Synthesis, Biological Evaluation, and Pharmacokinetic Study of Novel Liguzinediol Prodrugs

Abstract
:1. Introduction

2. Results and Discussion
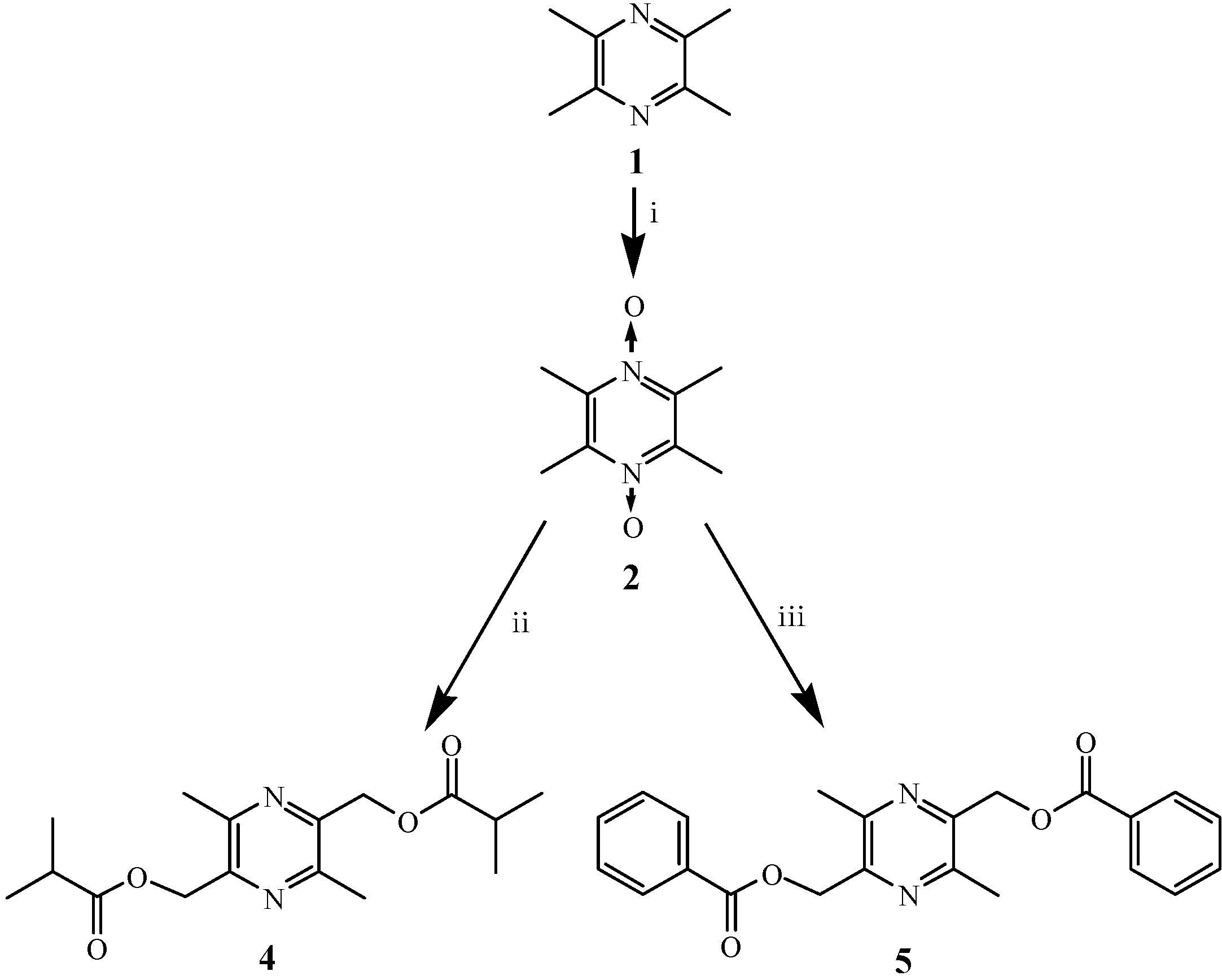
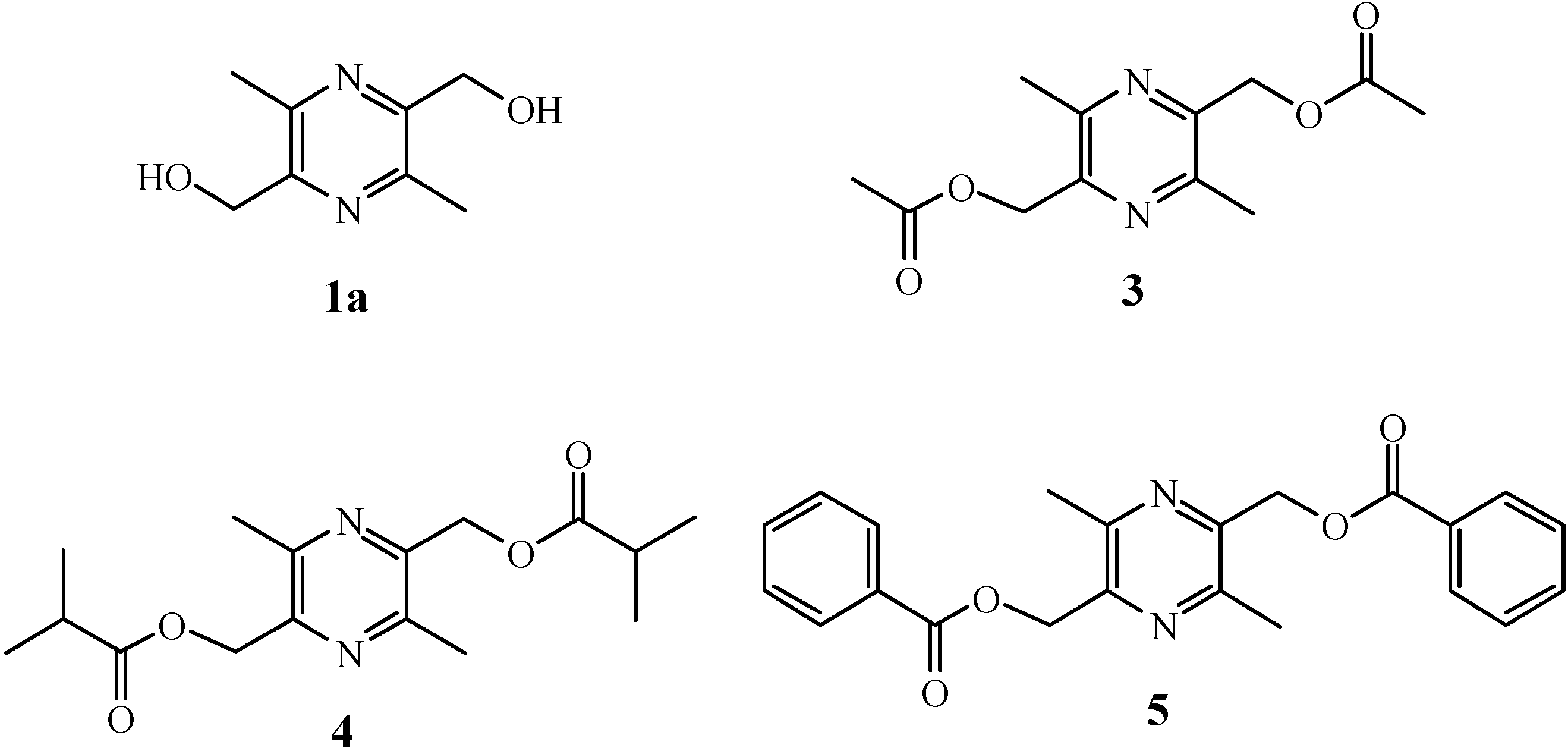
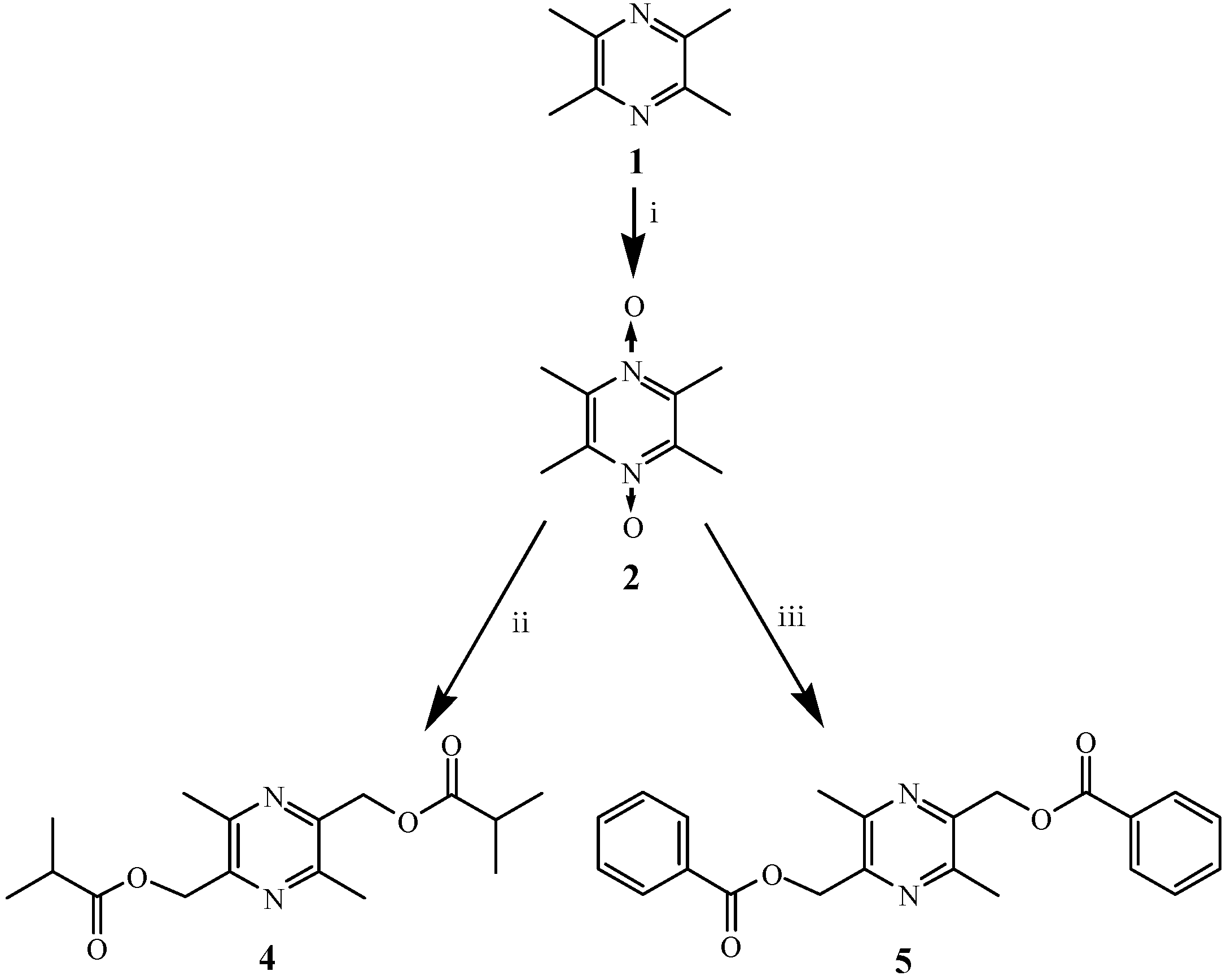
2.1. Chemistry
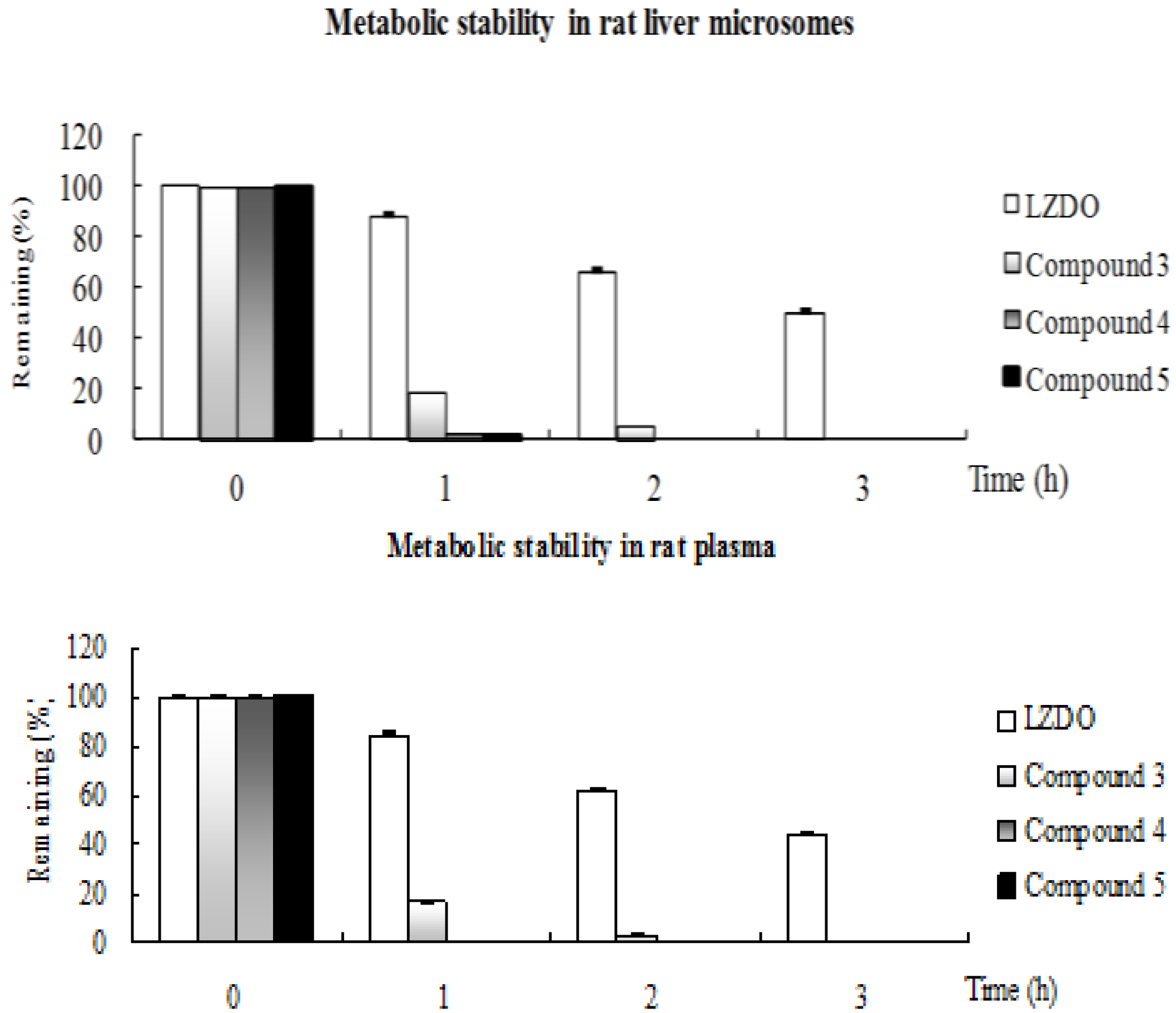
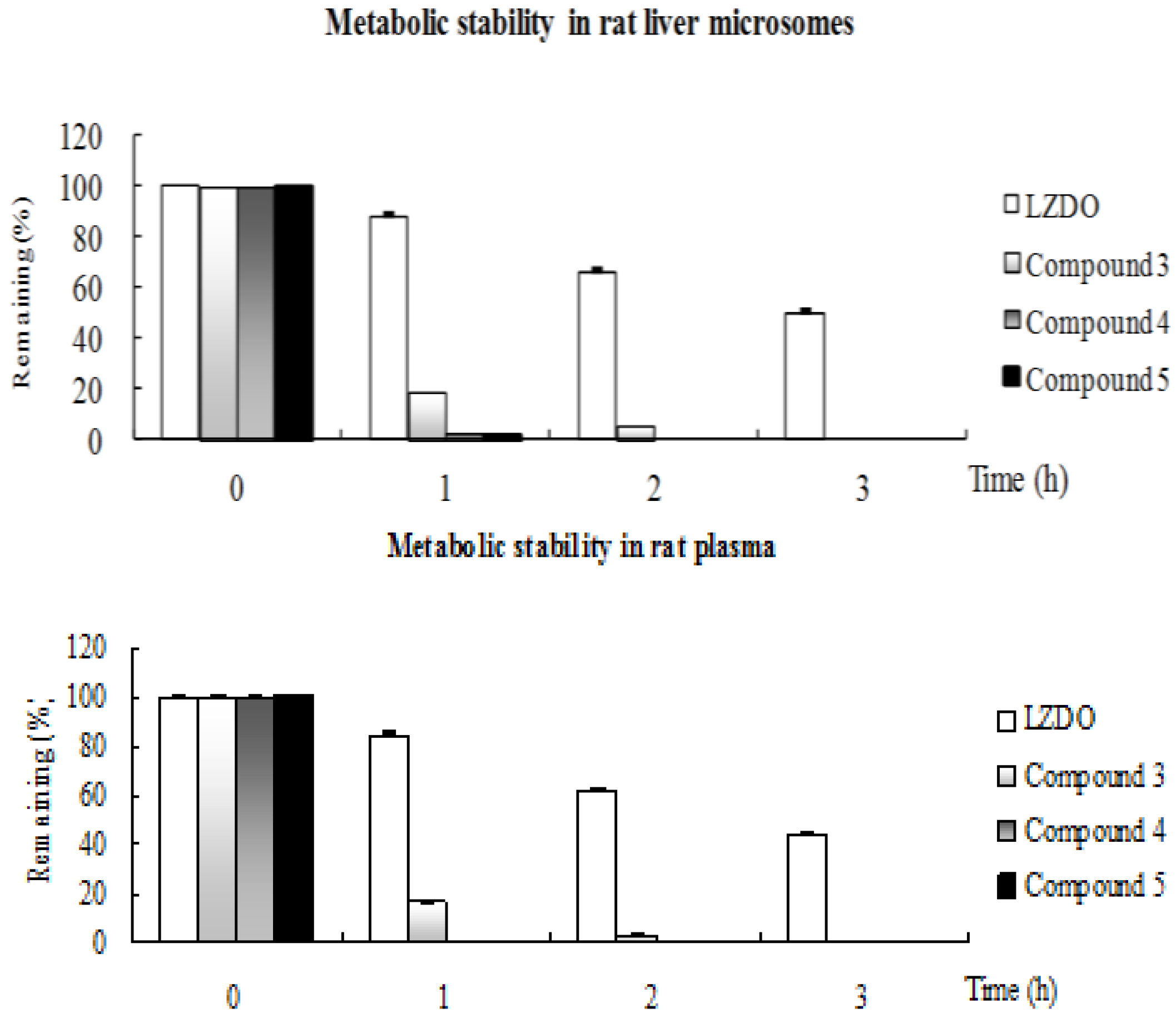
2.2. Metabolic Stability
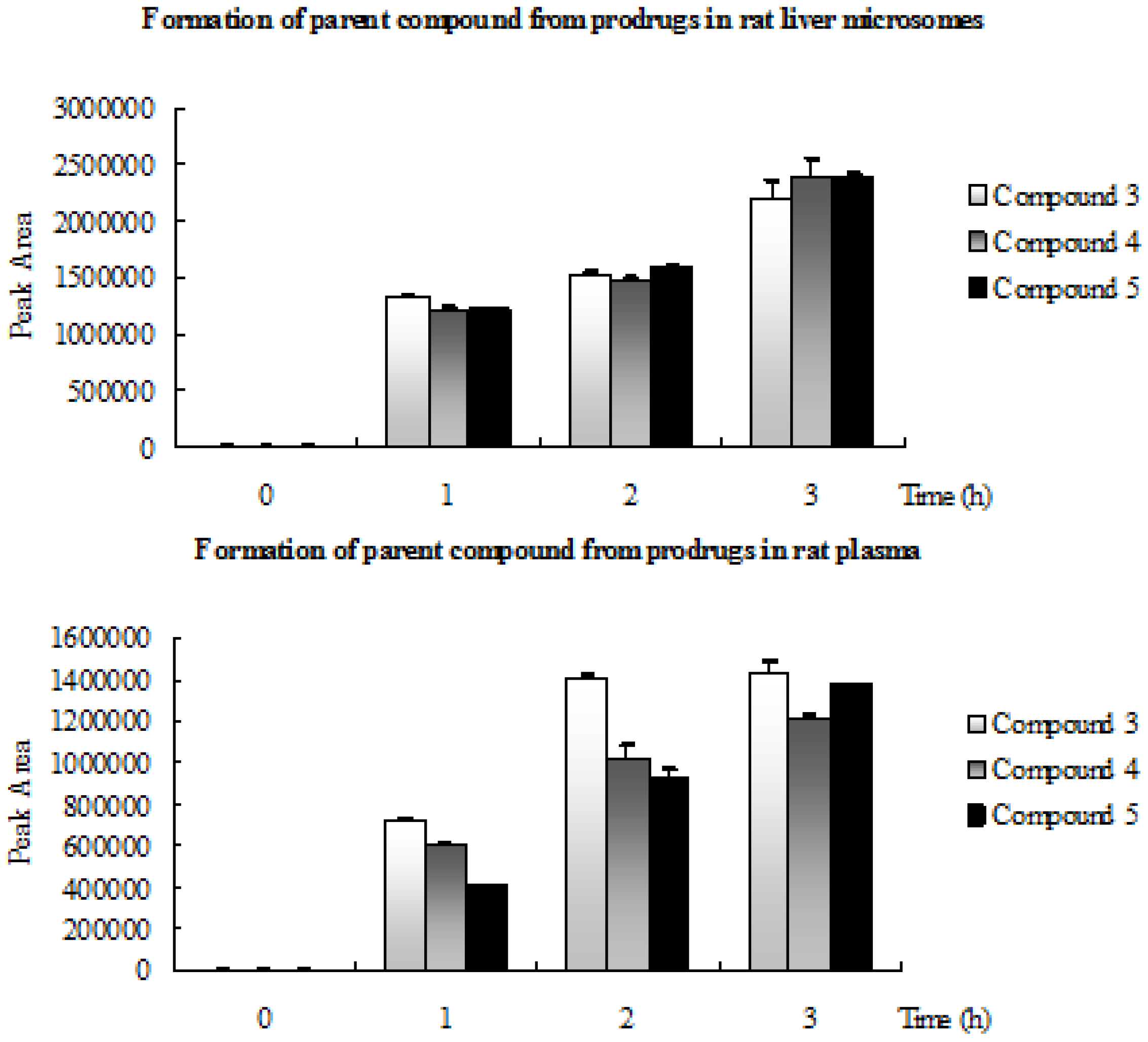
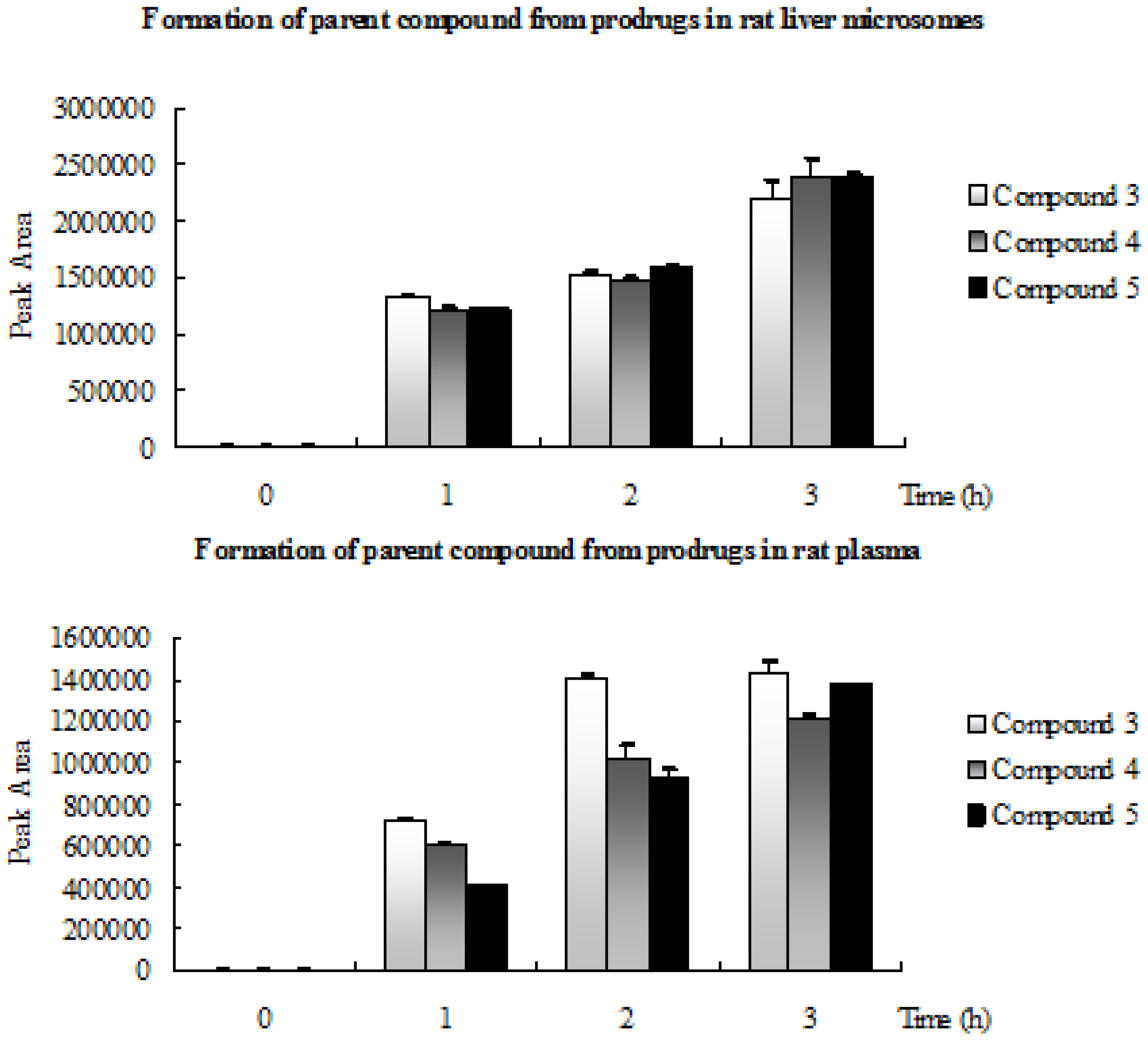
2.3. Formation of Parent Compound from Prodrugs in Rat Liver Microsomes and Rat Plasma
2.4. Cardiac Function Screening
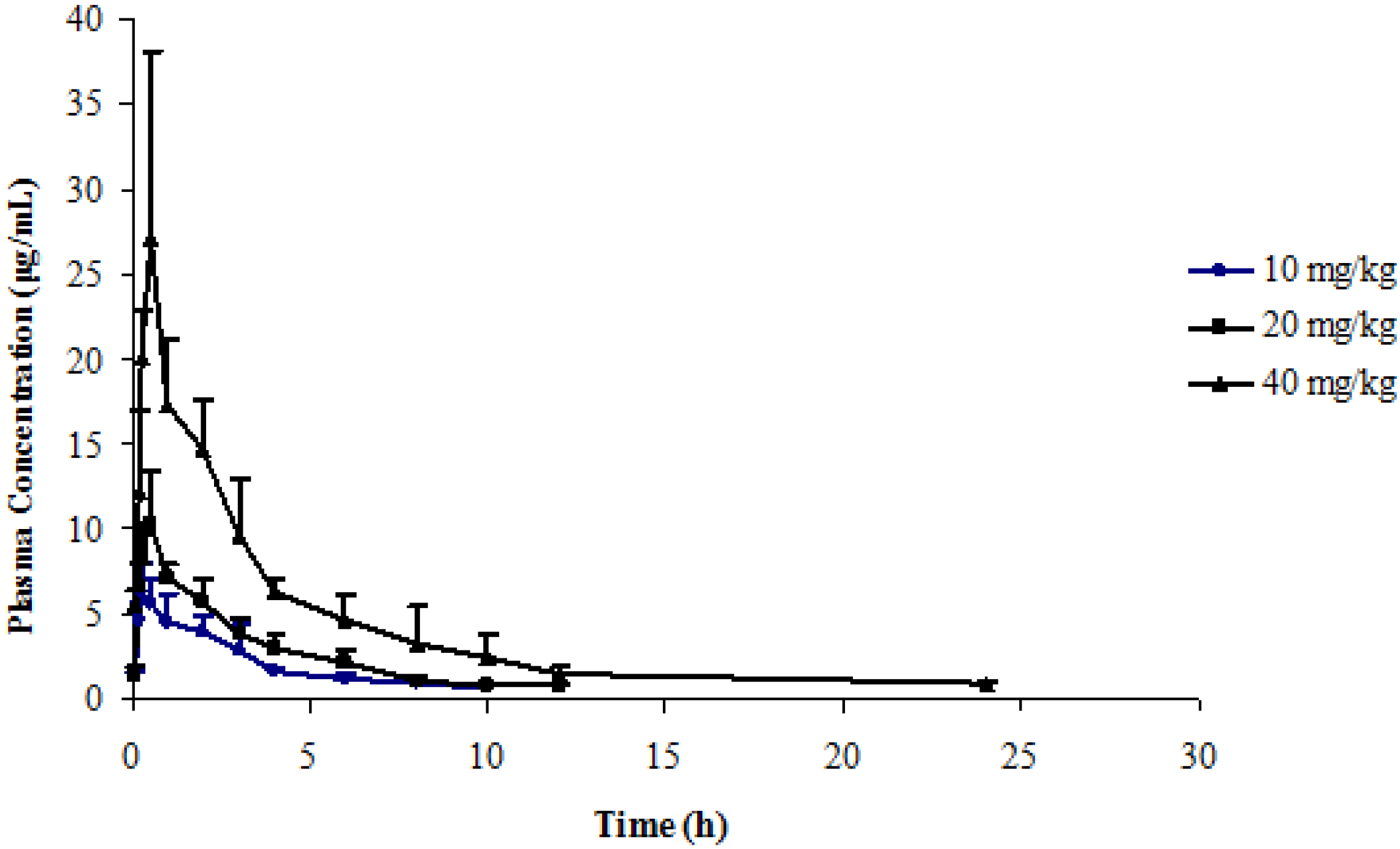
2.5. Application to a Pharmacokinetic Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | Group | LVDP | ∆% | +dP/dtmax | ∆% | −dP/dtmax | ∆% | HR |
|---|---|---|---|---|---|---|---|---|
| (μM) | (mmHg) | (mmHg/s) | (mmHg/s) | (beat·min−1) | ||||
| 1a | 0 | 50 ± 2.8 | 1,685 ± 147 | −1,058 ± 81 | 183 ± 22 | |||
| 1 | 60 ± 5 b | 19.4 ± 11.0 | 1,836 ± 121 | 9.4 ± 8.1 | −1,175 ± 125 | 11.1 ± 10.0 | 186 ± 29 | |
| 10 | 73 ± 4 bd | 45.6 ± 9.0 | 2,125 ± 242 bc | 26.5 ± 13.4 | −1,371 ± 186 bc | 30.3 ± 0.3 | 209 ± 21 | |
| 100 | 88 ± 3 bdf | 76.3 ± 12.3 | 2,702 ± 287 bdf | 60.9 ± 17.3 | −1,911 ± 200 bdf | 82.0 ± 27.9 | 193 ± 15 | |
| 3 | 0 | 55 ± 2.8 | 1,611 ± 27 | −1,086 ± 38 | 207 ± 8 | |||
