Multiplexed Analysis of Cage and Cage Free Chicken Egg Fatty Acids Using Stable Isotope Labeling and Mass Spectrometry

Abstract
:1. Introduction
- (1)
- SID can only be used to quantify metabolites where a commercially available isotopic analog exists.
- (2)
- The isotopic analogs should not contain deuterium, since such analogs will exhibit a small chromatographic separation from the endogenous protium from which can lead to differential suppression of ionization.
- (3)
- Many common metabolites do not sufficiently ionize in conventional MS, with or without isotopic atoms present, and thus are not quantified in a SID approach.
2. Results and Discussion
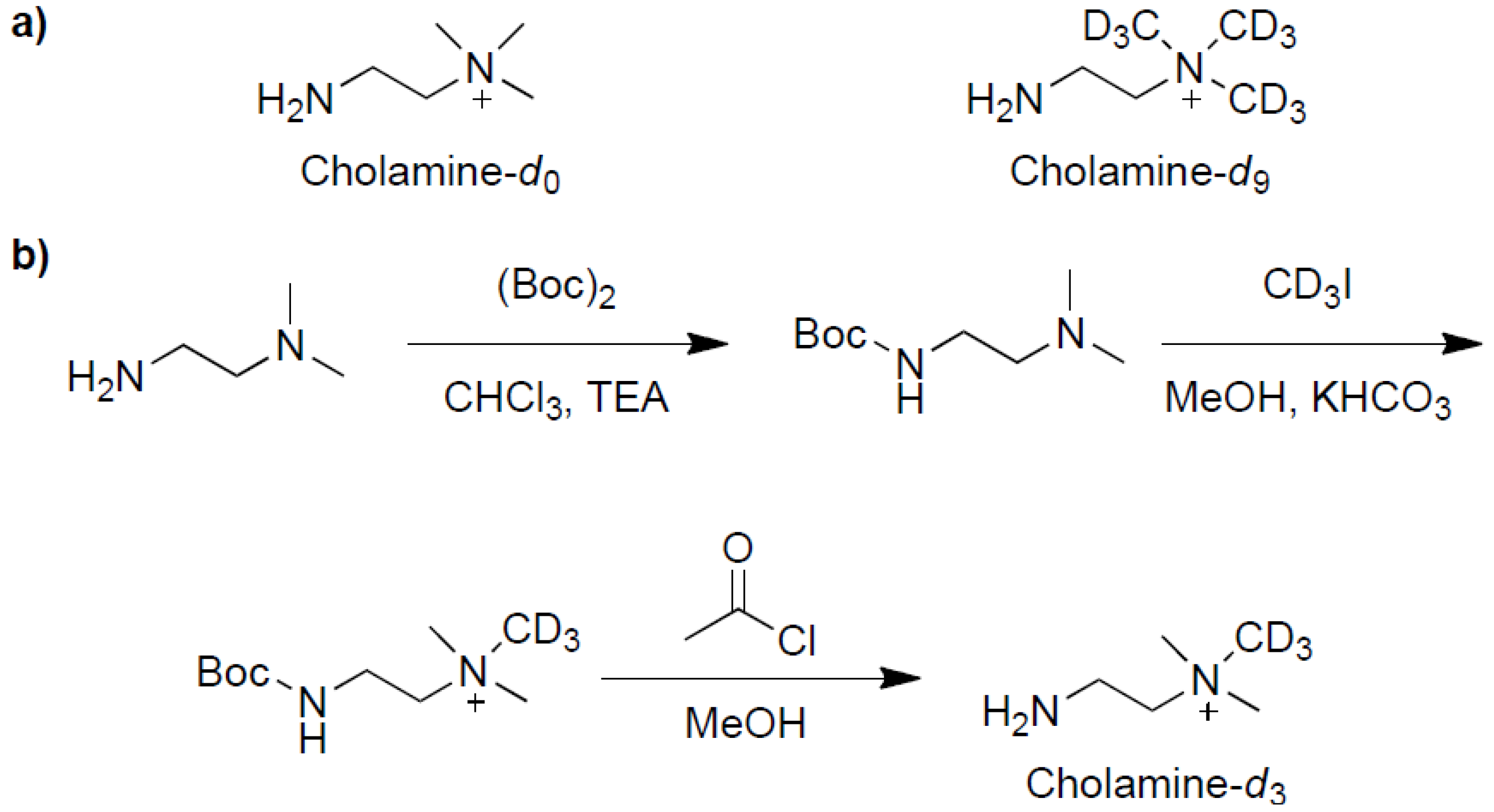
2.1. Synthesis and Evaluation of the Three Isotopic Labeling Cholamines
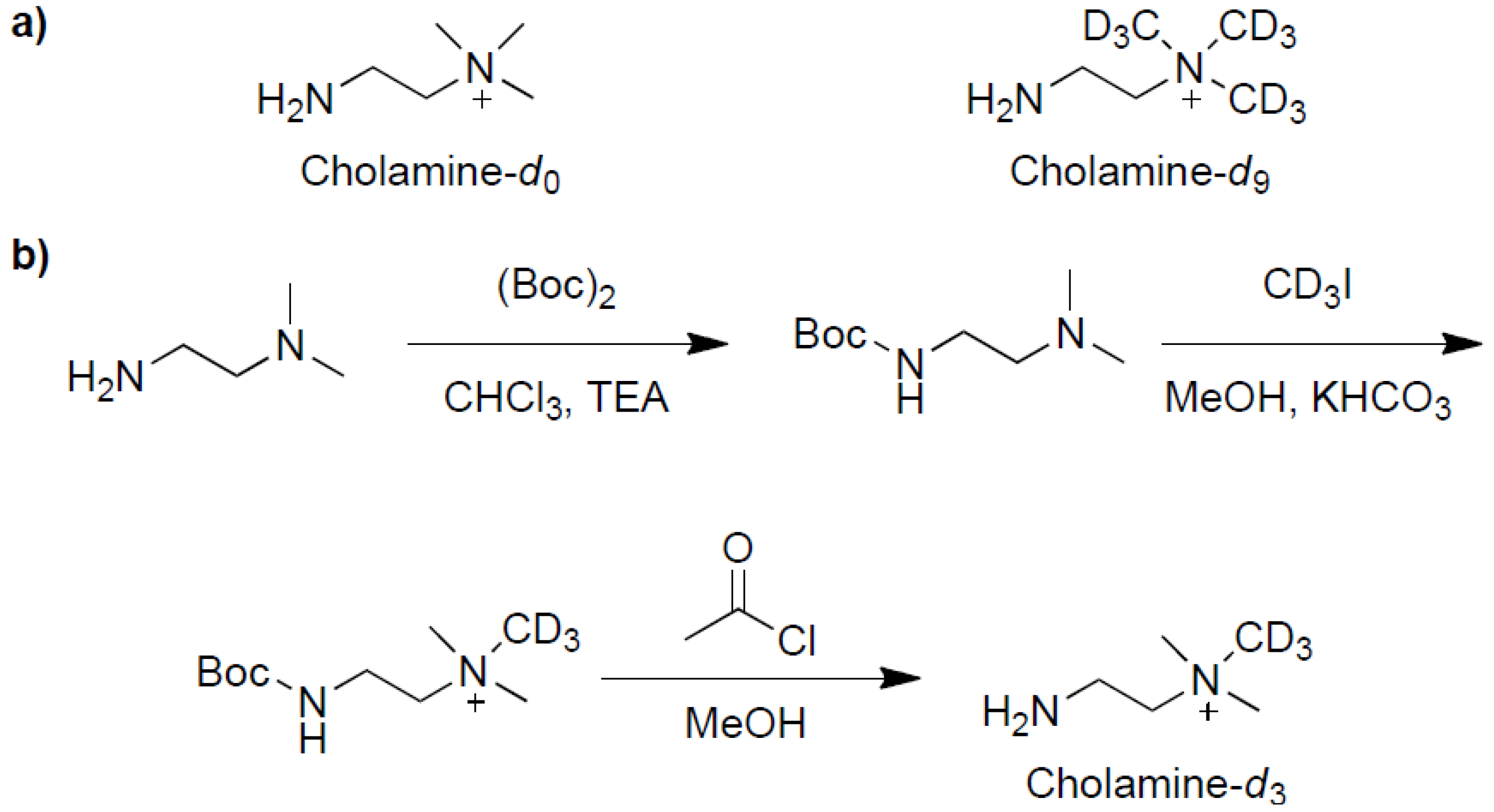
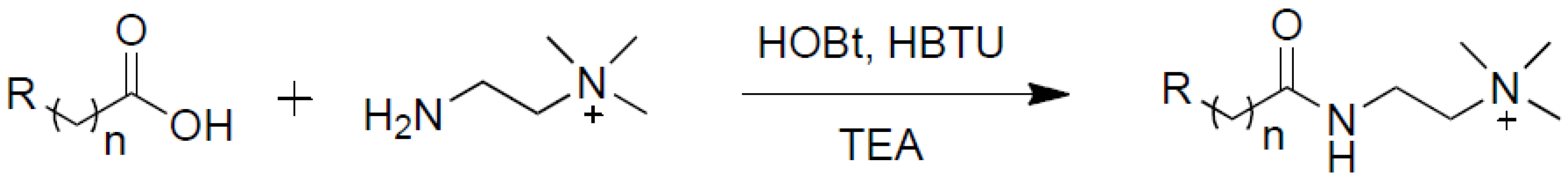
2.2. Simulated Multiplex Quantification of Lauric Acid
2.3. Simulated Multiplex Fatty Acid Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fatty acid a | Experimental ratio | Accuracy | |||
|---|---|---|---|---|---|
| d0 | d3 | d9 | (% error) | ||
| Myrisitc acid | 14:0 | 1.01 | 1.01 | 0.98 | 1.9 |
| Myristoleic acid | 14:1 | 1.03 | 0.95 | 1.02 | 4.1 |
| Palmitic acid | 16:0 | 0.98 | 1.03 | 0.99 | 2.7 |
| Palmitoleic acid | 16:1 | 0.95 | 0.96 | 1.09 | 8.0 |
| Stearic acid | 18:0 | 0.97 | 1.02 | 1.00 | 2.4 |
| Oleic acid | 18:1 | 1.08 | 0.95 | 0.96 | 7.2 |
