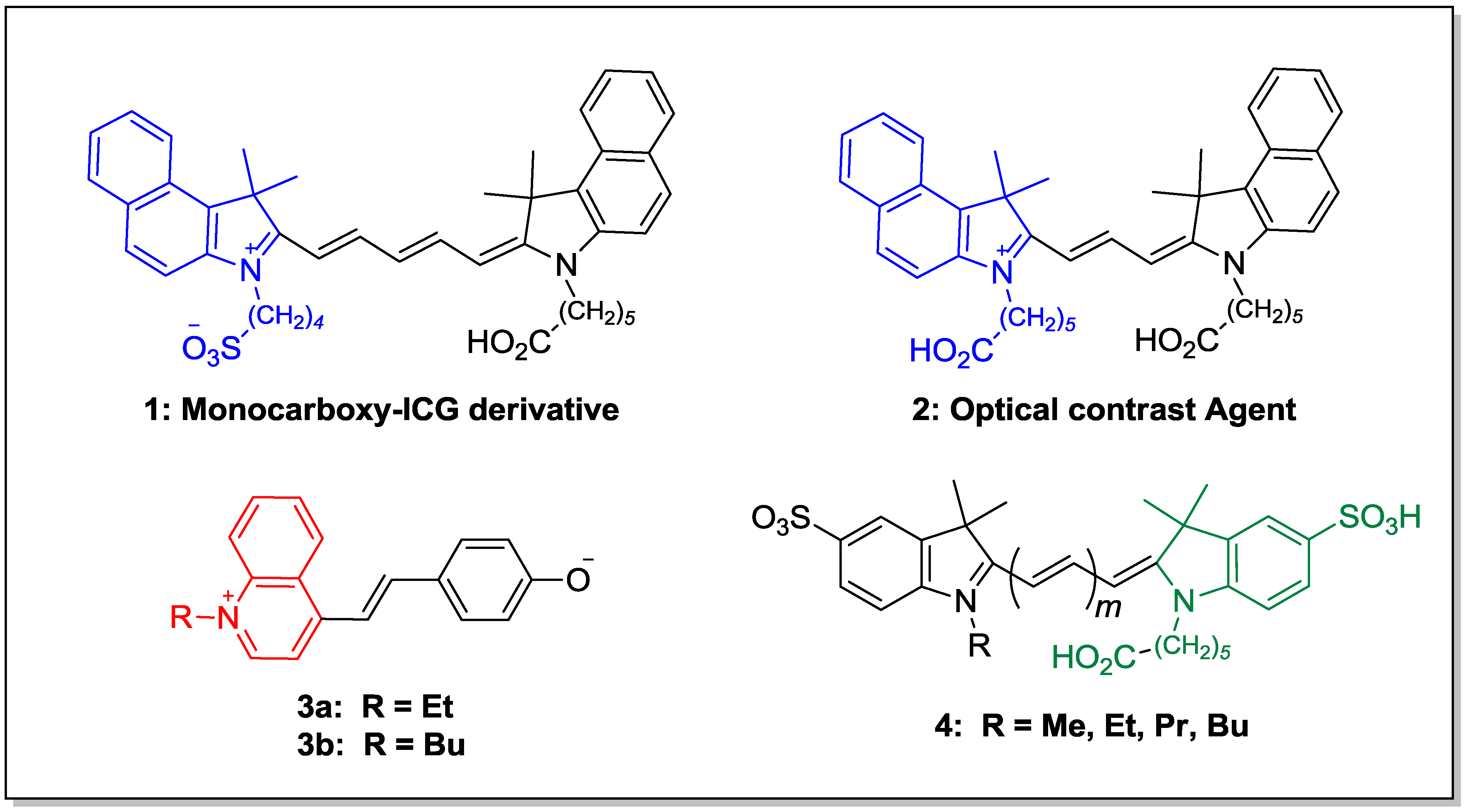
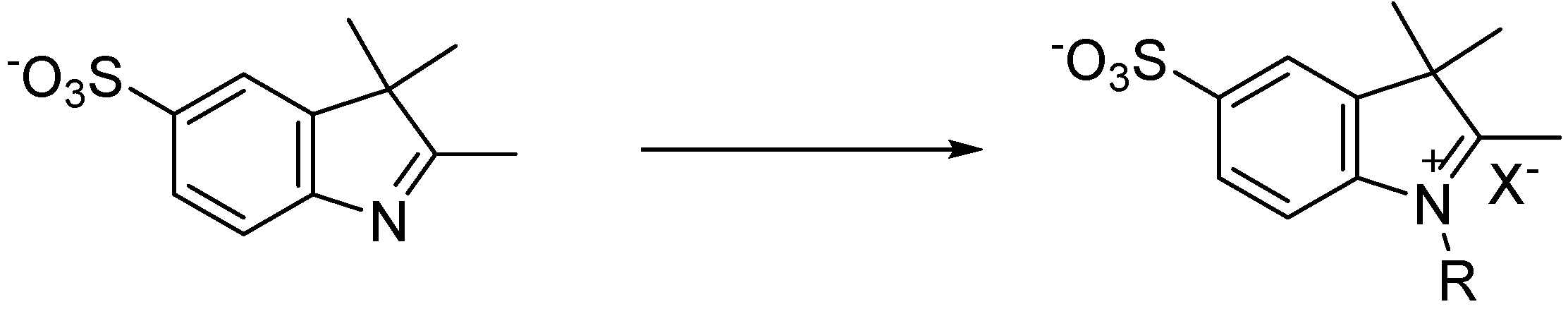3. Experimental
3.1. General
All microwave reactions were conducted using either the single mode Biotage Initiator 2.0 (Biotage, Uppsala, Sweden); benzoindolinine and quinolinium salts) or Discover SP CEM microwave oven (CEM Corporation, Matthews, NC, USA); sulfoindolenium salts. 1H and 13C-NMR spectra were obtained in DMSO-d6 (obtained from Cambridge Isotope Laboratories (Cambridge Isotope Laboratories, Tewksbury, MA, USA) using a Bruker Advance 400 MHz NMR and were recorded at 400 MHz and 100 MHz, respectively. Melting points are uncorrected. All reagents and chemicals were obtained from Aldrich Chemical Company (St. Louis, MO, USA) and Alfa Aesar (Ward Hill, MA, USA) and were used without further purification.
3.2. Synthesis of Trimethylbenz[e]indolium Salts
1,2,3,3-Tetramethylbenz[e]indolium iodide (1): 2,3,3-Trimethybenzolindolenine (0.30 g, 1.4 mmol) and iodomethane (0.18 mL, 3.0 mmol) were added to a microwave reaction vial. The microwave oven was set to 120 °C for 25 min with a ramp time of 2 min. After cooling, the green solid was filtered and washed with cold ether and dried in vacuo, yielding a grey solid in 80% yield. MP 220–225 °C; 1H-NMR δ (ppm): 8.38 (d, J = 8.5 Hz, 1H), 8.31 (d, J = 8.9 Hz, 1H), 8.23 (d, J = 7.8 Hz, 1H), 8.12 (d, J = 8.9 Hz, 1H), 7.78 (m, 2H), 4.11 (s, 3H), 2.89 (s, 3H), 1.76 (s, 6H); 13C-NMR δ (ppm): 195.8 (C), 139.4 (CH), 136.4 (CH), 132.9 (CH), 130.4 (CH), 129.7 (C), 129.3 (C), 127.0 (C), 123.3 (CH), 113.1 (CH), 55.2 C), 35.1(CH3), 21.2 (CH3), 14.0 (CH3).
