Application Scope and Limitations of TADDOL-Derived Chiral Ammonium Salt Phase-Transfer Catalysts


Abstract
:
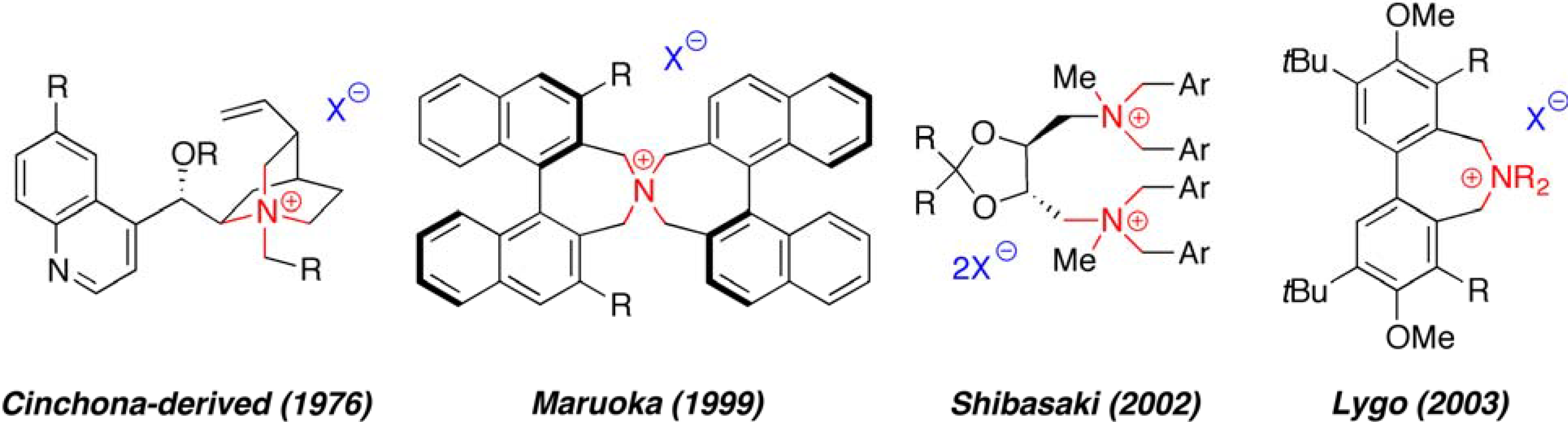
1. Introduction

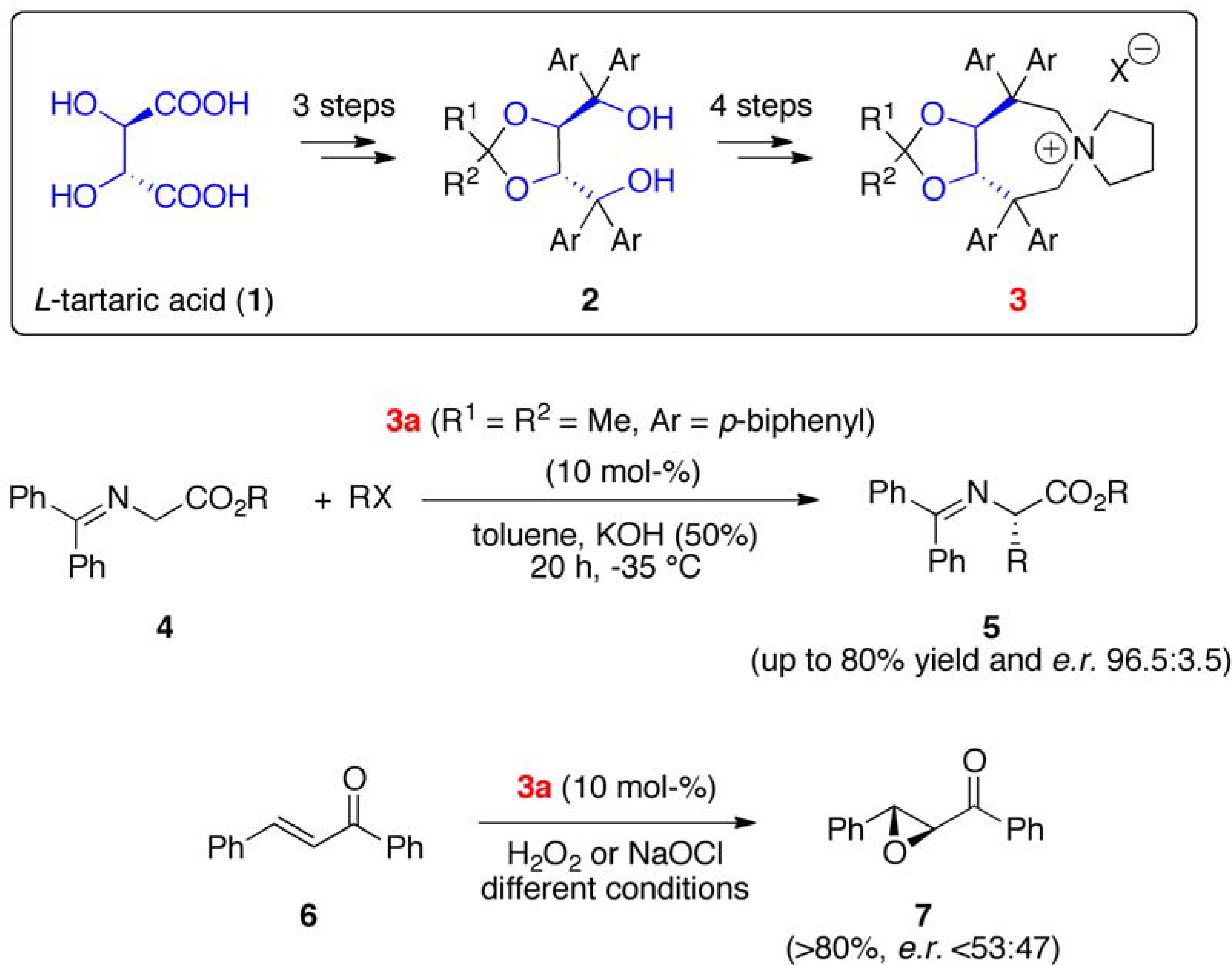
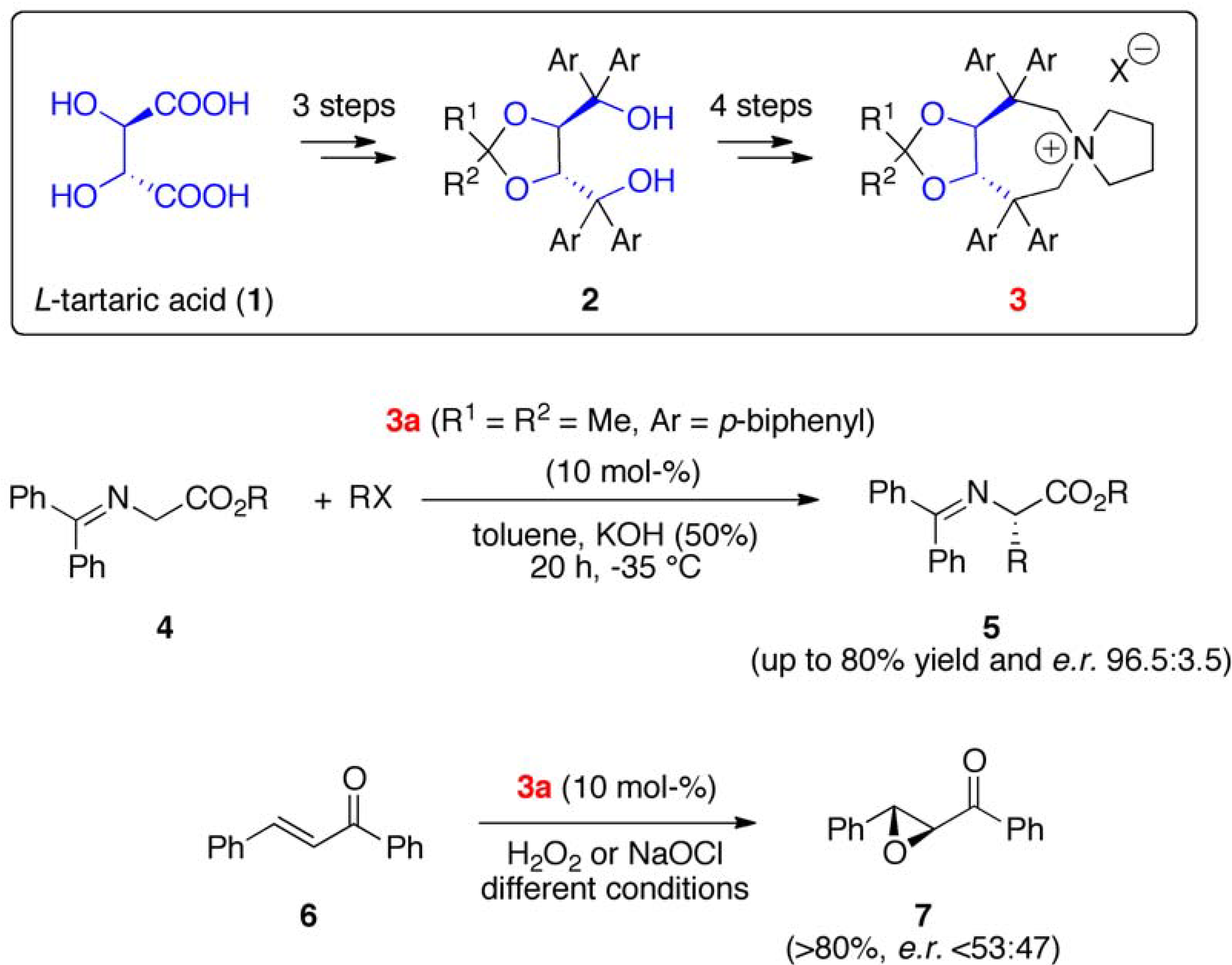
2. Results and Discussion
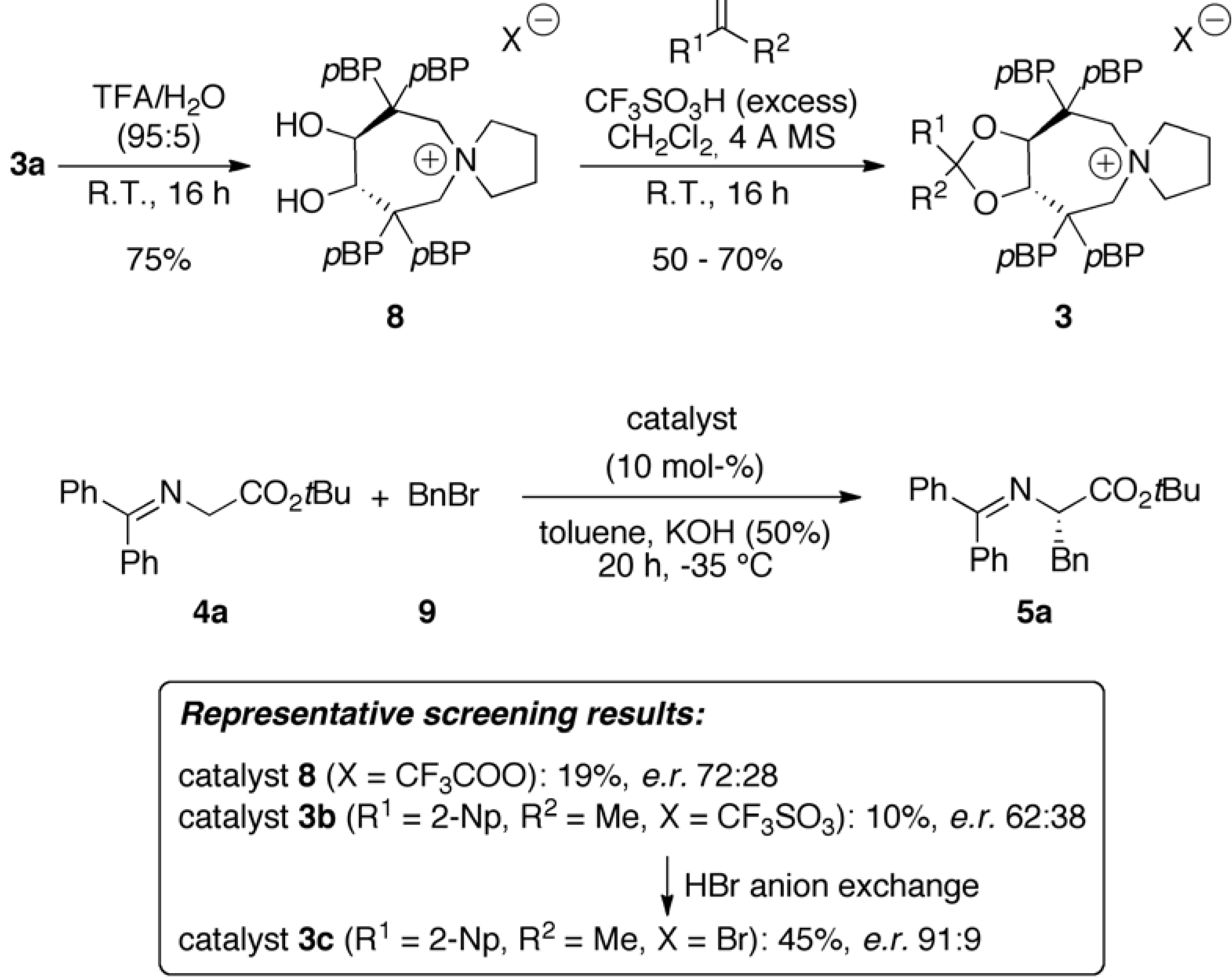
2.1. Late Stage Catalyst Modification

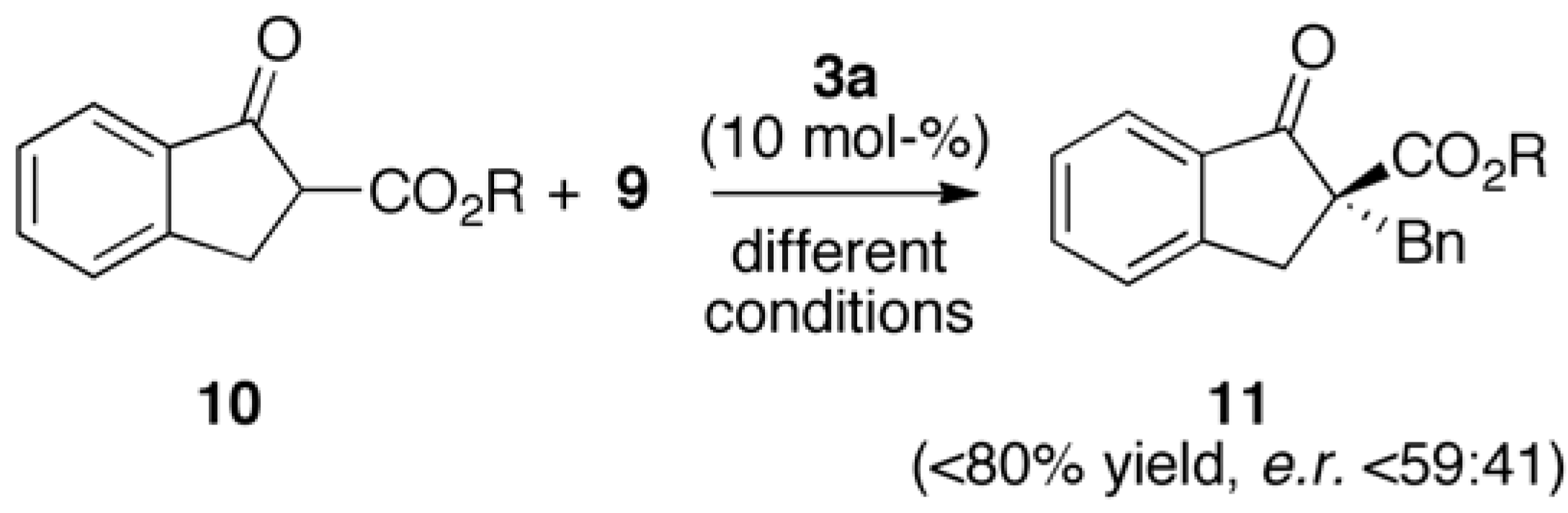
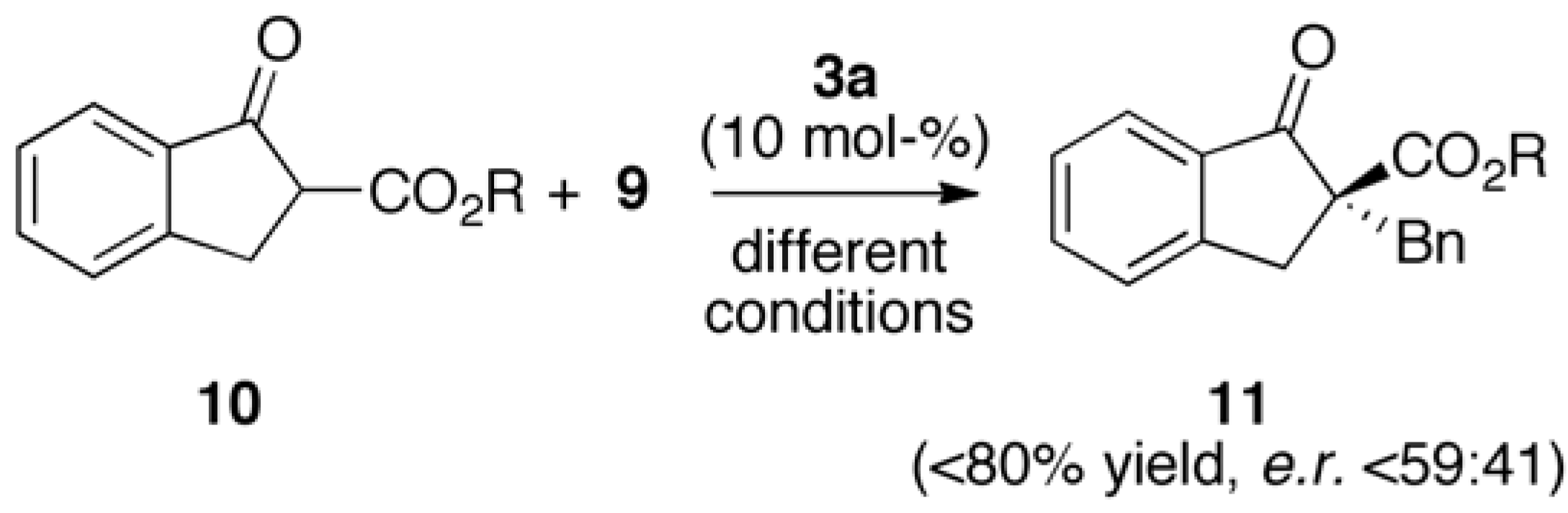
2.2. Asymmetric α-Alkylation of β-Keto Esters
2.3. Asymmetric Michael Addition Reactions of Glycine Schiff Bases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry a | Cat. (mol%) | 4 | 12 | Solv. | Base (eq.) | T [°C] | 13 | Yield b [%] | e.r. c (conf.) d |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3a (10%) | 4a | 12a | toluene | KOH (50%) (25×) | 0 | 13a | 94 | 51:49 (S) |
| 2 | 3a (10%) | 4a | 12a | toluene | KOH (50%) (1×) | 0 | 13a | 88 | 50:50 |
| 3 | 3a (10%) | 4a | 12a | toluene | KOH (s) (20×) | 0 | 13a | 76 | 50:50 |
| 4 | 3a (10%) | 4a | 12a | toluene | K3PO4 (50%) (10×) | 0 | 13a | 34 | 50:50 |
| 5 | 3a (10%) | 4a | 12a | toluene | K2CO3 (50%) (10×) | 0 | 13a | 18 | 52:48 (S) |
| 6 | 3a (10%) | 4a | 12a | toluene | Cs2CO3 (70%) (10×) | 0 | 13a | 10 | 50:50 |
| 7 | 3a (10%) | 4a | 12a | toluene | Cs2CO3 (s) (20×) | 0 | 13a | 73 | 66:34 (S) |
| 8 | 3a (10%) | 4a | 12a | toluene | Cs2CO3 (s) (20×) | −20 | 13a | 56 | 71:29 (S) |
| 9 e | 3a (10%) e | 4a | 12a | toluene | Cs2CO3 (s) (20×) | −20 | 13a | 62 | 61:39 (S) |
| 10 | 3d (10%) | 4a | 12a | toluene | Cs2CO3 (s) (20×) | −20 | 13a | 14 | 62:38 (S) |
| 11 | 3e (10%) | 4a | 12a | toluene | Cs2CO3 (s) (20×) | −20 | 13a | 82 | 64:36 (S) |
| 12 | 3a (10%) | 4a | 12a | benzene | Cs2CO3 (s) (20×) | 0 | 13a | 72 | 58:42 (S) |
| 13 | 3a (10%) | 4a | 12a | fluorobenzene | Cs2CO3 (s) (20×) | 0 | 13a | 89 | 54:46 (S) |
| 14 | 3a (10%) | 4a | 12a | mesitylene | Cs2CO3 (s) (20×) | 0 | 13a | 74 | 69:31 (S) |
| 15 f | 3a (10%) | 4a | 12a | mesitylene | Cs2CO3 (s) (20×) | 0 | 13a | 33 | 57:43 (S) |
| 16 g | 3a (10%) | 4a | 12a | mesitylene | Cs2CO3 (s) (20×) | 0 | 13a | 76 | 51:49 (S) |
| 17 | 3a (10%) | 4b | 12a | mesitylene | Cs2CO3 (s) (20×) | 0 | 13b | 66 | 75:25 (S) |
| 18 | 3a (10%) | 4c | 12a | mesitylene | Cs2CO3 (s) (20×) | 0 | 13c | 81 | 78:22 |
