Design and Synthesis of C-Terminal Modified Cyclic Peptides as VEGFR1 Antagonists

Abstract
:1. Introduction
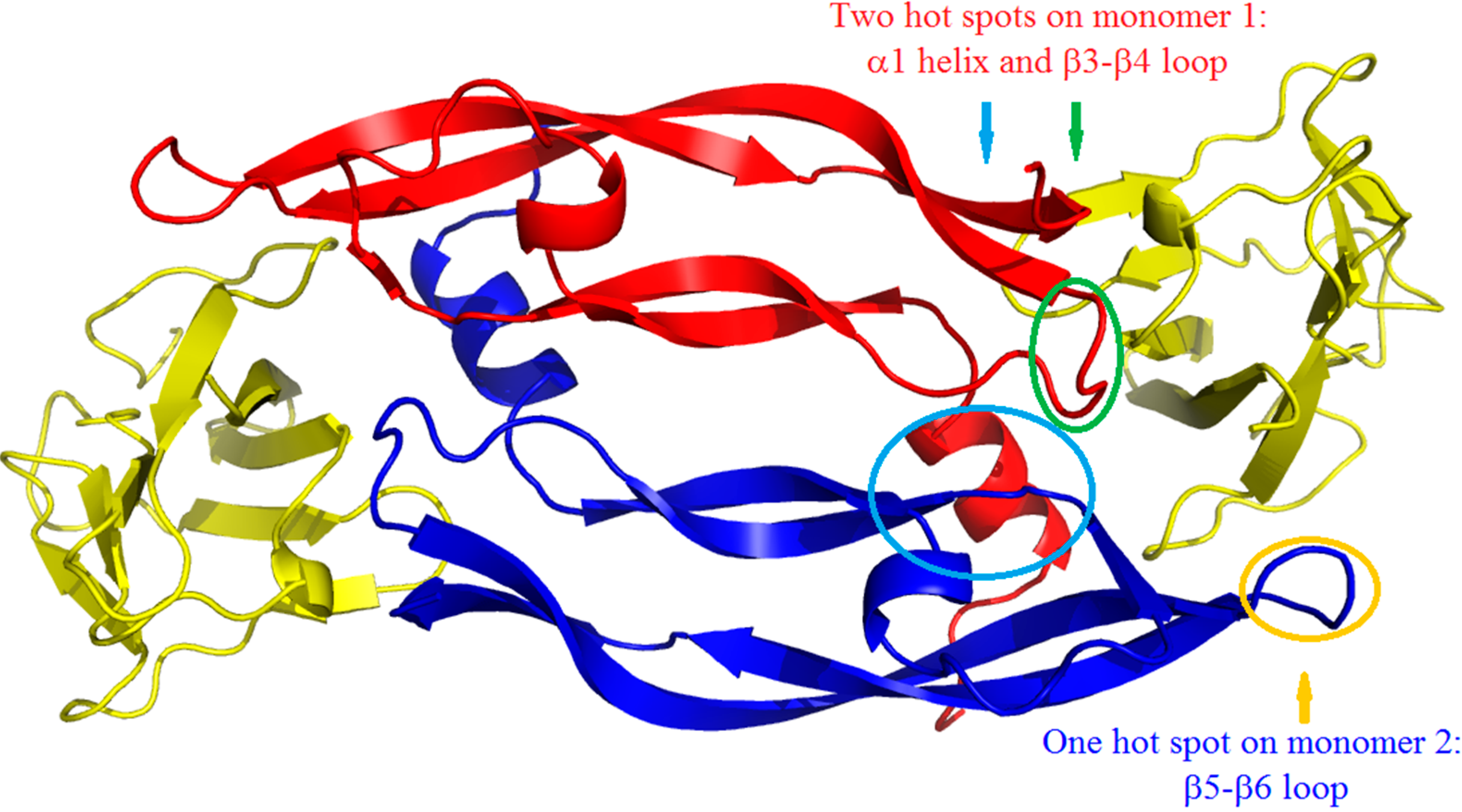
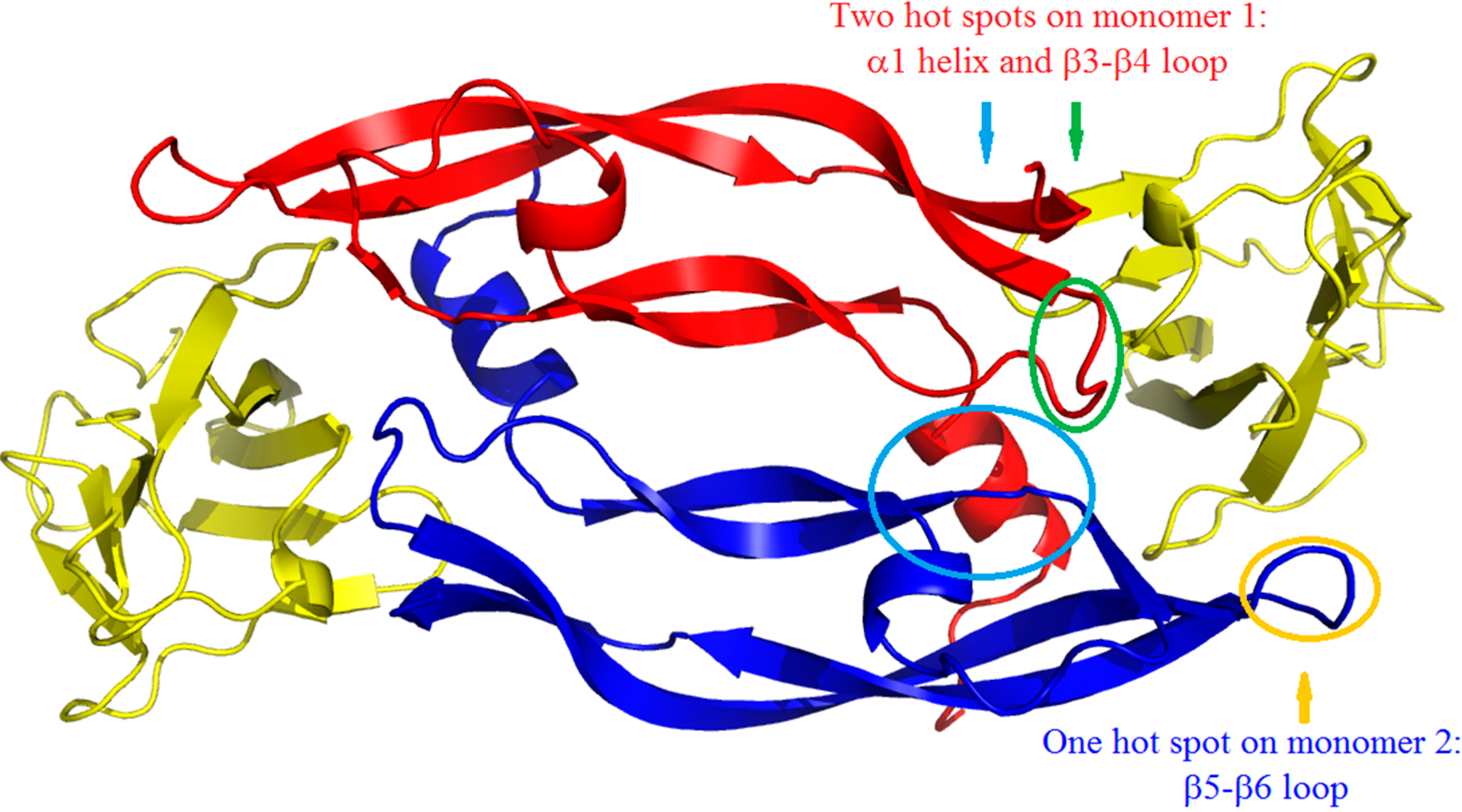
2. Results and Discussion
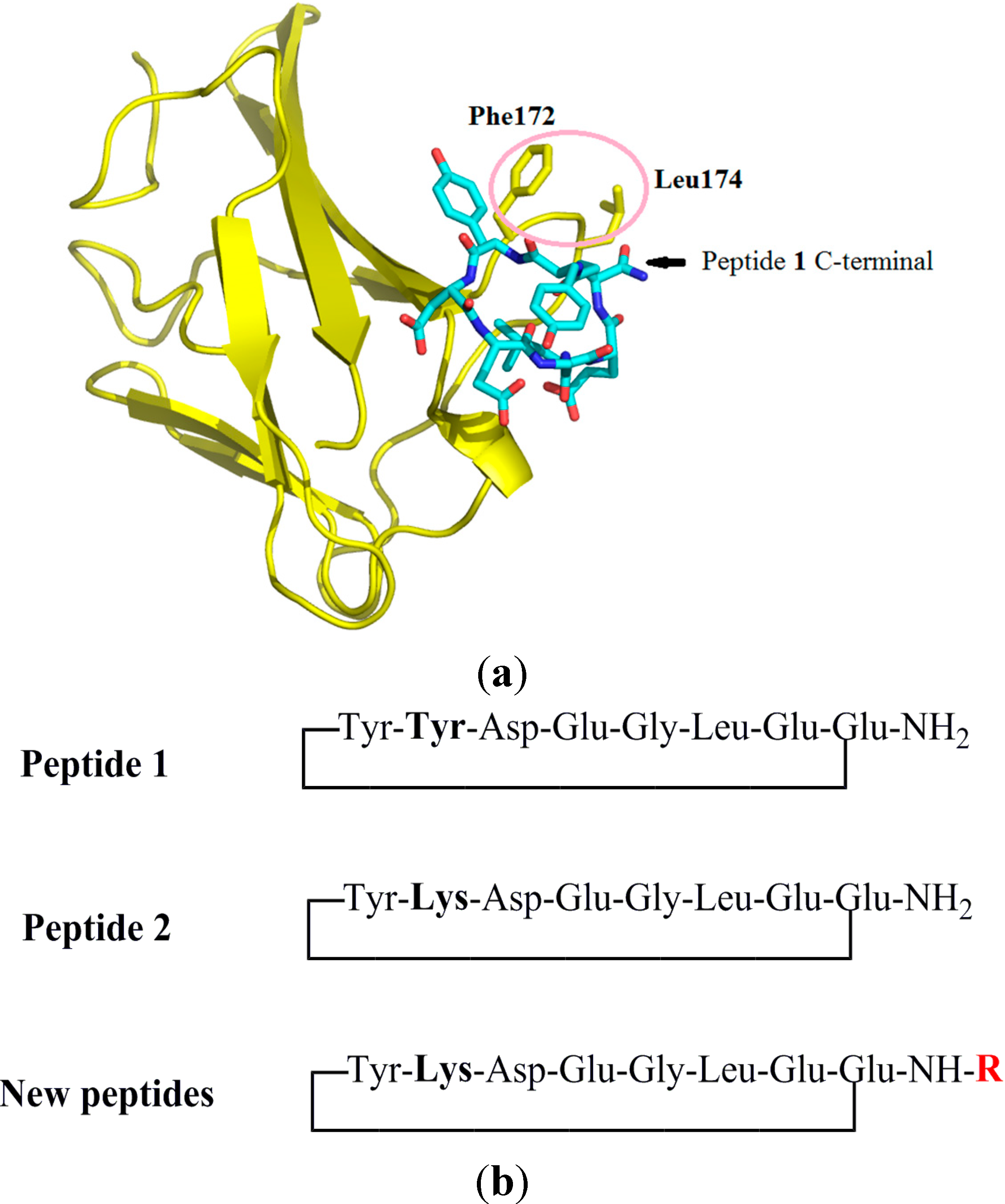
2.1. Design of Peptides
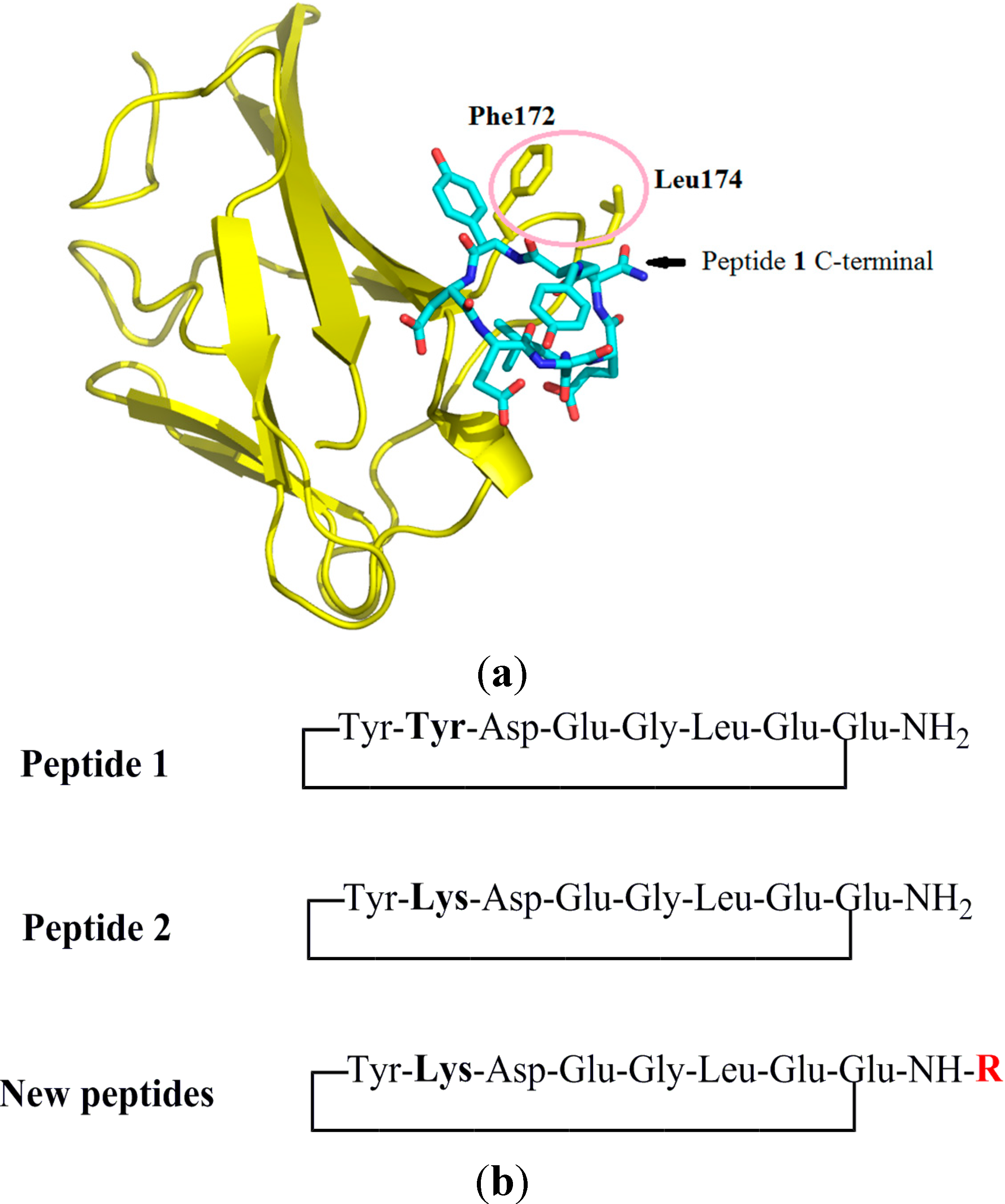
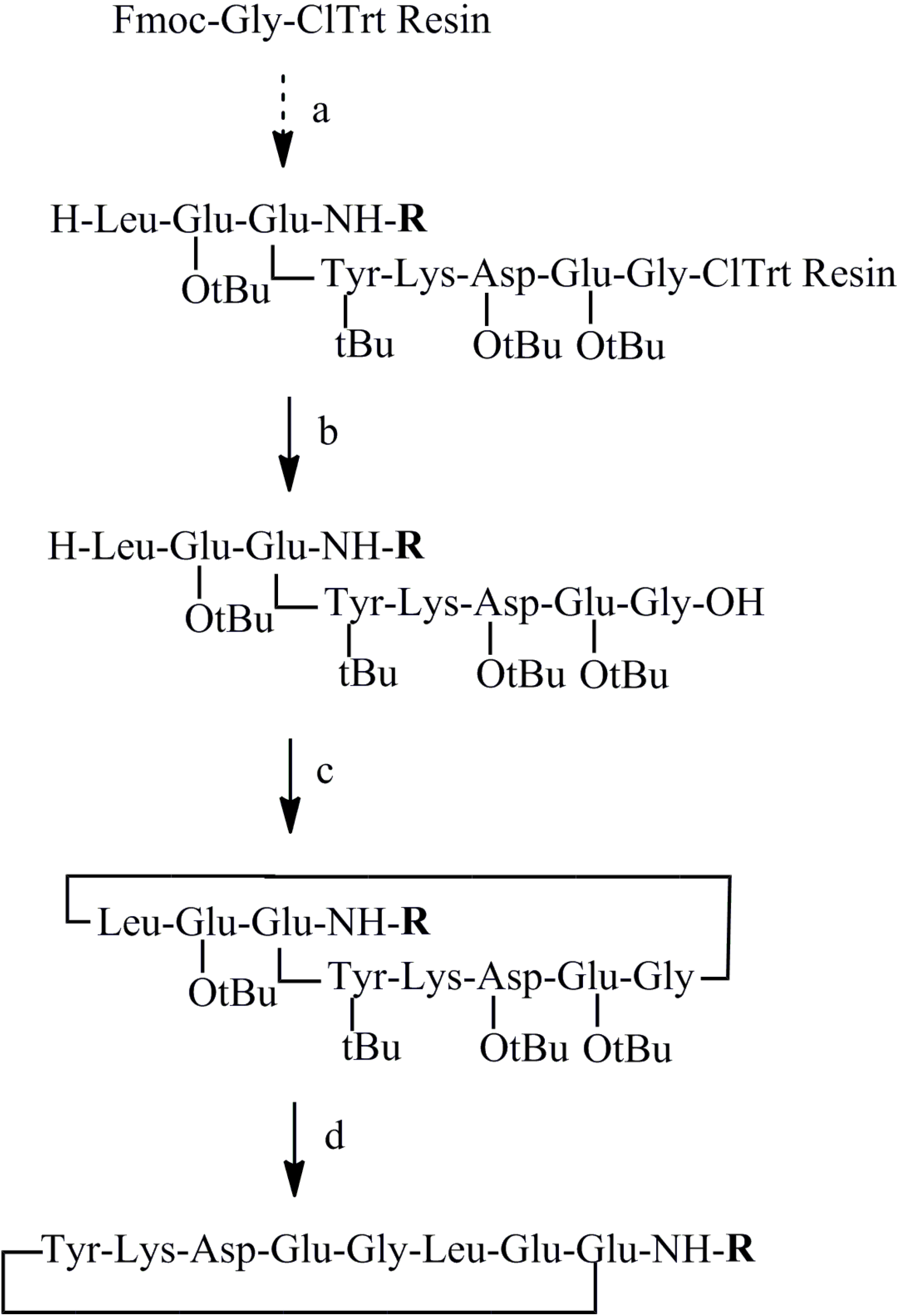
2.2. Synthesis of Peptides

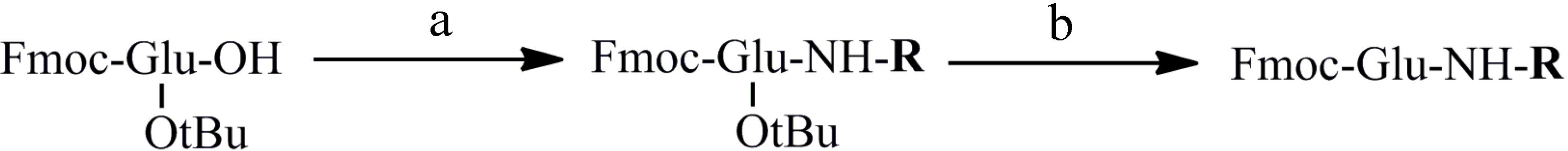

2.3. Evaluation of the Inhibitory Effect of Peptides on the VEGF-VEGFR1 Interaction

{kind=link}
{kind=link}
{kind=link}
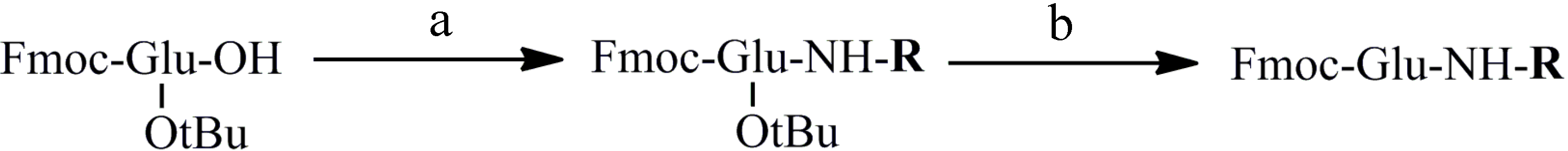{kind=link}
{kind=link}

| Peptide | R | Displacement 100 µM (%) | Peptide | R | Displacement 100 µM (%) |
|---|---|---|---|---|---|
| 2 | H [26] | 12 | 11 | -CH2-CH2-CH2-Ph | 12 |
| 3 | pentyl | 19 | 12 | -CH2-CH(Ph)2 | 20 |
| 4 | isobutyl | 11 | 13 | (1-naphthalene)methyl | 24 |
| 5 | allyl | 35 | 14 |  | 40 |
| 6 | (2-hydroxy)ethyl | 23 | 15 | 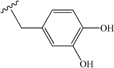 | 58 |
| 7 | cyclohexyl | 9 | 16 | 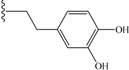 | 53 |
| 8 | (cyclohexyl)methyl | 7 | 17 |  | 42 |
| 9 | -CH2-Ph (benzyl) | 39 | 18 | 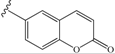 | 14 |
| 10 | -CH2-CH2-Ph | 43 | 19 | 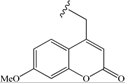 | 68 |
| Peptide | Displacement (%) | ||
|---|---|---|---|
| 100 µM | 50 µM | 30 µM | |
| 15 | 58 | 37 | 14 |
| 16 | 53 | 31 | NA |
| 19 | 68 | 41 | 11 |
3. Experimental Section
3.1. Preparation of 4-Aminomethyl-7-methoxy-chromen-2-one Hydrochloride
3.2. General Method for the Preparation of Substituted Glutamic Amides (Compounds 1–17)
3.3. New Synthesis of Reference Peptide 2
3.4. Synthesis of C-Terminal Substituted Cyclic Peptides (Peptides 3–19)