1-Ethyl-2,3,3-trimethylbenz[e]indolium iodide (2): 2,3,3-Trimethybenzolindolenine (0.30 g, 1.4 mmol) and iodoethane (0.52 mL, 6.5 mmol) were added to a microwave reaction vial. The microwave oven was set to 135 °C for 30 min with a ramp time of 2 min. After cooling, the white-blue solid was filtered and washed with cold ether and dried in vacuo, yielding 80% of pure product. MP 225–230 °C; 1H-NMR δ (ppm): 8.39 (d, J = 8.3 Hz, 1H), 8.32 (d, J = 8.9 Hz, 1H), 8.24 (d, J = 8.0 Hz, 1H), 8.18 (d, J = 8.9 Hz, 1H), 7.78 (m, 2H), 4.64 (q, J = 7.3 Hz, 2H), 2.96 (s, 3H), 1.77 (s, 6H), 1.52 (t, J = 7.3 Hz, 3H); 13C-NMR δ (ppm): 195.9 (C), 138.1 (CH), 136.9 (CH), 132.9 (CH), 130.6 (CH), 129.6 (C), 128.3 (C), 127.23 (C), 127.20 (C), 123.3 (C), 113.1 (CH), 55.4 (C), 43.3 (CH2), 21.4 (CH3), 13.6 (CH3), 12.8 (CH3).
1-Propyl-2,3,3-trimethylbenz[e]indolium iodide (3): 2,3,3-Trimethybenzolindolenine (0.30 g, 1.4 mmol) and iodopropane (0.54 mL, 5.5 mmol) were added to a microwave reaction vial. The microwave oven was set to 140 °C for 10 min with a ramp time of 2 min. After cooling, the product was stirred in acetone and ether. The yellow solid was filtered and washed with cold ether and dried in vacuo, yielding 83% pure product. MP 142-145 °C; 1H-NMR δ (ppm): 8.40 (d, J = 8.4 Hz, 1H), 8.32 (d, J = 8.9 Hz, 1H), 8.24 (d, J = 8.0 Hz, 1H), 8.21 (d, J = 8.9 Hz, 1H), 7.79 (m, 2H), 4.60 (t, J = 7.4 Hz, 2H), 2.98 (s, 3H), 1.95 (m, 2H), 1.79 (s, 6H), 1.04 (t, J = 7.4 Hz, 3H); 13C-NMR δ (ppm): 196.4 (C), 138.5 (CH), 136.9 (CH), 132.9 (CH), 130.6 (CH), 129.6 (C), 128.3 (C), 127.2 (C), 127.1 (C), 123.4 (C), 113.3 (CH), 55.4 (C), 49.0 (CH2), 21.6 (CH2), 21.0 (CH3), 13.9 (CH3), 10.7 (CH3).
1-Butyl-2,3,3-trimethylbenz[e]indolium iodide (4): 2,3,3-Trimethybenzolindolenine (0.30 g, 1.4 mmol) and iodobutane (1.0 mL, 8.7 mmol) were added to a microwave reaction vial. The microwave oven was set to 140 °C for 15 min with a ramp time of 2 min. After cooling, the product was stirred in acetone and ether. The yellow solid was filtered and washed with cold ether and dried in vacuo, yielding 82% pure product. MP 120–124 °C; 1H-NMR δ (ppm): 8.40 (d, J = 8.3 Hz, 1H), 8.31 (d, J = 8.9 Hz, 1H), 8.24 (d, J = 7.9 Hz, 1H), 8.18 (d, J = 8.9 Hz, 1H), 7.77 (m, 2H), 4.61 (t, J = 7.7 Hz, 2H), 2.98 (s, 3H), 1.89 (m, 2H), 1.78 (s, 6H), 1.49 (m, 2H), 0.97 (t, J = 7.4 Hz, 3H); 13C-NMR δ (ppm): 196.2 (C), 138.4 (CH), 136.9 (CH), 133.0 (CH), 130.6 (CH), 129.6 (C), 128.3 (C), 127.2 (C), 123.4 (C), 113.3 (CH), 55.4 (C), 47.7 (CH2), 29.4 (CH2), 21.6 (CH2), 19.3 (CH3), 13.9 (CH3), 13.6 (CH3).
