Cellular Transport Mechanisms of Cytotoxic Metallodrugs: An Overview beyond Cisplatin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Transport Processes of Metal-Based Compounds
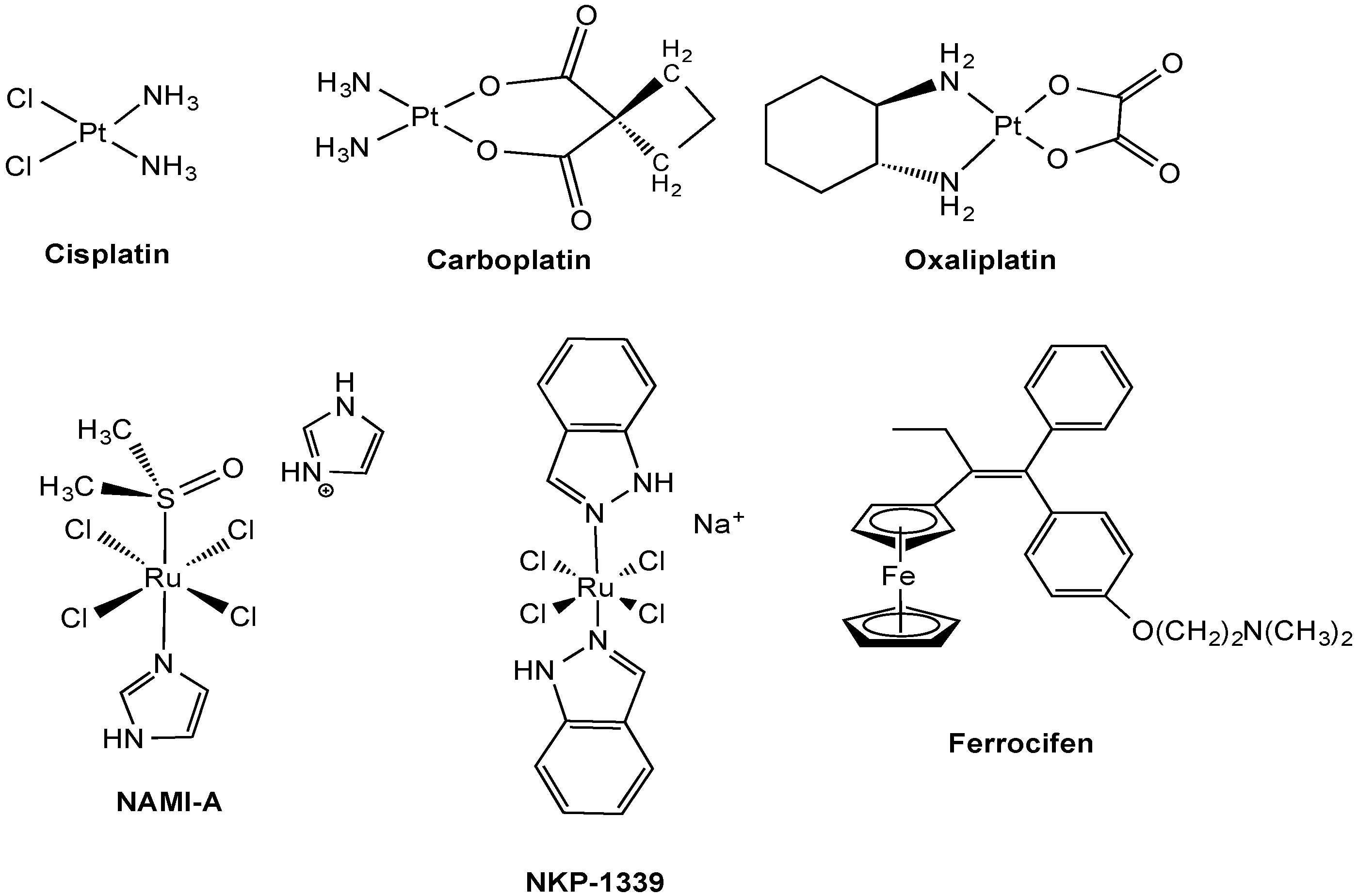
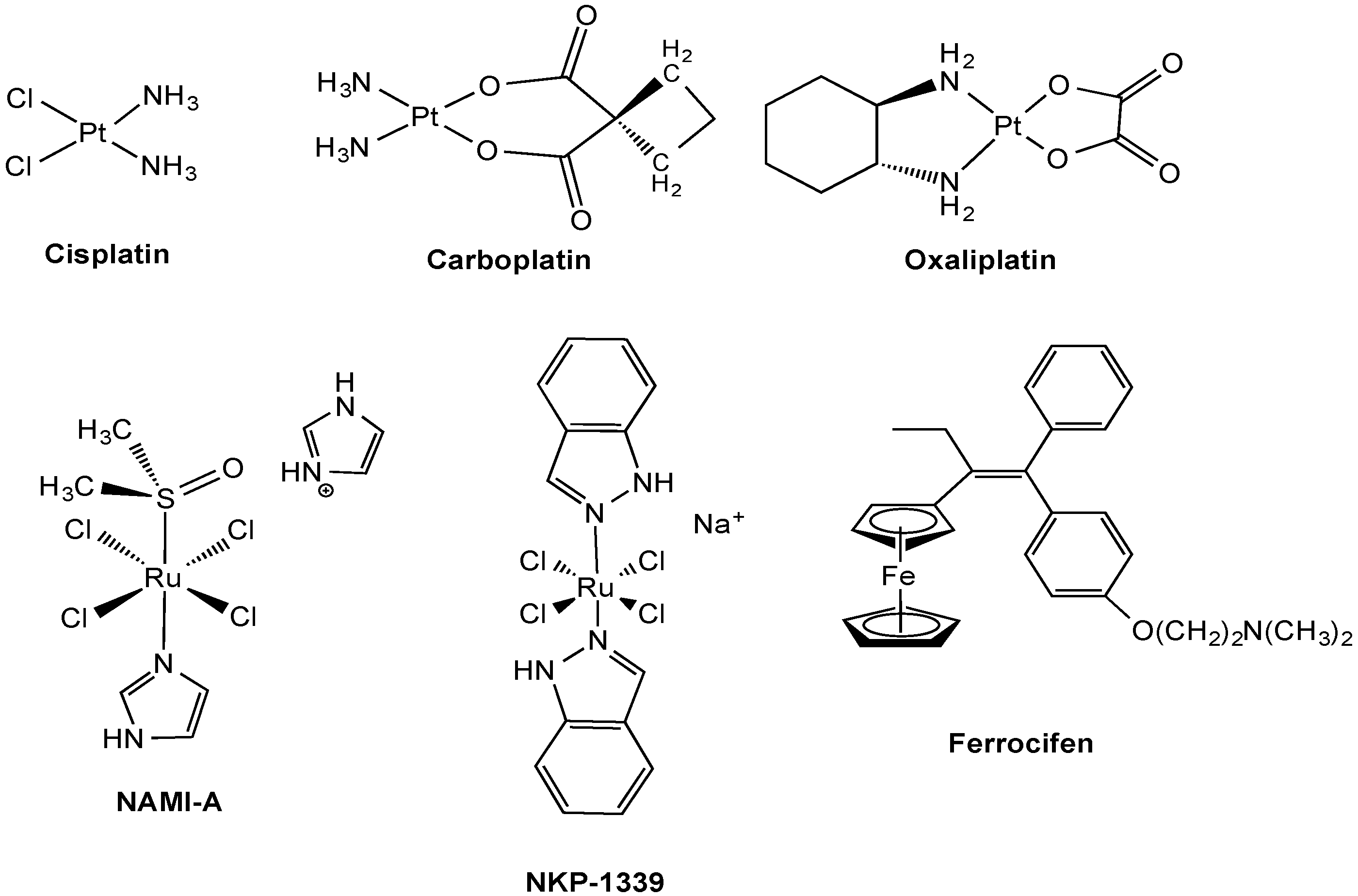
2.1. Anticancer Pt Drugs
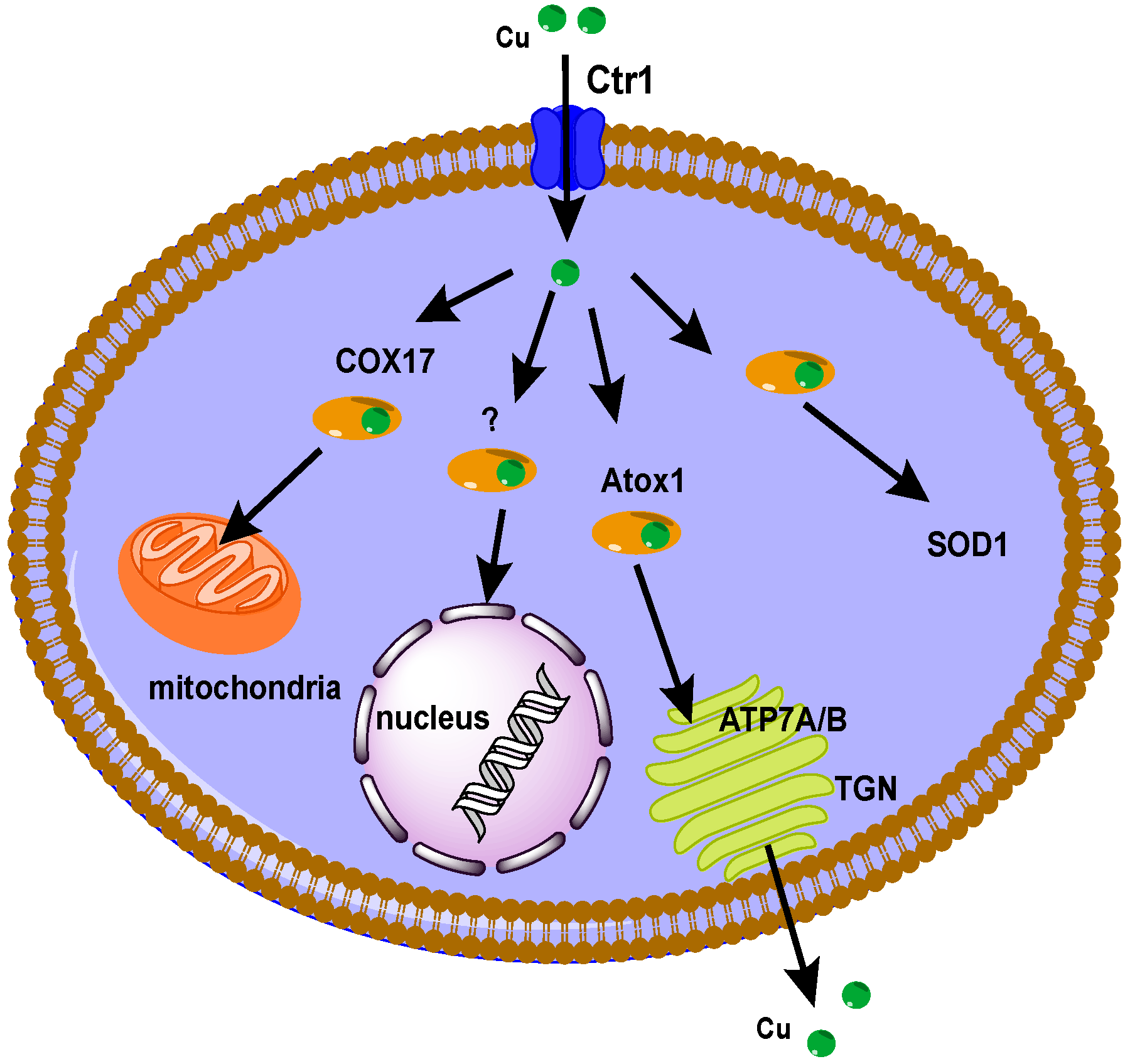
2.1.1. Cu Transporters

2.1.2. Organic Cation Transporters (OCTs) and Multidrug and Toxin Extrusion Proteins (MATEs)
2.2. Experimental Anticancer Metal Compounds
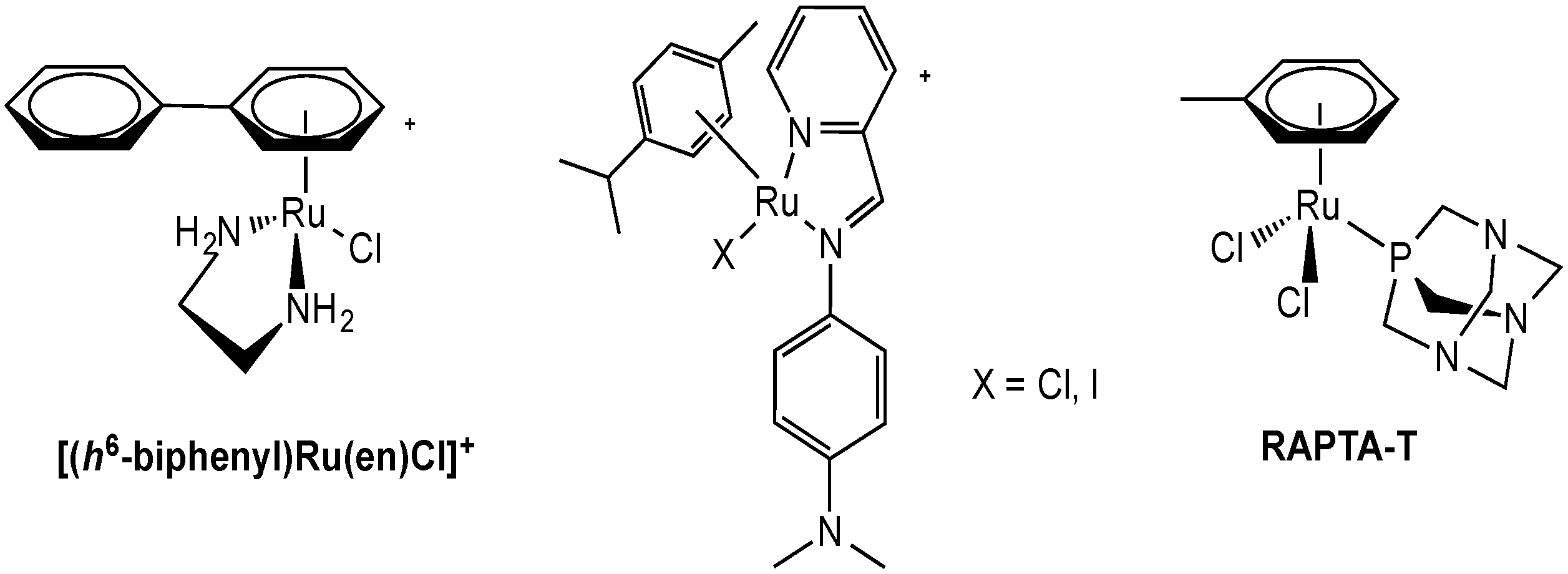
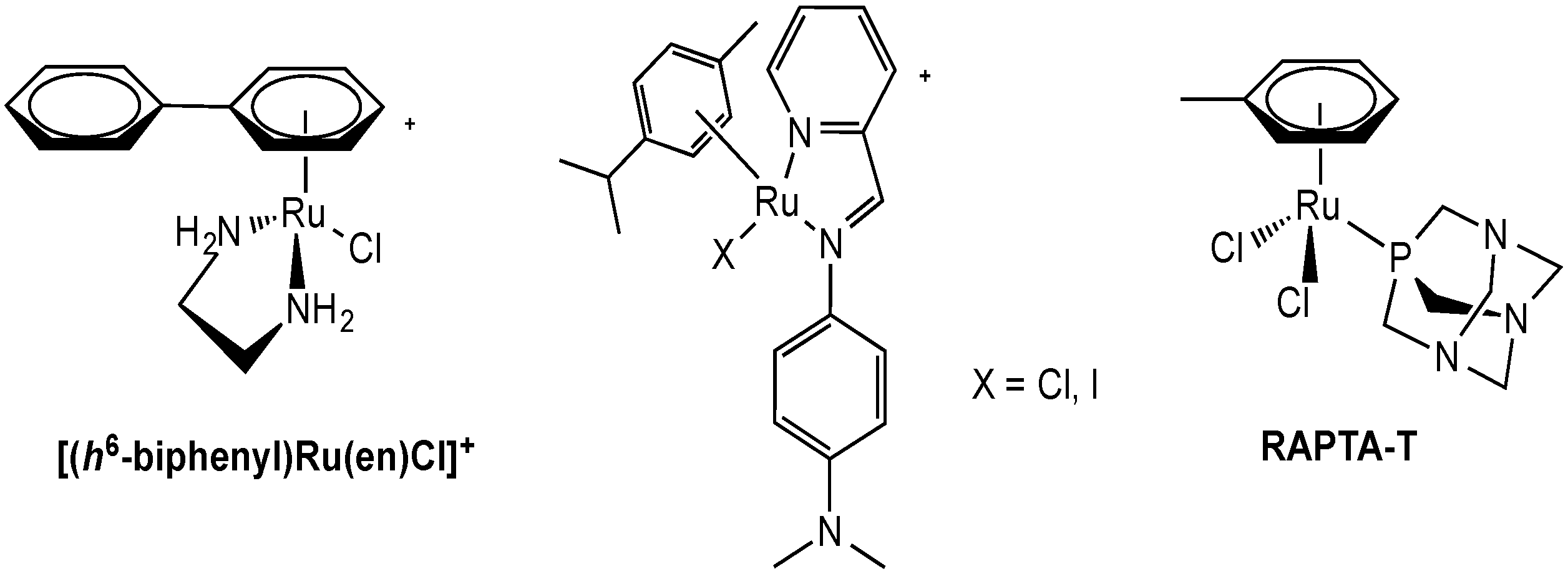
2.2.1. Ruthenium Complexes
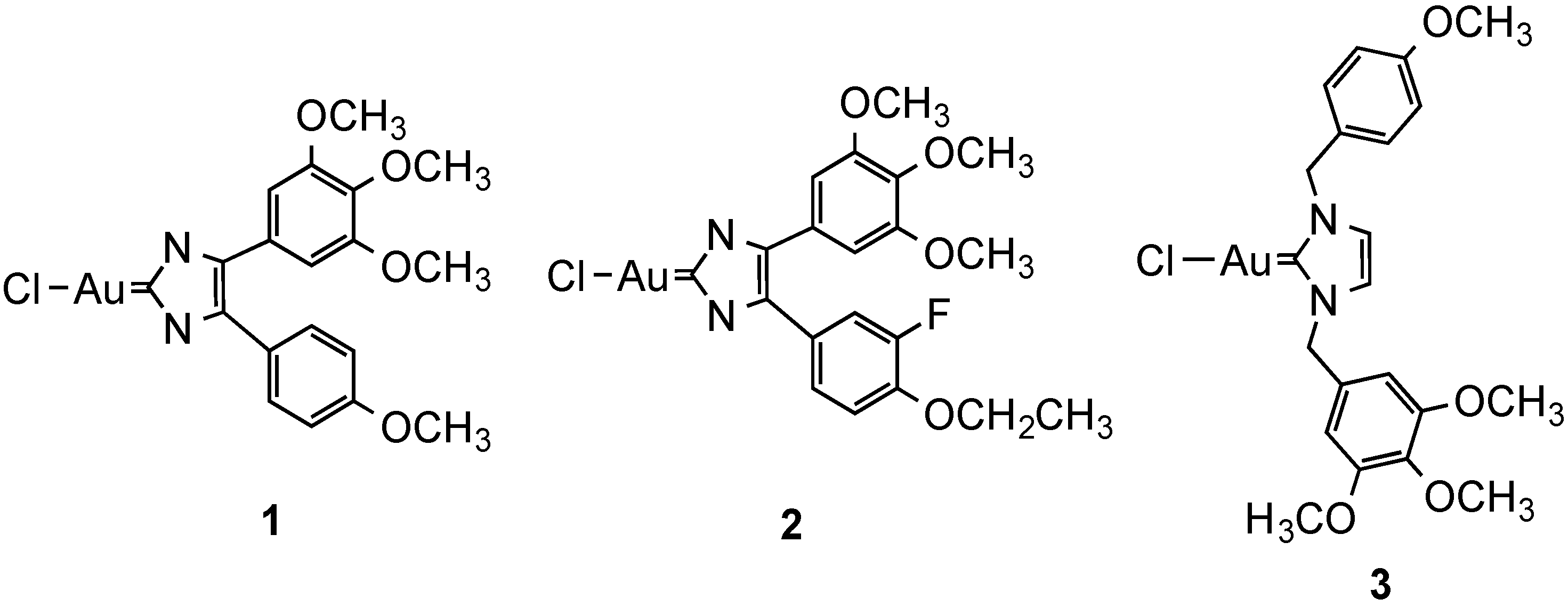
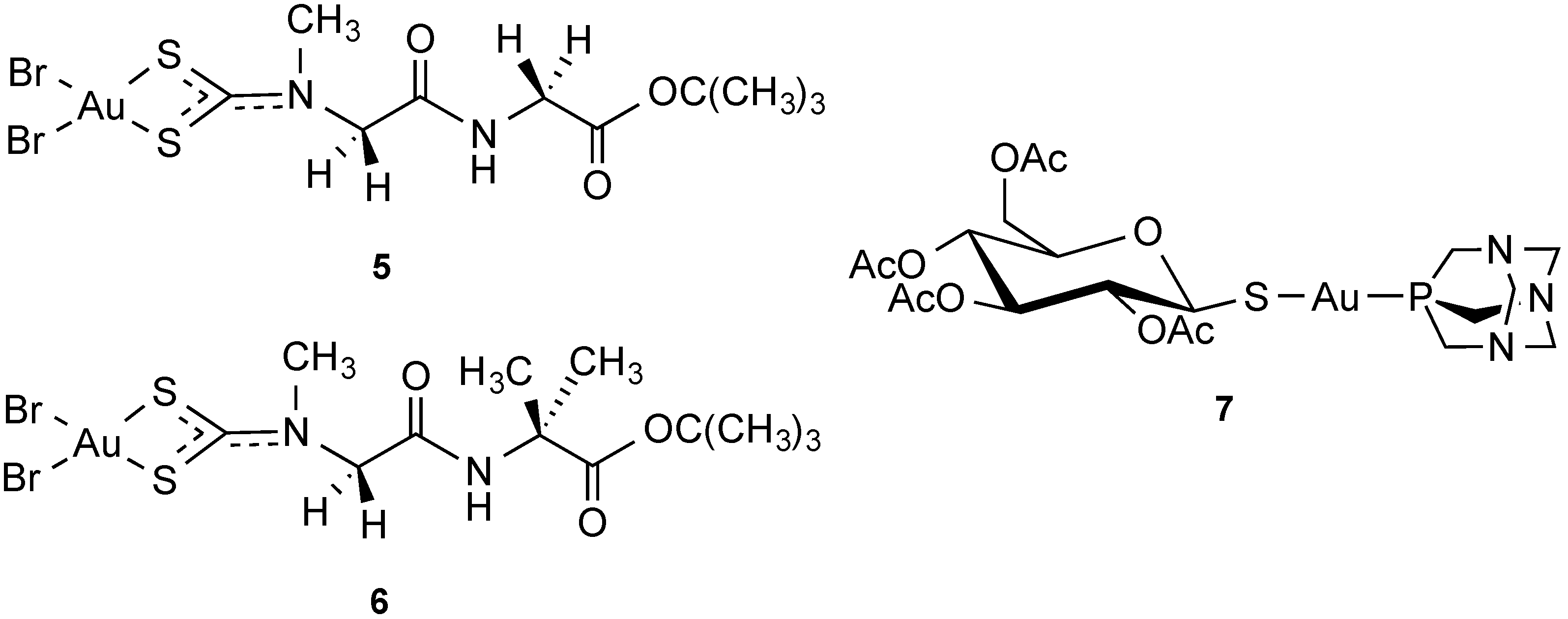
2.2.2. Gold Complexes
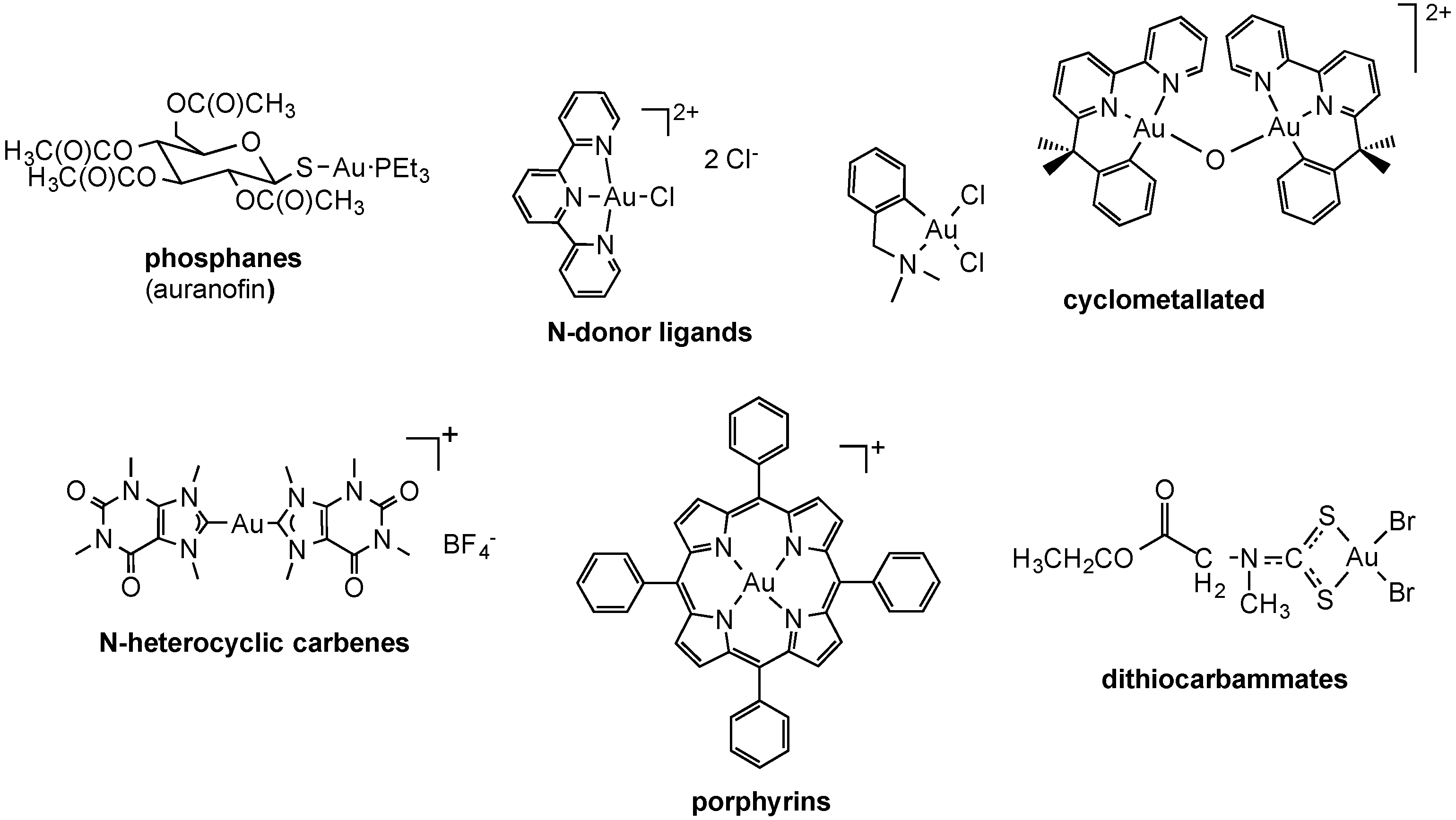

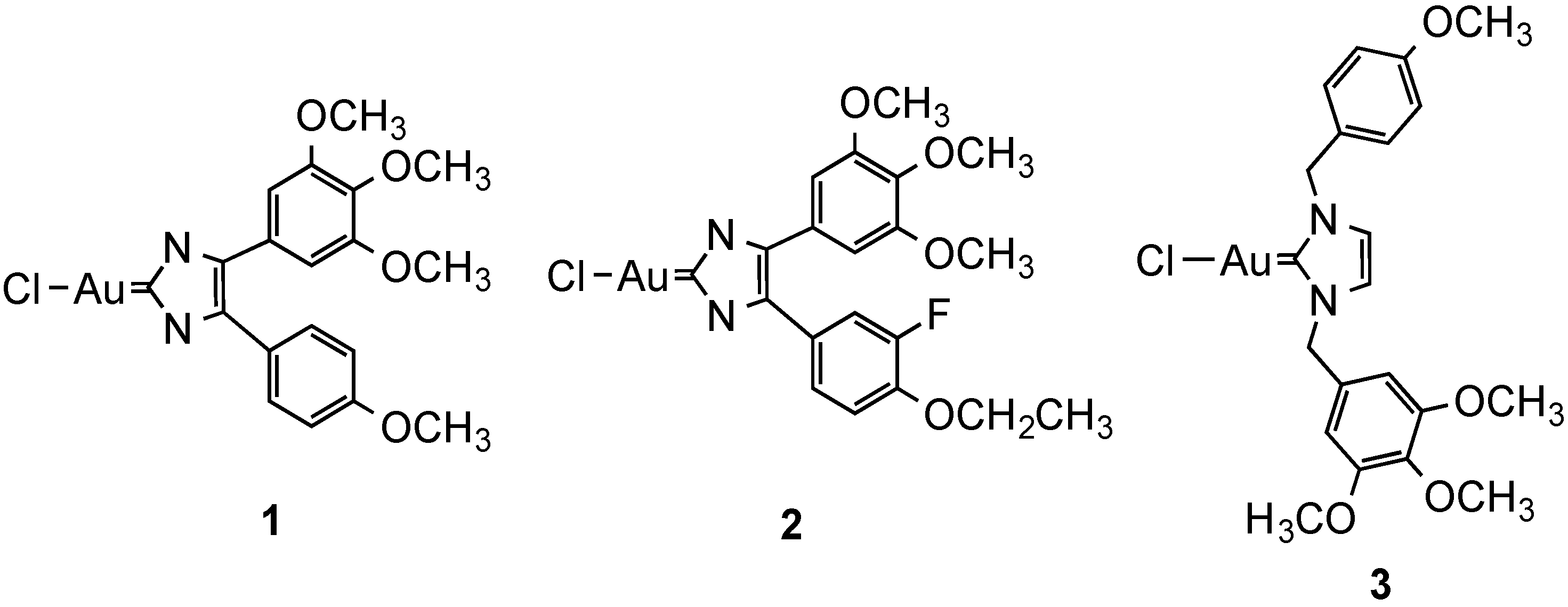
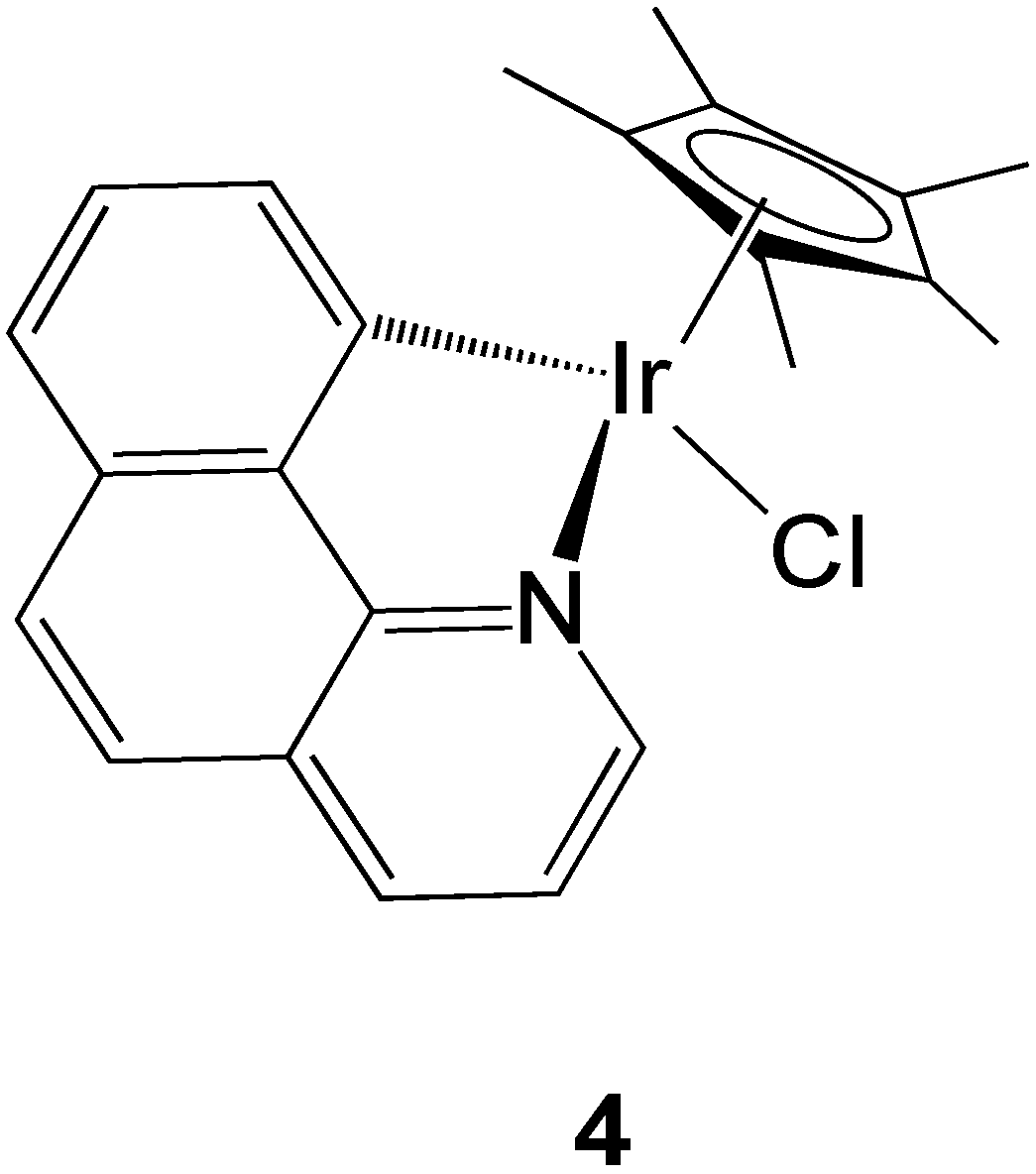
2.2.3. Iridium Complexes
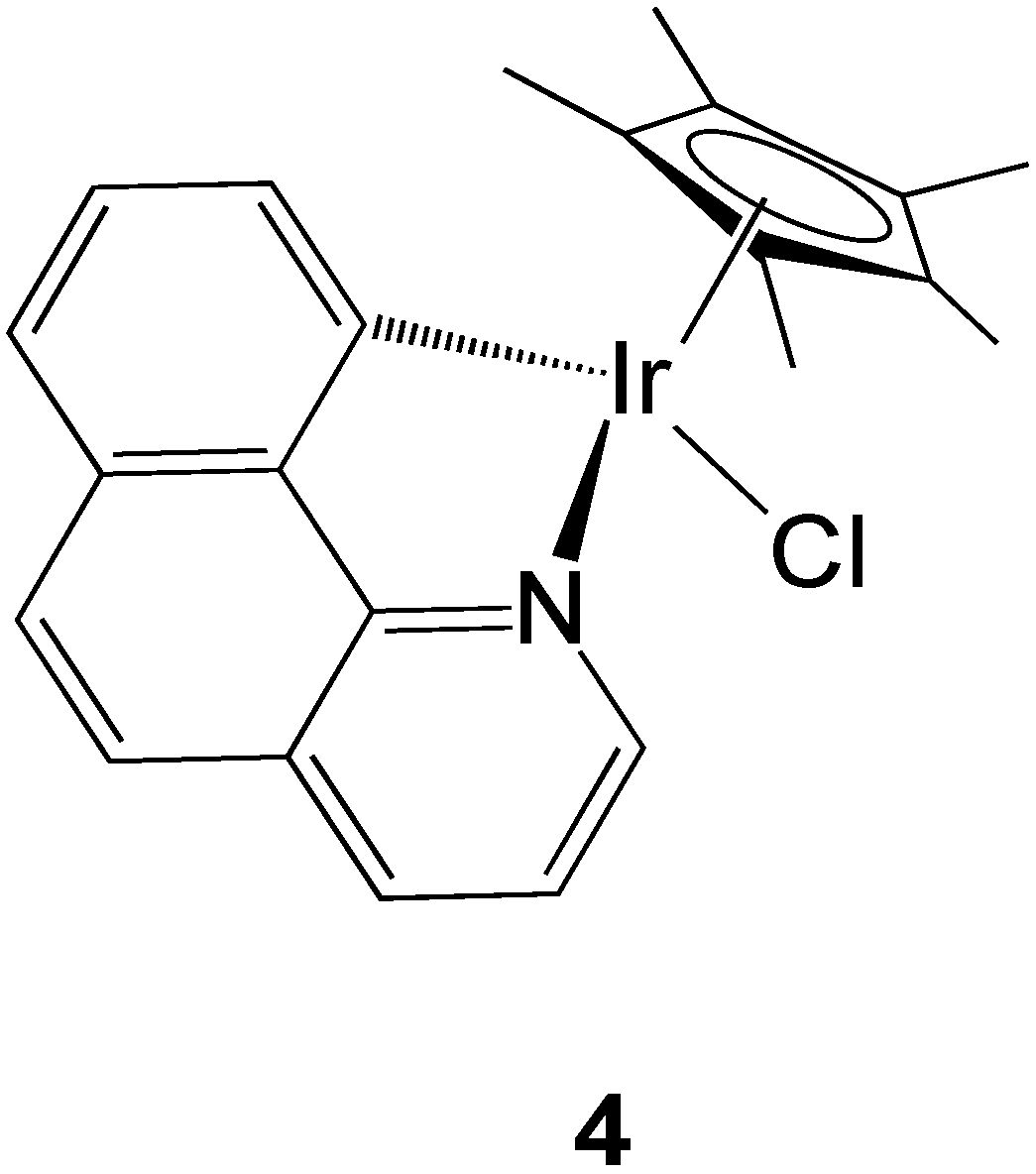
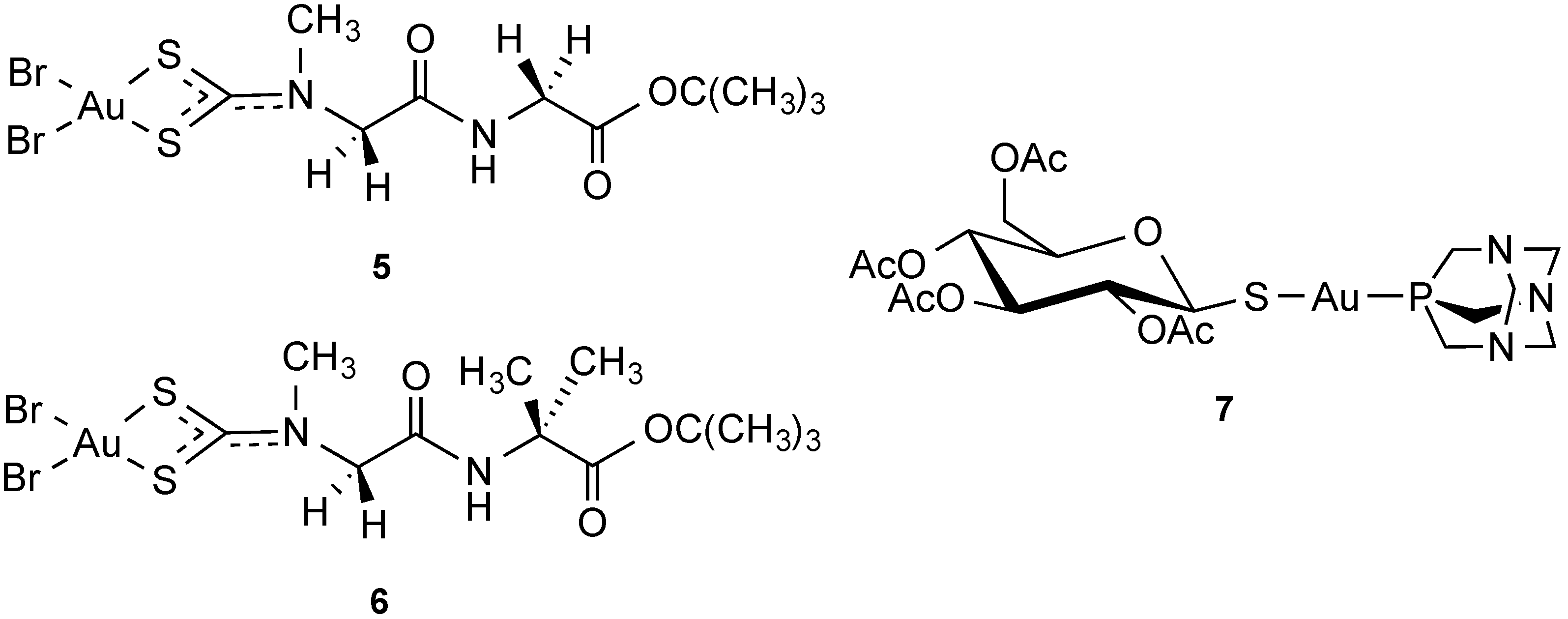
2.2.4. Transporter-Targeted Anticancer Metal Compounds
3. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar]
- Bergamo, A.; Sava, G. Ruthenium complexes can target determinants of tumour malignancy. Dalton Trans. 2007, 13, 1267–1272. [Google Scholar]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014. [Google Scholar] [CrossRef]
- Mjos, K.D.; Orvig, C. Metallodrugs in Medicinal Inorganic Chemistry. Chem. Rev. 2014, 114, 4540–4563. [Google Scholar]
- Nobili, S.; Mini, E.; Landini, I.; Gabbiani, C.; Casini, A.; Messori, L. Gold Compounds as Anticancer Agents: Chemistry, Cellular Pharmacology, and Preclinical Studies. Med. Res. Rev. 2010, 30, 550–580. [Google Scholar]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic Anticancer Compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [Green Version]
- Fregona, D.; Ronconi, L.; Aldinucci, D. Groundbreaking gold(III) anticancer agents. Drug Discov. Today 2009, 14, 1075–1076. [Google Scholar]
- Ott, I. On the medicinal chemistry of gold complexes as anticancer drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar]
- Nguyen, A.; Top, S.; Vessieres, A.; Pigeon, P.; Huche, M.; Hillard, E.A.; Jaouen, G. Organometallic analogues of tamoxifen: Effect of the amino side-chain replacement by a carbonyl ferrocenyl moiety in hydroxytamoxifen. J. Organomet. Chem. 2007, 692, 1219–1225. [Google Scholar]
- Cohen, S.M.; Lippard, S.J. Cisplatin: From DNA damage to cancer chemotherapy. Prog. Nucleic Acid Res. Mol. Biol. 2001, 67, 93–130. [Google Scholar]
- Casini, A.; Guerri, A.; Gabbiani, C.; Messori, L. Biophysical characterisation of adducts formed between anticancer metallodrugs and selected proteins: New insights from X-ray diffraction and mass spectrometry studies. J. Inorg. Biochem. 2008, 102, 995–1006. [Google Scholar]
- De Almeida, A.; Oliveira, B.L.; Correia, J.D.G.; Soveral, G.; Casini, A. Emerging protein targets for metal-based pharmaceutical agents: An update. Coord. Chem. Rev. 2013, 257, 2689–2704. [Google Scholar]
- Casini, A.; Reedijk, J. Interactions of anticancer Pt compounds with proteins: An overlooked topic in medicinal inorganic chemistry? Chem. Sci. 2012, 3, 3135–3144. [Google Scholar]
