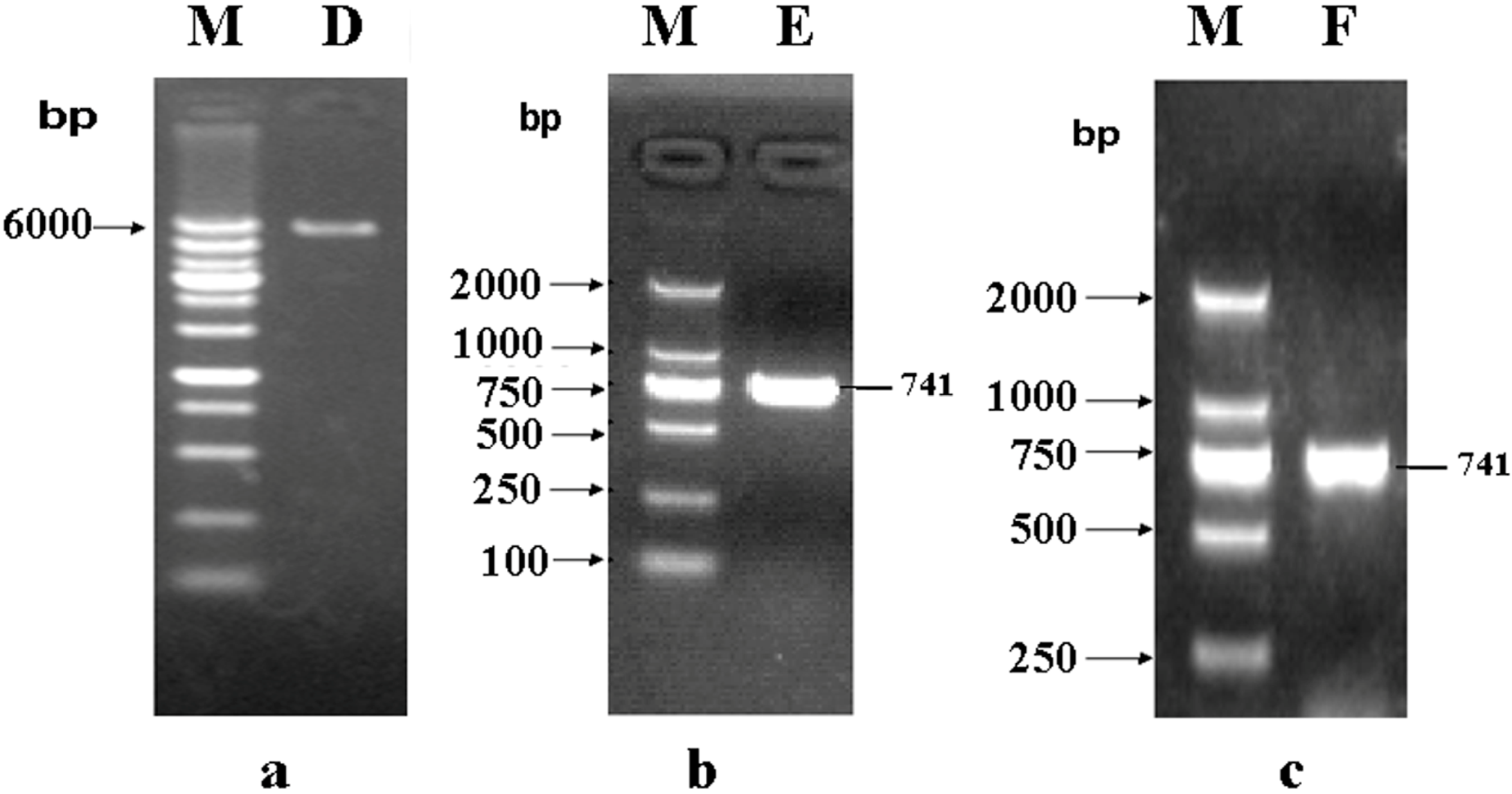
2.2. Construction of a pET32a (+) S-phoN2 Recombinant Plasmid
| ATG | AAA | ACC | AAA | AAC | TTT | CTT | CTT | TTT | TGT | ATT | GCT | ACA | AAT | ATG | ATT | TTT | ATC | CCC | TCA |
| M | K | T | K | N | F | L | L | F | C | I | A | T | N | M | I | F | I | P | S |
| GCA | AAT | GCT | CTG | AAG | GCA | GAA | GGT | TTT | CTC | ACT | CAA | CAA | ACT | TCA | CCA | GAC | AGT | TTG | |
| A | N | A | L | K | A | E | G | F | L | T | Q | Q | T | S | P | D | S | L | |
| TCA | ATA | CTT | CCG | CCG | CCT | CCG | GCA | GAG | GAT | TCA | GTA | GTA | TTT | CTG | GCT | GAC | AAA | GCT | CAT |
| S | I | L | P | P | P | P | A | E | D | S | V | V | F | L | A | D | K | A | H |
| TAT | GAA | TTC | GGC | CGC | TCG | CTC | CGG | GAT | GCT | AAT | CGT | GTA | CGT | CTC | GCT | AGC | GAA | GAT | GCA |
| Y | E | F | G | R | S | L | R | D | A | N | R | V | R | L | A | S | E | D | A |
