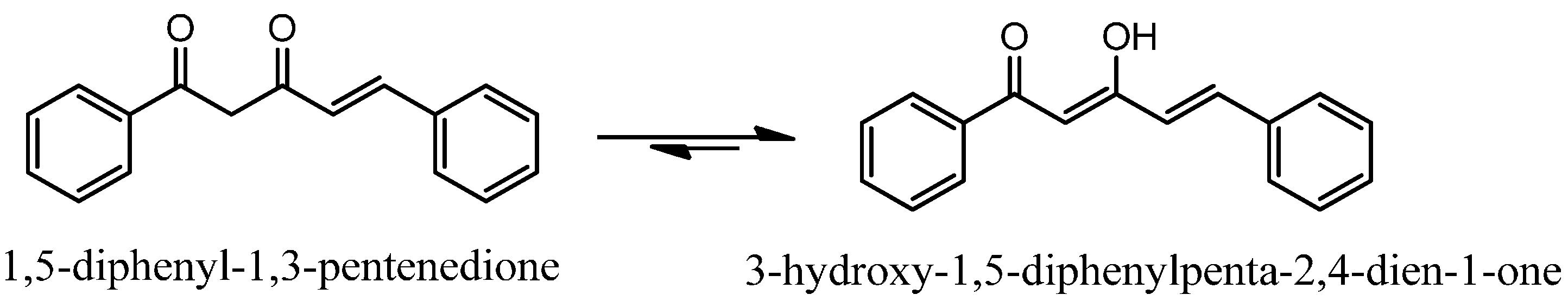
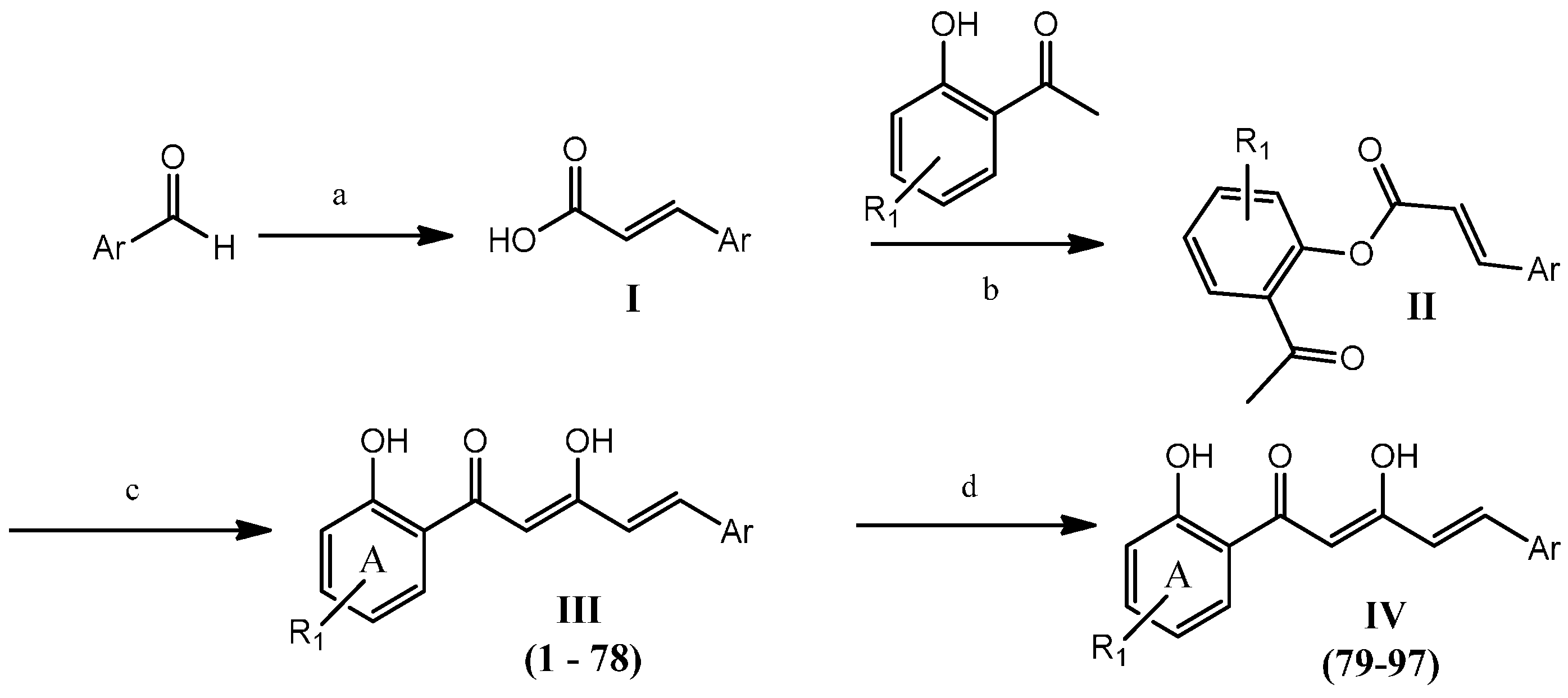
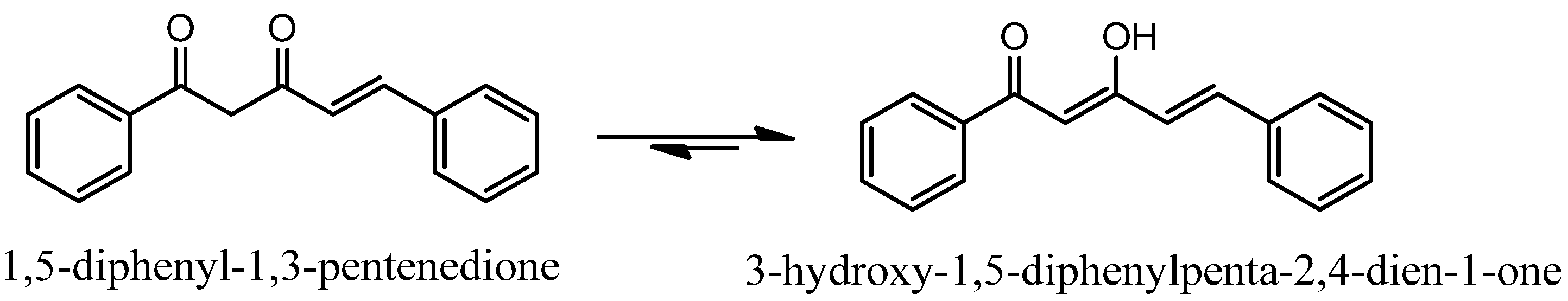
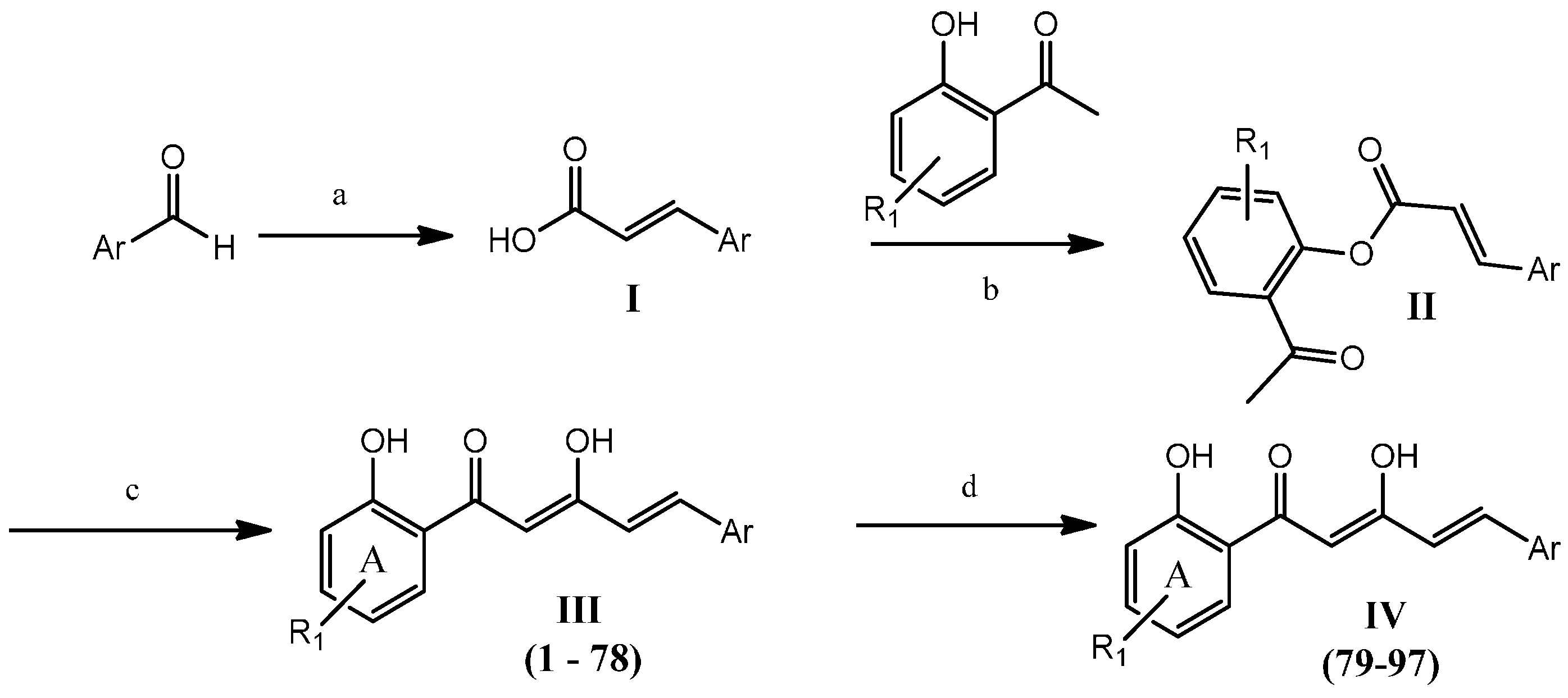
2.1. Chemistry
Synthesis of ninety-seven (
1–
97) analogues of 1,5-diphenyl-1,3-pentenedione was carried out through a series of reactions which included Knoevenagel reaction, phenolic esterification, Baker-Venkataraman rearrangements, and demethylation (
Scheme 1). Eighty-seven of the synthesized compounds are new. The new and known compounds are differentiated by the presence of a reference melting point as presented in
Table S1 in Supplementary Data. The respective commercially available aromatic aldehydes were reacted with malonic acid in the presence of a catalytic amount of piperidine in pyridine, under reflux to afford the respective acrylic acids
I [
24]. Aqueous workup of the crude products afforded the desired compounds without any further purification. All benzaldehydes and naphthaldehydes were successfully converted into the respective acrylic acids with greater than 85% conversion. The presence of an electron withdrawing group in the aromatic rings improved the yield by up to 96%, regardless of their substitution pattern. However, the product yields for the five-membered heterocyclic aldehydes were found to be relatively low (33%–65%) compared with the benzaldehydes and naphthaldehydes. This may be due to the electron-rich character of heterocyclic rings.
Scheme 1.
General synthetic steps for compounds 1–971.
Scheme 1.
General synthetic steps for compounds 1–971.
Notes: 1 Reagents and conditions: (a) malonic acid, pyridine, reflux (4 h); (b) POCl3, pyridine, RT (overnight); (c) KOH, pyridine, RT (overnight); (d) BBr3, CH2Cl2, 0 °C (8 h).
The resulting acrylic acid intermediates were then reacted with the selected 2'-hydroxyacetophenones at room temperature to provide the phenolic esters (
II), by employing phosphoryl chloride as
in situ chlorinating reagent of the cinnamic acid [
25]. Pyridine was chosen as the reaction medium due to its ability to remove the hydrochloric acid produced from the reaction. The desired phenolic esters were obtained in high yields (>90%) regardless of their ring substituents.
The phenolic ester intermediates were subjected to Baker-Venkataraman rearrangements by stirring with potassium hydroxide in pyridine at ambient temperature to produce the desired respective Products III (1–78). The product yields obtained were in the range of 11% to 82%. In general, higher yields were obtained when an electron-deficient aromatic ring was present. Surprisingly, intermediates with a thiofuran moiety, an electron-rich heterocyclic ring, gave better yields (greater than 70%) of the expected products.