| 19 | 3a (10%) | 4c | 12a | mesitylene | Cs2CO3 (s) (20×) | −20 | 13c | 35 | 85:15 |
| 20 | 3a (20%) | 4c | 12a | mesitylene | Cs2CO3 (s) (20×) | −20 | 13c | 71 | 90:10 |
| 21 | 3a (20%) | 4c | 12b | mesitylene | Cs2CO3 (s) (20×) | −20 | 13d | 68 | 89:11 |
| 22 | 3a (20%) | 4c | 12c | mesitylene | Cs2CO3 (s) (20×) | −20 | 13e | n.r. | n.d. |
| 23 | 3a (20%) | 4c | 12d | mesitylene | Cs2CO3 (s) (20×) | −20 | 13f | 81 | 87:13 |
| 24 | 3f (20%) | 4c | 12a | mesitylene | Cs2CO3 (s) (20×) | −20 | 13c | 51 | 80:20 |
| 25 | 3g (20%) | 4c | 12a | mesitylene | Cs2CO3 (s) (20×) | −20 | 13c | 56 | 86:14 |
| 26 | 3h (20%) | 4c | 12a | mesitylene | Cs2CO3 (s) (20×) | −20 | 13c | 68 | 91:9 |

| Entry | 4 | Acceptor (eq.) | Solv. | Base (eq.) | T [°C] | t [h] | Prod. | Yield a [%] | e.r. b |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4a | 14a (2×) | mesitylene | Cs2CO3 (s) (20×) | 0 | 20 | 16a | 32 | 50:50 |
| 2 | 4a | 14a (2×) | mesitylene | Cs2CO3 (s) (1×) | 0 | 20 | 16a | 11 | 50:50 |
| 3 | 4a | 14a (2×) | toluene | K2CO3 (s) (1×) | 0 | 20 | 16a | n.r. | n.d. |
| 4 | 4a | 14a (2×) | toluene | K2HPO4 (s) (1×) | 0 | 20 | 16a | n.r. | n.d. |
| 5 | 4a | 14a (2×) | toluene | KOH (s) (1×) | 0 | 20 | 16a | 55 | 56:44 |
| 6 | 4a | 14a (2×) | toluene | KOH (50%) (25×) | 0 | 20 | 16a | 62 | 65:35 |
| 7 | 4a | 14a (2×) | toluene | CsOH (50%) (25×) | 0 | 20 | 16a | 55 | 66:34 |
| 8 | 4a | 14a (2×) | toluene | RbOH (50%) (25×) | 0 | 20 | 16a | 69 | 69:31 |
| 9 | 4a | 14a (2×) | mesitylene | RbOH (50%) (25×) | 0 | 20 | 16a | 62 | 60:40 |
| 10 | 4a | 14a (2×) | toluene | RbOH (50%) (25×) | −20 | 48 | 16a | 65 | 75:25 |
| 11 | 4a | 14b (2×) | toluene | RbOH (50%) (25×) | −20 | 48 | 16b | 81 | 60:40 |
| 12 | 4c | 14b (2×) | toluene | RbOH (50%) (25×) | −20 | 48 | 16c | 64 | 57:43 |
| 13 | 4c | 15 (2×) | toluene | RbOH (50%) (25×) | −20 | 48 | 17 | 65 | 67:33 (S) c |
| 14 | 4c | 15 (1.5×) | mesitylene | Cs2CO3 (s) (20×) | −20 | 48 | 17 | 97 | 77:23 (S) c |
3. Experimental
3.1. General
3.2. Conditions A: General Procedure for the Phase-Transfer Catalysed Michael-Reaction under Liquid/Solid Phase-Transfer Conditions
3.3. Conditions B: General Procedure for the Phase-Transfer Catalysed Michael-Reaction under Liquid/Liquid Phase-Transfer Conditions