- Dobson, P.D.; Kell, D.B. Opinion—Carrier-mediated cellular uptake of pharmaceutical drugs: An exception or the rule? Nat. Rev. Drug Discov. 2008, 7, 205–220. [Google Scholar]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.Y.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar]
- Hillgren, K.M.; Keppler, D.; Zur, A.A.; Giacomini, K.M.; Stieger, B.; Cass, C.E.; Zhang, L.; Consortium, I.T. Emerging Transporters of Clinical Importance: An Update from the International Transporter Consortium. Clin. Pharmacol. Ther. 2013, 94, 52–63. [Google Scholar]
- Brunschweiger, A.; Hall, J. A Decade of the Human Genome Sequence—How Does the Medicinal Chemist Benefit? ChemMedChem 2012, 7, 194–203. [Google Scholar]
- Hall, M.D.; Okabe, M.; Shen, D.W.; Liang, X.J.; Gottesman, M.M. The role of cellular accumulation in determining sensitivity to platinum-based chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 495–535. [Google Scholar]
- Howell, S.B.; Safaei, R.; Larson, C.A.; Sailor, M.J. Copper Transporters and the Cellular Pharmacology of the Platinum-Containing Cancer Drugs. Mol. Pharmacol. 2010, 77, 887–894. [Google Scholar]
- Burger, H.; Loos, W.J.; Eechoute, K.; Verweij, J.; Mathijssen, R.H.J.; Wiemer, E.A.C. Drug transporters of platinum-based anticancer agents and their clinical significance. Drug Resist. Updates 2011, 14, 22–34. [Google Scholar]
- Ikari, A.; Nagatani, Y.; Tsukimoto, M.; Harada, H.; Miwa, M.; Takagi, K. Sodium-dependent glucose transporter reduces peroxynitrite and cell injury caused by cisplatin in renal tubular epithelial cells. Biochim. Biophys. Acta-Biomembr. 2005, 1717, 109–117. [Google Scholar]
- Schneider, V.; Krieger, M.L.; Bendas, G.; Jaehde, U.; Kalayda, G.V. Contribution of intracellular ATP to cisplatin resistance of tumor cells. J. Biol. Inorg. Chem. 2013, 18, 165–174. [Google Scholar]
- Hediger, M.A.; Romero, M.F.; Peng, J.B.; Rolfs, A.; Takanaga, H.; Bruford, E.A. The ABCs of solute carriers: Physiological, pathological and therapeutic implications of human membrane transport proteins—Introduction. Pflug. Arch.-Eur. J. Physiol. 2004, 447, 465–468. [Google Scholar]
- Ohrvik, H.; Thiele, D.J. The role of Ctr1 and Ctr2 in mammalian copper homeostasis and platinum-based chemotherapy. J. Trace Elem. Med. Biol. 2014. [Google Scholar] [CrossRef]
- Kuo, M.T.; Chen, H.H.W.; Song, I.S.; Savaraj, N.; Ishikawa, T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007, 26, 71–83. [Google Scholar]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar]
- Song, I.-S.; Savaraj, N.; Siddik, Z.H.; Liu, P.; Wei, Y.; Wu, C.J.; Kuo, M.T. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol. Cancer Ther. 2004, 3, 1543–1549. [Google Scholar]
- Puig, S.; Lee, J.; Lau, M.; Thiele, D.J. Biochemical and genetic analyses of yeast and human high affinity copper transporters suggest a conserved mechanism for copper uptake. J. Biol. Chem. 2002, 277, 26021–26030. [Google Scholar]