| linoleic acid | 18:2 | 0.96 | 1.00 | 1.04 | 3.7 |
| α-linolenic acid | 18:3 | 1.04 | 0.93 | 1.03 | 6.4 |
| Eicosatrienoic acid | 20:3 | 0.90 | 1.10 | 1.00 | 10.4 |
| Arachidonic acid | 20:4 | 0.90 | 1.10 | 1.00 | 9.6 |
| Docosapentaenoic acid | 22:5 | 0.93 | 1.05 | 1.02 | 6.0 |
| Docosahexaenoic acid | 22:6 | 0.99 | 1.02 | 0.99 | 2.1 |
| Average | 0.97 | 1.01 | 1.01 | 5.4 | |
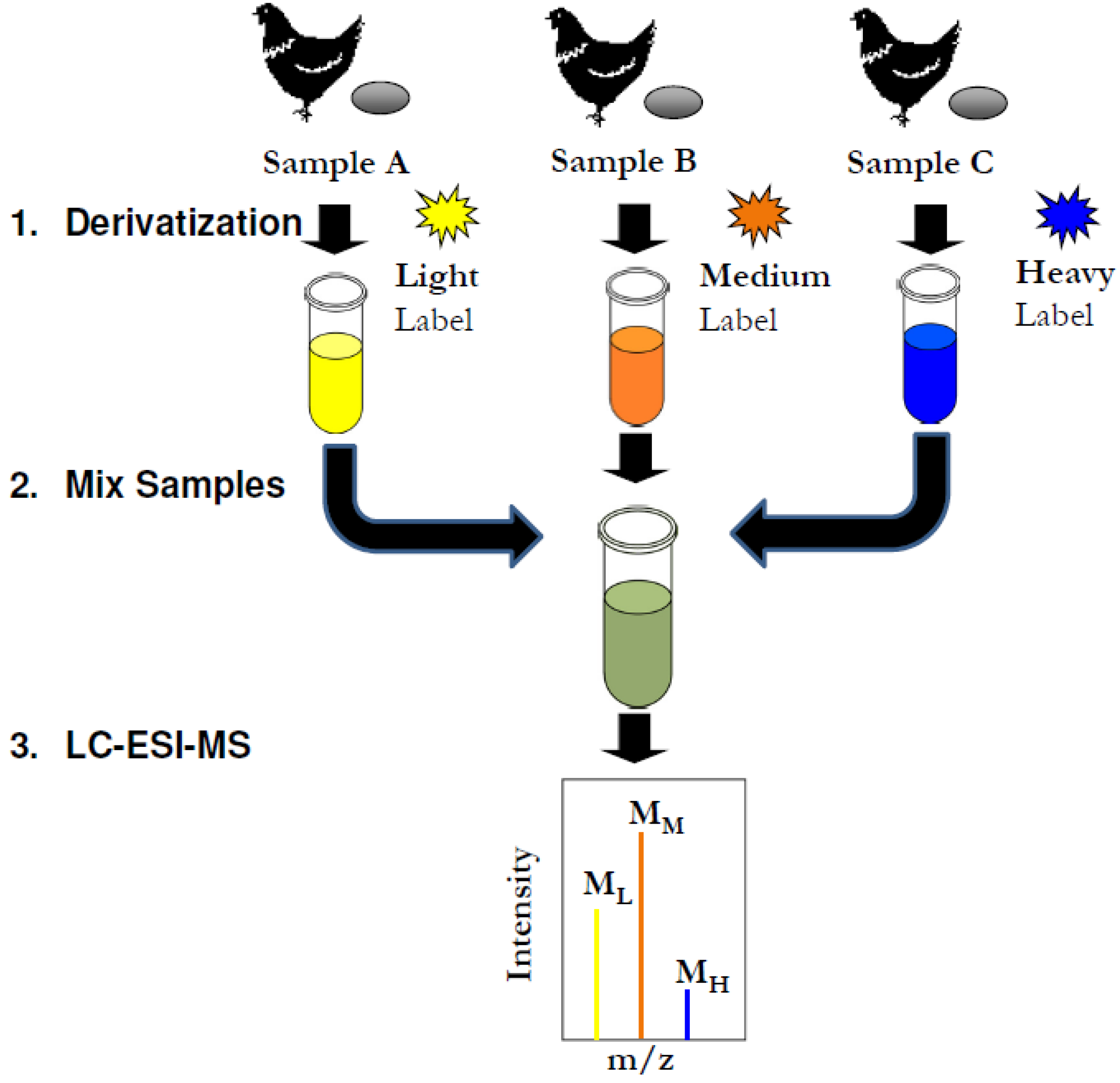
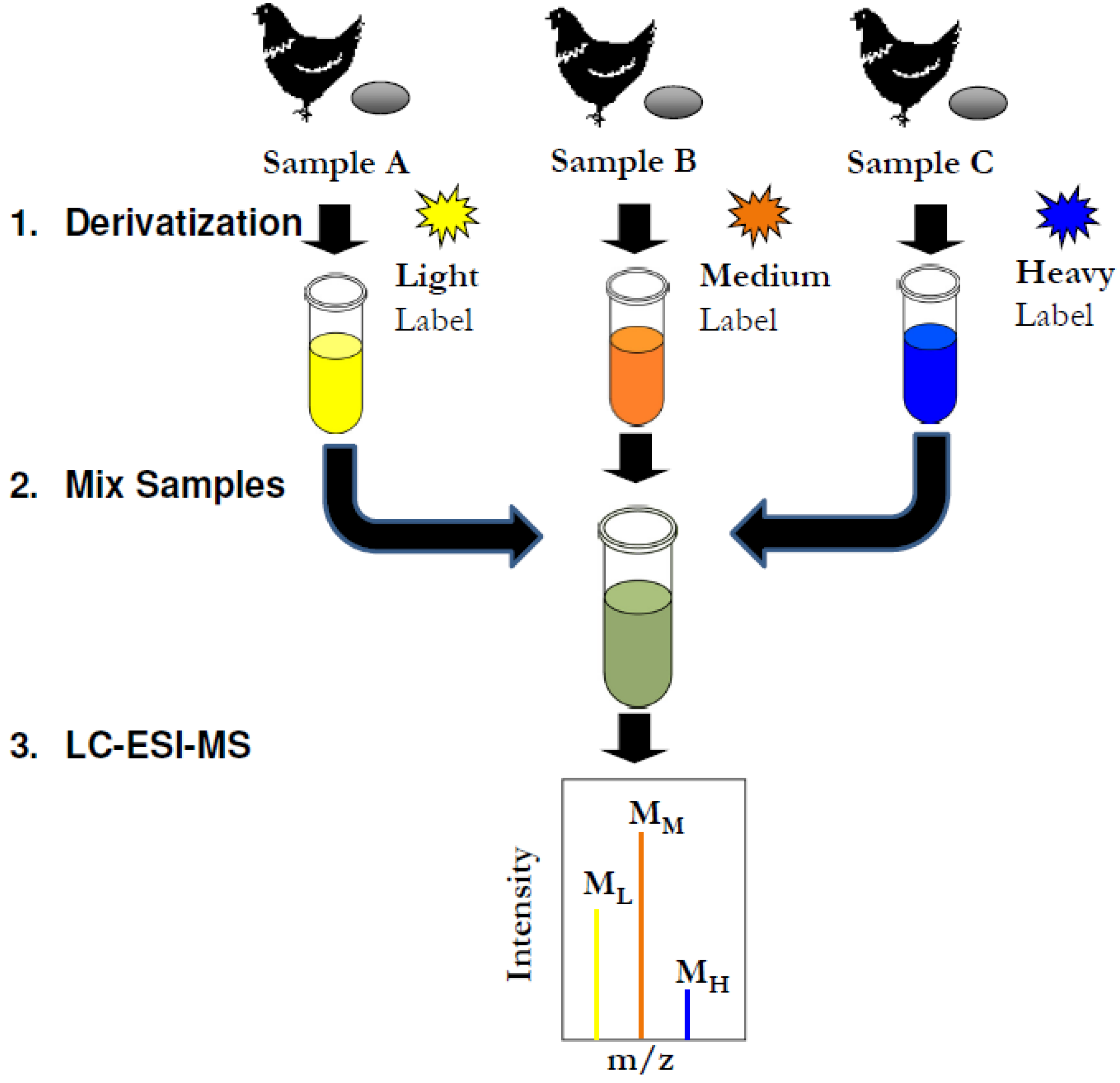
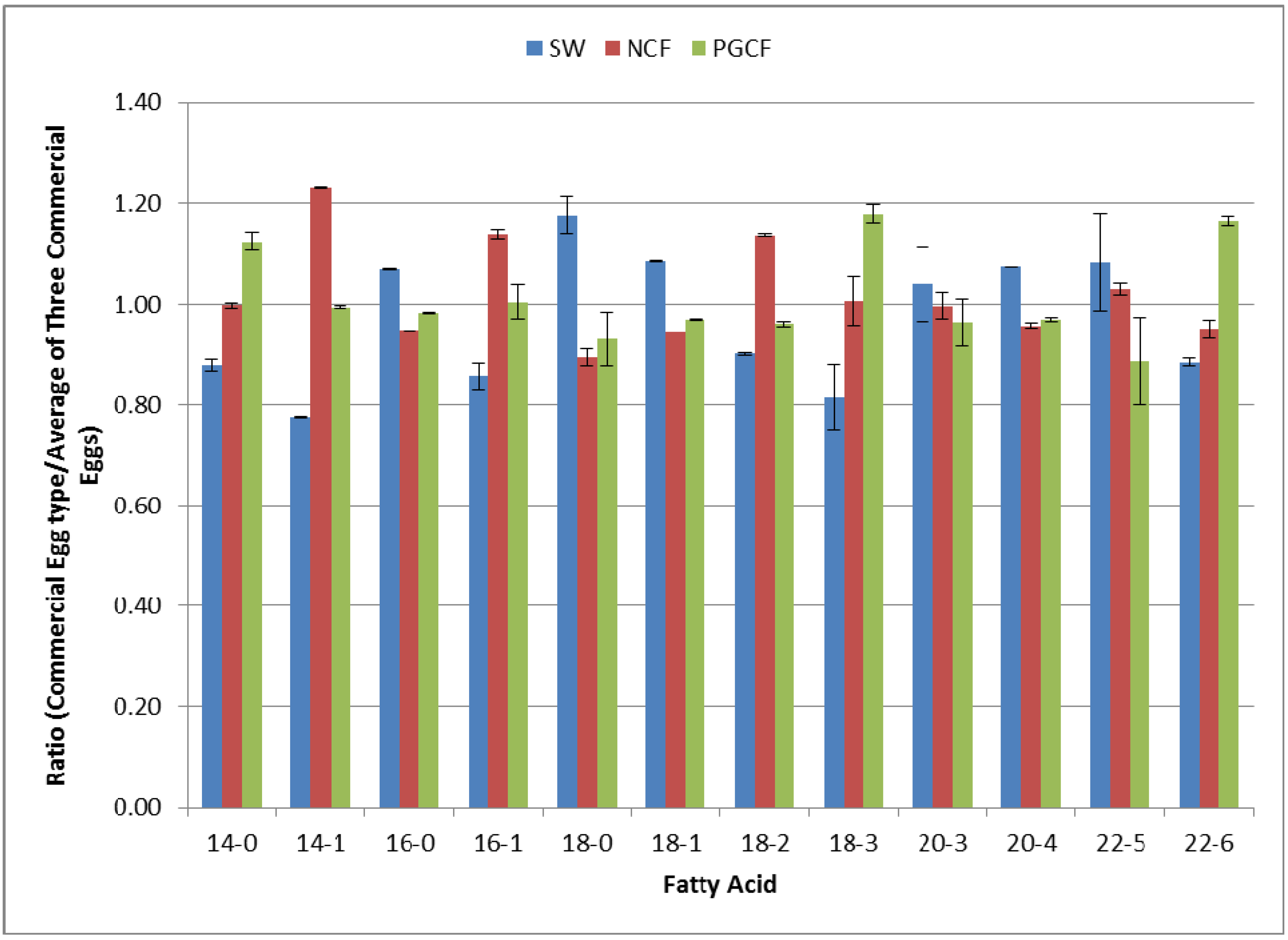
2.4. Multiplex Quantification of Fatty Acid Content in Three Different Commercial Egg Sources
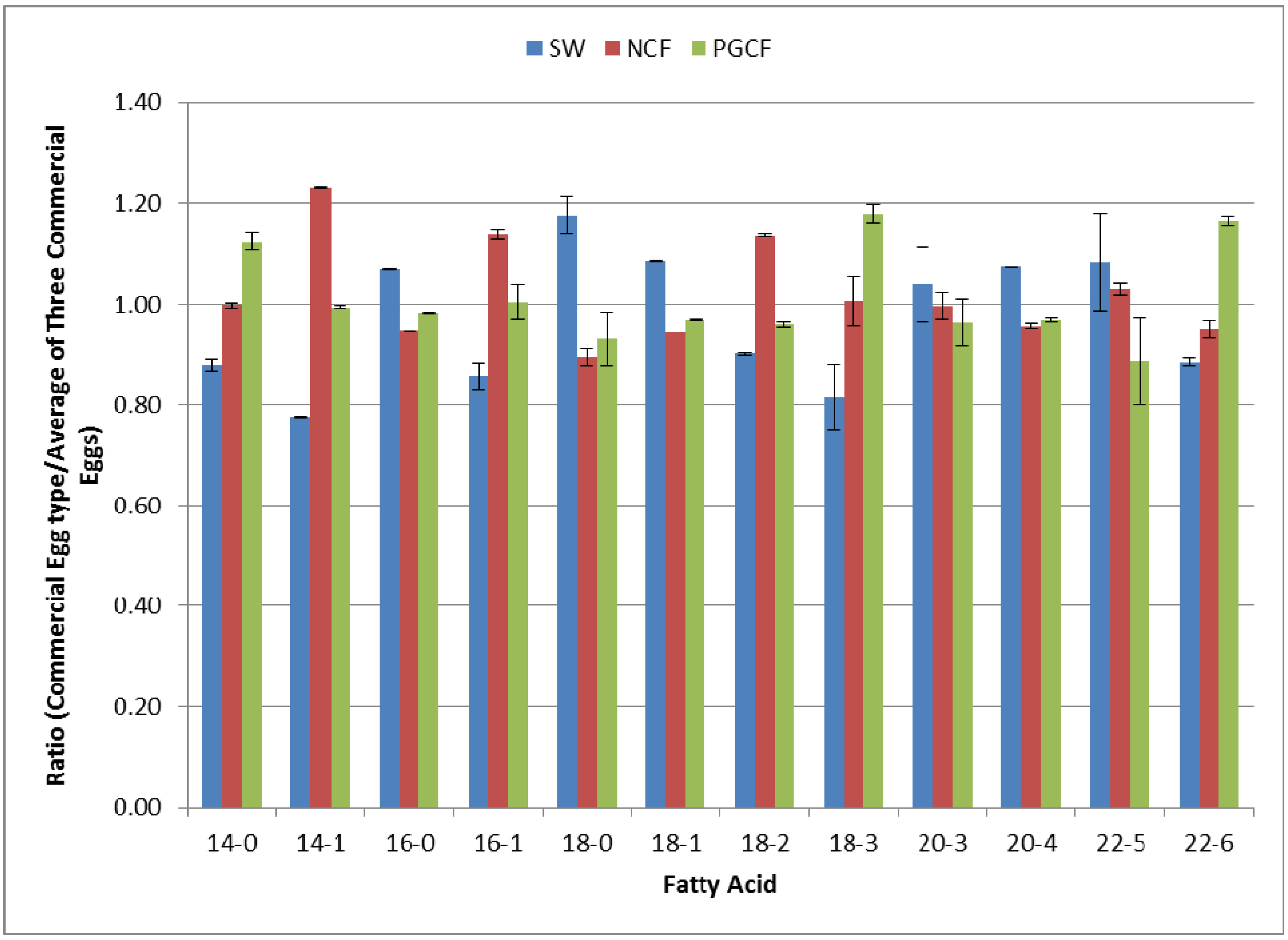
2.5. Discussion
3. Experimental
3.1. General Information
3.2. Synthesis of Cholamine-d3
3.3. Labeling of Lauric Acid Standard with Cholamine
3.4. Lipid Extraction from Egg Yolk
3.5. Labeling of Chicken Egg Fatty Acids with Cholamine
3.6. HPLC-ESI-MS/MS HCD Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Nielsen, J.; Jewett, M.C. The role of metabolomics in systems biology. Top. Curr. Genet. 2007, 18, 1–10. [Google Scholar] [CrossRef]
- Lei, Z.; Huhman, D.V.; Sumner, L.W. Mass spectrometry strategies in metabolomics. J. Biol. Chem. 2011, 286, 25435–25442. [Google Scholar] [CrossRef]
- Krone, N.; Hughes, B.A.; Lavery, G.G.; Stewart, P.M.; Arlt, W.; Shackleton, C.H.L. Gas chromatography/mass spectrometry (gc/ms) remains a pre-eminent discovery tool in clinical steroid investigations even in the era of fast liquid chromatography tandem mass spectrometry (lc/ms/ms). J. Steroid Biochem. Mol. Biol. 2010, 121, 496–504. [Google Scholar] [CrossRef]