| 1 | 63 ± 2 b | 15.0 ± 5.5 | 1,641 ± 29 | 1.9 ± 0.5 | −1,139 ± 76 | 4.9 ± 5.9 | 203 ± 15 | |
| 10 | 69 ± 1.1 bd | 26.3 ± 7.7 | 1,768 ± 91 bd | 9.8 ± 7.0 | −1,184 ± 75 a | 9.0 ± 5.1 | 204 ± 14 | |
| 100 | 79 ± 1.6 bdf | 44.7 ± 6.6 | 2,123 ± 82 bdf | 31.8 ± 5.3 | −1,313 ± 70 bdf | 20.9 ± 5.1 | 205 ± 26 | |
| 4 | control | 54 ± 5 | 1,634 ± 100 | −1,044 ± 84 | 196 ± 21 | |||
| 1 | 63 ± 3 b | 17.4 ± 9.8 | 1,669 ± 103 | 2.2 ± 3.4 | −1,101 ± 102 | 5.6 ± 8.3 | 213 ± 46 | |
| 10 | 60 ± 4 a | 14.2 ± 10.0 | 1,765 ± 52 a | 4.9 ± 5.4 | −1,196 ± 54 bc | 9.9 ± 9.5 | 196 ± 16 | |
| 100 | 73 ± 2.3 bdf | 20.2 ± 15.2 | 1,952 ± 49 bdf | 9.0 ± 8.9 | −1,399 ± 61 bdf | 16.7± 15.2 | 191 ± 29 | |
| 5 | control | 52 ± 4 | 1,671 ± 93 | −1,081 ± 40 | 209 ± 47 | |||
| 1 | 55 ± 5 | 5.3 ± 3.3 | 1,688 ± 95 | 1.0 ± 1.1 | −1,095 ± 45 | 1.3 ± 0.8 | 189 ± 30 | |
| 10 | 59 ± 2.5 b | 13.9 ± 7.4 | 1,831 ± 14 bd | 9.9 ± 5.9 | −1,245 ± 48 bd | 15.3 ± 2.8 | 196 ± 40 | |
| 100 | 64 ± 2.4 bde | 24.5 ± 11.2 | 1,962 ± 70 bdf | 17.6 ± 6.8 | −1,314 ± 63 bde | 21.7 ± 7.2 | 213 ± 41 |
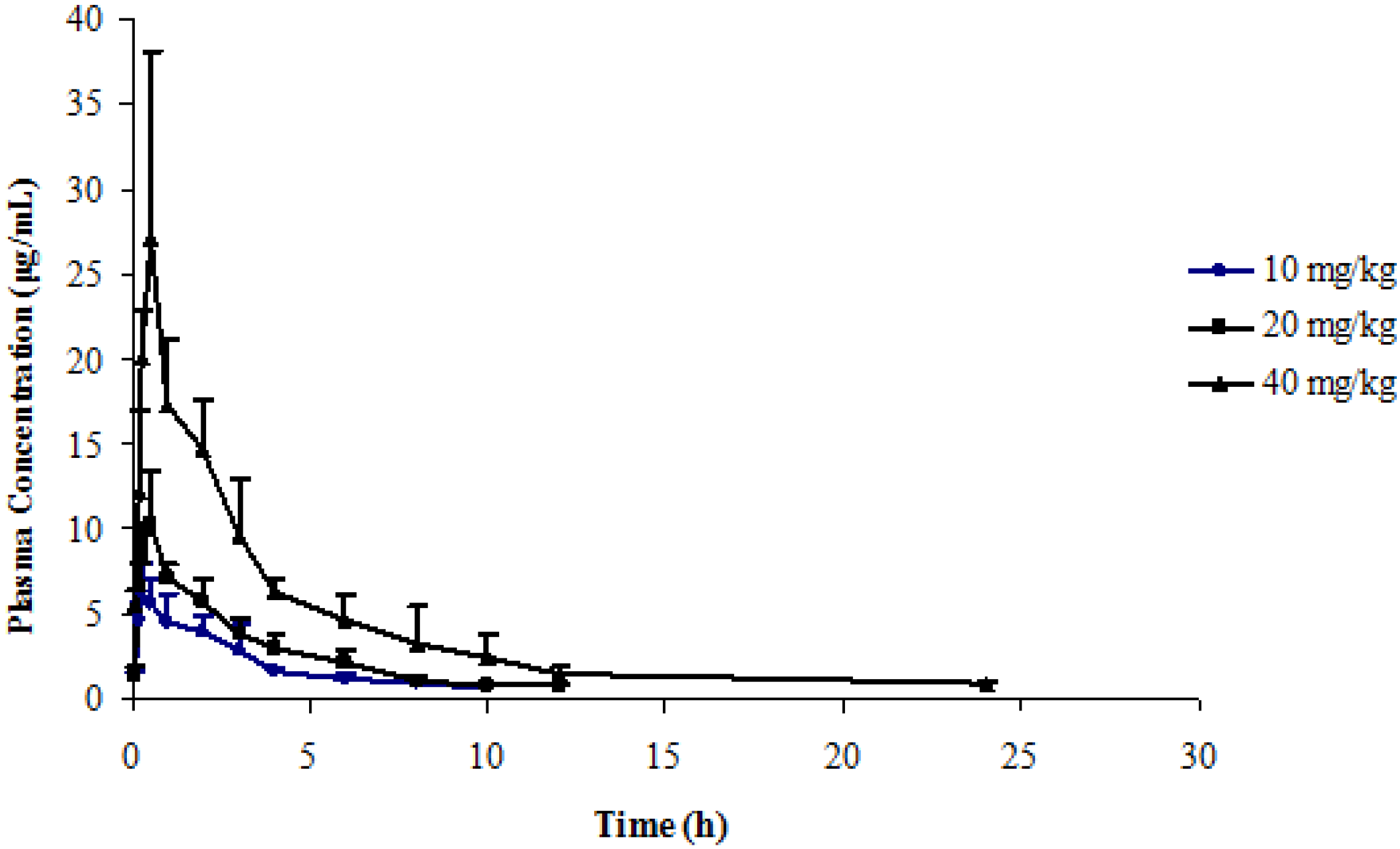
| Dose | 10 mg/kg | 20 mg/kg | 40 mg/kg |
|---|---|---|---|
| Tmax (h) | 0.42 ± 0.30 | 0.42 ± 0.13 | 0.42 ± 0.13 |
| Cmax (mg/L) | 6.50 ± 1.71 | 10.49 ± 3.14 | 28.39 ± 9.56 |
| t1/2 (h) | 4.01 ± 1.36 | 4.24 ± 1.52 | 4.39 ± 1.64 |
| CL/F (L/h·kg) | 0.40 ± 0.06 | 0.52 ± 0.08 | 0.41 ± 0.06 |