| TAC | TAC | GAG | AAT | TTT | GGT | CTT | GCA | TTT | TCA | GAT | GCT | TAT | GGC | ATG | GAT | ATT | TCA | AGG | GAA |
| Y | Y | E | N | F | G | L | A | F | S | D | A | Y | G | M | D | I | S | R | E |
| AAT | ACC | CCA | ATC | TTA | TAT | CAG | TTG | TTA | ACA | CAA | GTA | CTA | CAG | GAT | AGC | CAT | GAT | TAC | GCC |
| N | T | P | I | L | Y | Q | L | L | T | Q | V | L | Q | D | S | H | D | Y | A |
| GTG | CGT | AAC | GCC | AAA | GAA | TAT | TAT | AAA | AGA | GTT | CGT | CCA | TTC | GTT | ATT | TAT | AAA | GAC | GCA |
| V | R | N | A | K | E | Y | Y | K | R | V | R | P | F | V | I | Y | K | D | A |
| ACC | TGT | ACA | CCT | GAT | AAA | GAT | GAG | AAA | ATG | GCT | ATC | ACT | GGC | TCT | TAT | CCC | TCT | GGT | |
| T | C | T | P | D | K | D | E | K | M | A | I | T | G | S | Y | P | S | G | |
| CAT | GCA | TCC | TTT | GGT | TGG | GCA | GTA | GCA | CTG | ATA | CTT | GCG | GAG | ATT | AAT | CCT | CAA | CGT | AAA |
| H | A | S | F | G | W | A | V | A | L | I | L | A | E | I | N | P | Q | R | K |