4. Conclusions
Supplementary Materials
Acknowledgments
References
- O’Donnell, M.J. The enantioselective synthesis of α-amino acids by phase-transfer catalysis with achiral schiff base esters. Acc. Chem. Res. 2004, 37, 506–517. [Google Scholar] [CrossRef]
- Lygo, B.; Andrews, B.I. Asymmetric phase-transfer catalysis utilizing chiral quaternary ammonium salts: Asymmetric alkylation of glycine imines. Acc. Chem. Res. 2004, 37, 518–525. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maruoka, K. Recent development and application of chiral phase-transfer catalysts. Chem. Rev. 2007, 107, 5656–5682. [Google Scholar] [CrossRef]
- Ooi, T.; Maruoka, K. Recent advances in asymmetric phase-transfer catalysis. Angew. Chem. Int. Ed. 2007, 46, 4222–4266. [Google Scholar] [CrossRef]
- Shirakawa, S.; Maruoka, K. Recent developments in asymmetric phase-transfer reactions. Angew. Chem. Int. Ed. 2013. [Google Scholar] [CrossRef]
- Novacek, J.; Waser, M. Bifunctional chiral quaternary ammonium salt catalysts: A rapidly emerging class of powerful asymmetric catalysts. Eur. J. Org. Chem. 2013, 2013, 637–648. [Google Scholar] [CrossRef]
- Helder, R.; Hummelen, J.C.; Laane, R.W.P.M.; Wiering, J.S.; Wynberg, H. Catalytic asymmetric induction in oxidation reactions. The synthesis of optically active epoxides. Tetrahedron Lett. 1976, 17, 1831–1834. [Google Scholar] [CrossRef]
- Dolling, U.H.; Davis, P.; Grabowski, E.J.J. Efficient catalytic asymmetric alkylations. 1. Enantioselective synthesis of (+)-indacrinone via chiral phase-transfer catalysis. J. Am. Chem. Soc. 1984, 106, 446–447. [Google Scholar]
- O’Donnell, M.J.; Bennett, W.D.; Wu, S. The stereoselective synthesis of α-amino acids by phase-transfer catalysis. J. Am. Chem. Soc. 1989, 111, 2353–2355. [Google Scholar] [CrossRef]
- O’Donnell, M.J.; Wu, S.; Huffman, J.C. A new active catalyst species for enantioselective alkylation by phase-transfer catalysis. Tetrahedron 1994, 50, 4507–4518. [Google Scholar]
- Lygo, B.; Wainwright, P.G. A new class of asymmetric phase-transfer catalysts derived from Cinchona alkaloids—Application in the enantioselective synthesis of α-amino acids. Tetrahedron Lett. 1997, 38, 8595–8598. [Google Scholar]