| AUC0–t (mg·h/L) | 21.51 ± 2.82 | 33.90 ± 5.71 | 93.94 ± 13.76 |
| AUC0–∞ (mg·h/L) | 25.56 ± 3.82 | 38.86 ± 5.72 | 98.30 ± 15.37 |
| Dose | 10 mg·kg−1 i.g. | 20 mg·kg−1 i.g. | 50 mg·kg−1 i.g. |
|---|---|---|---|
| Tmax (h) | 0.29 ± 0.10 | 0.29 ± 0.10 | 0.29 ± 0.10 |
| Cmax (mg/L) | 8.58 ± 2.66 | 17.02 ± 5.50 | 49.33 ± 10.50 |
| t1/2 (h) | 2.21 ± 1.11 | 2.57 ± 2.14 | 1.78 ± 0.57 |
| CL/F (L/h·kg) | 0.79 ± 0.51 | 0.86 ± 0.57 | 0.98 ± 1.01 |
| AUC0–t (mg·h/L) | 16.74 ± 8.90 | 34.84 ± 24.64 | 92.85 ± 64.10 |
| AUC0–∞ (mg·h/L) | 17.08 ± 8.99 | 35.93 ± 24.08 | 94.82 ± 66.88 |
3. Experimental
3.1. General
3.2. Chemistry
3.3. Metabolic Stability Analysis
3.3.1. Rat Plasma
In Vitro Physiological Stability of Prodrugs 3–5 and LZDO (1a) in Rat Plasma
3.3.2. Rat Liver Microsomes
3.3.2.1. Materials
3.3.2.2. Assay Procedure
3.4. Ex Vivo Langendorff-Perfused Rat Heart [25]
3.5. Application to a Pharmacokinetic Study in Rats
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Lubsen, J.; Just, H.; Hjalmarsson, A.C.; Framboise, D.L.; Remme, W.J.; Heinrich-Nols, J.; Dumont, J.M.; Seed, P. Effect of pimobendan on exercise capacity in patients with heart failure: Main results from the pimobendan in congestive heart failure (PICO) Trial. Heart 1996, 76, 223–231. [Google Scholar] [CrossRef]