1-Pentyl-2,3,3-trimethylbenz[e]indolium iodide (5): 2,3,3-Trimethybenzolindolenine (0.30 g, 1.4 mmol) and iodopentane (1.2 mL, 9.1 mmol) were added to a microwave reaction vial. The microwave oven was set to 145 °C for 10 min with a ramp time of 2 min. After cooling, the product was stirred in acetone and ether. The orange solid was filtered and washed with cold ether and dried in vacuo, yielding 82% pure product. MP 137–139 °C; 1H-NMR δ (ppm): 8.40 (d, J = 8.3 Hz, 1H), 8.31 (d, J = 8.9 Hz, 1H), 8.24 (d, J = 7.9 Hz, 1H), 8.18 (d, J = 8.9 Hz, 1H), 7.77 (m, 2H), 4.61 (t, J = 7.7 Hz, 2H), 2.98 (s, 3H), 1.89 (m, 2H), 1.78 (s, 6H), 1.49 (m, 2H), 0.97 (t, J = 7.4 Hz, 3H); 13C-NMR δ (ppm): 196.2 (C), 138.4 (CH), 136.9 (CH), 132.9 (CH), 130.6 (CH), 129.6 (C), 128.3 (C), 127.2 (C), 127.1 (C), 127.0 (C), 123.4 (C), 113.3 (CH), 55.4 (C), 47.8 (CH2), 27.9 (CH2), 27.1 (CH2), 21.8 (CH2), 21.6 (CH3), 13.8 (CH3), 13.7 (CH3).
1-(2-Hydroxyethyl)-2,3,3-trimethylbenz[e]indolium bromide (6): 2,3,3-Trimethybenzolindolenine (0.30 g, 1.4 mmol) and 2-bromoethanol (0.2 mL, 2.8 mmol) were added to a microwave reaction vial. The microwave oven was set to 120 °C for 15 min with a ramp time of 2 min. After cooling, the product was stirred in acetonitrile, acetone and ether. The grey solid was filtered and washed with cold ether and dried in vacuo, yielding 74% pure product. MP 160–164 °C; 1H-NMR δ (ppm): 8.40 (d, J = 8.4 Hz, 1H), 8.30 (d, J = 8.9 Hz, 1H), 8.23 (d, J = 7.7 Hz, 1H), 8.19 (d, J = 8.9 Hz, 1H), 7.79 (m, 2H), 4.76 (t, J = 4.9 Hz, 2H), 3.95 (t, J = 4.8 Hz, 2H), 2.96 (s, 3H), 1.79 (s, 6H); 13C-NMR δ (ppm): 197.3 (C), 138.6 (CH), 136.7 (CH), 132.9 (CH), 130.4 (CH), 129.6 (C), 128.3 (C), 127.1 (C), 123.3 (C), 113.5 (CH), 57.9 (C), 55.5 (CH2), 50.4 (CH2), 21.5 (CH3), 14.3 (CH3).
1-(5-Carboxypentyl)-2,3,3-trimethylbenz[e]indolium iodide (7): 2,3,3-Trimethybenzolindolenine (0.30 g, 1.4 mmol), 6-bromohexanoic acid (0.60 g, 3.0 mmol) and 1,2-dichlorobenzene (1.5 mL 13.2 mmol) were added to a microwave reaction vial. The microwave oven was set to 145 °C for 30 min with a ramp time of 2 min. After cooling, the product was stirred in ether. The yellow solid was filtered and washed with cold ether and dried in vacuo, yielding 85% pure product. MP 174–177 °C; 1H-NMR δ (ppm): 8.40 (d, J = 8.2 Hz, 1H), 8.31 (d, J = 8.9 Hz, 1H), 8.24 (d, J = 8.1 Hz, 1H), 8.20 (d, J = 9.0 Hz, 1H), 7.77 (m, 2H), 4.62 (t, 7.5 Hz, 2H), 2.98 (s, 3H), 2.25 (t, J = 7.1 Hz, 2H), 1.92 (m, 2H), 1.78 (s, 6H), 1.60 (m, 2H), 1.49 (m, 2H); 13C-NMR δ (ppm): 196.3 (C), 174.3 (C), 138.4 (CH), 136.9 (CH), 132.9 (CH), 130.6 (CH), 129.6 (C), 128.3 (C), 127.2 (C), 127.1 (C), 123.4 (C), 113.3 (CH), 55.4 (C), 47.6 (CH2), 33.3 (CH2), 27.1 (CH2), 25.3 (CH2), 24.0 (CH2), 21.7 (CH3), 14.4 (CH3) 13.8 (CH3).
