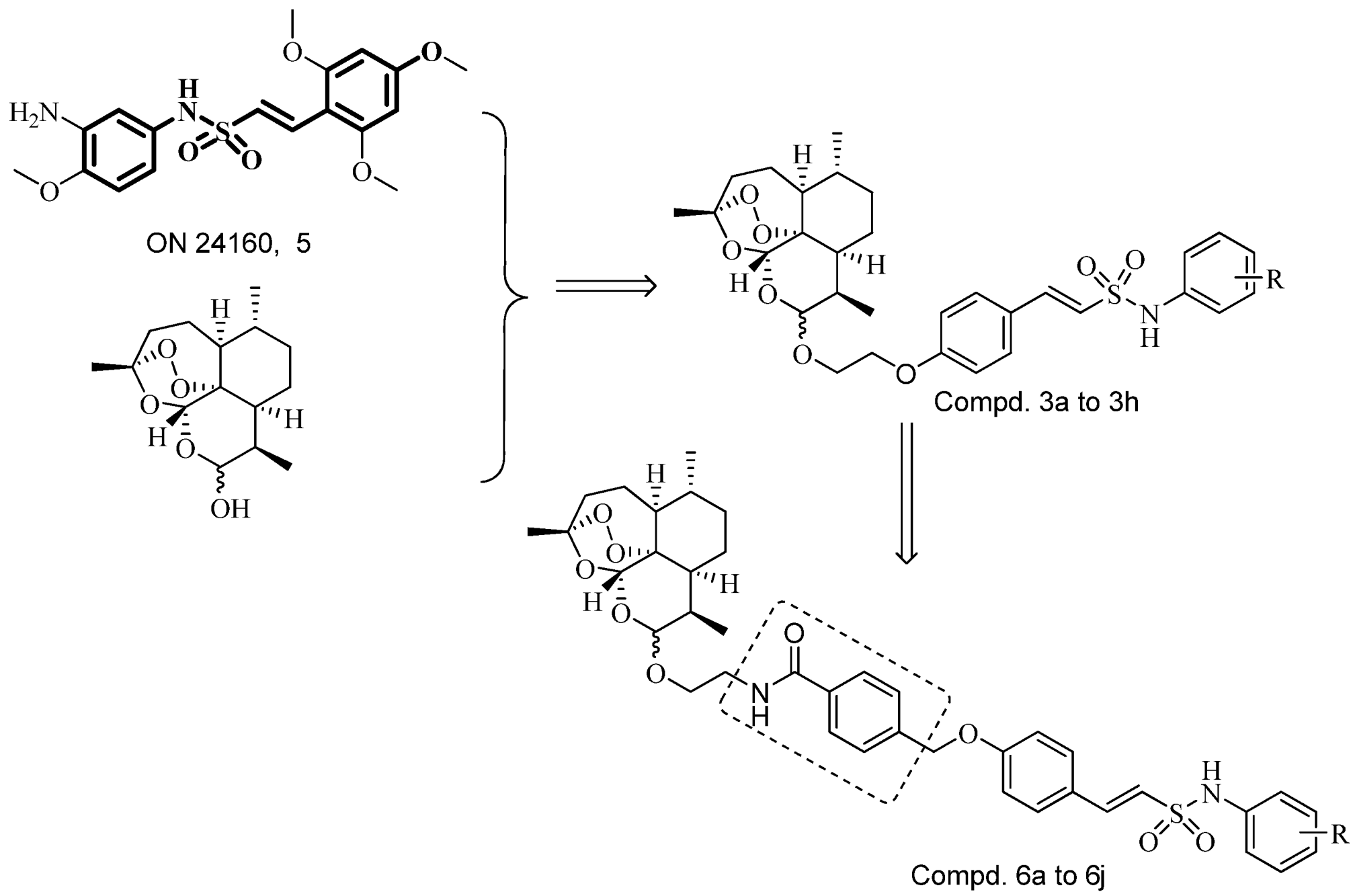
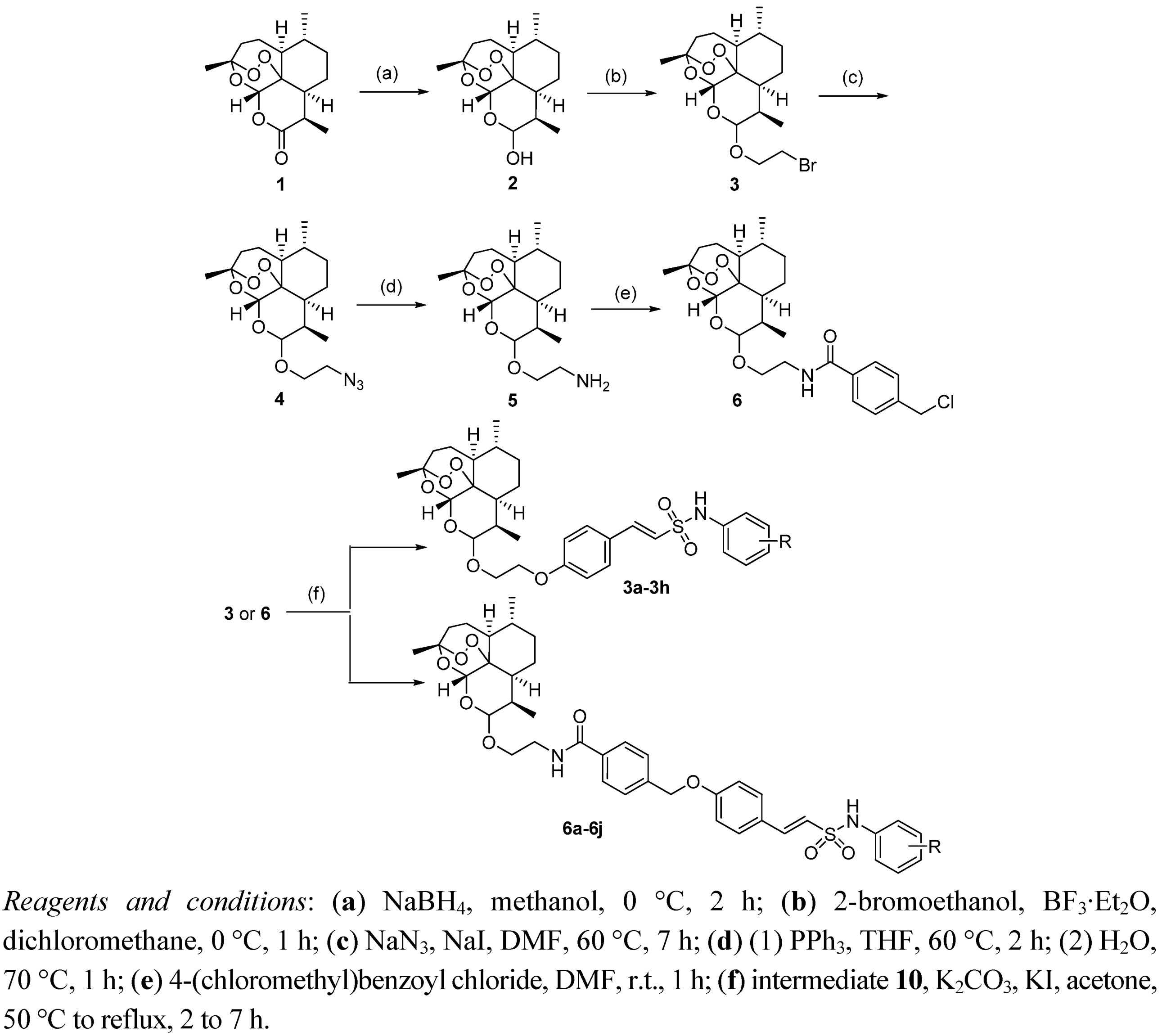
3.5. General Procedure for the Preparation of Target Compounds 6a–j
A mixture of compound 10 (0.024 mol), K2CO3 (4.2 g, 0.03 mol) and acetone (100 mL) was stirred at room temperature for 10 min, and then compound 6 (9.6 g, 0.02 mol) and KI (0.4 g, 2.6 mmol) was added while the reaction refluxed for 7–9 h. The acetone was removed under vacuum, and CH2Cl2 (100 mL) was poured into the residue. The solution was washed with water, dried with Na2SO4. The solvent was evaporated and the residue was purified by column chromatography (silica-gel, 1%–5% petroleum ether/ethyl acetate) to afford pure compounds 6a–j.