- Holzer, A.K.; Samimi, G.; Katano, K.; Naerdemann, W.; Lin, X.J.; Safaei, R.; Howell, S.B. The copper influx transporter human copper transport protein 1 regulates the uptake of cisplatin in human ovarian carcinoma cells. Mol. Pharmacol. 2004, 66, 817–823. [Google Scholar]
- Beretta, G.L.; Gatti, L.; Tinelli, S.; Corna, E.; Colangelo, D.; Zunino, F.; Perego, P. Cellular pharmacology of cisplatin in relation to the expression of human copper transporter CTR1 in different pairs of cisplatin-sensitive and -resistant cells. Biochem. Pharmacol. 2004, 68, 283–291. [Google Scholar]
- Holzer, A.K.; Manorek, G.H.; Howell, S.B. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol. Pharmacol. 2006, 70, 1390–1394. [Google Scholar]
- Holzer, A.K.; Katano, K.; Klomp, L.W.J.; Howell, S.B. Cisplatin rapidly down-regulates its own influx transporter hCTR1 in cultured human ovarian carcinoma cells. Clin. Cancer Res. 2004, 10, 6744–6749. [Google Scholar]
- Holzer, A.K.; Howell, S.B. The internalization and degradation of human copper transporter 1 following cisplatin exposure. Cancer Res. 2006, 66, 10944–10952. [Google Scholar]
- Chen, H.H.W.; Song, I.S.; Hossain, A.; Choi, M.K.; Yamane, Y.; Liang, Z.D.; Lu, J.; Wu, L.Y.H.; Siddik, Z.H.; Klomp, L.W.J.; et al. Elevated glutathione levels confer cellular sensitization to cisplatin toxicity by up-regulation of copper transporter hCtr1. Mol. Pharmacol. 2008, 74, 697–704. [Google Scholar]
- Liang, Z.D.; Long, Y.; Tsai, W.B.; Fu, S.Q.; Kurzrock, R.; Gagea-Iurascu, M.; Zhang, F.; Chen, H.H.W.; Hennessy, B.T.; Mills, G.B.; et al. Mechanistic Basis for Overcoming Platinum Resistance Using Copper Chelating Agents. Mol. Cancer Ther. 2012, 11, 2483–2494. [Google Scholar]
- Fu, S.Q.; Naing, A.; Fu, C.; Kuo, M.T.; Kurzrock, R. Overcoming Platinum Resistance through the Use of a Copper-Lowering Agent. Mol. Cancer Ther. 2012, 11, 1221–1225. [Google Scholar]
- Xu, X.J.; Duan, L.; Zhou, B.T.; Ma, R.; Zhou, H.H.; Liu, Z.Q. Genetic polymorphism of copper transporter protein 1 is related to platinum resistance in Chinese non-small cell lung carcinoma patients. Clin. Exp. Pharmacol. Physiol. 2012, 39, 786–792. [Google Scholar]
- Kim, E.S.; Tang, X.; Peterson, D.R.; Kilari, D.; Chow, C.-W.; Fujimoto, J.; Kalhor, N.; Swisher, S.G.; Stewart, D.J.; Wistuba, I.I.; et al. Copper transporter CTR1 expression and tissue platinum concentration in non-small cell lung cancer. Lung Cancer 2014, 85, 88–93. [Google Scholar]
- Pabla, N.; Murphy, R.F.; Liu, K.B.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol.-Ren. Physiol. 2009, 296, F505–F511. [Google Scholar]
- Guo, Y.; Smith, K.; Petris, M.J. Cisplatin stabilizes a multimeric complex of the human Ctr1 copper transporter—Requirement for the extracellular methionine-rich clusters. J. Biol. Chem. 2004, 279, 46393–46399. [Google Scholar]
- Arnesano, F.; Scintilla, S.; Natile, G. Interaction between platinum complexes and a methionine motif found in copper transport proteins. Angew. Chem. Int. Ed. 2007, 46, 9062–9064. [Google Scholar]