- Lygo, B.; Crosby, J.; Peterson, J.A. Enantioselective synthesis of bis-α-amino acid esters via asymmetric phase-transfer catalysis. Tetrahedron Lett. 1999, 40, 1385–1388. [Google Scholar] [CrossRef]
- Corey, E.J.; Xu, F.; Noe, M.C. A rational approach to catalytic enantioselective enolate alkylation using a structurally rigidified and defined chiral quaternary ammonium salt under phase transfer conditions. Am. Chem. Soc. 1997, 119, 12414–12415. [Google Scholar] [CrossRef]
- Corey, E.J.; Noe, M.C.; Xu, F. Highly enantioselective synthesis of cyclic and functionalized α-amino acids by means of a chiral phase transfer catalyst. Tetrahedron Lett. 1998, 39, 5347–5350. [Google Scholar] [CrossRef]
- Liu, Y.; Provencher, B.A.; Bartelson, K.J.; Deng, L. Highly enantioselective asymmetric Darzens reactions with a phase transfer catalyst. Chem. Sci. 2011, 2, 1301–1304. [Google Scholar] [CrossRef]
- Provencher, B.A.; Bartelson, K.J.; Liu, Y.; Foxman, B.M.; Deng, L. Structural study-guided development of versatile phase-transfer catalysts for asymmetric conjugate additions of cyanide. Angew. Chem. Int. Ed. 2011, 50, 10565–10569. [Google Scholar] [CrossRef]
- Bella, M.; Kobbelgaard, S.; Jørgensen, K.A. Organocatalytic regio- and asymmetric C-selective SNAr reactions—stereoselective synthesis of optically active spiro-pyrrolidone-3,3'-oxoindoles. J. Am. Chem. Soc. 2005, 127, 3670–3671. [Google Scholar]
- Poulsen, T.B.; Bernardi, L.; Aleman, J.; Overgaard, J.; Jørgensen, K.A. Organocatalytic asymmetric direct α-alkynylation of cyclic β-ketoesters. J. Am. Chem. Soc. 2007, 129, 441–449. [Google Scholar] [CrossRef]
- Maciver, E.E.; Knipe, P.C.; Cridland, A.P.; Thompson, A.L.; Smith, M.D. Catalytic enantioselective electrocyclic cascades. Chem. Sci. 2012, 3, 537–540. [Google Scholar] [CrossRef]
- Bernardi, L.; Indrigo, E.; Pollicino, S.; Ricci, A. Organocatalytic trifluoromethylation of imines using phase-transfer catalysis with phenoxides. A general platform for catalytic additions of organosilanes to imines. Chem. Commun. 2012, 48, 1428–1430. [Google Scholar]
- Fiandra, C.D.; Piras, L.; Fini, F.; Disetti, P.; Moccia, M.; Adamo, M.F.A. Phase transfer catalyzed enantioselective cyclopropanation of 4-nitro-5-styrylisoxazoles. Chem. Commun. 2012, 48, 3863–3865. [Google Scholar]