| Peptide | Yield (%) | MS Found | Rt (Minutes) |
|---|---|---|---|
| 2 | 10 (8.8 [26]) | 963 (M+H+) | 12.5 (10%–60% B in 30 min) |
| 3 | 29.0 | 1034 (M+H+) | 14.8 (20%–80% B in 30 min) |
| 4 | 12.7 | 1020 (M+H+) | 15.1 (20%–80% B in 30 min) |
| 5 | 3.5 | 1004 (M+H+) | 10.2 (20%–70% B in 20 min) |
| 6 | 7.8 | 1008 (M+H+) | 11.0 (10%–60% B in 20 min) |
| 7 | 23.4 | 1046 (M+H+) | 18.0 (10%–60% B in 20 min) |
| 8 | 10.5 | 1060 (M+H+) | 15.4 (20%–70% B in 20 min) |
| 9 | 6.5 | 1054 (M+H+) | 13.0 (20%–70% B in 20 min) |
| 10 | 30.9 | 1067 (M+H+) | 14.0 (20%–80% B in 30 min) |
| 11 | 15.5 | 1081 (M+H+) | 15.7 (20%–70% B in 20 min) |
| 12 | 11.8 | 1144 (M+H+) | 17.9 (20%–70% B in 20 min) |
| 13 | 37 | 1125 (M+Na+) | 18.5 (20%–80% B in 30 min) |
| 14 | 20.3 | 1083 (M+H+) | 13.7 (20%–80% B in 30 min) |
| 15 | 11.1 | 1085 (M+H+) | 13.5 (10%–60% B in 20 min) |
| 16 | 30.8 | 1099 (M+H+) | 14.4 (10%–60% B in 20 min) |
| 17 | 29.2 | 1098 (M+H+) | 12.8 (20%–70% B in 20 min) |
| 18 | 3.3 | 1108 (M+H+) | 12.2 (20%–70% B in 20 min) |
| 19 | 26.6 | 1152 (M+H+) | 13.5 (20%–70% B in 20 min) |
3.5. ELISA VEGF-VEGFR1 Binding Inhibition Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar]
- Kowanetz, M.; Ferrara, N. Vascular endothelial growth factor signaling pathways: Therapeutic perspective. Clin. Cancer Res. 2006, 17, 5018–5022. [Google Scholar]
- Kieran, M.W.; Kalluri, R.; Cho, Y.J. The VEGF pathway in cancer and disease: Responses, resistance, and the path forward. CSH-Perspect. Med. 2012, 5. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a therapeutic target. Nature 2005, 438, 967–974. [Google Scholar]
- Shibuya, M. VEGF-VEGFR signals in health and disease. Biomol. Ther. 2014, 22, 1–9. [Google Scholar]