| GCG | GAA | ATA | CTT | CGA | CGT | GGA | TAT | GAG | TTT | GGA | GAA | AGT | CGG | GTC | ATC | TGC | GGT | GCG | |
| A | E | I | L | R | R | G | Y | E | F | G | E | S | R | V | I | C | G | A | |
| CAT | TGG | CAA | AGC | GAT | GTA | GAG | GCT | GGG | CGT | TTA | ATG | GGA | GCA | TCG | GTT | GTT | GCA | GTA | |
| H | W | Q | S | D | V | E | A | G | R | L | M | G | A | S | V | V | A | V | |
| CTT | CAT | AAT | ACA | CCT | GAA | TTT | ACC | AAA | AGC | CTT | AGC | GAA | GCC | AAA | AAA | GAG | TTT | GAA | |
| L | H | N | T | P | E | F | T | K | S | L | S | E | A | K | K | E | F | E | |
| GAA | TTA | AAT | ACT | CCT | ACC | AAT | GAA | CTG | ACC | CCA | TAA | ||||||||
| E | L | N | T | P | T | N | E | L | T | P | R | ||||||||
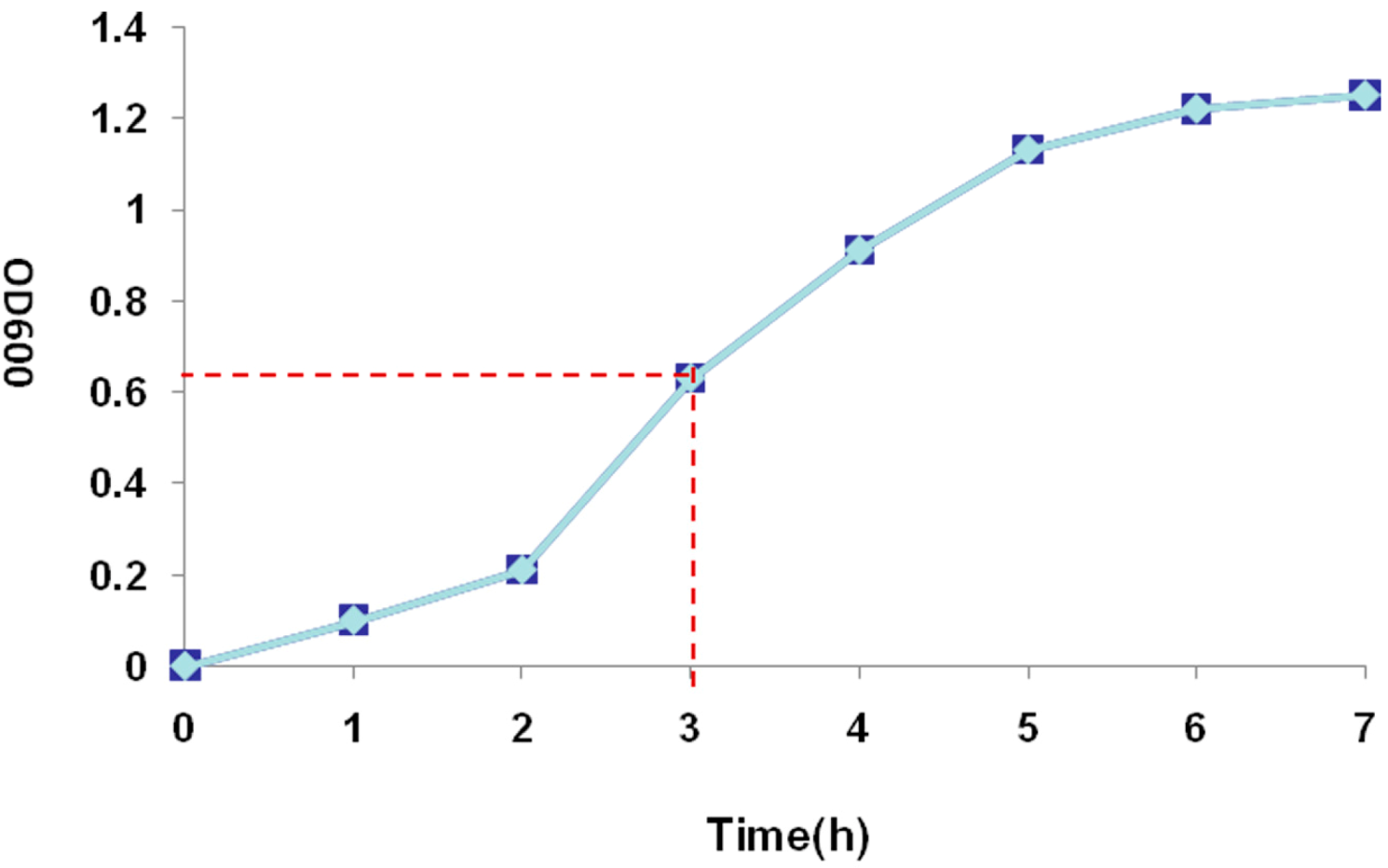
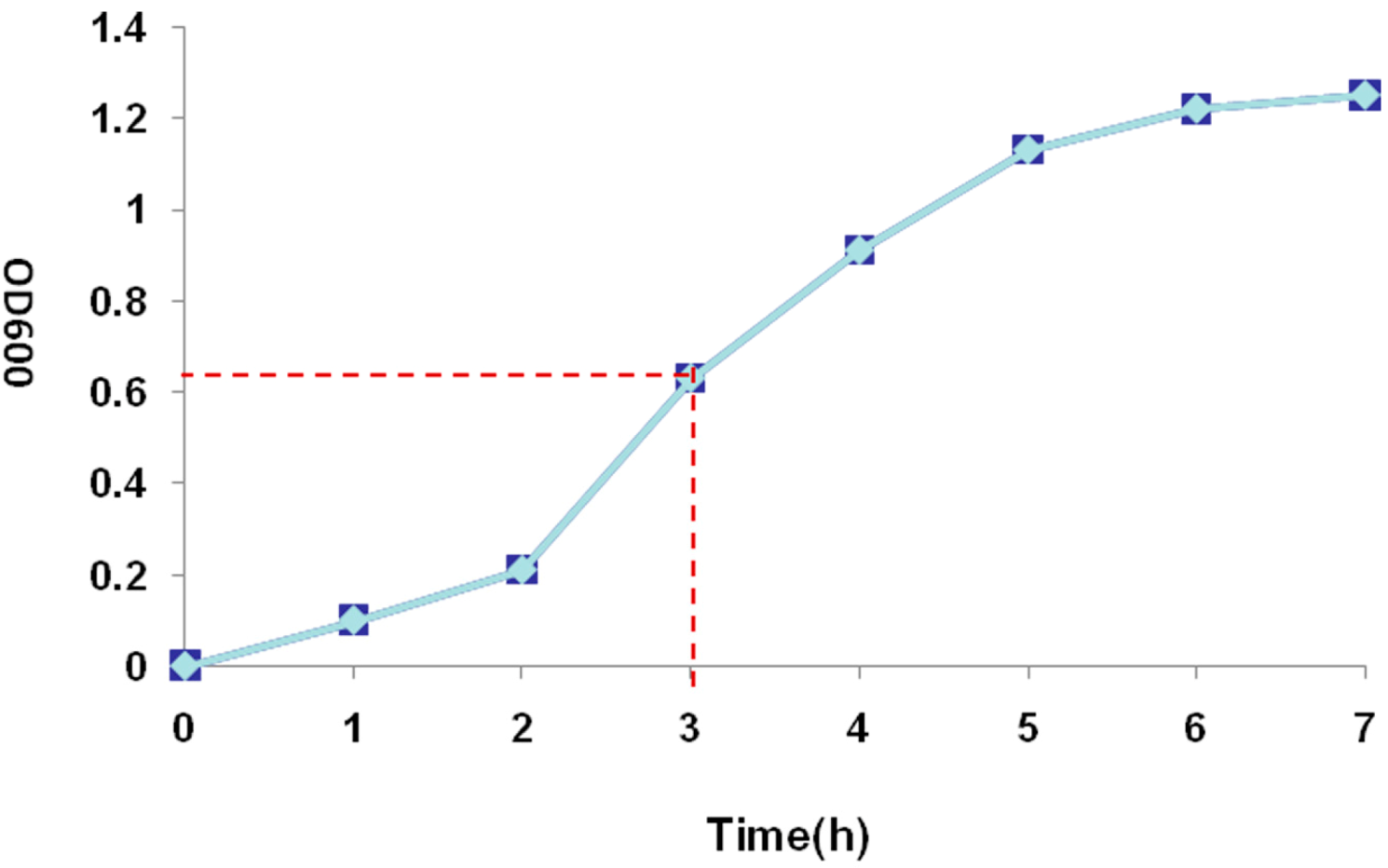
2.3. Recombinant Protein Expression
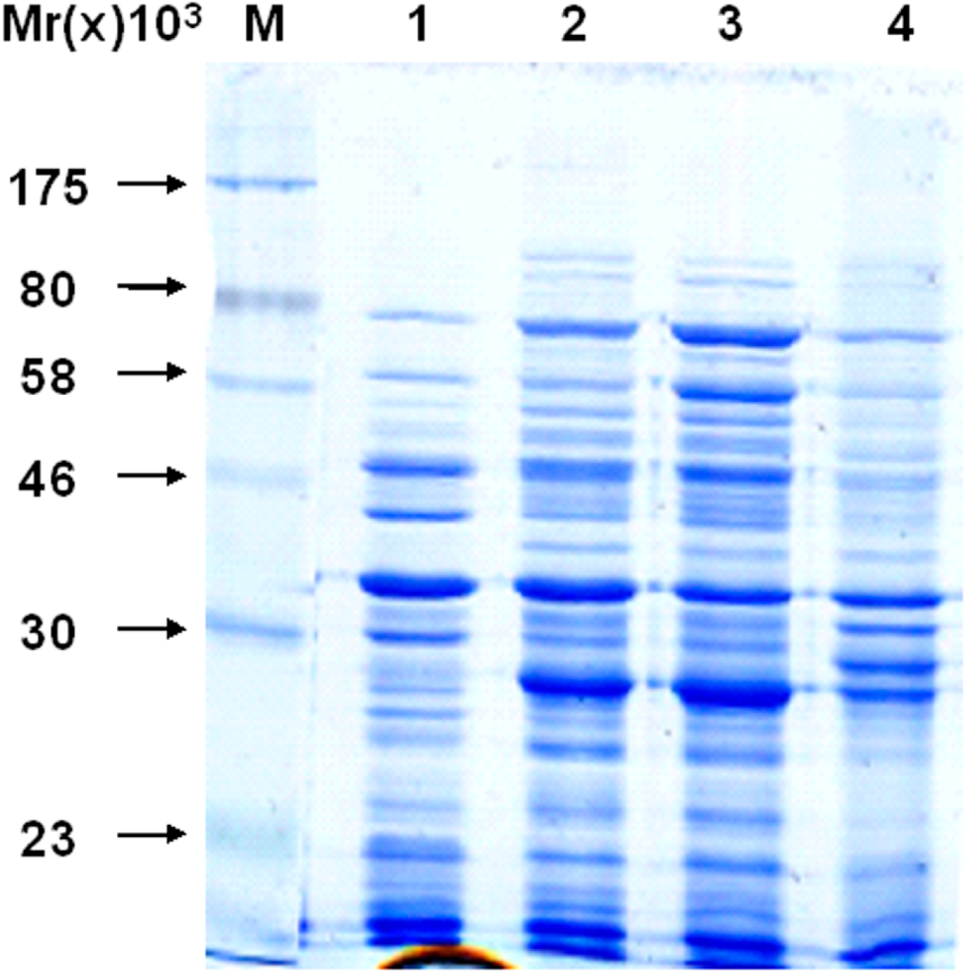
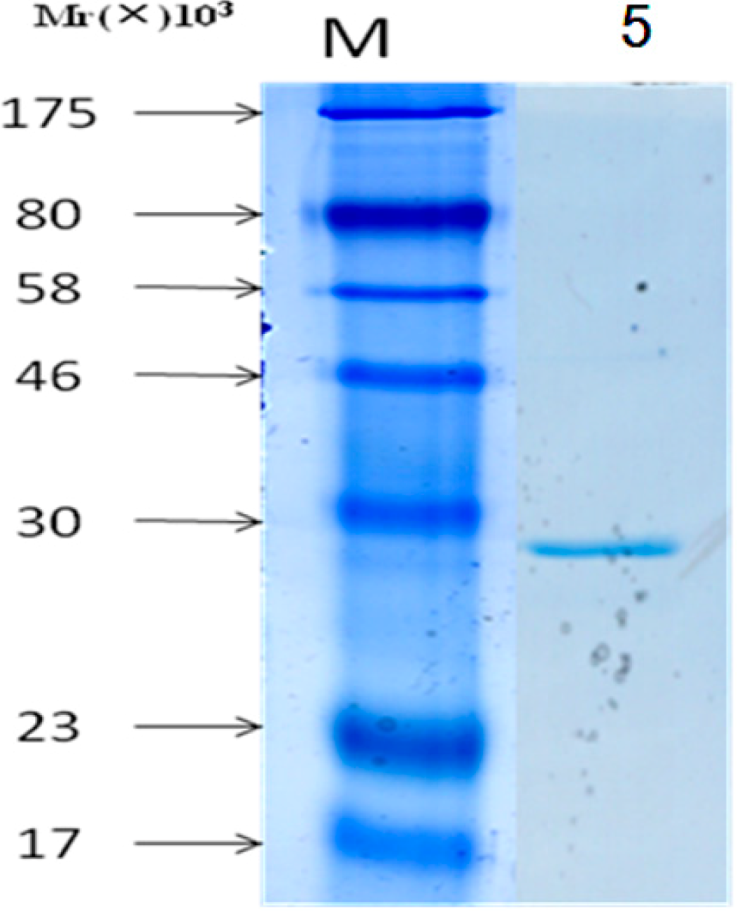