Methoxy-containing diarylpentanoids were further demethylated using boron tribromide in dichloromethane at 0 °C to produce polyhydroxylated diarylpentenedione Analogues
IV (
79–
97) [
26]. The yields obtained (15% to 46%) were inversely proportional to the number of methoxy groups present. All the purified diarylpentenediones were characterized by
1H-NMR,
13C-NMR, (
Figure S1 in Supplementary Data) and mass spectrometry. The spectrometric data is presented in the
Table S1 in Supplementary Data. The
1H-NMR spectra of all diarylpentenediones exhibited an intense and sharp singlet at 14–15 ppm, indicative of the chelated hydroxyl groups. It is thus concluded that these compounds are more stable in the keto-enol rather than in their diketo forms. In addition, the large coupling constant (
J) values of the double bond signals (15–16 Hz) indicated that all the compounds existed as the
trans isomer. All purified compounds used for bioassay were of 95% to 99% purity based on their respective high performance liquid chromatography (HPLC) profiles.
2.2. NO Suppression in IFN-γ/LPS-Stimulated Macrophages
The synthesized compounds were screened for NO suppression activity in IFN-γ/LPS-stimulated RAW 264.7 macrophages at a 50 µM test concentration. Fifty-seven compounds were found to significantly inhibit NO production, suggesting that the diarylpentenedione system possesses anti-inflammatory properties and is an interesting candidate for further investigations. The IC
50 values of the fifty-seven bioactive compounds were determined and compared to that of the positive control, curcumin. An MTT assay was also carried out to confirm that NO inhibition was not due to cytotoxicity. Compounds exhibiting significant bioactivities are listed in
Table 1,
Table 2,
Table 3 and
Table 4, grouped according to their functional or structural features. The complete bioassay results of all the compounds are provided as
Table S2 in Supplementary Data.
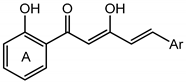
Table 1.
Nitric oxide (NO) suppression activity and cytotoxicity of active compounds in diarylpentenedione series on RAW 264.7 cells.
![Molecules 19 16058 i001]()
Table 1.
Nitric oxide (NO) suppression activity and cytotoxicity of active compounds in diarylpentenedione series on RAW 264.7 cells. ![Molecules 19 16058 i001]()
| Compounds | Ar (Ring B) | NO Inhibition at 50 µM (%) ± S.E.M | NO Inhibition IC50 (µM) ± S.E.M | Cytotoxicity IC50 (µM) ± S.E.M |
|---|
| Curcumin | - | 99.3 ± 0.2 | 14.7 ± 0.2 | 28.8 ± 0.8 |
| 1 | phenyl | 94.7 ± 1.2 | 22.6 ± 0.5 | 56.2 ± 1.1 |
| 2 | 2-chlorophenyl | 91.0 ± 2.9 | 27.3 ± 0.2 | >100 |
| 3 | 3-chlorophenyl | 93.0 ± 3.4 | 26.7 ± 0.7 | >100 |
| 4 | 3-bromophenyl | 88.4 ± 3.2 | 29.4 ± 0.7 | >100 |
| 5 | 2-methoxyphenyl | 91.8 ± 1.5 | 25.7 ± 0.6 | >100 |
| 6 | 3-methoxyphenyl | 81.6 ± 4.0 | 31.6 ± 0.6 | >100 |
| 8 | 3,4-dimethoxyphenyl | 94.7 ± 1.7 | 24.6 ± 0.7 | >100 |
| 9 | 3,4,5-trimethoxyphenyl | 96.6 ± 1.4 | 16.6 ± 1.1 | >100 |
| 12 | furan-2-yl | 82.0 ± 3.2 | 49.0 ± 1.0 | >100 |
| 14 | 2,5-dimethoxyphenyl | 92.9 ± 0.2 | 28.4 ± 0.4 | >100 |
| 16 | thiophen-2-yl | 79.1 ± 4.6 | 42.6 ± 0.1 | >100 |
| 17 | 5-chlorothiophen-2-yl | 63.8 ± 2.6 | 36.4 ± 1.8 | 67.9 ± 2.9 |
| 18 | 5-methylthiophen-2-yl | 58.9 ± 5.8 | 39.6 ± 4.1 | >100 |
| 19 | naphthalen-1-yl | 85.8 ± 3.9 | 23.0 ± 1.7 | >100 |
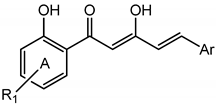
Table 2.
NO suppression activity and cytotoxicity of active compounds in halogenated diarylpentenedione series on RAW 264.7 cells.
![Molecules 19 16058 i002]()
Table 2.
NO suppression activity and cytotoxicity of active compounds in halogenated diarylpentenedione series on RAW 264.7 cells. ![Molecules 19 16058 i002]()
| Compounds | R1 (Ring A) | Ar (Ring B) | NO inhibition at 50 µM (%) ± S.E.M | NO inhibition IC50 (µM) ± S.E.M | Cytotoxicity IC50 (µM) ± S.E.M |
|---|
| 21 | Cl | Phenyl | 85.3 ± 6.8 | 47.6 ± 1.1 | >100 |
| 22 | Cl | 2-chlorophenyl | 82.5 ± 4.3 | 30.0 ± 2.1 | >100 |
| 23 | Cl | 3-chlorophenyl | 66.6 ± 4.1 | 36.2 ± 0.9 | 68.4 ± 2.2 |
| 24 | Cl | 2-methoxyphenyl | 70.4 ± 1.3 | 36.0 ± 0.5 | >100 |
| 25 | Cl | 3-methoxyphenyl | 76.3 ± 6.5 | 17.4 ± 0.4 | >100 |
| 28 | Cl | 3,4,5-trimethoxyphenyl | 89.5 ± 5.9 | 13.6 ± 0.5 | >100 |
| 35 | Cl | thiophen-2-yl | 68.8 ± 4.2 | 32.2 ± 0.2 | >100 |
| 36 | Cl | 5-chlorothiophen-2-yl | 71.6 ± 3.2 | 25.3 ± 1.7 | >100 |
| 37 | Cl | 5-methylthiophen-2-yl | 56.0 ± 2.6 | 62.4 ± 2.3 | >100 |
| 38 | Cl | 5-methylfuran-2-yl | 73.8 ± 1.1 | 25.5 ± 1.0 | >100 |
| 41 | Br | Phenyl | 79.9 ± 8.7 | 20.8 ± 0.5 | >100 |
| 43 | Br | 3-chlorophenyl | 81.2 ± 5.2 | 19.8 ± 0.9 | >100 |
Table 3.
NO suppression activity and cytotoxicity of active compounds in methoxylated diarylpentenedione series on RAW 264.7 cells.
![Molecules 19 16058 i003]()
Table 3.
NO suppression activity and cytotoxicity of active compounds in methoxylated diarylpentenedione series on RAW 264.7 cells. ![Molecules 19 16058 i003]()
| Compounds | R1 (Ring A) | Ar (Ring B) | NO inhibition at 50 µM (%) ± S.E.M | NO inhibition IC50 (µM) ± S.E.M | Cytotoxicity IC50 (µM) ± S.E.M |
|---|
| 45 | 4-OMe | phenyl | 87.9 ± 5.1 | 29.5 ± 0.5 | >100 |
| 46 | 4-OMe | 2-chlorophenyl | 76.2 ± 4.6 | 58.5 ± 2.5 | >100 |
| 48 | 4-OMe | 2-methoxyphenyl | 66.3 ± 6.8 | 39.0 ± 1.2 | 88.6 ± 2.5 |
| 49 | 4-OMe | 3-methoxyphenyl | 57.5 ± 6.5 | 45.8 ± 0.8 | >100 |
| 51 | 4-OMe | 3,4-dimethoxyphenyl | 58.9 ± 2.8 | 21.7 ± 0.5 | >100 |
| 52 | 4-OMe | 3,4,5-trimethoxyphenyl | 96.4 ± 0.4 | 29.3 ± 0.3 | >100 |
| 58 | 5-OMe | phenyl | 92.8 ± 1.8 | 25.9 ± 0.3 | >100 |
| 59 | 5-OMe | 3-chlorophenyl | 89.1 ± 3.6 | 35.7 ± 0.8 | >100 |
| 61 | 5-OMe | 2-methoxyphenyl | 65.8 ± 2.4 | 27.4 ± 0.2 | >100 |
| 62 | 5-OMe | 3-methoxyphenyl | 92.6 ± 1.2 | 35.1 ± 0.2 | >100 |
| 63 | 5-OMe | 3,4-dimethoxyphenyl | 92.2 ± 0.8 | 19.8 ± 0.4 | >100 |
| 64 | 5-OMe | 3,4,5-trimethoxyphenyl | 95.2 ± 1.1 | 18.4 ± 0.2 | >100 |
| 67 | 5-OMe | furan-2-yl | 72.1 ± 3.1 | 71.5 ± 2.5 | >100 |
| 68 | 5-OMe | 2,4-dimethoxyphenyl | 79.5 ± 4.7 | 33.4 ± 0.7 | >100 |
| 71 | 5-OMe | thiophen-2-yl | 85.0 ± 2.7 | 35.6 ± 0.6 | >100 |
Table 4.
NO suppression activity and cytotoxicity of active polyphenolic diarylpentenedione analogues on RAW 264.7 cells.
![Molecules 19 16058 i004]()
Table 4.
NO suppression activity and cytotoxicity of active polyphenolic diarylpentenedione analogues on RAW 264.7 cells. ![Molecules 19 16058 i004]()
| Compounds | R1 (Ring A) | Ar (Ring B) | NO inhibition at 50 µM (%) ± S.E.M | NO inhibition IC50 (µM) ± S.E.M | Cytotoxicity IC50 (µM) ± S.E.M |
|---|
| 79 | H | 2-hydroxyphenyl | 95.5 ± 0.6 | 28.9 ± 1.5 | 56.4 ± 1.2 |
| 80 | H | 3-hydroxyphenyl | 97.2 ± 0.7 | 30.8 ± 0.7 | 67.3 ± 0.6 |
| 81 | H | 4-hydroxyphenyl | 91.5 ± 3.9 | 19.1 ± 0.6 | >100 |
| 83 | H | 2,5-dihydroxyphenyl | 94.3 ± 1.8 | 16.7 ± 0.6 | >100 |
| 84 | 5-Cl | 2-hydroxyphenyl | 95.2 ± 0.9 | 15.9 ± 0.9 | 53.1 ± 1.0 |
| 85 | 5-Cl | 3-hydroxyphenyl | 95.9 ± 0.5 | 26.9 ± 0.8 | 89.1 ± 1.1 |
| 86 | 5-Cl | 4-hydroxyphenyl | 90.3 ± 2.7 | 18.7 ± 0.8 | 66.0 ± 1.2 |
| 87 | 5-Cl | 2,5-dihydroxyphenyl | 96.5 ± 1.4 | 32.3 ± 0.9 | 49.5 ± 1.7 |
| 88 | 5-Cl | 3,4-dihydroxyphenyl | 99.7 ± 1.2 | 4.9 ± 0.3 | 40.9 ± 1.6 |
| 89 | 5-OH | 2-hydroxyphenyl | 83.6 ± 2.0 | 31.0 ± 2.0 | 72.6 ± 2.6 |
| 90 | 5-OH | 3-hydroxyphenyl | 90.9 ± 2.0 | 35.7 ± 0. 9 | 85.0 ± 1.4 |
| 91 | 5-OH | furan-2-yl | 83.6 ± 2.8 | 75.0 ± 4.9 | 94.1 ±0.9 |
| 93 | 4-OH | phenyl | 94.7 ± 2.3 | 29.9 ± 0.4 | 59.5 ± 0.5 |
| 94 | 4-OH | 3-hydroxyphenyl | 95.4 ± 1.5 | 24.8 ± 0.4 | 70.1 ± 1.8 |
| 95 | 4-OH | thiophen-2-yl | 60.4 ± 2.0 | 44.8 ± 1.6 | >100 |
| 97 | H | 3,4-dihydroxyphenyl | 98.9 ± 1.6 | 9.6 ± 0.5 | >100 |
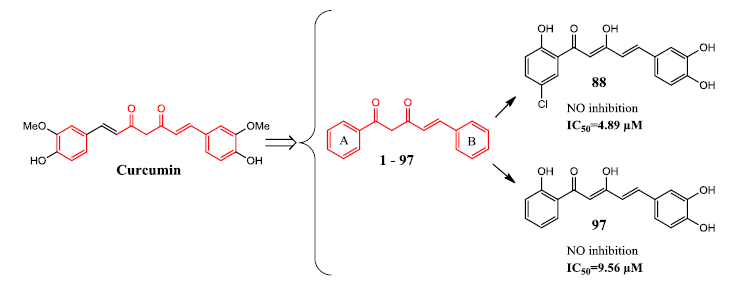
Two compounds (88 and 97) were found to exhibit the most potent anti-inflammatory activity, giving IC50 values of 4.9 μM and 9.6 μM, respectively. Meanwhile ten other compounds (9, 25, 28, 43, 63, 64, 81, 83, 84, and 86) exhibited comparable NO inhibitory activity to curcumin, with IC50 values of less than 20 µM. Based on these results, it appears that polyphenolic and poly(methoxyphenyl) moieties are important for NO suppression activity. The presence of a catechol moiety, as represented by compounds 88 and 97, appeared to be an important contributing factor since it resulted in a two- to four-fold improvement in bioactivity.