- Cohn, J.N.; Goldstein, S.O.G.; Geenberg, B.H.; Lorell, B.H.; Bourge, R.C.; Jaski, B.E.; Gottlieb, S.O.; Iii, F.K.; Demets, D.L.; White, B.G. A dose-dependent increased inmortality with vesnarinone among patients with serve heart failure. N. Engl. J. Med. 1998, 339, 1810–1816. [Google Scholar] [CrossRef]
- Xamoterol in Severe Heart Failure Study Group. Xamoterol in severe heart failure. Lancet 1990, 336, 126.
- Hampton, J.R.; Veldhusisen, D.J.V.; Kleber, F.X.; Cowley, A.J.; Ardia, A.; Block, P.; Cortina, A.; Cserhalmi, L.; Follath, F.; Jensen, G.; et al. Randomized study of effect of ibopamine on survival in patients with advanced heart failure. Lancet 1997, 349, 971–977. [Google Scholar] [CrossRef]
- Van Veldhuisen, D.J.; Man in’t Veld, A.J.; Dunselman, P.H.; Lok, D.J.; Dohmen, H.J.; Poortermans, J.C.; Withagen, A.J.; Pasteuning, W.H.; Brouwer, J.; et al. Double-blind placebo-controlled study ibopamine and digoxin in patients with mild to moderate heart failure: Results of the Dutch Ibopamine Multicenter Trial (DIMT). J. Am. Coll. Cardiol. 1993, 22, 1564–1573. [Google Scholar] [CrossRef]
- Oliva, F.; Latini, R.; Politi, A.; Staszewsky, L.; Maggioni, A.P.; Nicolis, E.; Mauri, F. Intermittent 6-month low dose dobutamine infusion in severe heart failure: DICE multicenter trial. Am. Heart J. 1999, 138, 247–253. [Google Scholar] [CrossRef]