- Koal, T.; Deigner, H.P. Challenges in mass spectrometry based targeted metabolomics. Curr. Mol. Med. 2010, 10, 216–226. [Google Scholar] [CrossRef]
- Theodoridis, G.A.; Gika, H.G.; Want, E.J.; Wilson, I.D. Liquid chromatography-mass spectrometry based global metabolite profiling: A review. Anal. Chim. Acta 2012, 711, 7–16. [Google Scholar] [CrossRef]
- Cubbon, S.; Antonio, C.; Wilson, J.; Thomas-Oates, J. Metabolomic applications of HILIC-LC-MS. Mass Spectrom. Rev. 2010, 29, 671–684. [Google Scholar] [CrossRef]
- Ballardini, R.; Benevento, M.; Arrigoni, G.; Pattini, L.; Roda, A. Massuntangler: A novel alignment tool for label-free liquid chromatography-mass spectrometry proteomic data. J. Chromatogr. A 2011, 1218, 8859–8868. [Google Scholar] [CrossRef]
- Zhang, Z.-M.; Liang, Y.-Z.; Lu, H.-M.; Tan, B.-B.; Xu, X.-N.; Ferro, M. Multiscale peak alignment for chromatographic datasets. J. Chromatogr. A 2012, 1223, 93–106. [Google Scholar] [CrossRef]
- Nadeau, J.S.; Wilson, R.B.; Hoggard, J.C.; Wright, B.W.; Synovec, R.E. Study of the interdependency of the data sampling ratio with retention time alignment and principal component analysis for gas chromatography. J. Chromatogr. A 2011, 1218, 9091–9101. [Google Scholar] [CrossRef]