This observation is consistent with previous reports in which it was shown that the presence of a catechol moiety in flavonoids [
27], curcuminoids [
28], and aurones [
29] enhanced the NO inhibition activity significantly. Furthermore, it has also been reported that catechol moiety containing compounds are frequently bioactive against various inflammatory enzymes, such as COX-2 and iNOS, through inhibition of NF-κB activation [
30]. Notably, the unexpected synergistic effect of catechol and α,β-unsaturated carbonyl moieties on NF-κB inactivation was presented by Chiang and co-workers [
31]. On this account, compounds
88 and
97, the catechol and α,β-unsaturated carbonyl moieties containing diarylpentenediones, may represent new candidates for further investigation of their effects towards inflammatory mediators including the NF-κB (LPS-induced pathway) and Jak-STAT (IFN-γ-induced pathway) inactivation analysis and direct modulation of iNOS.
Comparisons between compounds within the same and between different groups were conducted to identify correlations between the structural features with NO suppression activity. For the diarylpentenedione series (
Table 1), the trimethoxylated compound
9, exhibited highest NO suppression which suggests that high electron density in ring-B is an important factor in improving NO inhibition. A similar trend was also observed for the halogenated and methoxylated diarylpentenedione series listed in
Table 2 and
Table 3, respectively. In the respective groups, the trimethoxylated compounds
28 and
64 exhibited the highest NO suppression. In contrast, analogues with low electron density ring-B, as in the dihalogenated diaryl analogues (
15,
37 and
70), further supported our conclusion.
Further structure-activity comparison between all the series of analogues showed that low electron density of ring-A was important in enhancing the NO inhibition activity. This was clearly demonstrated by compounds 6, 25, 49, and 62, in which the presence of an electron donating group (methoxy) in ring-A was accompanied by reduced NO suppression, while the presence of an electron withdrawing group (chloro) enhanced the bioactivity. The same conclusion could also be drawn based on the bioactivity displayed by the Analogues 9, 28, 52 and 64, where the diarylpentenedione analogue with halogenated ring-A (28) exhibited the highest activity. In contrast, the methoxylated compounds 49 and 52 exhibited the weakest activity. Further comparison between the Analogues 23, 25, 59 and 62 further supported the electron density-related effect.
Demethylation of methoxylated diarylpentenediones to form the hydroxylated analogues increased the bioactivity significantly. Comparison of compounds
7 (See
Table 2 in Supplementary Data),
14 and
27 with their respective demethylated analogues (compounds
81,
83 and
88) showed that NO inhibition improved very significantly consistent with the decrease in the electron density of ring-B. This could be rationalized by the higher affinity for hydrogen bonding by the hydroxyl groups as compared to methoxyl. This observation suggested that hydrogen bonding is an additional contributing factor for the bioactivity enhancement along with electron density. Thus, the polyphenolic analogues, with their higher hydrogen bonding capacity, are the more potent group in this class of compounds.
Apart from this, it has also been found that the substitution position of functionalities at ring-B plays a pivotal role in influencing the NO inhibitory activity. The comparison of compounds 79, 80, 81, 82, 83 and 97 shows that the substitution of hydroxyl groups at both meta- and para-positions is essential for bioactive molecules as compound 97, a meta- and para-hydroxylated analogue has displayed much better activity than any other mono- or di-hydroxylated derivative. The suggested trend was further supported by similar observations made in the comparisons of compounds 84, 85, 86, 87 and 88 of which compound 88 with di-substitution at both meta- and para-positions possessed the highest activity. Interestingly, the same conclusion can also been drawn from the comparisons of poly(methoxyphenyl) containing analogues. Compounds with a meta- and para-dimethoxylated phenyl ring (8, 9, 28, 51, 52, 63 and 64) exhibited significantly better activity than those with other substitution patterns. Thus, it is confirmed that the substitution at both meta- and para-positions contributes to NO inhibitory activity. In contrast, replacing the aryl ring-B with heterocyclic aromatic rings such as thiophene and furan (see compounds 16 and 31) was found to be undesirable. Although higher in electron density, it dramatically decreased NO suppression activity.
2.3. Quantitative Structure Activity Relationship (QSAR) Analysis
Quantitative structure activity relationship (QSAR) analysis is a statistical approach commonly used to explore, explain, and rationalize the significant correlation between the experimental biological activity or chemical reactivity of a series of drugs with their molecular geometry and physicochemical properties. Currently, there are six types of QSAR models including 1D, 2D, 3D, 4D, 5D, and 6D QSAR with 2D and 3D QSAR being the most commonly used models. In the present study, 2D and 3D QSAR analyses were employed to understand the physicochemical properties and molecular descriptors or features, which could contribute to the NO suppression activity.
2.3.1. 2D-QSAR
Genetic function approximation (GFA) analysis is one of the most common algorithms used in QSAR models. Sivakumar and co-workers reported that GFA analysis had been successfully applied to study the anti-tuberculosis properties of selected chalcones and flavonoids with excellent predictive models [
32]. The r
2 (conventional correlation coefficient) and q
2 (cross-validation correlation coefficient) values achieved by the group were between 0.85 to 0.97 and, between 0.79 to 0.94, respectively. These values imply that GFA analysis is very accurate in predicting the bioactivity of small molecules. Thus, in this study, GFA was selected as the analytical method to establish a 2D QSAR model for the anti-inflammatory activity of diarylpentenedione system as a family of small molecules.
GFA analysis was carried out by using Discovery Studio 3.1 based on thermodynamic, constitutional, and topological descriptors including ALogP (lipophilicity), EPFP_6 (fingerprint), number of hydrogen donors and molecular fractional polar surface area (MFPSA) where pIC
50 (−log IC
50) is the dependent variable. GFA equation represented the best equation generated by GFA analysis and it was further evaluated with randomization test to ensure its reliability as presented in
Table 5.
GFA equation:
Parameter values generated: n = 57; r
2 = 0.8639; r
2 (adj) = 0.8432; r
2 (pred) = 0.8152; q
2 = 0.558; RMS Residual Error = 0.07841; Friedman L.O.F. = 0.01223
where: n = number of compounds involve in analysis; r
2 = coefficient of determination; r
2 (adj) = r
2 adjusted = the number of terms in the model; r
2 (pred) = prediction (PRESS) r
2, and equivalent to q
2 from a leave-1-out cross-validation; q
2 = cross-validation correlation coefficient; RMS = root mean square residual error; Friedman L.O.F. = Friedman lack-of-fit score.
Table 5.
Randomization test of genetic function approximation (GFA) equation.
Table 5.
Randomization test of genetic function approximation (GFA) equation.
| Eq. No. | (1) |
|---|
| r2 from nonrandom model | 0.8639 |
| Confidence level | 90% | 95% | 98% |
| Total trials | 9 | 19 | 49 |
| Nonrandom r2 < random r2 | 0 | 0 | 0 |
| Mean value of r2 from random trials | 0.105 | 0.129 | 0.138 |
| Standard deviation | ±0.070 | ±0.121 | ±0.131 |
Analysis based on this equation found the r
2 value to be 0.8639 with low Friedman L.O.F value of 0.01223, thus indicating that the model is acceptable. The Friedman L.O.F value is a measurement that estimates the most appropriate number of features, resists overfitting, and allows control over the smoothness of fit. The lower the value of Friedman L.O.F, the less likely it is that the GFA model is overfitting the data, thus the more reliable is the model. Further randomization test at 90%, 95%, and 98% confidence level were performed to confirm the equation’s reliability. The randomization was done by repeatedly permuting the activity values of the training set to obtain subsequent GFA scores. If the score of the original GFA model is better than those from the permuted data sets, the model is considered statistically significant. All equations using the scrambled data notably displayed lower r
2 values (
Table 5) compared to the non-random model, which increased the significance and confidence in the training compounds and suggested that the descriptors used were highly selective.
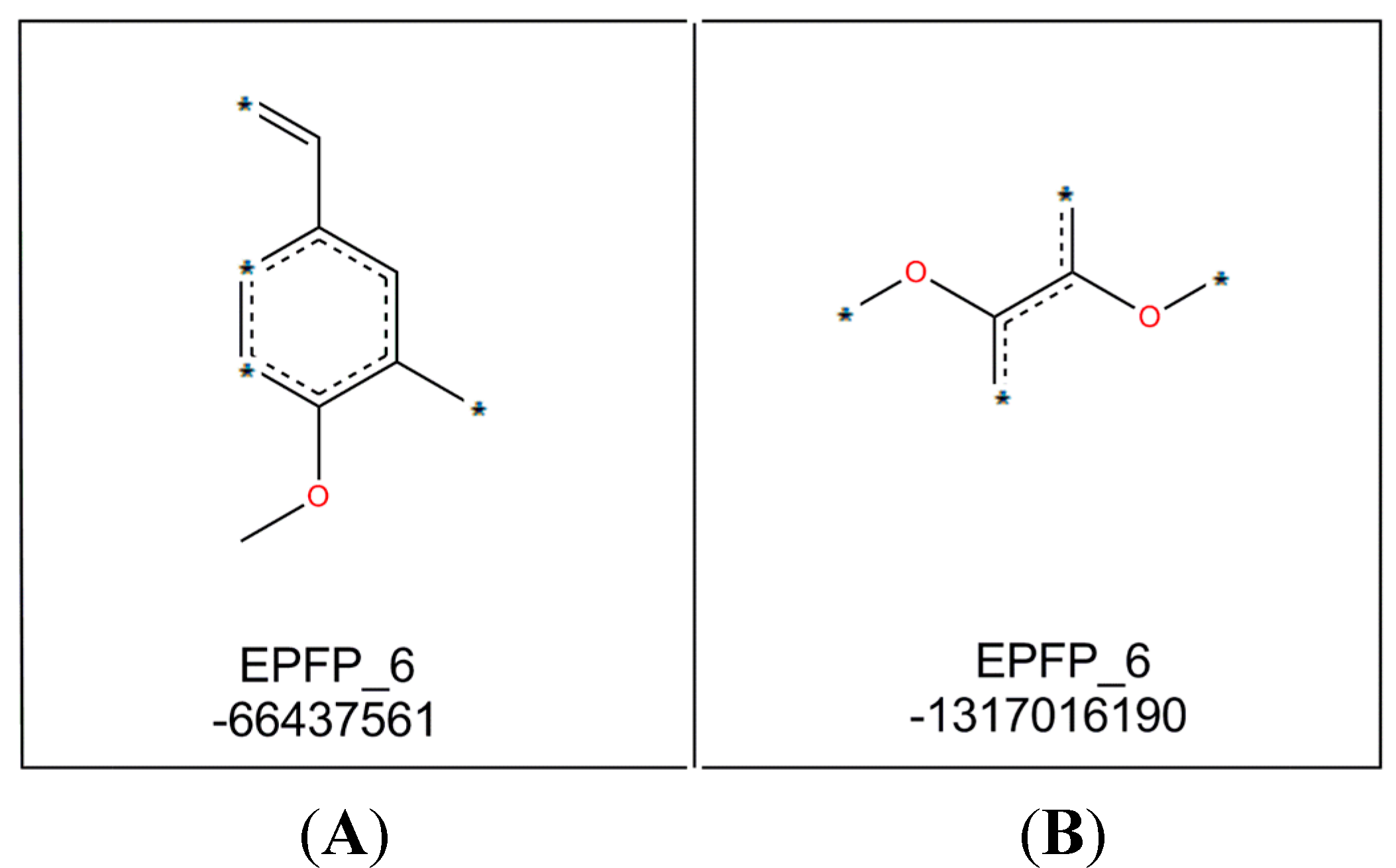
As stated in the GFA equation, two important molecular fragments (as shown in
Figure 3) were determined by this model. Molecular fragment B (EPFP_6: −1317016190) is important for activity of compounds while molecular fragment A (EPFP_6: −66437561) tend to reduce it.