- Johnson, K.M.; Rattley, M.S.; Sladojevich, F.; Barber, D.M.; Nunez, M.G.; Goldys, A.M.; Dixon, D.J. A new family of cinchona-derived bifunctional asymmetric phase-transfer catalysts: Application to the enantio- and diastereoselective nitro-Mannich reaction of amidosulfones. Org. Lett. 2012, 14, 2492–2495. [Google Scholar]
- Bernal, P.; Fernández, R.; Lassaletta, J.M. Organocatalytic asymmetric cyanosilylation of nitroalkenes. Chem. Eur. J. 2010, 16, 7714–7718. [Google Scholar] [CrossRef]
- Haraguchi, N.; Ahamed, P.; Parvez, M.; Itsuno, S. Synthesis of main-chain chiral quaternary ammonium polymers for asymmetric catalysis using quaternization polymerization. Molecules 2012, 17, 7569–7583. [Google Scholar] [CrossRef]
- Hintermann, L.; Dittmer, C. Asymmetric ion-pairing catalysis of the reversible cyclization of 2-hydroxychalcone to flavanone: Asymmetric catalysis of an equilibrating reaction. Eur. J. Org. Chem. 2012, 5573–5584. [Google Scholar] [CrossRef]
- Herchl, R.; Waser, M. Asymmetric cyclopropanation of chalcones using chiral phase-transfer catalysts. Tetrahedron Lett. 2013, 54, 2472–2475. [Google Scholar] [CrossRef]
- Kamlar, M.; Putaj, P.; Vesely, J. Organocatalytic alkynylation of densely functionalized monofluorinated derivatives: C(sp3)–C(sp) coupling. Tetrahedron Lett. 2013, 54, 2097–2100. [Google Scholar] [CrossRef]
- Denmark, S.E.; Weintraub, R.C. Deconstructing quinine. Part 1. Toward an understanding of the remarkable performance of cinchona alkaloids in asymmetric phase-transfer catalysis. Heterocycles 2011, 82, 1527–1540. [Google Scholar]
- Tanzer, E.-M.; Schweizer, W.B.; Ebert, M.-O.; Gilmour, R. Designing fluorinated cinchona alkaloids for enantioselective catalysis: Controlling internal rotation by a fluorine-ammonium ion gauche effect (φNCCF). Chem. Eur. J. 2012, 18, 2006–2013. [Google Scholar]
- Ooi, T.; Kameda, M.; Maruoka, K. Molecular design of a C2-symmetric chiral phase-transfer catalyst for practical asymmetric synthesis of α-amino acids. J. Am. Chem. Soc. 1999, 121, 6519–6520. [Google Scholar] [CrossRef]
- Shirakawa, S.; Liu, K.; Ito, H.; Maruoka, K. Catalytic asymmetric synthesis of 1,1-disubstituted tetrahydro-β-carbolines by phase-transfer catalyzed alkylations. Chem. Commun. 2011, 47, 1515–1517. [Google Scholar] [CrossRef]