- Rabik, C.A.; Maryon, E.B.; Kasza, K.; Shafer, J.T.; Bartnik, C.M.; Dolan, M.E. Role of copper transporters in resistance to platinating agents. Cancer Chemother. Pharmacol. 2009, 64, 133–142. [Google Scholar]
- Wu, Z.Y.; Liu, Q.; Liang, X.; Yang, X.L.; Wang, N.Y.; Wang, X.H.; Sun, H.Z.; Lu, Y.; Guo, Z.J. Reactivity of platinum-based antitumor drugs towards a Met- and His-rich 20mer peptide corresponding to the N-terminal domain of human copper transporter 1. J. Biol. Inorg. Chem. 2009, 14, 1313–1323. [Google Scholar]
- Crider, S.E.; Holbrook, R.J.; Franz, K.J. Coordination of platinum therapeutic agents to met-rich motifs of human copper transport protein1. Metallomics 2010, 2, 74–83. [Google Scholar]
- Wang, X.H.; Li, H.Y.; Du, X.B.; Harris, J.; Guo, Z.J.; Sun, H.Z. Activation of carboplatin and nedaplatin by the N-terminus of human copper transporter 1 (hCTR1). Chem. Sci. 2012, 3, 3206–3215. [Google Scholar]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar]
- Boal, A.K.; Rosenzweig, A.C. Structural Biology of Copper Trafficking. Chem. Rev. 2009, 109, 4760–4779. [Google Scholar]
- Boal, A.K.; Rosenzweig, A.C. Crystal Structures of Cisplatin Bound to a Human Copper Chaperone. J. Am. Chem. Soc. 2009, 131, 14196–14197. [Google Scholar]
- Mercer, J.F.B. The molecular basis of copper-transport diseases. Trends Mol. Med. 2001, 7, 64–69. [Google Scholar]
- Komatsu, M.; Sumizawa, T.; Mutoh, M.; Chen, Z.S.; Terada, K.; Furukawa, T.; Yang, X.L.; Gao, H.; Miura, N.; Sugiyama, T.; et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000, 60, 1312–1316. [Google Scholar]
- Katano, K.; Kondo, A.; Safaei, R.; Holzer, A.; Samimi, G.; Mishima, M.; Kuo, Y.M.; Rochdi, M.; Howell, S.B. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002, 62, 6559–6565. [Google Scholar]
- Mangala, L.S.; Zuzel, V.; Schmandt, R.; Leshane, E.S.; Haider, J.B.; Armaiz-Pena, G.N.; Spannuth, W.A.; Tanaka, T.; Shahzad, M.M.K.; Lin, Y.G.; et al. Therapeutic Targeting of ATP7B in Ovarian Carcinoma. Clin. Cancer Res. 2009, 15, 3770–3780. [Google Scholar]
- Katano, K.; Safaei, R.; Samimi, G.; Holzer, A.; Rochdi, M.; Howell, S.B. The copper export pump ATP7B modulates the cellular pharmacology of carboplatin in ovarian carcinoma cells. Mol. Pharmacol. 2003, 64, 466–473. [Google Scholar]
- Samimi, G.; Katano, K.; Holzer, A.K.; Safaei, R.; Howell, S.B. Modulation of the cellular pharmacology of cisplatin and its analogs by the copper exporters ATP7A and ATP7B. Mol. Pharmacol. 2004, 66, 25–32. [Google Scholar]
- Nakagawa, T.; Inoue, Y.; Kodama, H.; Yamazaki, H.; Kawai, K.; Suemizu, H.; Masuda, R.; Iwazaki, M.; Yamada, S.; Ueyama, Y.; et al. Expression of copper-transporting P-type adenosine triphosphatase (ATP7B) correlates with cisplatin resistance in human non-small cell lung cancer xenografts. Oncol. Rep. 2008, 20, 265–270. [Google Scholar]
- Martinez-Balibrea, E.; Martinez-Cardus, A.; Musulen, E.; Gines, A.; Manzano, J.L.; Aranda, E.; Plasencia, C.; Neamati, N.; Abad, A. Increased levels of copper efflux transporter ATP7B are associated with poor outcome in colorectal cancer patients receiving oxaliplatin-based chemotherapy. Int. J. Cancer 2009, 124, 2905–2910. [Google Scholar]