- Goncalves, V.; Gautier, B.; Lenoir, C.; Garbay, C.; Vidal, M.; Inguimbert, N. Peptides as antagonists of the VEGF receptors. Pharmachem. 2006. Available online: http://www.b5srl.com/articles_pharmachem.php?search_ok=1&year_search=32042&OrderFld=3&ot=R&az=A&author_search=2590&company_search=+&num_search=32142&topic_search=31655&x=17&y=10 (accessed on 23 September 2014).
- Mullard, A. Protein-protein interaction inhibitors get into the groove. Nat. Rev. Drug Discov. 2012, 11, 173–175. [Google Scholar]
- Aeluri, M.; Chamakuri, S.; Dasari, B.; Guduru, S.K.R.; Jimmidi, R.; Jogula, S.; Arya, P. Small molecule modulators of protein-protein interactions: Selected case studies. Chem. Rev. 2014, 114, 4640–4694. [Google Scholar]
- Wiesmann, C.; Fuh, G.; Christinger, H.W.; Eigenbrot, C.; Wells, J.A.; de Vos, A.M. Crystal structure at 1.7 Å resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell 1997, 91, 695–704. [Google Scholar]
- Christinger, H.W.; Germaine, F.; de Vos, A.M.; Wiesmann, C. Crystal structure of PlGF in complex with domain 2 of VEGFR1. J. Biol. Chem. 2003, 279, 10382–10388. [Google Scholar]
- Iyer, S.; Darley, P.I.; Acharya, K.R. Structural insights into the binding of vascular endothelial growth factor-B by VEGFR-1(D2): Recognition and specificity. J. Biol. Chem. 2010, 285, 23779–23789. [Google Scholar]
- Leppänen, V.M.; Prota, A.E.; Jeltsch, M.; Anisimov, A.; Kalkkinen, N.; Strandin, T.; Lankinen, H.; Goldman, A.; Ballmer-Hofer, K.; Alitalo, K. Structural determinants of growth factor binding and specificity by VEGF receptor 2. Proc. Natl. Acad. Sci. USA 2010, 107, 2425–2430. [Google Scholar]