Figure 3.
Molecular fragments generated by GFA analysis.
Figure 3.
Molecular fragments generated by GFA analysis.
The positive effects of fragment B may be explained by comparing the structures and bioactivities of compounds 7–9, which indicated that a higher number of methoxyl groups in the ring increases the NO suppression activity of the diarylpentenedione system. This trend is also observed for the Analogues 62, 63 and 64 where bioactivity for the 3,4,5-trimethoxylated compound 64 was the most active. The significant role of fragment B was also revealed when by the bioactivities of Analogues 83 and 87 were compared to those of 88 and 97. The ortho-dihydroxylated compounds 88 and 97 were seven- and two-fold better inhibitors of NO production than their para-hydroxylated counterparts 87 and 83, respectively, which indicated that the presence of the group was much more important than non-adjacent dihydroxylated analogues.
The decrease in NO inhibition due to the presence of molecular fragment A may be represented by the analogues containing a single methoxyl group, especially that with p-methoxylated phenyl ring. This trend is clearly demonstrated by compounds 5–7, of which compound 7 with p-methoxy group in ring-B exhibited the weakest activity. A similar trend is also detected in compounds 45–56, where the NO inhibition of these p-methoxylated ring-A containing analogues are generally weaker.
The number of H-donors present in the molecule is also an important factor influencing the activity. GFA analysis suggested that a higher number of H-donors present in the diarylpentenedione system increases the bioactivity of the compounds. This correlation was clearly demonstrated by comparing the bioactivities of Analogues 21, 86 and 88, which showed that the increase in the number of hydrogen donors led to higher NO inhibition. The same trend was also shown by the analogue Series 1, 80 and 97.
Apart from number of H-donors, AlogP and MFPSA are two other important factors influencing bioactivity of the analogues. AlogP represents lipophilicity of the respective compound while MFPSA represents the fraction of polar surface to total areas. Based on the GFA equation (1), higher lipophilicity and lower MFPSA are preferable for potency of the compounds. Thus, the analogues containing a heterocyclic aromatic ring were found to gradually decrease in bioactivity due to their lower lipophilicity and higher MFPSA characteristics. All of the calculated parameters are listed in
Table 6.
Table 6.
IC50, pIC50, ALogP, number of hydrogen donors and molecular fractional polar surface area (MFPSA) values of selected active compounds.
Table 6.
IC50, pIC50, ALogP, number of hydrogen donors and molecular fractional polar surface area (MFPSA) values of selected active compounds.
| Compounds | IC50 | pIC50 | ALogP | Number of Hydrogen Donors | MFPSA |
|---|
| 1 | 22.6 ± 0.5 | 4.65 | 3.4 | 2 | 0.2 |
| 2 | 27.3 ± 0.2 | 4.56 | 4.1 | 2 | 0.2 |
| 3 | 26.7 ± 0.7 | 4.57 | 4.1 | 2 | 0.2 |
| 4 | 29.4 ± 0.7 | 4.53 | 4.1 | 2 | 0.2 |
| 5 | 25.7 ± 0.6 | 4.59 | 3.4 | 2 | 0.2 |
| 6 | 31.6 ± 0.6 | 4.5 | 3.4 | 2 | 0.2 |
| 8 | 24.6 ± 0.7 | 4.61 | 3.4 | 2 | 0.2 |
| 9 | 16.6 ± 1.1 | 4.78 | 3.3 | 2 | 0.2 |
| 12 | 49.0 ± 1.0 | 4.31 | 2.8 | 2 | 0.3 |
| 14 | 28.4 ± 0.4 | 4.55 | 3.4 | 2 | 0.2 |
| 16 | 42.6 ± 0.1 | 4.37 | 3.3 | 2 | 0.3 |
| 17 | 36.4 ± 1.8 | 4.44 | 3.8 | 2 | 0.3 |