- Moreno-Smith, M.; Halder, J.B.; Meltzer, P.S.; Gonda, T.A.; Mangala, L.S.; Rupaimoole, R.; Lu, C.H.; Nagaraja, A.S.; Gharpure, K.M.; Kang, Y.; et al. ATP11B mediates platinum resistance in ovarian cancer. J. Clin. Investig. 2013, 123, 2119–2130. [Google Scholar]
- Safaei, R.; Holzer, A.K.; Katano, K.; Samimi, G.; Howell, S.B. The role of copper transporters in the development of resistance to Pt drugs. J. Inorg. Biochem. 2004, 98, 1607–1613. [Google Scholar]
- Tadini-Buoninsegni, F.; Bartolommei, G.; Moncelli, M.R.; Inesi, G.; Galliani, A.; Sinisi, M.; Losacco, M.; Natile, G.; Arnesano, F. Translocation of Platinum Anticancer Drugs by Human Copper ATPases ATP7A and ATP7B. Angew. Chem. Int. Ed. 2014, 53, 1297–1301. [Google Scholar]
- Ciarimboli, G. Membrane Transporters as Mediators of Cisplatin Side-effects. Anticancer Res. 2014, 34, 547–550. [Google Scholar]
- Wensing, K.U.; Ciarimboli, G. Saving ears and kidneys from cisplatin. Anticancer Res. 2013, 33, 4183–4188. [Google Scholar]
- Ciarimboli, G.; Ludwig, T.; Lang, D.F.; Pavenstadt, H.; Koepsell, H.; Piechota, H.J.; Haier, J.; Jaehde, U.; Zisowsky, J.; Schlatter, E. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am. J. Pathol. 2005, 167, 1477–1484. [Google Scholar]
- Ciarimboli, G.; Deuster, D.; Knief, A.; Sperling, M.; Holtkamp, M.; Edemir, B.; Pavenstadt, H.; Lanvers-Kaminsky, C.; Zehnhoff-Dinnesen, A.A.; Schinkel, A.H.; et al. Organic Cation Transporter 2 Mediates Cisplatin-Induced Oto- and Nephrotoxicity and Is a Target for Protective Interventions. Am. J. Pathol. 2010, 176, 1169–1180. [Google Scholar]
- Katsuda, H.; Yamashita, M.; Katsura, H.; Yu, J.; Waki, Y.; Nagata, N.; Sai, Y.; Miyamoto, K. Protecting Cisplatin-Induced Nephrotoxicity with Cimetidine Does Not Affect Antitumor Activity. Biol. Pharm. Bull. 2010, 33, 1867–1871. [Google Scholar]
- Yonezawa, A.; Masuda, S.; Yokoo, S.; Katsura, T.; Inui, K. Cisplatin and Oxaliplatin, but Not Carboplatin and Nedaplatin, Are Substrates for Human Organic Cation Transporters (SLC22A1-3 and Multidrug and Toxin Extrusion Family). J. Pharmacol. Exp. Ther. 2006, 319, 879–886. [Google Scholar]
- Li, Q.; Peng, X.; Yang, H.; Rodriguez, J.-A.; Shu, Y. Contribution of organic cation transporter 3 to cisplatin cytotoxicity in human cervical cancer cells. J. Pharm. Sci. 2012, 101, 394–404. [Google Scholar]
- Yokoo, S.; Masuda, S.; Yonezawa, A.; Terada, T.; Katsura, T.; Inui, K. Significance of Organic Cation Transporter 3 (SLC22A3) Expression for the Cytotoxic Effect of Oxaliplatin in Colorectal Cancer. Drug Metab. Dispos. 2008, 36, 2299–2306. [Google Scholar]
- Yonezawa, A.; Inui, K. Importance of the multidrug and toxin extrusion MATE/SLC47A family to pharmacokinetics, pharmacodynamics/toxicodynamics and pharmacogenomics. Br. J. Pharm. 2011, 164, 1817–1825. [Google Scholar]
- Ciarimboli, G. Membrane Transporters as Mediators of Cisplatin Effects and Side Effects. Scientifica 2012. [Google Scholar] [CrossRef]