- D’Andrea, L.D.; Iaccarino, G.; Fattorusso, R.; Sorriento, D.; Carannante, C.; Capasso, D.; Trimarco, B.; Pedone, C. Targeting angiogenesis: Structural characterization and biological properties of a de novo engineered VEGF mimicking peptide. Proc. Natl. Acad. Sci. USA. 2005, 102, 14215–14220. [Google Scholar]
- Goncalves, V.; Gautier, B.; Garbay, C.; Vidal, M.; Inguimbert, N. Structure-based design of a bicyclic peptide antagonist of the vascular endothelial growth factor receptors. J. Pept. Sci. 2008, 14, 767–772. [Google Scholar]
- García-Aranda, M.I.; González-López, S.; Santiveri, C.M.; Gagey-Eilstein, N.; Reille-Seroussi, M.; Martín-Martínez, M.; Inguimbert, N.; Vidal, M.; García-López, M.T.; Jiménez, M.A.; et al. Helical peptides from VEGF and Vammin hotspots for modulating the VEGF-VEGFR interaction. Org. Biomol. Chem. 2013, 11, 1896–1905. [Google Scholar]
- Vicari, D.; Foy, K.C.; Liotta, E.M.; Kaumaya, P.T. Engineered conformation-dependent VEGF peptide mimics are effective in inhibiting VEGF signaling pathways. J. Biol. Chem. 2011, 286, 13612–13625. [Google Scholar]
- Zilberberg, L.; Shinkaruk, S.; Lequin, O.; Rousseau, B.; Hagedorn, M.; Costa, F.; Caronzolo, D.; Balke, M.; Canron, X.; Convert, O.; et al. Structure and inhibitory effects on angiogenesis and tumor development of a new vascular endothelial growth inhibitor. J. Biol. Chem. 2003, 278, 35564–35573. [Google Scholar]
- García-Aranda, M.I.; Marrero, P.; Gautier, B.; Martín-Martínez, M.; Inguimbert, N.; Vidal, M.; García-López, M.T.; Jiménez, M.A.; González-Muñiz, R.; Pérez de Vega, M.J. Parallel solid-phase synthesis of a small library of linear and hydrocarbon-bridged analogues of VEGF(81–91): Potential biological tools for studying the VEGF/VEGFR-1 interaction. Bioorg. Med. Chem. 2011, 19, 1978–1986. [Google Scholar]