- King, R.; Bonfiglio, R.; Fernandez-Metzler, C.; Miller-Stein, C.; Olah, T. Mechanistic investigation of ionization suppression in electrospray ionization. J. Am. Soc. Mass Spectrom. 2000, 11, 942–950. [Google Scholar] [CrossRef]
- Shah Sumit, J.; Yu Kenneth, H.; Sangar, V.; Parry Samuel, I.; Blair Ian, A. Identification and quantification of preterm birth biomarkers in human cervicovaginal fluid by liquid chromatography/tandem mass spectrometry. J. Proteome Res. 2009, 8, 2407–2417. [Google Scholar] [CrossRef]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, silac, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Huang, Y.; Khartulyari, S.; Morales, M.E.; Stanislawska-Sachadyn, A.; Von Feldt, J.M.; Whitehead, A.S.; Blair, I.A. Quantification of key red blood cell folates from subjects with defined mthfr 677c>t genotypes using stable isotope dilution liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 2403–2412. [Google Scholar] [CrossRef]
- Bluck, L.J.C. Recent progress in stable isotope methods for assessing vitamin metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 495–500. [Google Scholar] [CrossRef]
- Shortreed, M.R.; Lamos, S.M.; Frey, B.L.; Phillips, M.F.; Patel, M.; Belshaw, P.J.; Smith, L.M. Ionizable isotopic labeling reagent for relative quantification of amine metabolites by mass spectrometry. Anal. Chem. 2006, 78, 6398–6403. [Google Scholar] [CrossRef]