- Singh, K.; Communal, C.; Sawyer, D.B.; Colucci, W.S. Adrenergic regulation of myocardial apoptosis. Cardiovasc. Res. 2000, 45, 713–719. [Google Scholar] [CrossRef]
- Packer, M.; Medina, N.; Yushak, M. Hemodynamic and clinical limitations of long-term inotropic therapy with amrinone in patients with severe chronic heart failure. Circulation 1984, 70, 1038–1047. [Google Scholar] [CrossRef]
- Moiseyev, V.S.; Poder, P.; Andrejevs, N.; Ruda, M.Y.; Golikov, A.P.; Lazebnik, L.B.; Kobalava, Z.D.; Lehtonen, L.A.; Laine, T.; Nieminen, M.S.; et al. Safety and efficacy of a novel calcium ensitizer, levosimendan, In patients with left ventricular failure due to an acute myocardial infarction: A randomized, Placebo-controlled, Double-blind study (RUSSLAN). Eur. Heart J. 2002, 23, 1422–1432. [Google Scholar] [CrossRef]
- Antila, S.; Kivikko, M.; Lehtonen, L.; Eha, J.; Heikkilä, A.; Pohjanjousi, P.; Pentikäinen, P.J. Pharmacokinetics of levosimendan and its circulating metabolites in patients with heart failure after an extended continuous infusion of levosimendan. Br. J. Clin. Pharmacol. 2004, 57, 412–415. [Google Scholar] [CrossRef]
- Chen, S.Y.; Hsiao, G.; Hwang, H.R.; Cheng, P.Y.; Lee, Y.M. Tetramethylpyrazine induces heme oxygenase-1 expression and attenuates myocardial ischemia/reperfusion injury in rats. J. Biomed. Sci. 2006, 13, 731–740. [Google Scholar] [CrossRef]