- García-Aranda, M.I.; Mirassou, Y.; Gautier, B.; Martín-Martínez, M.; Inguimbert, N.; Vidal, M.; García-López, M.T.; Jiménez, M.A.; González-Muñiz, R.; Pérez de Vega, M.J. Disulfide and amide-bridged cyclic peptide analogues of the VEGF81–91 fragment: Synthesis, conformational analysis and biological evaluation. Bioorg. Med. Chem. 2011, 19, 7526–7533. [Google Scholar]
- Diana, D.; Basile, A.; de Rosa, L.; di Stasi, R.; Auriemma, S.; Arra, C.; Pedone, C.; Turco, M.C.; Fattorusso, R.; D’Andrea, L.D. β-Hairpin peptide that targets vascular endothelial growth factor (VEGF) receptors: Design, NMR characterization, and biological activity. J. Biol. Chem. 2011, 286, 41680–41691. [Google Scholar]
- Bello, L.; Lucini, V.; Costa, F.; Pluderi, M.; Giussani, C.; Acerbi, F.; Carrabba, G.; Pannacci, M.; Caronzolo, D.; Grosso, S.; et al. Combinatorial administration of molecules that simultaneously inhibit angiogenesis and invasion leads to increased therapeutic efficacy in mouse models of malignant glioma. Clin. Cancer Res. 2004, 10, 4527–4537. [Google Scholar]
- Basile, A.; Del Gatto, A.; Diana, D.; di Stasi, R.; Falco, A.; Festa, M.; Rosati, A.; Barbieri, A.; Franco, R.; Arra, C.; et al. Characterization of a designed vascular endothelial growth factor receptor antagonist helical peptide with antiangiogenic activity in vivo. J. Med. Chem. 2011, 54, 1391–1400. [Google Scholar]
- Finetti, F.; Basile, A.; Capasso, D.; di Gaetano, S.; di Stasi, R.; Pascale, M.; Turco, C.M.; Ziche, M.; Morbidelli, L.; D’Andrea, L.D. Functional and pharmacological characterization of a VEGF mimetic peptide on reparative angiogenesis. Biochem. Pharmacol. 2012, 84, 303–311. [Google Scholar]