- Lamos, S.M.; Shortreed, M.R.; Frey, B.L.; Belshaw, P.J.; Smith, L.M. Relative quantification of carboxylic acid metabolites by liquid chromatography-mass spectrometry using isotopic variants of cholamine. Anal. Chem. 2007, 79, 5143–5149. [Google Scholar] [CrossRef]
- Yang, W.-C.; Mirzaei, H.; Liu, X.; Regnier, F.E. Enhancement of amino acid detection and quantification by electrospray ionization mass spectrometry. Anal. Chem. 2006, 78, 4702–4708. [Google Scholar] [CrossRef]
- Huang, Y.-Q.; Liu, J.-Q.; Gong, H.; Yang, J.; Li, Y.; Feng, Y.-Q. Use of isotope mass probes for metabolic analysis of the jasmonate biosynthetic pathway. Analyst 2011, 136, 1515–1522. [Google Scholar] [CrossRef]
- Guo, K.; Bamforth, F.; Li, L. Qualitative metabolome analysis of human cerebrospinal fluid by 13c-/12c-isotope dansylation labeling combined with liquid chromatography fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 2011, 22, 339–347. [Google Scholar] [CrossRef]
- Yang, W.-C.; Regnier, F.E.; Sliva, D.; Adamec, J. Stable isotope-coded quaternization for comparative quantification of estrogen metabolites by high-performance liquid chromatography-electrospray ionization mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 870, 233–240. [Google Scholar] [CrossRef]
- Blair, I.A. Analysis of estrogens in serum and plasma from postmenopausal women: Past present, and future. Steroids 2010, 75, 297–306. [Google Scholar] [CrossRef]