- Yokoo, S.; Yonezawa, A.; Masuda, S.; Fukatsu, A.; Katsura, T.; Inui, K.I. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem. Pharmacol. 2007, 74, 477–487. [Google Scholar]
- Li, Q.; Guo, D.; Dong, Z.Q.; Zhang, W.; Zhang, L.; Huang, S.M.; Polli, J.E.; Shu, Y. Ondansetron can enhance cisplatin-induced nephrotoxicity via inhibition of multiple toxin and extrusion proteins (MATEs). Toxicol. Appl. Pharmacol. 2013, 273, 100–109. [Google Scholar]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar]
- Timerbaev, A.R.; Hartinger, C.G.; Aleksenko, S.S.; Keppler, B.K. Interactions of antitumor metallodrugs with serum proteins: Advances in characterization using modern analytical methodology. Chem. Rev. 2006, 106, 2224–2248. [Google Scholar]
- Li, H.; Qian, Z.M. Transferrin/transferrin receptor-mediated drug delivery. Med. Res. Rev. 2002, 22, 225–250. [Google Scholar]
- Heffeter, P.; Pongratz, M.; Steiner, E.; Chiba, P.; Jakupec, M.A.; Elbling, L.; Marian, B.; Körner, W.; Sevelda, F.; Micksche, M.; et al. Intrinsic and Acquired Forms of Resistance against the Anticancer Ruthenium Compound KP1019 [Indazolium trans-[tetrachlorobis(1H-indazole)ruthenate (III)] (FFC14A). J. Pharm. Exp. Ther. 2005, 312, 281–289. [Google Scholar]
- Fojo, T.; Bates, S. Strategies for reversing drug resistance. Oncogene 2003, 22, 7512–7523. [Google Scholar]
- Ang, W.H.; Casini, A.; Sava, G.; Dyson, P.J. Organometallic ruthenium-based antitumor compounds with novel modes of action. J. Organomet. Chem. 2011, 696, 989–998. [Google Scholar]
- Peacock, A.F.A.; Sadler, P.J. Medicinal Organometallic Chemistry: Designing Metal Arene Complexes as Anticancer Agents. Chem. Asian J. 2008, 3, 1890–1899. [Google Scholar]
- Yan, Y.K.; Melchart, M.; Habtemariam, A.; Sadler, P.J. Organometallic chemistry, biology and medicine: Ruthenium arene anticancer complexes. Chem. Commun. 2005, 4764–4776. [Google Scholar]
- Casini, A.; Gabbiani, C.; Sorrentino, F.; Rigobello, M.P.; Bindoli, A.; Geldbach, T.J.; Marrone, A.; Re, N.; Hartinger, C.G.; Dyson, P.J.; et al. Emerging Protein Targets for Anticancer Metallodrugs: Inhibition of Thioredoxin Reductase and Cathepsin B by Antitumor Ruthenium (II)-Arene Compounds. J. Med. Chem. 2008, 51, 6773–6781. [Google Scholar]
- Romero-Canelon, I.; Pizarro, A.M.; Habtemariam, A.; Sadler, P.J. Contrasting cellular uptake pathways for chlorido and iodido iminopyridine ruthenium arene anticancer complexes. Metallomics 2012, 4, 1271–1279. [Google Scholar]
- Ronconi, L.; Fregona, D. The Midas touch in cancer chemotherapy: From platinum- to gold-dithiocarbamato complexes. Dalton Trans. 2009, 48, 10670–10680. [Google Scholar]
- Messori, L.; Casini, A. Molecular mechanism and proposed targets for selected anticancer gold compounds. Curr. Top. Med. Chem. 2011, 11, 2647–2660. [Google Scholar]
- Bertrand, B.; Casini, A. A golden future in medicinal inorganic chemistry: The promise of anticancer gold organometallic compounds. Dalton Trans. 2014, 43, 4209–4219. [Google Scholar]