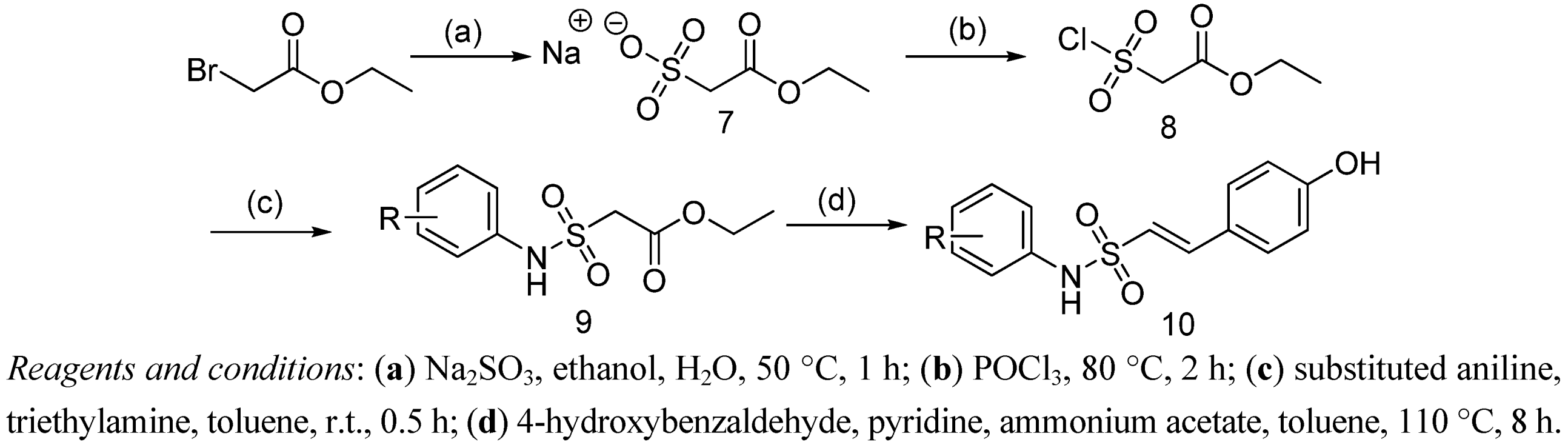
(E)-2-(4-(2-(10β-Dihydroartemisinoxy)ethoxy)phenyl)-N-phenylethenesulfonamide (3a). Light yellow solid (38% yield); m.p.: 136–138 °C; MS (ESI) m/z: 584.2 (M-H)–; IR (KBr) cm−1: 3422.1, 2923.1, 1603.8, 1514.4, 1343.4, 1147.5, 1027.3, 983.5, 696.9, 597.1; 1H-NMR (300 MHz, DMSO-d6) δ: 7.54 (d, J = 7.2 Hz, 2H), 7.35 (m, 5H), 7.15 (d, J = 15.2 Hz, 1H), 7.05 (d, J = 15.2 Hz, 1H), 6.79 (d, J = 7.5 Hz, 2H), 5.22 (s, 1H), 4.60 (s, 1H), 3.80 (m, 2H), 3.70 (m, 2H), 3.28 (m, 1H), 2.83 (m, 2H), 2.49 (m, 1H), 2.33 (m, 1H), 2.16 (m, 1H), 1.99 (m, 1H), 1.76 (m, 1H), 1.47 (m, 1H), 1.28 (m, 2H), 1.26 (s, 3H), 1.08 (s, 1H), 0.88 (d, J = 4.8 Hz, 3H), 0.75 (d, J = 7.1 Hz, 3H). Anal. Calcd. for C31H39NO8S: C 63.57, H 6.71, N 2.39. Found: C 63.52, H 6.50, N 2.41.
(E)-2-(4-(2-(10β-Dihydroartemisinoxy)ethoxy)phenyl)-N-(3-chlorophenyl)ethenesulfonamide (3b). Light yellow solid (44% yield); m.p.: 137–138 °C; MS (ESI) m/z: 618.0 (M−H)–; IR (KBr) cm−1: 3477.3, 2923.4, 1603.4, 1514.3, 1343.9, 1148.9, 1027.6, 983.4, 788.6, 597.7; 1H-NMR (300 MHz, DMSO-d6) δ 10.12(br, 1H), 7.58 (d, J = 8.6 Hz, 2H), 7.45 (m, 4H), 7.19 (d, J = 15.3 Hz, 1H), 7.08 (d, J = 15.3 Hz, 1H), 6.80 (d, J = 8.6 Hz, 2H), 5.22 (s, 1H), 4.60 (d, J = 3.1 Hz, 1H), 3.83 (m, 2H), 3.71 (m, 2H), 3.04 (s, 1H), 2.32 (m, 2H), 2.13 (d, J = 10.0 Hz, 1H), 2.00 (t, J = 13.9 Hz, 1H), 1.76 (m, 1H), 1.45 (m, 2H), 1.25 (d, J = 8.7 Hz, 5H), 1.11 (d, J = 18.8 Hz, 2H), 0.88 (d, J = 6.2 Hz, 3H), 0.72 (d, J = 7.4 Hz, 3H). Anal. Calcd. for C31H38ClNO8S: C 60.04, H 6.18, N 2.26. Found: C 59.98, H 6.21, N 2.19.