- Kano, T.; Yamamoto, A.; Song, S.; Maruoka, K. Catalytic asymmetric syntheses of isoxazoline-N-oxides under phase-transfer conditions. Chem. Commun. 2011, 47, 4358–4360. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Sakata, K.; Maruoka, K. Phase-transfer-catalyzed olefin isomerization/α-alkylation of α-alkynylcrotonates as a route for 1,4-enynes. Adv. Synth. Catal. 2010, 352, 1653–1656. [Google Scholar] [CrossRef]
- Lan, Q.; Wang, X.; Shirakawa, S.; Maruoka, K. Phase-transfer catalyzed asymmetric conjugate additions of β-ketoesters to acetylenic ketones. Org. Process Res. Dev. 2010, 14, 684–686. [Google Scholar] [CrossRef]
- Shirakawa, S.; Liu, K.; Maruoka, K. Catalytic asymmetric synthesis of axially chiral o-iodoanilides by phase-transfer catalyzed alkylations. J. Am. Chem. Soc. 2012, 134, 916–919. [Google Scholar] [CrossRef]
- Shibuguchi, T.; Fukuta, Y.; Akachi, Y.; Sekine, A.; Ohshima, T.; Shibasaki, M. Development of new asymmetric two-center catalysts in phase-transfer reactions. Tetrahedron Lett. 2002, 43, 9539–9543. [Google Scholar]
- Okada, A.; Shibuguchi, T.; Ohshima, T.; Masu, H.; Yamaguchi, K.; Shibasaki, M. Enantio- and diastereoselective catalytic Mannich-type reaction of a glycine schiff base using a chiral two-center phase-transfer catalyst. Angew. Chem. Int. Ed. 2005, 44, 4564–4567. [Google Scholar] [CrossRef]
- Shibuguchi, T.; Mihara, H.; Kuramochi, A.; Ohshima, T.; Shibasaki, M. Catalytic asymmetric phase-transfer Michael reaction and Mannich-type reaction of glycine schiff bases with tartrate-derived diammonium Salts. Chem. Asian. J. 2007, 2, 794–801. [Google Scholar]
- Lygo, B.; Allbutta, B.; James, S.R. Identification of a highly effective asymmetric phase-transfer catalyst derived from α-methylnaphthylamine. Tetrahedron Lett. 2003, 44, 5629–5632. [Google Scholar] [CrossRef]
- Lygo, B.; Allbutta, B.; Kirton, E.H.M. Asymmetric Michael addition of glycine imines via quaternary ammonium ion catalysis. Tetrahedron Lett. 2005, 46, 4461–4464. [Google Scholar] [CrossRef]
- Arai, S.; Tsuji, R.; Nishida, A. Phase-transfer-catalyzed asymmetric Michael reaction using newly-prepared chiral quaternary ammonium salts derived from L-tartrate. Tetrahedron Lett. 2002, 43, 9535–9537. [Google Scholar] [CrossRef]