- Chapman, H.M.; Schutt, K.L.; Dieter, E.M.; Lamos, S.M. Relative quantification of biomarkers using mixed-isotope labeling coupled with ms. Bioanalysis 2012, 4, 2525–2541. [Google Scholar] [CrossRef]
- DeSouza, L.V.; Krakovska, O.; Darfler, M.M.; Krizman, D.B.; Romaschin, A.D.; Colgan, T.J.; Siu, K.W.M. Mtraq-based quantification of potential endometrial carcinoma biomarkers from archived formalin-fixed paraffin-embedded tissues. Proteomics 2010, 10, 3108–3116. [Google Scholar] [CrossRef]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. [Google Scholar] [CrossRef]
- Jun, B.-H.; Kang, H.; Lee, Y.-S.; Jeong, D.H. Fluorescence-based multiplex protein detection using optically encoded microbeads. Molecules 2012, 17, 2474–2490. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Chen, C.-Y.; Wang, G.-S. Analysis of steroid estrogens in water using liquid chromatography/tandem mass spectrometry with chemical derivatizations. Rapid Commun. Mass Spectrom. 2007, 21, 1973–1983. [Google Scholar] [CrossRef]
- Johnson, D.W. Ketosteroid profiling using girard t derivatives and electrospray ionization tandem mass spectrometry: Direct plasma analysis of androstenedione, 17-hydroxyprogesterone and cortisol. Rapid Commun. Mass Spectrom. 2005, 19, 193–200. [Google Scholar] [CrossRef]
- Zhang, R.; Sioma, C.S.; Thompson, R.A.; Xiong, L.; Regnier, F.E. Controlling deuterium isotope effects in comparative proteomics. Anal. Chem. 2002, 74, 3662–3669. [Google Scholar] [CrossRef]