(E)-2-(4-(2-(10β-Dihydroartemisinoxy)ethoxy)phenyl)-N-(4-trifluoromethoxyphenyl)ethenesulfonamide (3c). Yellow solid (42% yield); m.p.: 139–142 °C; MS (ESI) m/z: 692.1 (M+Na)+; IR (KBr) cm−1: 3416.7, 2924.6, 1604.1, 1510.7, 1346.0, 1258.8, 1224.3, 1027.9, 983.6, 838.8, 504.3; 1H-NMR (300 MHz, DMSO-d6) δ 10.06 (br, 1H), 7.56 (d, J = 8.5 Hz, 2H), 7.48 (d, J = 8.9 Hz, 2H), 7.37 (d, J = 8.5 Hz, 2H), 7.19 (d, J = 15.3 Hz, 1H), 7.08 (d, J = 15.3 Hz, 1H), 6.79 (d, J = 8.6 Hz, 2H), 5.21 (s, 1H), 4.60 (d, J = 3.3 Hz, 1H), 3.82 (m, 2H), 3.76 (m, 2H), 2.32 (m, 1H), 2.15 (m, 1H), 2.00 (m, 1H), 1.76 (m, 1H), 1.46 (m, 2H), 1.41 (m, 1H), 1.26 (m, 6H), 1.09 (m, 2H), 0.87 (m, 3H), 0.70 (d, J = 7.3 Hz, 3H). Anal. Calcd. for C32H38F3NO9S: C 57.39, H 5.72, N 2.09. Found: C 57.41, H 5.68, N 2.11.
(E)-2-(4-(2-(10β-Dihydroartemisinoxy)ethoxy)phenyl)-N-(2-fluoro-5-methylphenyl)ethenesulfonamide (3d). Light yellow solid (36% yield); m.p.: 138–140 °C; MS (ESI) m/z: 640.1 (M+Na)+; IR (KBr) cm−1: 3424.5, 2923.5, 1604.1, 1510.7, 1343.8, 1148.7, 1028.1, 984.6, 872.5, 601.1; 1H-NMR (300 MHz, DMSO-d6) δ 10.06 (br, 1H), 7.56 (d, J = 8.5 Hz, 2H), 7.24 (m, 2H), 7.18 (m, 2H), 7.09 (d, J = 15.3 Hz, 1H), 6.81 (d, J = 8.6 Hz, 2H), 5.22 (s, 1H), 4.59 (d, J = 3.2 Hz, 1H), 3.72 (m, 4H), 2.26 (m, 1H), 2.15 (s, 3H), 2.10 (m, 1H), 2.00 (m, 1H), 1.77 (m, 1H), 1.40 (m, 3H), 1.28 (m, 6H), 1.09 (m, 2H), 0.87 (d, J = 5.9 Hz, 3H), 0.71 (d, J = 7.3 Hz, 3H). Anal. Calcd. for C32H40FNO8S: C 62.22, H 6.53, N 2.27. Found: C 62.19, H 6.55, N 2.29.
(E)-2-(4-(2-(10β-Dihydroartemisinoxy)ethoxy)phenyl)-N-(2-methylphenyl)ethenesulfonamide (3e). Light brown solid (26% yield); m.p.: 136–138 °C; MS (ESI) m/z: 598.2 (M−H)–; IR (KBr) cm−1: 3414.8, 2923.1, 1604.2, 1514.7, 1342.5, 1146.2, 1027.5, 983.6, 872.4, 603.3; 1H-NMR (300 MHz, DMSO-d6) δ 7.59 (d, J = 8.5 Hz, 2H), 7.28 (m, 6H), 6.80 (d, J = 8.6 Hz, 2H), 5.15(s, 1H), 4.62 (d, J = 3.3 Hz, 1H), 4.45 (d, J = 5.7 Hz, 1H), 3.82 (m, 2H), 3.70 (m, 2H), 2.35 (s, 3H), 2.07–1.87 (m, 1H), 1.75 (m, 1H), 1.42 (m, 2H), 1.26 (s, 3H), 1.25 (m, 4H), 1.06 (t, J = 7.0 Hz, 2H), 0.86 (d, J = 5.9 Hz, 3H), 0.73 (d, J = 7.3 Hz, 3H). Anal. Calcd. for C32H41NO8S: C 64.09, H 6.89, N 2.34. Found: C 64.13, H 6.81, N 2.39.
(E)-2-(4-(2-(10β-Dihydroartemisinoxy)ethoxy)phenyl)-N-(4-chlorophenyl)ethenesulfonamide (3f). Light brown solid (44% yield); m.p.: 138–141 °C; MS (ESI) m/z: 618.1 (M−H)–; IR (KBr) cm−1: 3411.5, 2925.1, 1601.5, 1490.3, 1343.2, 1149.3, 1054.2, 1027.0, 1007.0, 822.9, 760.8; 1H-NMR (300 MHz, DMSO-d6) δ 7.56 (d, J = 8.3 Hz, 1H), 7.43 (m, 3H), 7.18 (m, 2H), 7.01 (d, J = 8.8 Hz, 1H), 6.80 (d, J = 7.6 Hz, 1H), 6.67 (d, J = 7.0 Hz, 1H), 6.50 (d, J = 9.0 Hz, 1H), 5.17 (s, 1H), 4.76 (d, J = 3.5 Hz, 1H), 4.35 (m, 2H), 3.82 (m, 2H), 3.62 (m, 2H), 2.05 (s, 3H), 1.56 (m, 2H), 1.35 (m, 2H), 1.24 (s, 3H), 1.10 (m, 3H), 0.97 (d, J = 6.0 Hz, 3H), 0.80 (d, J =7.4 Hz, 3H). Anal. Calcd. for C31H38ClNO8S: C 60.04, H 6.18, N 2.26. Found: C 60.11, H 6.12, N 2.30.