| 18 | 39.6 ± 4.1 | 4.4 | 3.5 | 2 | 0.3 |
| 19 | 23.0 ± 1.7 | 4.64 | 4.3 | 2 | 0.2 |
| 21 | 47.6 ± 1.1 | 4.32 | 4.1 | 2 | 0.2 |
| 22 | 30.0 ± 2.1 | 4.52 | 4.7 | 2 | 0.2 |
| 23 | 36.2 ± 0.9 | 4.44 | 4.7 | 2 | 0.2 |
| 24 | 36.0 ± 0.5 | 4.44 | 4.0 | 2 | 0.2 |
| 25 | 17.4 ± 0.4 | 4.76 | 4.0 | 2 | 0.2 |
| 28 | 13.6 ± 0.5 | 4.87 | 4.0 | 2 | 0.2 |
| 35 | 32.2 ± 0.2 | 4.49 | 4.0 | 2 | 0.3 |
| 36 | 25.3 ± 1.7 | 4.6 | 4.5 | 2 | 0.3 |
| 37 | 62.4 ± 2.3 | 4.2 | 4.2 | 2 | 0.3 |
| 38 | 25.5 ± 1.0 | 4.59 | 3.6 | 2 | 0.2 |
| 41 | 20.8 ± 0.5 | 4.68 | 4.1 | 2 | 0.2 |
| 43 | 19.8 ± 0.9 | 4.7 | 4.8 | 2 | 0.2 |
| 45 | 29.5 ± 0.5 | 4.53 | 3.4 | 2 | 0.2 |
| 46 | 58.5 ± 2.4 | 4.23 | 4.0 | 2 | 0.2 |
| 48 | 39.0 ± 1.2 | 4.41 | 3.4 | 2 | 0.2 |
| 49 | 45.8 ± 0.8 | 4.34 | 3.4 | 2 | 0.2 |
| 51 | 21.7 ± 0.5 | 4.66 | 3.3 | 2 | 0.2 |
| 52 | 29.3 ± 0.3 | 4.53 | 3.3 | 2 | 0.2 |
| 58 | 25.9 ± 0.3 | 4.59 | 3.4 | 2 | 0.2 |
| 59 | 35.7 ± 0.8 | 4.45 | 4.0 | 2 | 0.2 |
| 61 | 27.4 ± 0.2 | 4.56 | 3.4 | 2 | 0.2 |
| 62 | 35.1 ± 0.2 | 4.46 | 3.4 | 2 | 0.2 |
| 63 | 19.8 ± 0.4 | 4.7 | 3.3 | 2 | 0.2 |
| 64 | 18.4 ± 0.2 | 4.74 | 3.3 | 2 | 0.2 |
| 67 | 71.5 ± 2.5 | 4.15 | 2.8 | 2 | 0.3 |
| 68 | 33.4 ± 0.7 | 4.48 | 3.3 | 2 | 0.2 |
| 71 | 35.6 ± 0.6 | 4.45 | 3.3 | 2 | 0.3 |
| 79 | 28.9 ± 1.5 | 4.54 | 3.1 | 3 | 0.3 |
| 80 | 30.8 ± 0.7 | 4.51 | 3.1 | 3 | 0.3 |
| 81 | 19.1 ± 0.6 | 4.72 | 3.1 | 3 | 0.3 |
| 83 | 16.7 ± 0.6 | 4.78 | 2.9 | 4 | 0.3 |
| 84 | 15.9 ± 0.9 | 4.8 | 3.8 | 3 | 0.3 |
| 85 | 26.9 ± 0.8 | 4.57 | 3.8 | 3 | 0.3 |
| 86 | 18.7 ± 0.8 | 4.73 | 3.8 | 3 | 0.3 |
| 87 | 32.3 ± 0.9 | 4.49 | 3.6 | 4 | 0.3 |
| 88 | 4.9 ± 0.3 | 5.31 | 3.6 | 4 | 0.3 |
| 89 | 35.7 ± 0.9 | 4.51 | 2.9 | 4 | 0.3 |
| 90 | 31.0 ± 2.0 | 4.45 | 2.9 | 4 | 0.3 |
| 91 | 75.0 ± 4.9 | 4.13 | 2.5 | 3 | 0.3 |
| 93 | 29.9 ± 0.4 | 4.52 | 3.1 | 3 | 0.3 |
| 94 | 24.8 ± 0.4 | 4.61 | 2.9 | 4 | 0.3 |
| 95 | 44.8 ± 1.6 | 4.35 | 3.1 | 3 | 0.4 |
| 97 | 9.6 ± 0.5 | 5.02 | 2.9 | 4 | 0.3 |
2.3.2. 3D-QSAR
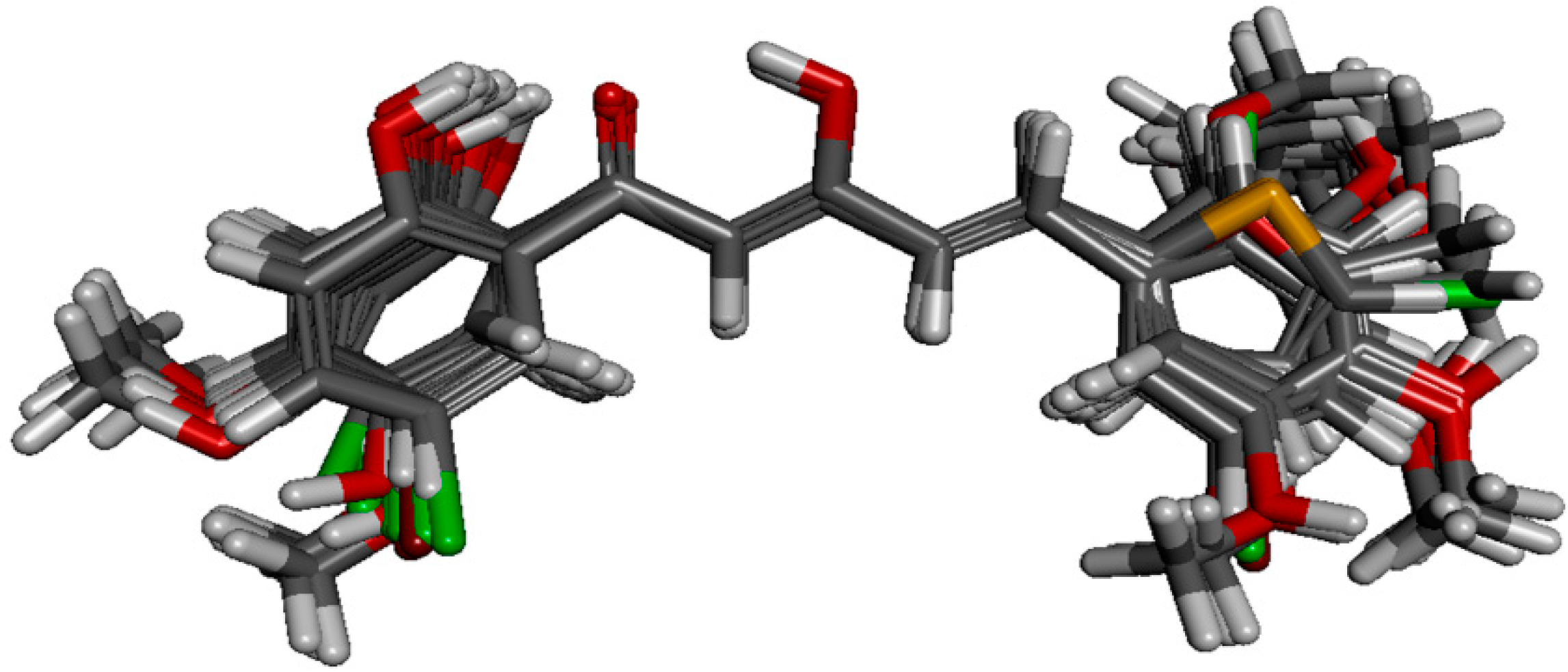
Comparative molecular field analysis (CoMFA) was employed to investigate the correlation between NO suppression activity of the diarylpentenedione analogues to their 3D structures, as well as to their electrostatic and steric grid map. Based on their pIC
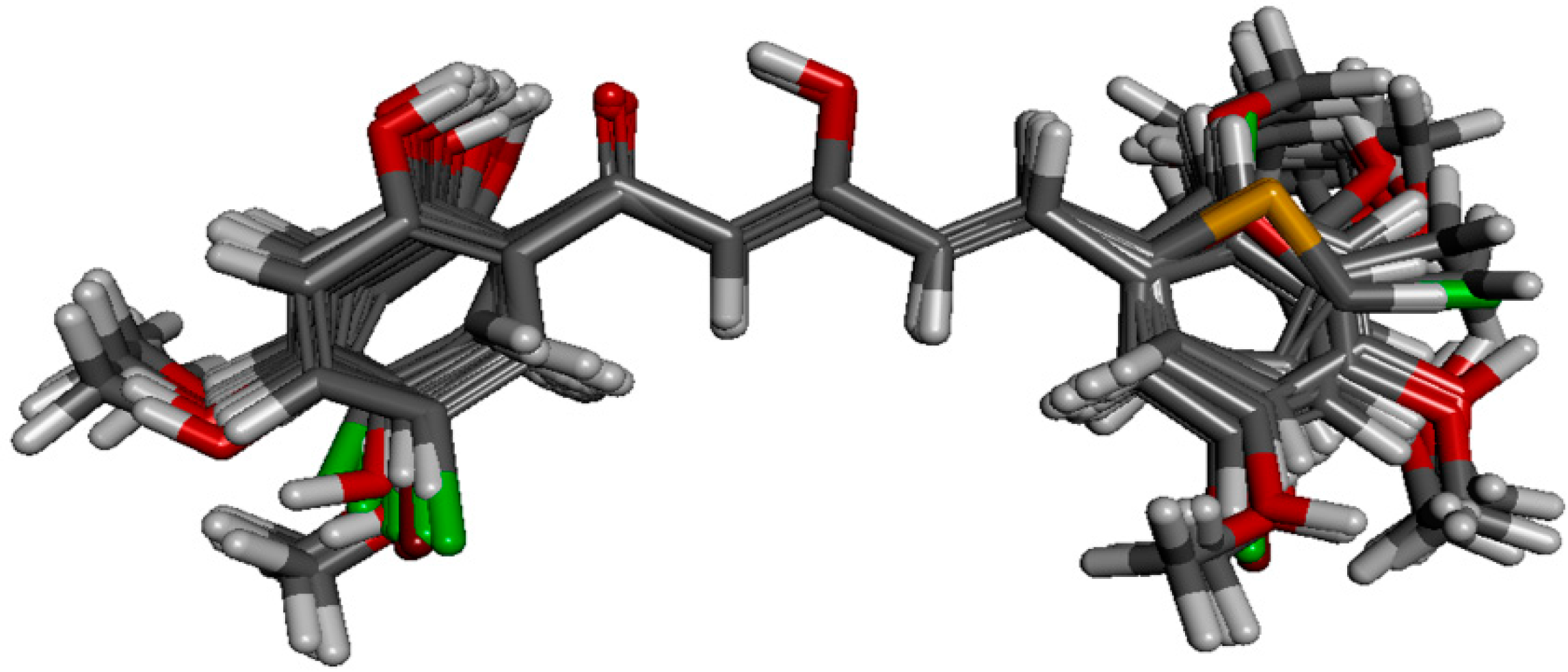
50 values, fifty-seven of the analogues were selected as model data set. Structural alignment is critical in determining the accuracy of results and reliability in CoMFA analysis so that the best compound could be selected as template molecule. Compound
88 was chosen due to its highest potency in inhibiting NO production. The alignment of the selected analogues at the common α,β-unsaturated β-diketone fragment with compound
88 is shown in
Figure 4.
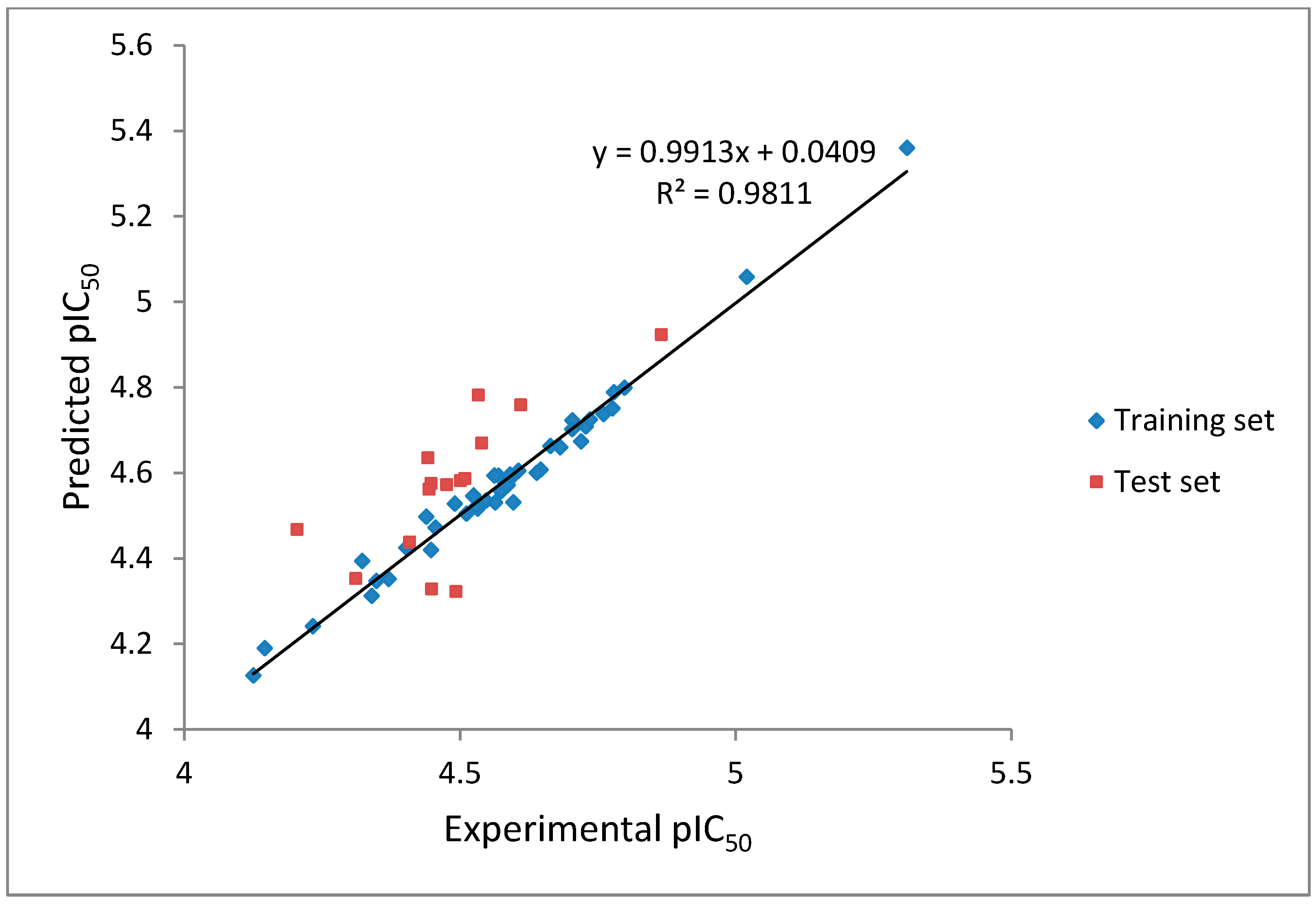
CoMFA analysis is considered valid if and only if the r
2 and q
2 values are greater than 0.8 and 0.5, respectively. The r
2 and q
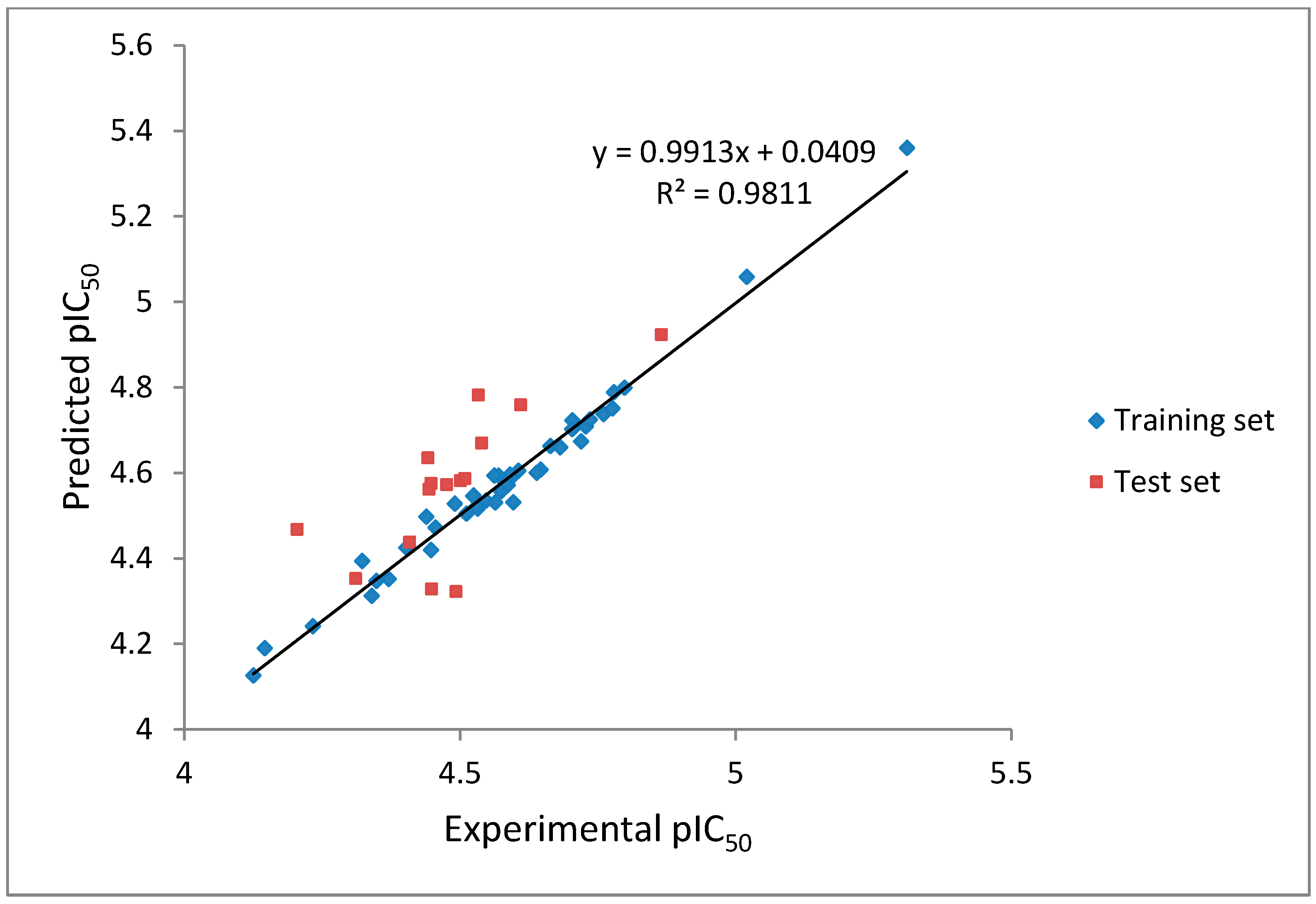
2 values of our CoMFA model were found to be 0.981 and 0.562, respectively, which implied that the generated correlations were acceptable. A plot depicting the experimental
versus predicted pIC
50 values for training and test set compounds are shown in
Figure 5. The CoMFA contour maps generated were presented together with the most potent compound (
88) in
Figure 6A,B representing the van der Waals and electrostatic contour maps, respectively.