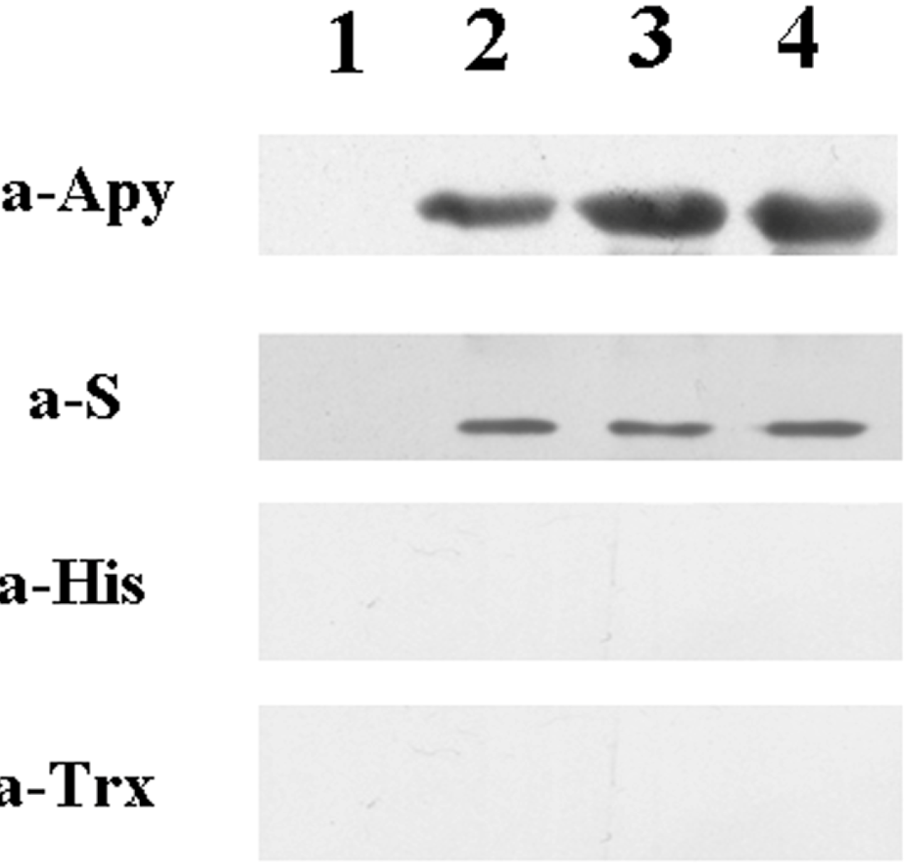
2.4. Detecting of the Specificity of Expressing Bacteria

3. Experimental Section
3.1. Materials
3.2. Vector Construction
- SF: 5' GGTTTCTTTCATATGTATATCTCCTTCTTAAAGTTAAACA 3'
- SR: 5' ATGAAAGAAACCGCTGCTGCTAAATTCG 3'

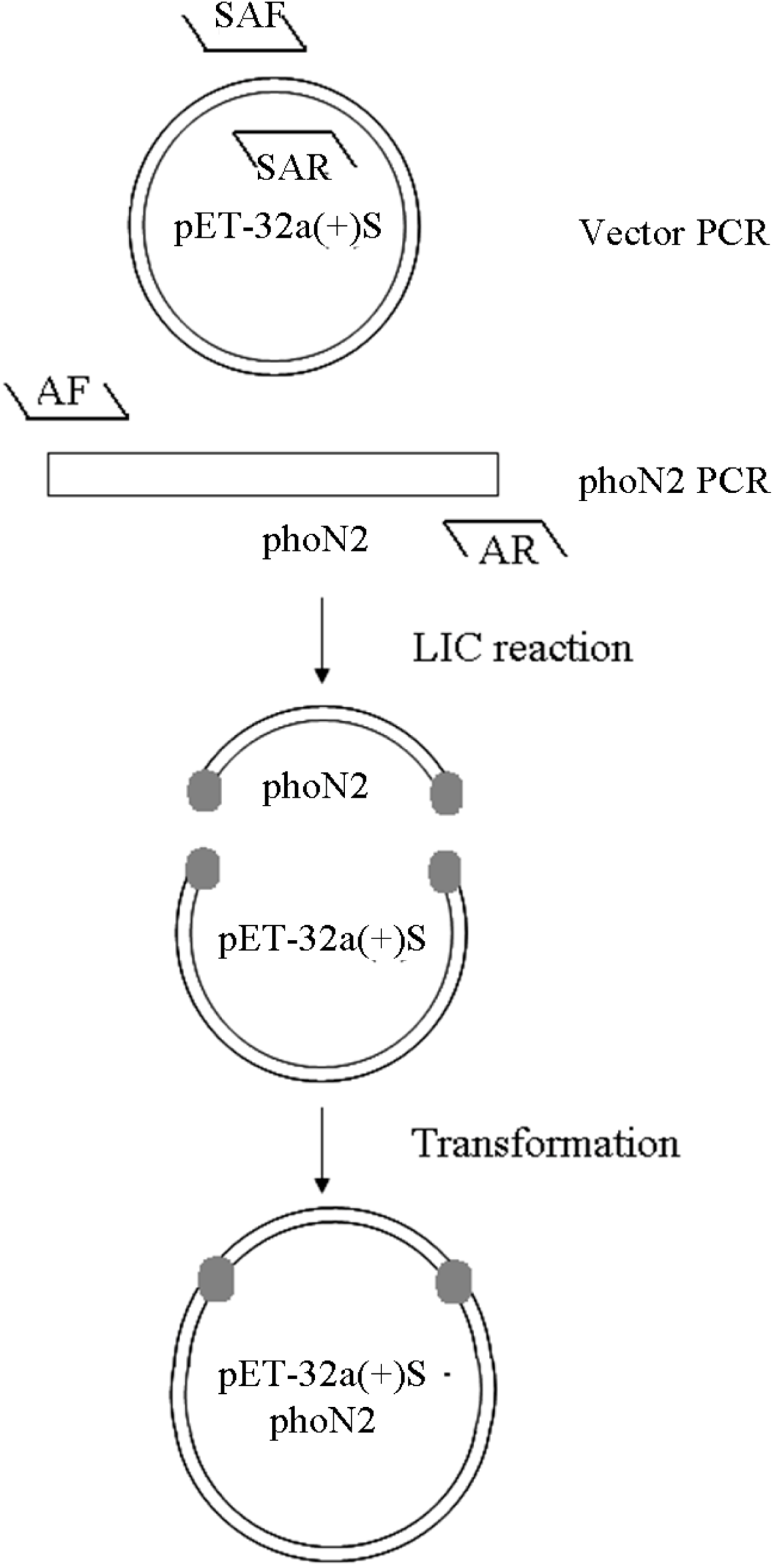
3.3. Construction of pET32a (+) S-Apyrase (Figure 8)
- SAF: 5' GAAAGTTTTTGGTTTTCATGGATCCGATATCAGCCATGG 3'
- SAR: 5' CCAATGAACTGACCCCATAACTCGAGCACCACCACCACCA 3'
3.4. Recombinant Protein Expression
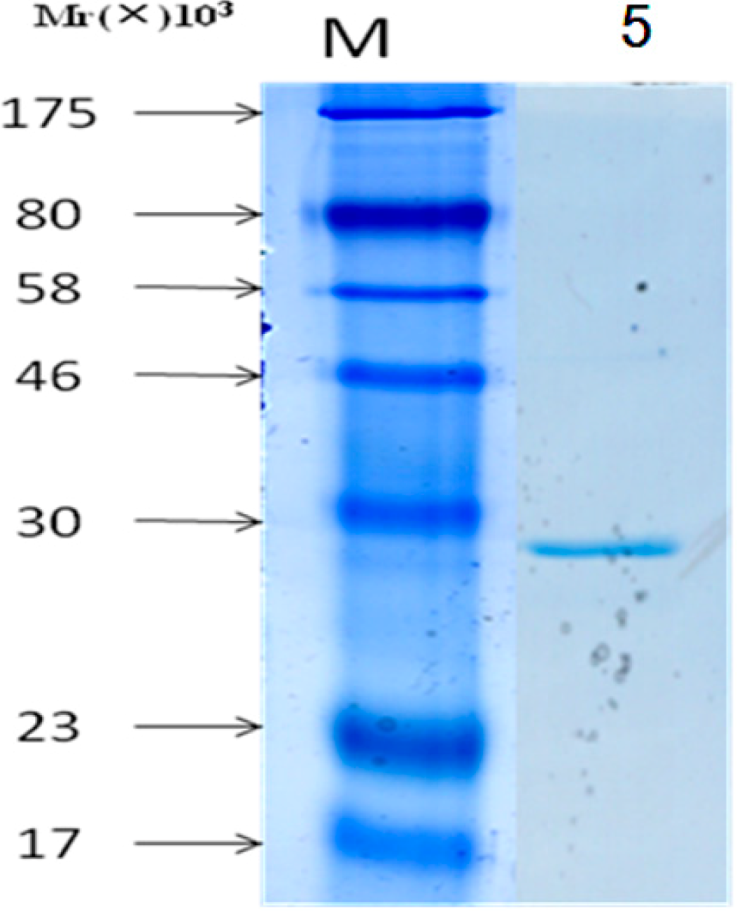
3.5. Purification of Proteins
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tenno, T.; Goda, N.; Tateishi, Y.; Tochio, H.; Mishima, M.; Hayashi, H.; Shirakawa, M.; Hiroaki, H. High-throughput construction method for expression vector of peptides for NMR study suited fo risotopic labeling. Protein Eng. Des. Sel. 2004, 17, 305–314. [Google Scholar]