- Esberg, L.B.; Ren, J. The oxygen radical generator pyrogallol impairs cardiomyocyte contractile function via a superoxide and p38 MAP kinase-dependent pathway: protection by anisodamine and tetramethylpyrazine. Cardiovasc. Toxicol. 2004, 4, 375–384. [Google Scholar] [CrossRef]
- Hintz, K.K.; Ren, J. Tetramethylpyrazine elicits disparate responses in cardiac contraction and intracellular Ca2+ transients in isolated adult rat ventricular myocytes. Vascul. Pharmacol. 2003, 40, 213–217. [Google Scholar] [CrossRef]
- Zhao, H.P.; Lü, D.; Zhang, W.; Zhang, L.; Wang, S.M.; Ma, C.M.; Qin, C.; Zhang, L.F. Protective action of tetramethylpyrazine phosphate against dilated cardiomyopathy in cTnT (R141W) transgenic mice. Acta Pharmacol. Sin. 2010, 31, 281–288. [Google Scholar] [CrossRef]
- Zhou, Y.; Hu, C.P.; Deng, P.Y.; Deng, H.W.; Li, Y.J. The protective effects of ligustrazine on ischemia-reperfusion and DPPH free radical–induced myocardial injury in isolated rat hearts. Planta Med. 2004, 70, 818–822. [Google Scholar] [CrossRef]
- Zou, L.Y.; Hao, X.M.; Zhang, G.Q.; Zhang, M.; Guo, J.H.; Liu, T.F. Effect of tetramethyl pyrazine on l-type calcium channel in rat ventricular myocytes. Can. J. Physiol. Pharmacol. 2001, 79, 621–626. [Google Scholar] [CrossRef]