Figure 4.
Structural alignment of the derivatives by template-based method according to the core of compound 88.
Figure 4.
Structural alignment of the derivatives by template-based method according to the core of compound 88.
Figure 5.
The experimental pIC50 versus predicted activity plot of training set and test set compounds.
Figure 5.
The experimental pIC50 versus predicted activity plot of training set and test set compounds.
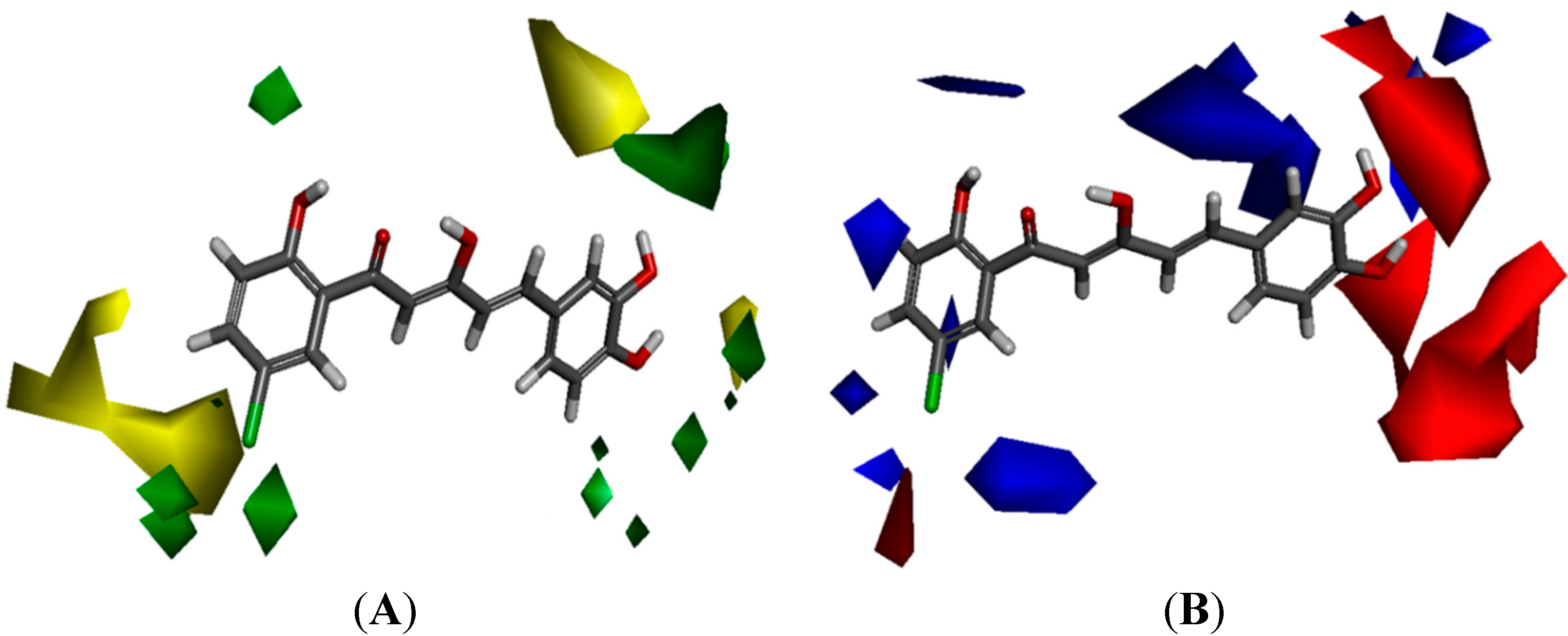
The yellow regions in
Figure 6A indicate where a sterically bulkier moiety is unfavorable while the green regions indicate where a sterically bulkier moiety is preferable in enhancing the bioactivity. For the electrostatic contour maps, the red regions represent the space where a hydrogen acceptor is preferable, while the blue regions represent the space where a hydrogen donor is preferable in improving potency.
Based on the contour map in
Figure 6A, bulkier groups were more preferred at the C-2' and C-5' position of ring-A while they were undesirable at the
para position of ring-A. This explaines why compound
88, possessing these features, is a potent NO inhibitor while the
m-methoxylated ring-A analogues are relatively poor NO inhibitors. These 3D-QSAR predictions further support the conclusion that the presence of
p-methoxylation of the phenyl ring will lead to a reduction of potency. On the other hand, bulkier moieties are more favorable at the
meta- and
para-positions of ring-B which explaines the higher bioactivity exhibited by compound
9 in comparison with compounds
7 and
8. Interestingly, the yellow regions are also observed at the
meta- and
para-positions of ring-B, which seems to contradict our real experimental results where demethylation of the methoxyl groups leads to enhancement of potency (
84 and
86 vs. 24 and
26). As we have mentioned in the earlier section, this phenomenon could be rationalized by the increase in hydrogen bonding capacity after converting the methoxyl groups to hydroxyl. A hydroxyl group can act as both hydrogen bond donor and acceptor while a methoxyl group can only act as hydrogen bond acceptor.
Figure 6.
(A) Van der Waals contour maps generated by 3D-Quantitative structure activity relationship (QSAR) modeling. Green contours indicate the regions where bulky groups are favorable in enhancing activity, whereas yellow contours indicate regions where bulky groups are disfavorable and reduce activity. (B) Electrostatic contour maps generated by 3D-QSAR modeling. Blue contours indicate regions where electronegative groups are favorable in increasing activity, whereas red contours indicate regions where electropositive groups are favorable in improving activity. The most potent candidate, compound 88 was chosen as reference molecule.
Figure 6.
(A) Van der Waals contour maps generated by 3D-Quantitative structure activity relationship (QSAR) modeling. Green contours indicate the regions where bulky groups are favorable in enhancing activity, whereas yellow contours indicate regions where bulky groups are disfavorable and reduce activity. (B) Electrostatic contour maps generated by 3D-QSAR modeling. Blue contours indicate regions where electronegative groups are favorable in increasing activity, whereas red contours indicate regions where electropositive groups are favorable in improving activity. The most potent candidate, compound 88 was chosen as reference molecule.
The electrostatic contour maps (
Figure 6B) shows that H-donors are preferable on both the phenyl rings. Therefore, presence of multiple hydrogen donors is important in preparing more bioactive molecules. This observation is in agreement with the 2D-QSAR results, which indicates that the number of hydrogen donors is directly proportional to the potency of compounds. Comparison of the bioactivities of compounds
21,
86 and
88 revealed the importance of H-donor in bioactivity enhancement. Conversely, multiple hydrogen acceptors are only preferable at
meta- and
para-positions of ring-B. Thus, analogues with
o,
m-methoxylatedor,
o,
para-methoxylated ring-B (
68 and
69) gave low NO inhibition, while those with
m,
p-methoxylated ring-B (
63 and
64) showed stronger NO inhibition.
2.4. Pharmacophore Mapping
Pharmacophore mapping is an abstract concept to illustrate common interaction patterns of a series of active ligands with a biological receptor. It can also be defined as the common features which are responsible for pharmacological activities or chemical reactivity, possessed by a series of bioactive compounds [
33]. Pharmacophore mapping has been widely used in drug discovery programs due to its ability to provide important information in designing highly active lead compounds. Recent studies have integrated pharmacophore structures with molecular docking, which further strengthen the hypothesis on the pharmacophore [
34,
35]. Another study by Dong and coworkers has proven the rationale and feasibility of pharmacophore utilization in drug development [
36].
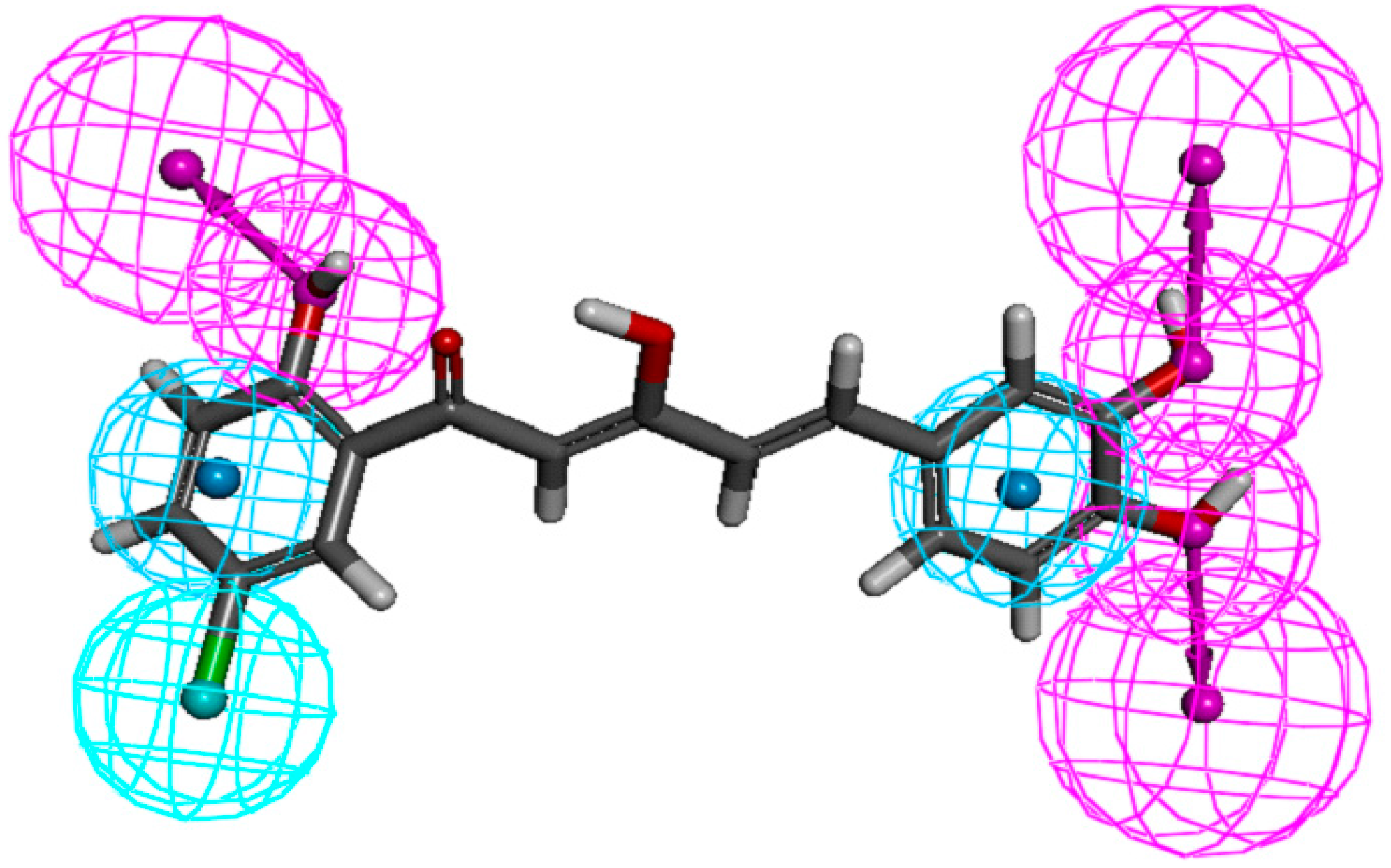
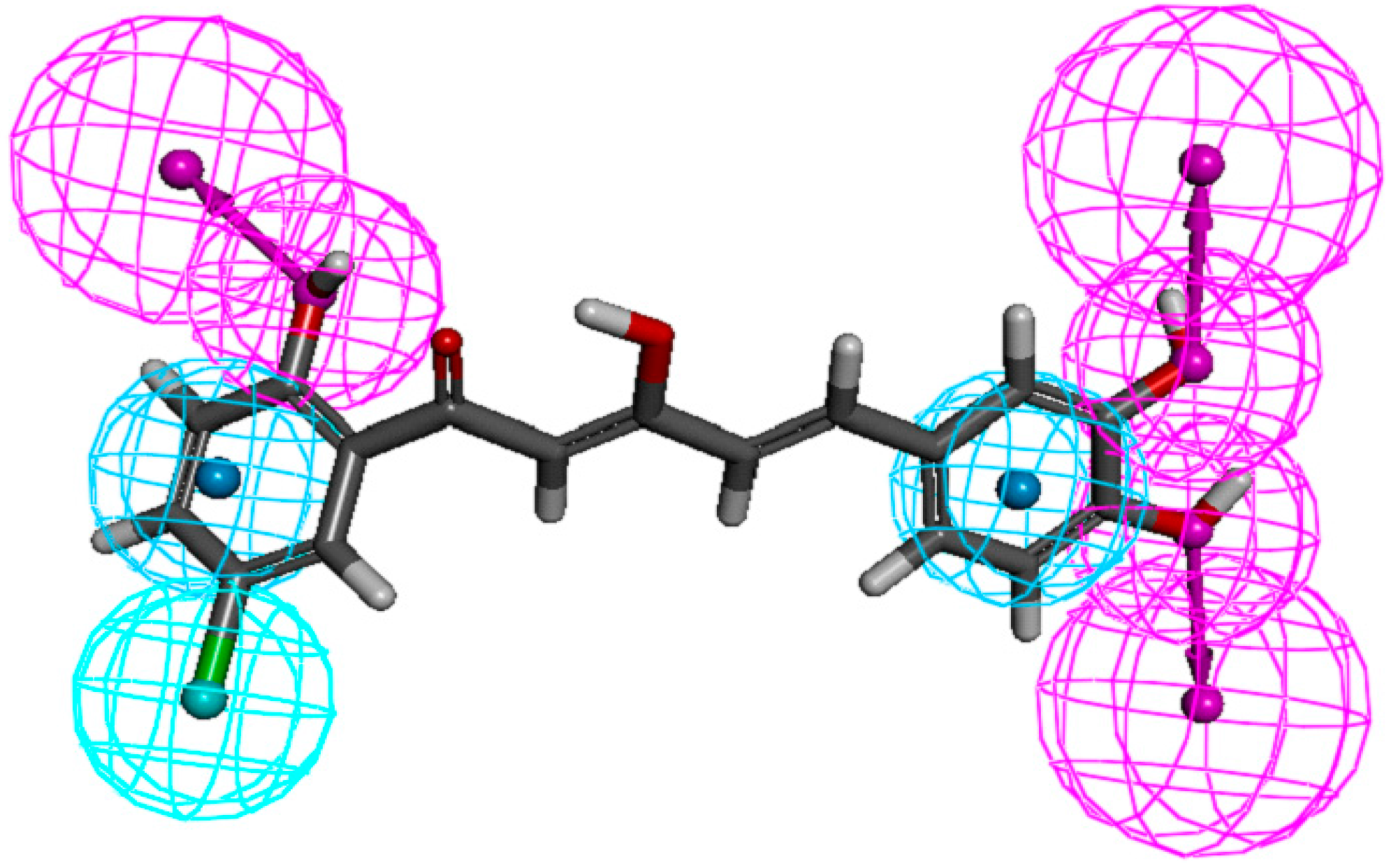
In the present study, pharmacophore mapping was performed using Discovery Studio 3.1, based on selected chemical features, such as hydrophobicity (HYP), hydrophobic aromatic ring (HA), hydrogen bond acceptor (HBA), and hydrogen bond donor (HBD), based on NO suppression activities and common features of ten selected analogues (
9,
12,
28,
37,
46,
67,
84,
88,
91 and
97). The pharmacophore map generated from this exercise is shown in
Figure 7, where cyan, blue, and red represent aromatic hydrophobic, hydrophobic, and hydrogen-bond donor regions, respectively. As shown in
Figure 7, important features of the diarylpentanoids, which were responsible for bioactivity, included the hydrogen-bond donor at
meta- and
para-positions of ring-B and
ortho position of ring-A, the hydrophobic region at 5'-position of ring-A and the aromatic hydrophobic regions on both phenyl rings.
Figure 7.
Pharmacophore mapping of compound 88. Features are color-coded as follows: hydrophobic aromatic (HA), blue; hydrophobic (HYP), cyan; hydrogen-bond donor (HBD), pink.
Figure 7.
Pharmacophore mapping of compound 88. Features are color-coded as follows: hydrophobic aromatic (HA), blue; hydrophobic (HYP), cyan; hydrogen-bond donor (HBD), pink.