- Goncalves, V.; Gautier, B.; Coric, P.; Bouaziz, S.; Lenoir, C.; Garbay, C.; Vidal, M.; Inguimbert, N. Rational design, structure, and biological evaluation of cyclic peptides mimicking the vascular endothelial growth factor. J. Med. Chem. 2007, 50, 5135–5146. [Google Scholar]
- Gautier, B.; Goncalves, V.; Diana, D.; di Stasi, R.; Teillet, F.; Lenoir, C.; Huguenot, F.; Garbay, C.; Fattorusso, R.; D’Andrea, L.D.; et al. Biochemical and structural analysis of the binding determinants of a vascular endothelial growth factor receptor peptidic antagonist. J. Med. Chem. 2010, 53, 4428–4440. [Google Scholar]
- Jia, H.; Aqil, R.; Cheng, L.; Chapman, C.; Shaikh, S.; Jarvis, A.; Chan, A.W.; Hartzoulakis, B.; Evans, I.M.; Frolov, A.; et al. N-terminal modification of VEGF-A C terminus-derived peptides delineates structural features involved in neuropilin-1 binding and functional activity. ChemBioChem 2014, 26, 1161–1170. [Google Scholar]
- Yamamoto, T.; Nair, P.; Davis, P.; Ma, S.W.; Navratilova, E.; Moye, S.; Tumati, S.; Lai, J.; Vanderah, T.W.; Yamamura, H.I.; et al. Design, synthesis, and biological evaluation of novel bifunctional C-terminal-modified peptides for delta/mu opioid receptor agonists and neurokinin-1 receptor antagonists. J. Med. Chem. 2007, 50, 2779–2786. [Google Scholar]
- Gaucher, J.F.; Reille-Seroussi, M.; Gagey-Eilstein, N.; Broussy, S.; Coric, P.; Seijo, B.; Lascombe, M.-B.; Gautier, B.; Liu, W.-Q.; Huguenot, F.; et al. Metal-Induced vascular Endothelial Growth Factor Receptor 1 Dimerization Competes with VEGF-A Binding. J. Biol. Chem. 2014. submitted. [Google Scholar]
- Johnson, T.; Liley, M.J.; Cheeseright, T.J.; Begum, F. Problems in the synthesis of cyclic peptides through use of the Dmab protecting group. J. Chem. Soc. Perkin Trans. 1 2000, 43, 2811–2820. [Google Scholar]