(E)-2-(4-(2-(10β-Dihydroartemisinoxy)ethoxy)phenyl)-N-(2-chlorophenyl)ethenesulfonamide (3g). Yellow solid (38% yield); m.p.: 141–143 °C; MS (ESI) m/z: 618.2 (M-H)–; IR (KBr) cm−1: 3412.1, 2926.3, 1604.2, 1493.5, 1340.3, 1145.7, 1052.8, 1022.4, 828.5, 772.4; 1H-NMR (300 MHz, DMSO-d6) δ 7.65 (d, J = 8.2 Hz, 2H), 7.45 (m, 2H), 7.28 (m, 2H), 7.20 (d, J = 15.1 Hz, 1H), 7.04 (d, J = 15.1 Hz, 1H), 6.81 (d, J = 8.3 Hz, 2H), 5.27 (s, 1H), 4.62 (d, J = 3.2 Hz, 1H), 3.82 (m, 2H), 3.76 (m, 2H), 3.30 (m, 2H), 2.81 (m, 1H), 2.53 (m, 1H), 2.37 (m, 1H), 2.20 (m, 1H), 2.00 (m, 1H), 1.85 (m, 1H), 1.51 (m, 1H), 1.32 (m, 2H), 1.27 (s, 3H), 1.09 (s, 1H), 0.85 (d, J = 6.8 Hz, 3H), 0.79 (d, J = 7.3 Hz, 3H). Anal. Calcd. for C31H38ClNO8S: C 60.04, H 6.18, N 2.26. Found: C 59.93, H 6.21, N 2.29.
(E)-2-(4-(2-(10β-Dihydroartemisinoxy)ethoxy)phenyl)-N-(2,6-dichlorophenyl)ethenesulfonamide (3h). Brown solid (41% yield); m.p.: 144–146 °C; MS (ESI) m/z: 653.1 (M-H)–; IR (KBr) cm−1: 3420.8, 2922.6, 1602.6, 1508.2, 1348.2, 1147.1, 1027.8, 984.7, 874.3, 541.5; 1H-NMR (300 MHz, DMSO-d6) δ 7.78 (d, J = 8.1 Hz, 2H), 7.28 (m, 2H), 7.15 (d, J = 15.3 Hz, 1H), 6.97 (m, 2H), 6.83 (d, J = 7.9 Hz, 2H), 5.21 (s, 1H), 4.62 (d, J = 3.3 Hz, 1H), 3.82 (m, 4H), 2.31 (m, 2H), 2.16 (s, 2H), 2.12 (m, 1H), 1.98 (m, 1H), 1.75 (m, 2H), 1.39 (m, 2H), 1.26 (m, 6H), 1.07 (m, 2H), 0.89 (d, J = 5.9 Hz, 3H), 0.78 (d, J = 7.4 Hz, 3H). Anal. Calcd. for C31H37Cl2NO8S: C 56.88, H 5.70, N 2.14. Found: C 56.92, H 5.75, N 2.17.
(E)-N-(2-(10β-Dihydroartemisinoxy)ethyl)-4((4-(2-(N-phenylsulfamoyl)vinyl)phenoxy)methyl)benzamide (6a). Light yellow solid (48% yield); m.p.: 142–145 °C; MS (ESI) m/z: 717.1 (M-H)–; IR (KBr) cm−1: 3413.1, 2924.1, 1604.2, 1513.9, 1336.6, 1146.1, 1020.8, 873.7, 801.7, 699.4, 603.3; 1H-NMR (300 MHz, DMSO-d6) δ 8.49 (br, 1H), 7.85 (d, J = 8.1 Hz, 2H), 7.62 (m, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 15.2 Hz, 1H), 7.24 (t, J = 7.6 Hz, 2H), 7.17 (d, J = 8.1 Hz, 2H), 7.0 (m, 4H), 5.26 (s, 1H), 5.21 (s, 2H), 4.69 (d, J = 3.2 Hz, 1H), 4.44 (d, J = 5.2 Hz, 1H), 4.39 (t, J = 3.9 Hz, 2H), 3.82 (m, 2H), 2.24 (m, 2H), 2.06 (m, 3H), 1.67 (m, 3H), 1.48 (m, 2H), 1.26 (s, 3H), 1.23 (s, 1H), 0.81 (d, J = 7.4 Hz, 3H), 0.68 (d, J = 5.6 Hz, 3H). Anal. Calcd. for C39H46N2O9S: C 65.16, H 6.45, N 3.90. Found: C 65.09, H 6.43, N 3.96.