2.5. ADMET Analysis
ADMET analysis refers to the absorption, distribution, metabolism, excretion, and toxicity prediction of a molecule within an organism based on its molecular structure. It is one of the crucial steps in computer-aided drug design (CADD) due to its ability to filter out low bioavailability and toxic candidates, thus improving the efficiency and reduce the cost of research and development.
ADMET analysis was performed using Discovery Studio 3.1, based on aqueous solubility (AS), human intestinal absorption (HIA), blood brain barrier (BBB), cytochrome P450 2D6 (CYP2D6), plasma protein binding (PPB), and hepatotoxicity (HT) descriptors of ten selected compounds. A summary of the results from this experiment is presented in
Table 7.
The poor bioavailability of oral drugs is related to their low solubility and low permeability. Therefore, sufficient levels of aqueous solubility and human intestinal absorption are important in improving drug delivery in the human body. From the data presented in this analysis, the Analogues 9, 64, 81, 83, 84, 86, 88 and 97 showed good aqueous solubility and intestinal absorption, indicating that they could be good candidates for oral drugs. Meanwhile, despite offering good human intestinal absorption property, the Analogues 25 and 28 have low aqueous solubility.
Table 7.
Results of absorption, distribution, metabolism, excretion, and toxicity (ADMET) predictions on six important parameters.
Table 7.
Results of absorption, distribution, metabolism, excretion, and toxicity (ADMET) predictions on six important parameters.
| Compounds | AS | HIA | BBB | CYP2D6 | PPB | HT |
|---|
| 9 | Good | Good | Medium | Non-inhibit | Bound | Hepatotoxin |
| 25 | Low | Good | High | Non-inhibit | Bound | Hepatotoxin |
| 28 | Low | Good | Medium | Non-inhibit | Bound | Hepatotoxin |
| 64 | Good | Good | Low | Non-inhibit | Bound | Hepatotoxin |
| 81 | Good | Good | Low | Non-inhibit | Bound | Non-hepatotoxin |
| 83 | Good | Good | Low | Non-inhibit | Bound | Hepatotoxin |
| 84 | Good | Good | Medium | Non-inhibit | Bound | Hepatotoxin |
| 86 | Good | Good | Medium | Non-inhibit | Bound | Hepatotoxin |
| 88 | Good | Good | Undefined | Non-inhibit | Bound | Hepatotoxin |
| 97 | Good | Good | Low | Non-inhibit | Bound | Non-hepatotoxin |
The blood brain barrier (BBB) descriptor relates to the ability of a compound to cross the blood brain barrier. High BBB penetration is a much sought-after property for central nervous system (CNS) targeted drugs, but for CNS unrelated diseases, it is undesirable. Therefore, the BBB descriptor can acts as a filter to improve efficiency of drug developments for CNS related diseases. As shown in
Table 7, the Analogues
9,
25,
28,
84 and
86 could be potential candidates for use against CNS inflammatory disorders due to their moderate to high BBB penetration, while
64,
81,
83,
88 and
97 could be more suitable for CNS unrelated diseases due to their low BBB penetration.
Cytochrome P450 2D6 (CYP2D6) encompasses a class of enzymes, which catalyze the oxidative metabolism of drugs in the liver. It can either metabolize a drug from its active form into its inactive metabolites or convert an inactive drug into its active metabolites. Therefore, CYP2D6 inhibitory factors should be considered in reducing toxicity caused by the inactive metabolites for the former case but it should be avoided to preserve drug efficiency for the latter. The ADMET analysis conducted on our diarylpentenedione analogues indicates that all the selected compounds are non-CYP2D6 inhibitors on account of their inactive behavior towards CYP2D6. On the other hand, plasma protein binding is an important factor that determines the drug efficiency since only the unbound fraction is responsible for pharmacological effects. As presented in
Table 7, all selected compounds were expected to be highly bound to protein plasma, thus implying that a high dosage might be required to achieve therapeutic concentration in treatments.
Lastly, the hepatotoxicity descriptor was used to predict potential organ toxicity caused by the compounds. As displayed in
Table 7, only two compounds (
81 and
97) were non-hepatotoxic while the rest were calculated as hepatotoxin. On account of these, more studies must be carried out to investigate the hepatotoxic effect of the Analogues
9,
25,
28,
64,
83,
84,
86 and
88 in addition to their optimal therapeutic dosage.
2.6. TOPKAT Analysis
Toxicity Prediction by Komputer Assisted Technology (TOPKAT) is a common method used to predict the ecotoxicity, toxicity, mutagenicity, and reproductive or developmental toxicity of selected candidates. At the early research stage, the utilization of TOPKAT predictions may be useful in prioritizing promising compounds for further development and investigation. Besides, it can also act as a factor to accelerate optimization of lead compounds in terms of their therapeutic ratios in both animal and human models.
In this study, the ten selected analogues from ADMET analysis were further screened for toxicity prediction including aerobic biodegradability, mutagenicity, rodent carcinogenicity, ocular irritancy, skin irritancy, and skin sensitization parameters. From the results, it may be generalized that all compounds were non-mutagenic, non-carcinogen and non-skin irritant. Analogues
9 and
64 were predicted as biodegradable while
28,
84,
86 and
88 were found to be ocular irritants. Compound
84 is the only candidate expected to be a non-skin sensitizer. Integration of both ADMET and TOPKAT analyses revealed that the Analogues
81 and
97 could be good candidates for further investigation based on their low toxicity and good aqueous solubility and intestinal absorption. Of the two,
97 appeared to be the most potent candidate as it exhibited two-fold better properties than
81. However, extensive toxicity studies should be carried out on
88 since it exhibited the strongest activity among the diarylpentenedione analogues. The results from this prediction exercise are presented in
Table 8.
Table 8.
Results of toxicity predictive test on six important parameters.
Table 8.
Results of toxicity predictive test on six important parameters.