- Reczek, J.J.; Rebolini, E.; Urbach, A.R. Solid-phase synthesis of peptide-Viologen conjugates. J. Org. Chem. 2010, 75, 2111–2114. [Google Scholar]
- Gausepohl, H.; Pieles, U.; Frank, R. Schiff base analog formation during in situ activation by HBTU and TBTU. In Peptides, Chemistry and Biology, Proceedings of the 12th American Peptide Symposium, Cambridge, MA, USA, 16–21 June 1991; Smith, J.A., Rivier, J.E., Eds.; ESCOM: Leiden, The Netherlands, 1992; pp. 523–524. [Google Scholar]
- Lim, N.C.; Pavlova, S.V.; Brückner, C. Squaramide hydroxamate-based chemidosimeter responding to iron(III) with a fluorescence intensity increase. Inorg. Chem. 2009, 48, 1173–1182. [Google Scholar]
- Chang, C.-D.; Waki, M.; Ahmad, M.; Meienhofer, J.; Lundell, E.O.; Haug, J.D. Preparation and properties of Nα-9-fluorenylmethyloxycarbonyl-amino acids bearing tert.-butyl side chain protection. Int. J. Pept. Protein Res. 1980, 15, 59–66. [Google Scholar]
- Ye, Y.-H.; Gao, X.-M.; Liu, M.; Tang, Y.-C.; Tian, G.-L. Studies on the synthetic methodology of head to tail cyclization of linear peptides. Lett. Pept. Sci. 2003, 10, 571–579. [Google Scholar]
- Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Oxyma: An efficient additive for peptide synthesis to replace the benzotriazole-based HOBt and HOAt with a lower risk of explosion. Chem. Eur. J. 2009, 15, 9394–9403. [Google Scholar]
- Spengler, J.; Fernandez-Llamazares, A.-I.; Albericio, F. Use of an internal reference for the quantitative HPLC-UV analysis of solid-phase reactions: A case study of 2-chlorotrityl chloride resin. ACS Comb. Sci. 2013, 15, 229–234. [Google Scholar]