(E)-N-(2-(10β-Dihydroartemisinoxy)ethyl)-4((4-(2-(N-(3-chlorophenyl)sulfamoyl)vinyl)phenoxy)methyl)benzamide (6b). Light yellow solid (45% yield); m.p.: 146–149 °C; MS (ESI) m/z: 751.0 (M-H)–; IR (KBr) cm−1: 3397.3, 2923.3, 1602.5, 1512.7, 1344.3, 1147.4, 1026.6, 983.7, 872.9, 791.7, 594.8; 1H-NMR (300 MHz, DMSO-d6) δ 8.40 (br, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.60 (d, J = 8.5 Hz, 2H), 7.50 (d, J = 13.1 Hz, 2H), 7.37 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 4.6 Hz, 2H), 7.26 (d, J = 8.1 Hz, 2H), 6.82 (d, J = 8.2 Hz, 2H), 5.25 (s, 1H), 4.92 (d, J = 13.4 Hz, 2H), 4.68 (d, J = 3.0 Hz, 1H), 3.82 (m, 2H), 3.53(m, 2H), 2.34 (m, 1H), 2.12 (m, 2H), 2.00 (m, 2H), 1.66 (m, 2H), 1.49 (m, 2H), 1.26 (m, 6H), 0.80 (d, J = 7.2 Hz, 3H), 0.65 (d, J = 5.7 Hz, 3H). Anal. Calcd. for C39H45ClN2O9S: C 62.18, H 6.02, N 3.72. Found: C 62.23, H 6.18, N 3.68.
(E)-N-(2-(10β-Dihydroartemisinoxy)ethyl)-4((4-(2-(N-(4-trifluoromethoxyphenyl)sulfamoyl)vinyl)phenoxy)methyl)benzamide (6c). Yellow solid (43% yield); m.p.: 149–151 °C; MS (ESI) m/z: 801.2 (M−H)–; IR (KBr) cm−1: 3396.9, 2924.0, 1603.0, 1507.9, 1346.4, 1360.2, 1146.2, 1025.4, 870.5, 801.6, 593.1; 1H-NMR (300 MHz, DMSO-d6) δ 8.38 (br, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.60 (d, J = 8.5 Hz, 2H), 7.47 (d, J = 9.0 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 7.34 (m, 4H), 6.82 (d, J = 8.2 Hz, 2H), 5.25 (s, 1H), 4.88 (s, 2H), 4.68 (d, J = 3.2 Hz, 1H), 4.45 (m, 1H), 4.35 (m, 1H), 3.82 (s, 2H), 3.62 (m, 1H), 2.35 (m, 1H), 2.11 (m, 1H), 1.96 (m, 1H), 1.65 (m, 3H), 1.51 (m, 1H), 1.25 (m, 7H), 0.79 (d, J = 7.2 Hz, 3H), 0.65 (d, J = 5.8 Hz, 3H). Anal. Calcd. for C40H45F3N2O10S: C 59.84, H 5.65, N 3.49. Found: C 59.93, H 5.69, N 3.52.
(E)-N-(2-(10β-Dihydroartemisinoxy)ethyl)-4((4-(2-(N-(2-fluoro-5-methylphenyl)sulfamoyl)vinyl)phenoxy)methyl)benzamide (6d) Light yellow solid (35% yield); m.p.: 135–137 °C; MS (ESI) m/z: 749.2 (M-H)–; IR (KBr) cm−1: 3412.2, 2924.6, 1603.9, 1509.1, 1343.9, 1146.1, 1025.8, 984.1, 872.7, 843.9, 597.9; 1H-NMR (300 MHz, DMSO-d6) δ 8.40 (br, 1H), 7.74 (d, J = 8.1 Hz, 2H), 7.60 (d, J = 8.4 Hz, 2H), 7.41(d, J = 8.2 Hz, 2H), 7.28 (s, 1H), 7.20 (m, 2H), 7.13 (m, 2H), 6.83 (d, J = 8.1 Hz, 2H), 5.26 (s, 1H), 4.78 (s, 2H), 4.69 (d, J = 3.3 Hz, 1H), 4.46 (d, J = 3.5 Hz, 1H), 4.39 (m, 1H), 3.82 (m, 2H), 3.52 (m, 2H), 2.34 (m, 1H), 2.19 (s, 3H), 2.10 (m, 1H), 1.94 (m, 1H), 1.66 (m, 2H), 1.52 (m, 1H), 1.38 (s, 1H), 1.25 (m, 6H), 0.80 (d, J = 7.3 Hz, 3H), 0.66 (d, J = 5.6 Hz, 3H). Anal. Calcd. for C40H47FN2O9S: C 63.98, H 6.31, N 3.73. Found: C 64.02, H 6.37, N 3.68.