- Ohsugi, T.; Kumasaka, T.; Urano, T. Construction of a full-length human T cell leukemia virus type I genome from MT-2 cells containing multiple defective proviruses using overlapping polymerase chain reaction. Anal. Biochem. 2004, 329, 281–288. [Google Scholar] [CrossRef]
- Holton, T.A.; Graham, M.W. A simple and efficient method for direct cloning of PCR products using ddT-tailed vectors. Nucleic Acids Res. 1991, 19, 1156. [Google Scholar] [CrossRef]
- Katzen, F. Gateway recombinational cloning: A biological operating system. Expert Opin. Drug Discov. 2007, 2, 571–589. [Google Scholar] [CrossRef]
- Hartley, J.L.; Temple, G.F.; Brasch, M.A. DNA cloning using in vitro site-specific recombination. Genome Res. 2000, 10, 1788–1795. [Google Scholar]
- Cohen, S.N.; Chang, A.C.; Boyer, H.W.; Helling, R.B. Construction of biologically functional bacterial plasmids in vitro. Proc. Natl. Acad. Sci. USA 1973, 70, 3240–3244. [Google Scholar] [CrossRef]
- Haun, R.S.; Serventi, I.M.; Moss, J. Rapid, reliable ligation-independent cloning of PCR products using modified plasmid vectors. Biotechniques 1992, 13, 515–518. [Google Scholar]
- Aslanidis, C.; de Jong, P.J. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 1990, 18, 6069–6074. [Google Scholar] [CrossRef]
- Barry, E.M.; Pasetti, M.F.; Sztein, M.B.; Fasano, A.; Karen, L.; Levine, K.M.M. Progress and pitfalls in Shigella vaccine research. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 245–255. [Google Scholar]
- Haun, R.S.; Moss, J. Ligation-independent cloning of glutathione Stransferase fusion genes for expression in Escherichia coli. Gene 1992, 112, 37–43. [Google Scholar] [CrossRef]
- Stevenson, J.; Krycer, J.R.; Phan, L.; Brown, A.J. A practical comparison of ligation-independent cloning techniques. PLoS One 2013, 8, e83888. [Google Scholar] [CrossRef]