- Goncalves, V.; Gautier, B.; Garbay, C.; Vidal, M.; Inguimbert, N. Development of a chemiluminescent screening assay for detection of vascular endothelial growth factor receptor 1 ligands. Anal. Biochem. 2007, 366, 108–110. [Google Scholar] [Green Version]
- Chen, Y.-Y.; Brown, N.J.; Jones, R.; Lewis, C.E.; Mujamammi, A.H.; Muthana, M.; Seed, M.P.; Barker, M.D. A peptide derived from TIMP-3 inhibits multiple angiogenic growth factor receptors and tumour growth and inflammatory arthritis in mice. Angiogenesis 2014, 17, 207–219. [Google Scholar]
- Djordjevic, S.; Driscoll, P.C. Targeting VEGF signaling via the neuropilin co-receptor. Drug Discov. Today 2013, 18, 447–455. [Google Scholar]
- Nakamura, K.; Hanna, I.H.; Cai, H.; Nishimura, Y.; Williams, K.M.; Guengerich, F.P. Coumarin substrates for cytochrome P450 2D6 fluorescence assays. Anal. Biochem. 2001, 292, 280–286. [Google Scholar]
- Sample Availability: Samples of the compounds and peptides are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Gagey-Eilstein, N.; Broussy, S.; Reille-Seroussi, M.; Huguenot, F.; Vidal, M.; Liu, W.-Q. Design and Synthesis of C-Terminal Modified Cyclic Peptides as VEGFR1 Antagonists. Molecules 2014, 19, 15391-15407. https://doi.org/10.3390/molecules191015391
Wang L, Gagey-Eilstein N, Broussy S, Reille-Seroussi M, Huguenot F, Vidal M, Liu W-Q. Design and Synthesis of C-Terminal Modified Cyclic Peptides as VEGFR1 Antagonists. Molecules. 2014; 19(10):15391-15407. https://doi.org/10.3390/molecules191015391
Chicago/Turabian StyleWang, Lei, Nathalie Gagey-Eilstein, Sylvain Broussy, Marie Reille-Seroussi, Florent Huguenot, Michel Vidal, and Wang-Qing Liu. 2014. "Design and Synthesis of C-Terminal Modified Cyclic Peptides as VEGFR1 Antagonists" Molecules 19, no. 10: 15391-15407. https://doi.org/10.3390/molecules191015391