(E)-N-(2-(10β-Dihydroartemisinoxy)ethyl)-4((4-(2-(N-(2-methylphenyl)sulfamoyl)vinyl)phenoxy)methyl)benzamide (6e) Light yellow solid (40% yield); m.p.: 143–145 °C; MS (ESI) m/z: 731.2 (M-H)–; IR (KBr) cm−1: 3397.2, 2923.2, 1603.4, 1510.1, 1341.8, 1143.7, 1027.5, 984.2, 874.6, 800.3, 594.8; 1H-NMR (300 MHz, DMSO-d6) δ 8.39 (s, 1H), 7.71 (d, J = 8.1 Hz, 2H), 7.61 (m, 4H), 7.34 (m, 4H), 7.15 (d, J = 8.2 Hz, 2H), 6.83 (d, J = 8.2 Hz, 2H), 5.29 (s, 1H), 4.87 (m, 1H), 4.69 (m, 2H), 3.83 (m, 2H), 3.47 (m, 2H), 2.40 (m, 2H), 2.31 (s, 1H), 2.15 (m, 1H), 2.07 (s, 3H), 1.95 (m, 2H), 1.70 (m, 2H), 1.51 (m, 1H), 1.27 (m, 4H), 1.04 (m, 2H), 0.80 (d, J = 7.3 Hz, 3H), 0.72 (d, J = 4.7 Hz, 3H). Anal. Calcd. for C40H48N2O9S: C 65.55, H 6.60, N 3.82. Found: C 65.48, H 6.57, N 3.85.
(E)-N-(2-(10β-Dihydroartemisinoxy)ethyl)-4((4-(2-(N-(4-chlorophenyl)sulfamoyl)vinyl)phenoxy)methyl)benzamide (6f). Yellow solid (39% yield); m.p.: 138–141 °C; MS (ESI) m/z: 751.1 (M-H)–; IR (KBr) cm−1: 3397.1, 2923.1, 1603.4, 1509.9, 1345.8, 1146.5, 1026.2, 982.5, 871.4, 800.5, 594.3; 1H-NMR (300 MHz, DMSO-d6) δ 8.39 (br, 1H), 7.72 (d, J = 8.2 Hz, 2H), 7.59 (d, J = 8.5 Hz, 2H), 7.48 (d, J = 13.1 Hz, 2H), 7.32 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 4.6 Hz, 2H), 7.17 (d, J = 8.1 Hz, 2H), 6.80 (d, J = 8.2 Hz, 2H), 5.24 (s, 1H), 4.83 (s, 2H), 4.62 (d, J = 3.2 Hz, 1H), 4.41 (m, 1H), 4.30 (m, 1H), 3.81 (s, 2H), 3.60 (m, 1H), 2.33 (m, 1H), 2.08 (m, 1H), 1.93 (m, 1H), 1.61 (m, 3H), 1.47 (m, 2H), 1.26 (m, 6H), 0.78 (d, J = 7.2 Hz, 3H), 0.64 (d, J = 5.8 Hz, 3H). Anal. Calcd. for C39H45ClN2O9S: C 62.18, H 6.02, N 3.72. Found: C 62.23, H 5.98, N 3.76.
(E)-N-(2-(10β-Dihydroartemisinoxy)ethyl)-4((4-(2-(N-(2-chlorophenyl)sulfamoyl)vinyl)phenoxy)methyl)benzamide (6g). Light yellow solid (38% yield); m.p.: 147–150 °C; MS (ESI) m/z: 775.2 (M+Na)+; IR (KBr) cm−1: 3394.1,2925.3, 1605.5, 1499.1, 1339.3, 1144.8, 1028.8, 979.5, 881.7, 806.7, 598.2; 1H-NMR (300 MHz, DMSO-d6) δ 7.59 (m, 2H), 7.40 (d, J = 8.6 Hz, 4H), 7.32 (m, 3H), 6.94 (s, 2H), 6.79 (m, 3H), 5.62 (s, 1H), 5.35 (d, J = 10.3 Hz, 1H), 4.80 (s, 2H), 4.68 (d, J = 3.1 Hz, 1H), 3.81 (m, 2H), 3.45 (m, 2H), 2.18 (m, 1H), 2.05 (m, 2H), 1.94 (m, 2H), 1.63 (m, 2H), 1.52 (m, 2H), 1.33 (s, 3H), 0.98 (m, 2H), 0.83 (d, J = 7.4 Hz, 3H), 0.71 (d, J = 5.7 Hz, 3H). Anal. Calcd. for C39H45ClN2O9S: C 62.18, H 6.02, N 3.72. Found: C 62.12, H 5.97, N 3.79.