- Li, W.; Chen, L.; Bian, H.M.; Wen, H.M.; Liu, Z. Application of 2,5-dihydroxymethyl-3,6-dimethylphrazine and its derivates in pharmacy. U.S. Patent 8,158,630, 17 April 2012. [Google Scholar]
- Chen, L.; Xu, Y.; Li, W.; Wu, H.; Luo, Z.K.; Li, X.H.; Huang, F.F.; Young, C.; Liu, Z.; Zhou, S.Y. The novel compound liguzinediol exerts positive inotropic effects in isolated rat heart via sarcoplasmic reticulum Ca2+ ATPase-dependent mechanism. Life Sci. 2012, 91, 402–408. [Google Scholar] [CrossRef]
- Shan, C.X. Studies on the pharmacokinetics and disposition of Liguzinediol in rats. Dissertation for Master Degree, Nanjing University of Chinese Medicine, Nanjing, China, 2012. [Google Scholar]
- Ettmayer, P.; Amidon, G.L.; Clement, B.; Testa, B. Lessons learned from marketed and investigational prodrugs. J. Med. Chem. 2004, 47, 2393–2404. [Google Scholar] [CrossRef]
- Ettmayer, P.; Amidon, G.L.; Gardner, I.; Dack, K. Design of ester prodrugs to enhance oral absorption of poorly permeable compounds: Challenges to the discovery scientist. Curr. Drug Metab. 2003, 4, 461–485. [Google Scholar] [CrossRef]
- Liu, J.J. Design, Synthesis and Bioactivity Studies of the Novel Ligustrazine Derivatives. Dissertation for Master Degree, Shandong University, Jinan, China, 2006. [Google Scholar]