- Oehninger, L.; Rubbiani, R.; Ott, I. N-Heterocyclic carbene metal complexes in medicinal chemistry. Dalton Trans. 2013, 42, 3269–3284. [Google Scholar]
- Berners-Price, S.J.; Filipovska, A. The design of gold-based, mitochondria-targeted chemotherapeutics. Aust. J. Chem. 2008, 61, 661–668. [Google Scholar]
- Humphreys, A.S.; Filipovska, A.; Berners-Price, S.J.; Koutsantonis, G.A.; Skelton, B.W.; White, A.H. Gold(I) chloride adducts of 1,3-bis(di-2-pyridylphosphino) propane: Synthesis, structural studies and antitumour activity. Dalton Trans. 2007, 43, 4943–4950. [Google Scholar]
- Trapp, S.; Horobin, R. A predictive model for the selective accumulation of chemicals in tumor cells. Eur. Biophys. J. 2005, 34, 959–966. [Google Scholar]
- Modica-Napolitano, J.S.; Aprille, J.R. Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv. Drug Deliv. Rev. 2001, 49, 63–70. [Google Scholar]
- Ross, M.F.; Kelso, G.F.; Blaikie, F.H.; James, A.M.; Cocheme, H.M.; Filipovska, A.; da Ros, T.; Hurd, T.R.; Smith, R.A.J.; Murphy, M.P. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry (Mosc.) 2005, 70, 222–230. [Google Scholar]
- Davis, S.; Weiss, M.J.; Wong, J.R.; Lampidis, T.J.; Chen, L.B. Mitochondrial and Plasma-Membrane Potentials Cause Unusual Accumulation and Retention of Rhodamine-123 by Human-Breast Adenocarcinoma-Derived Mcf-7 Cells. J. Biol. Chem. 1985, 260, 3844–3850. [Google Scholar]
- Summerhayes, I.C.; Lampidis, T.J.; Bernal, S.D.; Nadakavukaren, J.J.; Nadakavukaren, K.K.; Shepherd, E.L.; Chen, L.B. Unusual Retention of Rhodamine-123 by Mitochondria in Muscle and Carcinoma-Cells. Proc. Natl. Acad. Sci. USA 1982, 79, 5292–5296. [Google Scholar]
- Lampidis, T.J.; Castello, C.; Delgiglio, A.; Pressman, B.C.; Viallet, P.; Trevorrow, K.W.; Valet, G.K.; Tapiero, H.; Savaraj, N. Relevance of the Chemical Charge of Rhodamine Dyes to Multiple-Drug Resistance. Biochem. Pharmacol. 1989, 38, 4267–4271. [Google Scholar]
- Kurtoglu, M.; Lampidis, T.J. From delocalized lipophilic cations to hypoxia: Blocking tumor cell mitochondrial function leads to therapeutic gain with glycolytic inhibitors. Mol. Nutr. Food Res. 2009, 53, 68–75. [Google Scholar]
- Rubbiani, R.; Can, S.; Kitanovic, I.; Alborzinia, H.; Stefanopoulou, M.; Kokoschka, M.; Monchgesang, S.; Sheldrick, W.S.; Wolfl, S.; Ott, I. Comparative in Vitro Evaluation of N-Heterocyclic Carbene Gold(I) Complexes of the Benzimidazolylidene Type. J. Med. Chem. 2011, 54, 8646–8657. [Google Scholar]
- Snyder, R.M.; Mirabelli, C.K.; Crooke, S.T. Cellular-Association, Intracellular-Distribution, and Efflux of Auranofin via Sequential Ligand-Exchange Reactions. Biochem. Pharmacol. 1986, 35, 923–932. [Google Scholar]
- Kaps, L.; Biersack, B.; Muller-Bunz, H.; Mahal, K.; Munzner, J.; Tacke, M.; Mueller, T.; Schobert, R. Gold(I)-NHC complexes of antitumoral diarylimidazoles: Structures, cellular uptake routes and anticancer activities. J. Inorg. Biochem. 2012, 106, 52–58. [Google Scholar]
- Liu, Z.; Sadler, P.J. Organoiridium Complexes: Anticancer Agents and Catalysts. Acc. Chem. Res. 2014, 47, 1174–1185. [Google Scholar]