(E)-N-(2-(10β-Dihydroartemisinoxy)ethyl)-4((4-(2-(N-(2,6-dichlorophenyl)sulfamoyl)vinyl)phenoxy)methyl)benzamide (6h). Light yellow solid (40% yield); m.p.: 151–153 °C; MS (ESI) m/z: 784.9 (M-H)–; IR (KBr) cm−1: 3420.8, 2922.6, 1602.6, 1508.2, 1348.2, 1147.1, 1027.8, 984.7, 874.3, 541.5; 1H-NMR (300 MHz, DMSO-d6) δ 8.40 (br, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.62 (d, J = 8.3 Hz, 2H), 7.59 (m, 4H), 7.28 (m, 4H), 6.84 (d, J = 7.5 Hz, 2H), 5.26 (s, 1H), 4.80 (s, 2H), 4.69 (d, J = 3.3 Hz, 1H), 3.85 (m, 2H), 3.59 (m, 2H), 2.35 (m, 1H), 2.13 (m, 1H), 1.97 (m, 2H), 1.67 (m, 3H), 1.52 (m, 2H), 1.27 (s, 3H), 1.24 (s, 2H), 1.04 (m, 1H), 0.81 (d, J = 7.4 Hz, 3H), 0.69 (d, J = 4.5 Hz, 3H). Anal. Calcd. for C39H44Cl2N2O9S: C 59.46, H 5.63, N 3.56. Found: C 59.42, H 5.59, N 3.50.
(E)-N-(2-(10β-Dihydroartemisinoxy)ethyl)-4((4-(2-(N-(3-methoxyphenyl)sulfamoyl)vinyl)phenoxy)methyl)benzamide (6i). Light yellow solid (48% yield); m.p.: 144–145 °C; MS (ESI) m/z: 747.2 (M−H)–; IR (KBr) cm−1: 3392.7, 2923.7, 1603.2, 1512.6, 1340.0, 1142.2, 1025.9, 982.8, 871.7, 840.7, 596.3; 1H-NMR (300 MHz, DMSO-d6) δ 8.40 (br, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.60 (d, J = 8.5 Hz, 2H), 7.50 (s, 1H), 7.43 (s, 1H), 7.37 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 4.6 Hz, 2H), 7.26 (d, J = 8.1 Hz, 2H), 6.82 (d, J = 8.6 Hz, 2H), 5.25 (s, 1H), 4.92 (d, J = 5.4 Hz, 2H), 4.68 (m, 1H), 3.95(s, 3H), 3.82 (m, 2H), 3.51(m, 2H), 2.34 (m, 2H), 2.12 (m, 2H), 2.00 (m, 2H), 1.66 (m, 2H), 1.49 (m, 1H), 1.26 (s, 3H), 1.21 (m, 3H), 0.81 (d, J = 7.3 Hz, 3H), 0.69 (d, J = 5.2 Hz, 3H). Anal. Calcd. for C40H48N2O10S: C 64.15, H 6.46, N 3.74. Found: C 64.09, H 6.41, N 3.67.
(E)-N-(2-(10β-Dihydroartemisinoxy)ethyl)-4((4-(2-(N-(3-trifluoromethyl-4fluorophenyl)sulfamoyl)vinyl)phenoxy)methyl)benzamide (6j). Yellow solid (32% yield); m.p.: 152–154 °C; MS (ESI) m/z: 803.2 (M-H)–; IR (KBr) cm−1: 3401.8, 2924.7, 1602.3, 1503.7, 1328.9, 1145.3, 1026.0, 983.9, 845.8, 646.6, 542.5; 1H-NMR (300 MHz, DMSO-d6) δ 8.40 (br, 1H), 7.81 (m, 4H), 7.63 (m, 3H), 7.47 (d, J = 15.6 Hz, 1H), 7.37 (t, J = 8.4 Hz, 2H), 7.27 (d, J = 15.6 Hz, 1H), 6.82 (d, J = 8.6 Hz, 2H), 5.24 (s, 1H), 4.98 (s, 1H), 4.93 (s, 2H), 4.68 (d, J = 3.1 Hz, 1H), 3.83 (m, 2H), 3.51 (m, 2H), 2.34 (m, 1H), 2.16 (m, 2H), 1.94 (m, 1H), 1.65 (m, 2H), 1.49 (m, 1H), 1.26 (s, 4H), 1.14 (m, 2H), 1.05 (m, 1H), 0.79 (d, J = 7.3 Hz, 3H), 0.60 (d, J = 6.0 Hz, 3H). Anal. Calcd. for C40H44F4N2O9S: C 59.69, H 5.51, N 3.48. Found: C 59.74, H 5.46, N 3.41.




{kind=link}
{kind=link}
{kind=link}
{kind=link}