- Villas-Boas, S.G.; Smart, K.F.; Sivakumaran, S.; Lane, G.A. Alkylation or silylation for analysis of amino and non-amino organic acids by GC-MS? Metabolites 2011, 1, 3–20. [Google Scholar] [CrossRef]
- Khakimov, B.; Motawia, M.S.; Bak, S.; Engelsen, S.B. The use of trimethylsilyl cyanide derivatization for robust and broad-spectrum high-throughput gas chromatography-mass spectrometry based metabolomics. Anal. Bioanal. Chem. 2013, 405, 9193–9205. [Google Scholar] [CrossRef]
- Wei, R.; Li, G.; Seymour, A.B. High-throughput and multiplexed lc/ms/mrm method for targeted metabolomics. Anal. Chem. 2010, 82, 5527–5533. [Google Scholar] [CrossRef]
- Crews, B.; Wikoff, W.R.; Patti, G.J.; Woo, H.-K.; Kalisiak, E.; Heideker, J.; Siuzdak, G. Variability analysis of human plasma and cerebral spinal fluid reveals statistical significance of changes in mass spectrometry-based metabolomics data. Anal. Chem. 2009, 81, 8538–8544. [Google Scholar] [CrossRef]
- Antruejo, A.; Azcona, J.O.; Garcia, P.T.; Gallinger, C.; Rosmini, M.; Ayerza, R.; Coates, W.; Perez, C.D. Omega-3 enriched egg production: The effect of α-linolenic ω-3 fatty acid sources on laying hen performance and yolk lipid content and fatty acid composition. Br. Poult. Sci. 2011, 52, 750–760. [Google Scholar] [CrossRef]
- Pariza, M.W.; Park, Y.; Cook, M.E. The biologically active isomers of conjugated linoleic acid. Prog. Lipid Res. 2001, 40, 283–298. [Google Scholar] [CrossRef]