- Liu, X.Q.; Zhao, Y.; Ren, Y.L.; Wang, G.J.; Qian, Z.Y. Enzyme kinetics of nicardipinemetabolism in human liver microsomes. Acta Pharm. Sin. 2001, 36, 891–893. [Google Scholar]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Yang, B.C.; Nichols, W.W.; Mehta, J.L. Cardiac effect of acetylcholine in rat hearts: Role of endothelium-derived relaxing factor and prostaglandins. Am. J. Physiol. 1993, 264, 1388–1393. [Google Scholar]
- National Research Council, Guide for the Care and Use of Laboratory Animals, 1st ed.; National Academy Press: Washington, DC, USA, 1996; pp. 21–25.
- Sample Availability: Samples of the compounds (1a, 3, 4, 5) are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, Z.; Li, W.; Wen, H.-M.; Bian, H.-M.; Zhang, J.; Chen, L.; Chen, L.; Yang, K.-D. Synthesis, Biological Evaluation, and Pharmacokinetic Study of Novel Liguzinediol Prodrugs. Molecules 2013, 18, 4561-4572. https://doi.org/10.3390/molecules18044561
Liu Z, Li W, Wen H-M, Bian H-M, Zhang J, Chen L, Chen L, Yang K-D. Synthesis, Biological Evaluation, and Pharmacokinetic Study of Novel Liguzinediol Prodrugs. Molecules. 2013; 18(4):4561-4572. https://doi.org/10.3390/molecules18044561
Chicago/Turabian StyleLiu, Zheng, Wei Li, Hong-Mei Wen, Hui-Min Bian, Jing Zhang, Lei Chen, Long Chen, and Kun-Di Yang. 2013. "Synthesis, Biological Evaluation, and Pharmacokinetic Study of Novel Liguzinediol Prodrugs" Molecules 18, no. 4: 4561-4572. https://doi.org/10.3390/molecules18044561