| Compounds | AB | AM | RC | OI | SI | SS |
|---|
| 9 | Biodegradable | Non-mutagen | Non-carcinogen | Non-irritant | Non-irritant | Sensitizer |
| 25 | Non-biodegradable | Non-mutagen | Non-carcinogen | Non-irritant | Non-irritant | Sensitizer |
| 28 | Non-biodegradable | Non-mutagen | Non-carcinogen | Irritant | Non-irritant | Sensitizer |
| 64 | Biodegradable | Non-mutagen | Non-carcinogen | Non-irritant | Non-irritant | Sensitizer |
| 81 | Non-biodegradable | Non-mutagen | Non-carcinogen | Non-irritant | Non-irritant | Sensitizer |
| 83 | Non-biodegradable | Non-mutagen | Non-carcinogen | Non-irritant | Non-irritant | Sensitizer |
| 84 | Non-biodegradable | Non-mutagen | Non-carcinogen | Irritant | Non-irritant | Non-sensitizer |
| 86 | Non-biodegradable | Non-mutagen | Non-carcinogen | Irritant | Non-irritant | Sensitizer |
| 88 | Non-biodegradable | Non-mutagen | Non-carcinogen | Irritant | Non-irritant | Sensitizer |
| 97 | Non-biodegradable | Non-mutagen | Non-carcinogen | Non-irritant | Non-irritant | Sensitizer |
2.7. Chemical Stability Test
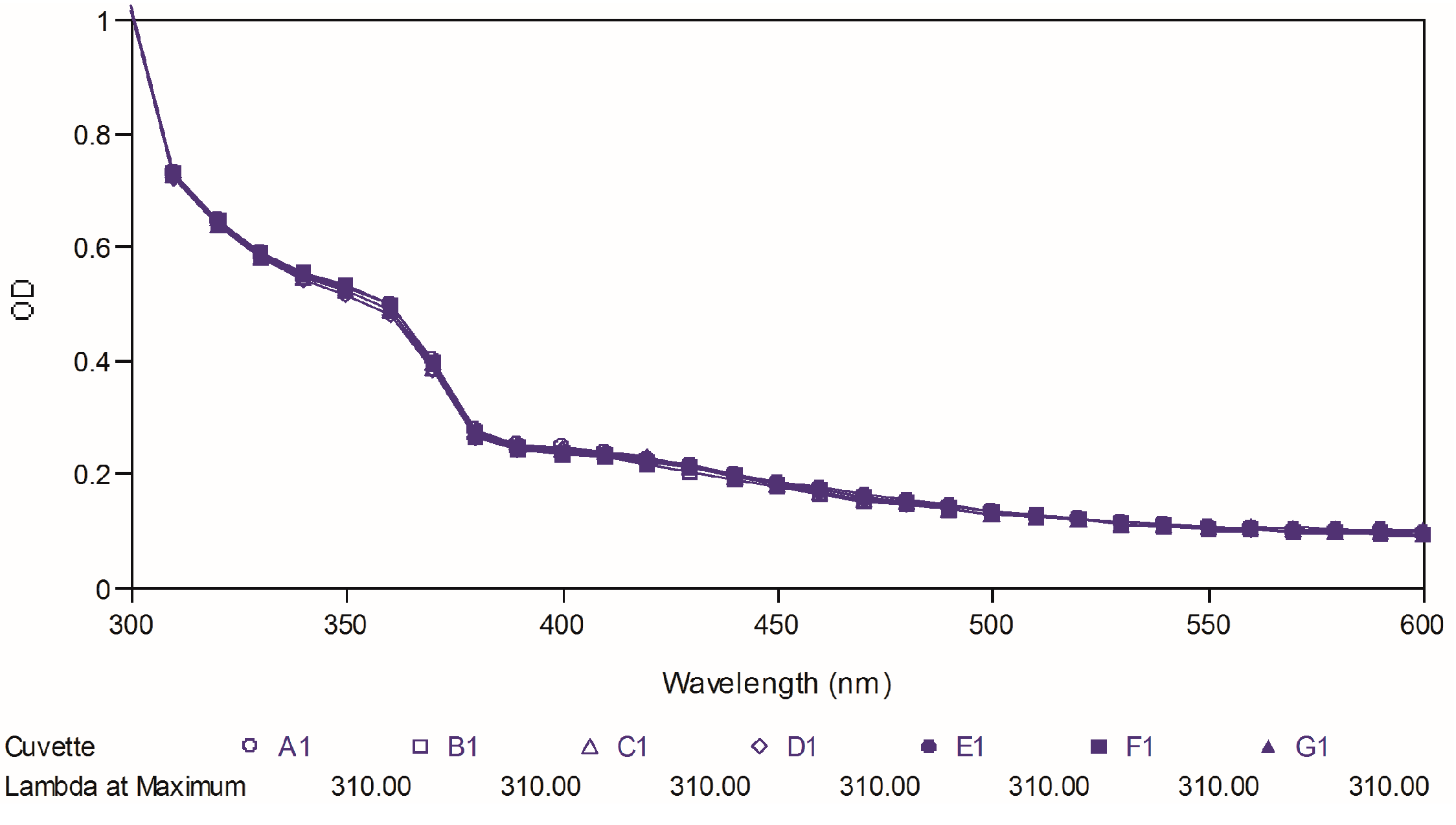
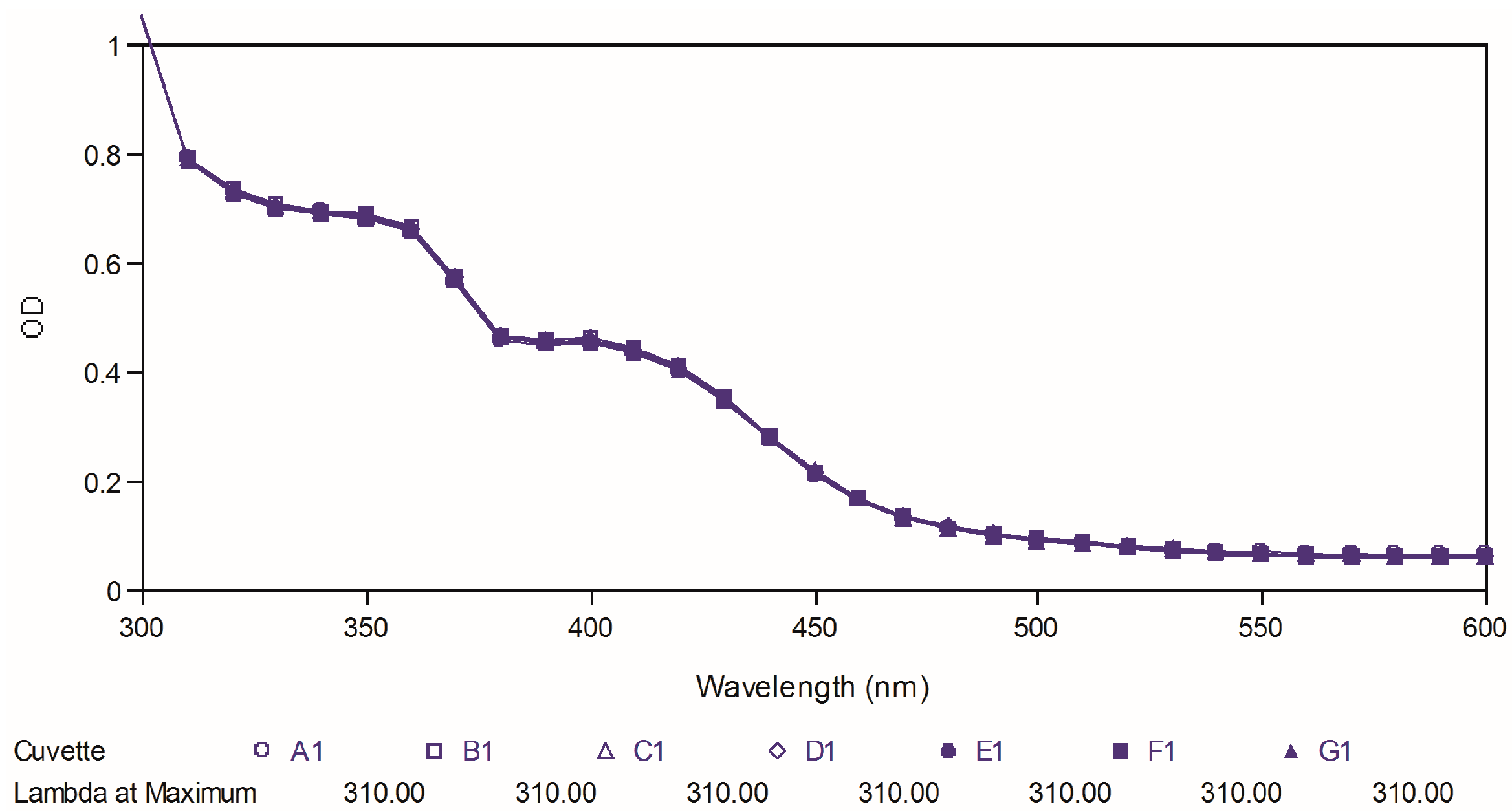
Poor chemical stability of curcumin has been proven as one of the important factors, which cause its low bioavailability. Therefore, in the present study, we further tested the two best compounds (
88 and
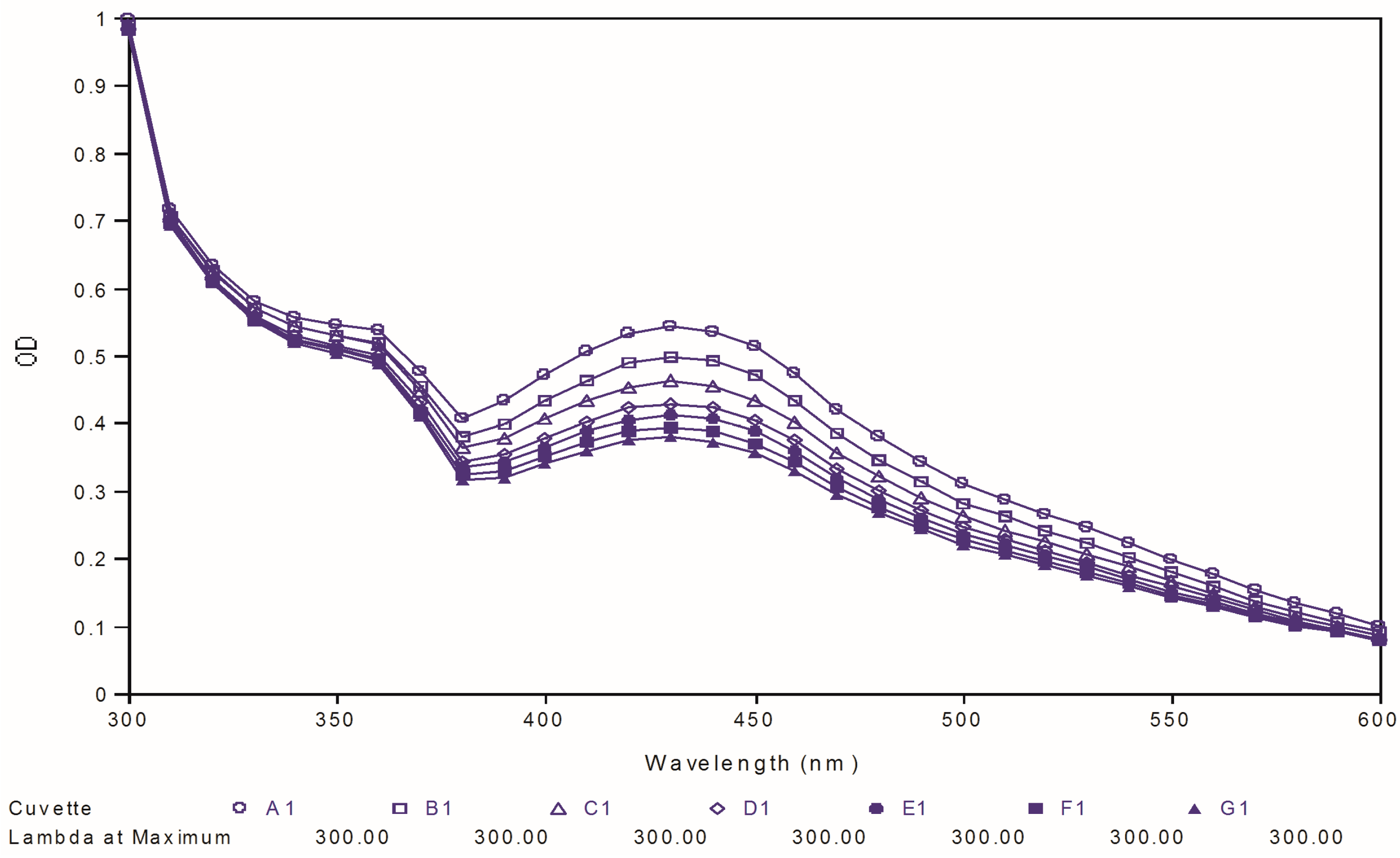
97) for their chemical stability at physiological pH and compared it to that of curcumin used as reference. The test was carried out by observing the ultraviolet spectral changes of the targeted compounds for 30 minutes with 5-minutes intervals in phosphate buffer (pH 7.4).
Figure 8,
Figure 9 and
Figure 10 depict the ultraviolet changes of curcumin, compound
88 and compound
97, respectively. As shown in
Figure 8, the maximal absorption peaks of curcumin gradually decreased over time. However, no significant changes were observed in the case of two best compounds, which indicated that our targeted compounds were chemically more stable than curcumin
in vitro.
Figure 8.
Ultraviolet-visible absorption spectra of curcumin.
Figure 8.
Ultraviolet-visible absorption spectra of curcumin.
Figure 9.
Ultraviolet-visible absorption spectra of compound 88.
Figure 9.
Ultraviolet-visible absorption spectra of compound 88.
Figure 10.
Ultraviolet-visible absorption spectra of compound 97.
Figure 10.
Ultraviolet-visible absorption spectra of compound 97.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}