- Anderson, K.E. Comparison of fatty acid, cholesterol, and vitamin a and e composition in eggs from hens housed in conventional cage and range production facilities. Poult. Sci. 2011, 90, 1600–1608. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The mediterranean diets: What is so special about the diet of greece? The scientific evidence. J. Nutr. 2001, 131, 3065S–3073S. [Google Scholar]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar]
- Sample Availability: Samples of the compounds cholamine-d0, -d3, and -d9 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Torde, R.G.; Therrien, A.J.; Shortreed, M.R.; Smith, L.M.; Lamos, S.M. Multiplexed Analysis of Cage and Cage Free Chicken Egg Fatty Acids Using Stable Isotope Labeling and Mass Spectrometry. Molecules 2013, 18, 14977-14988. https://doi.org/10.3390/molecules181214977
Torde RG, Therrien AJ, Shortreed MR, Smith LM, Lamos SM. Multiplexed Analysis of Cage and Cage Free Chicken Egg Fatty Acids Using Stable Isotope Labeling and Mass Spectrometry. Molecules. 2013; 18(12):14977-14988. https://doi.org/10.3390/molecules181214977
Chicago/Turabian StyleTorde, Richard G., Andrew J. Therrien, Michael R. Shortreed, Lloyd M. Smith, and Shane M. Lamos. 2013. "Multiplexed Analysis of Cage and Cage Free Chicken Egg Fatty Acids Using Stable Isotope Labeling and Mass Spectrometry" Molecules 18, no. 12: 14977-14988. https://doi.org/10.3390/molecules181214977