- Kowtoniuk, W.E.; MacFarland, D.K.; Grover, G.N. Combining chiral elements: A novel approach to asymmetric phase-transfer catalyst design. Tetrahedron Lett. 2005, 46, 5703–5705. [Google Scholar] [CrossRef]
- Kumar, S.; Ramachandram, U. The synthesis and applications of asymmetric phase-transfer catalysts derived from isomannide and isosorbide. Tetrahedron 2005, 61, 4141–4148. [Google Scholar] [CrossRef]
- Denmark, S.E.; Gould, N.D.; Wolf, L.M. A systematic investigation of quaternary ammonium ions as asymmetric phase-transfer catalysts. Synthesis of catalyst libraries and evaluation of catalyst activity. J. Org. Chem. 2011, 76, 4260–4336. [Google Scholar] [CrossRef]
- Denmark, S.E.; Gould, N.D.; Wolf, L.M. A systematic investigation of quaternary ammonium ions as asymmetric phase-transfer catalysts. Application of quantitative structure activity/selectivity relationships. J. Org. Chem. 2011, 76, 4337–4357. [Google Scholar] [CrossRef]
- Seebach, D.; Keck, A.B.; Heckel, A. TADDOLs, their derivatives, and TADDOL analogues: Versatile chiral auxiliaries. Angew. Chem. Int. Ed. 2001, 40, 92–138. [Google Scholar] [CrossRef]
- Pellissier, H. Use of TADDOLs and their derivatives in asymmetric synthesis. Tetrahedron 2008, 64, 10279–10317. [Google Scholar] [CrossRef]
- Waser, M.; Gratzer, K.; Herchl, R.; Müller, N. Design, synthesis, and application of tartaric acid derived N-spiro quaternary ammonium salts as chiral phase-transfer catalysts. Org. Biomol. Chem. 2012, 10, 251–254. [Google Scholar] [CrossRef]
- Gratzer, K.; Waser, M. Investigations concerning the syntheses of TADDOL-derived secondary amines and their use to access novel chiral organocatalysts. Synthesis 2012, 44, 3661–3670. [Google Scholar] [CrossRef]
- Chinchilla, R.; Mazon, P.; Najera, C.; Ortega, F.J. The counterion effect in the phase-transfer catalyzed asymmetric synthesis of α-amino acids promoted by anthryl-derived dimeric Cinchona ammonium salts. Tetrahedron Asymmetry 2004, 15, 2603–2607. [Google Scholar] [CrossRef]
- Ohshima, T.; Gnanadesikan, V.; Shibuguchi, T.; Fukuta, Y.; Nemoto, T.; Shibasaki, M. Enantioselective syntheses of Aeruginosin 298-A and its analogues using a catalytic asymmetric phase-transfer reaction and epoxidation. J. Am. Chem. Soc. 2003, 125, 11206–11207. [Google Scholar]