- Weeks, S.D.; Drinker, M.; Loll, P.J. Ligation independent cloning vectors for expression of SUMO fusions. Protein Expr. Purif. 2007, 53, 40–50. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.H. High-throughput T7 LIC vector for introducing Cterminal poly-histidine tags with variable lengths without extra sequences. Protein Expr. Purif. 2009, 63, 58–61. [Google Scholar] [CrossRef]
- Chanda, P.K.; Edris, W.A.; Kennedy, J.D. A set of ligation-independent expression vectors for co-expression of proteins in Escherichia coli. Protein Expr. Purif. 2006, 47, 217–224. [Google Scholar] [CrossRef]
- Qin, H.; Hu, J.; Hua, Y.Z.; Challa, S.V.; Cross, T.A.; Gao, F.P. Construction of a series of vectors for high throughput cloning and expression screening of membrane proteins from Mycobacterium tuberculosis. BMC Biotechnol. 2008, 8, 51. [Google Scholar] [CrossRef]
- Kato, T.; Thompson, J.R.; Park, E.Y. Construction of new ligation-independent cloning vectors for the expression and furifcation of recombinant proteins in silkworms using BmNPV bacmid system. PLoS One 2013, 8, e64007. [Google Scholar] [CrossRef]
- Wang, R.Y.; Shi, Z.-Y.; Guo, Y.-Y.; Chen, J.-C.; Chen, G.-Q. DNA fragments assembly based on nicking enzyme system. PLoS One 2013, 8, e57943. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Gong, G.-H.; Wei, C.-X.; Liang, L.; Zhang, B. Recombinant Expressed Vector pET32a (+) S Constructed by Ligation Independent Cloning. Molecules 2014, 19, 16179-16189. https://doi.org/10.3390/molecules191016179
Wang Y, Gong G-H, Wei C-X, Liang L, Zhang B. Recombinant Expressed Vector pET32a (+) S Constructed by Ligation Independent Cloning. Molecules. 2014; 19(10):16179-16189. https://doi.org/10.3390/molecules191016179
Chicago/Turabian StyleWang, Yu, Guo-Hua Gong, Cheng-Xi Wei, Long Liang, and Bin Zhang. 2014. "Recombinant Expressed Vector pET32a (+) S Constructed by Ligation Independent Cloning" Molecules 19, no. 10: 16179-16189. https://doi.org/10.3390/molecules191016179