- Novohradsky, V.; Liu, Z.; Vojtiskova, M.; Sadler, P.J.; Brabec, V.; Kasparkova, J. Mechanism of cellular accumulation of an iridium(iii) pentamethylcyclopentadienyl anticancer complex containing a C,N-chelating ligand. Metallomics 2014, 6, 682–690. [Google Scholar]
- Gibalova, L.; Seres, M.; Rusnak, A.; Ditte, P.; Labudova, M.; Uhrik, B.; Pastorek, J.; Sedlak, J.; Breier, A.; Sulova, Z. P-glycoprotein depresses cisplatin sensitivity in L1210 cells by inhibiting cisplatin-induced caspase-3 activation. Toxicol. In Vitro 2012, 26, 435–444. [Google Scholar]
- Sharp, S.Y.; Smith, V.; Hobbs, S.; Kelland, L.R. Lack of a role for MRP1 in platinum drug resistance in human ovarian cancer cell lines. Br. J. Cancer 1998, 78, 175–180. [Google Scholar]
- Monney, A.; Albrecht, M. Transition metal bioconjugates with an organometallic link between the metal and the biomolecular scaffold. Coord. Chem. Rev. 2013, 257, 2420–2433. [Google Scholar]
- Rubio-Aliaga, I.; Daniel, H. Peptide transporters and their roles in physiological processes and drug disposition. Xenobiotica 2008, 38, 1022–1042. [Google Scholar]
- Kouodom, M.N.; Ronconi, L.; Celegato, M.; Nardon, C.; Marchio, L.; Dou, Q.P.; Aldinucci, D.; Formaggio, F.; Fregona, D. Toward the Selective Delivery of Chemotherapeutics into Tumor Cells by Targeting Peptide Transporters: Tailored Gold-Based Anticancer Peptidomimetics. J. Med. Chem. 2012, 55, 2212–2226. [Google Scholar]
- Nardon, C.; Schmitt, S.M.; Yang, H.J.; Zuo, J.; Fregona, D.; Dou, Q.P. Gold(III)-Dithiocarbamato Peptidomimetics in the Forefront of the Targeted Anticancer Therapy: Preclinical Studies against Human Breast Neoplasia. PLoS One 2014, 9, e84248. [Google Scholar]
- Hartinger, C.G.; Nazarov, A.A.; Ashraf, S.M.; Dyson, P.J.; Keppler, B.K. Carbohydrate-Metal Complexes and Their Potential as Anticancer Agents. Curr. Med. Chem. 2008, 15, 2574–2591. [Google Scholar]
- Vergara, E.; Cerrada, E.; Clavel, C.; Casini, A.; Laguna, M. Thiolato gold(I) complexes containing water-soluble phosphane ligands: A characterization of their chemical and biological properties. Dalton Trans. 2011, 40, 10927–10935. [Google Scholar]
- De Graaf, I.A.M.; Olinga, P.; de Jager, M.H.; Merema, M.T.; de Kanter, R.; van de Kerkhof, E.G.; Groothuis, G.M.M. Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat. Protoc. 2010, 5, 1540–1551. [Google Scholar]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spreckelmeyer, S.; Orvig, C.; Casini, A. Cellular Transport Mechanisms of Cytotoxic Metallodrugs: An Overview beyond Cisplatin. Molecules 2014, 19, 15584-15610. https://doi.org/10.3390/molecules191015584
Spreckelmeyer S, Orvig C, Casini A. Cellular Transport Mechanisms of Cytotoxic Metallodrugs: An Overview beyond Cisplatin. Molecules. 2014; 19(10):15584-15610. https://doi.org/10.3390/molecules191015584
Chicago/Turabian StyleSpreckelmeyer, Sarah, Chris Orvig, and Angela Casini. 2014. "Cellular Transport Mechanisms of Cytotoxic Metallodrugs: An Overview beyond Cisplatin" Molecules 19, no. 10: 15584-15610. https://doi.org/10.3390/molecules191015584