- Lygo, B.; Beynon, C.; Lumley, C.; McLeod, M.C.; Wade, C.E. Co-catalyst enhancement of enantioselective PTC Michael additions involving glycine imines. Tetrahedron Lett. 2009, 50, 3363–3365. [Google Scholar] [CrossRef]
- Saito, S.; Tsubogo, T.; Kobayashi, S. Chiral calcium complexes as Brønsted base catalysts for asymmetric addition of α-amino acid derivatives to α,β-unsaturated carbonyl compounds. J. Am. Chem. Soc. 2007, 129, 5364–5365. [Google Scholar]
- Shibuguchi, T.; Mihara, H.; Kuramochi, A.; Sakuraba, S.; Ohshima, T.; Shibasaki, M. Short synthesis of (+)-Cylindricine C by using a catalytic asymmetric Michael reaction with a two-center organocatalyst. Angew. Chem. Int. Ed. 2006, 45, 4635–4637. [Google Scholar] [CrossRef]
- Arai, S.; Tokumaru, K.; Aoyama, T. Asymmetric Michael reaction promoted by new chiral phase-transfer catalysts. Chem. Pharm. Bul. 2004, 52, 646–648. [Google Scholar] [CrossRef]
- Akiyama, T.; Hara, M.; Fuchibe, K.; Sakamoto, S.; Yamaguchi, K. Synthesis of a novel crown ether derived from chiro-inositol and its catalytic activity on the asymmetric Michael addition. Chem. Commun. 2003, 1734–1735. [Google Scholar]
- Tsubogo, T.; Saito, S.; Seki, K.; Yamashita, Y.; Kobayashi, S. Development of catalytic asymmetric 1,4-addition and [3 + 2] cycloaddition reactions using chiral calcium complexes. J. Am. Chem. Soc. 2008, 130, 13321–13332. [Google Scholar]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gururaja, G.N.; Herchl, R.; Pichler, A.; Gratzer, K.; Waser, M. Application Scope and Limitations of TADDOL-Derived Chiral Ammonium Salt Phase-Transfer Catalysts. Molecules 2013, 18, 4357-4372. https://doi.org/10.3390/molecules18044357
Gururaja GN, Herchl R, Pichler A, Gratzer K, Waser M. Application Scope and Limitations of TADDOL-Derived Chiral Ammonium Salt Phase-Transfer Catalysts. Molecules. 2013; 18(4):4357-4372. https://doi.org/10.3390/molecules18044357
Chicago/Turabian StyleGururaja, Guddeangadi N., Richard Herchl, Antonia Pichler, Katharina Gratzer, and Mario Waser. 2013. "Application Scope and Limitations of TADDOL-Derived Chiral Ammonium Salt Phase-Transfer Catalysts" Molecules 18, no. 4: 4357-4372. https://doi.org/10.3390/molecules18044357