3-(4-Sulfobutyl)-1,1,2-trimethylbenz[e]indolium iodide (8): 2,3,3-Trimethybenzolindolenine (0.30 g, 1.4 mmol) and 1,4-butane sultone (0.70 mL, 6.8 mmol) were added to a microwave reaction vial. The microwave oven was set to 145 °C for 30 min with a ramp time of 2 min. After cooling, the product was stirred in acetone and ether. The purple solid was filtered and washed with cold ether and dried in an oven at 100 °C, yielding 83% pure product. MP 80–85 °C; 1H-NMR δ (ppm): 8.40 (d, J = 8.3 Hz, 1H), 8.31 (d, J = 8.9 Hz, 1H), 8.24 (d, J = 7.9 Hz, 1H), 8.18 (d, J = 8.9 Hz, 1H), 7.75 (m, 2H), 4.63 (t, J = 8.02 Hz, 2H), 2.96 (s, 3H), 2.57 (t, J = 7.19 Hz, 2H), 2.51 (m 2H), 2.04 (m, 2H), 1.75 (s, 6H); 13C-NMR δ (ppm): 196.3 (C), 138.5 (CH), 136.8 (CH), 132.9 (CH), 131 (CH), 130.6 (C), 129.6 (C), 127.1 (C), 123.3 (C), 113.4 (CH), 55.4 (C), 50.1 (CH2), 47.4 (CH2), 26.1 (CH2), 22.0 (CH2), 21.5 (CH3), 13.6 (CH3).
3.3. Synthesis of Quinolinium Salts
1,4-Dimethylquinolinium iodide (9): Lepidine (0.20 mL 1.4 mmol) and iodomethane (0.22 mL, 0.28 mmol) were added to a microwave reaction vial. The microwave oven was set to 130 °C for 5 min with a ramp time of 2 min. After cooling, the product was washed with cold ether and acetone and dried in vacuo, yielding a yellow-green solid in 98% yield. MP 130 °C; 1H-NMR δ (ppm): 9.36 (q, J = 7.33 Hz, 2H), 8.52 (t, J = 8.55 Hz, 1H), 8.28 (q, J = 8.48 Hz, 2H), 8.07 (d, J = 6.05 Hz, 1H), 4.58 (s, 3H), 3.41 (s, 3H); 13C-NMR δ (ppm): 158.0 (C), 148.8 (C), 137.5 (CH), 134.8 (CH), 129.5 (CH), 128.3 (CH), 126.6 (CH), 122.3 (CH), 119.4 (CH), 44.9 (CH2), 19.5 (CH3).
1-Ethyl-4-methylquinolinium iodide (10): Lepidine (0.20 mL, 1.4 mmol) and iodoethane (0.39 mL, 0.28 mmol) were added to a microwave reaction vial. The microwave oven was set to 130 °C for 5 min with a ramp time of 2 min. After cooling, the product was filtered and washed with cold ether and acetone and dried in vacuo, yielding a yellow solid in 70% yield. MP 126 °C; 1H-NMR δ (ppm): 9.43 (d, J = 7.33 Hz, 1H), 8.58 (dd, J = 8.55 Hz, 2H), 8.27 (t, J = 8.48 Hz, 1H), 8.07 (d, J = 6.05 Hz, 2H), 5.01 (q, J = 7.22 Hz, 2H), 3.01 (s, 3H), 1.59 (t, J = 7.22 Hz, 3H); 13C-NMR δ (ppm): 158.2 (C), 148.0 (C), 136.4 (CH), 134.9 (CH), 129.1 (CH), 128.8 (CH), 127.0 (CH), 122.6 (CH), 119.1 (CH), 52.4 (CH2), 19.5 (CH3), 15.1 (CH3).
1-Propyl-4-methylquinolinium iodide (11): Lepidine (0.20 mL, 1.4 mmol) and iodopropane (0.27 mL, 0.28 mmol) were added to a microwave reaction vial. The microwave oven was set to 130 °C for 5 min with a ramp time of 2 min. After cooling, the product was filtered and washed with cold ether and acetone and dried in vacuo, yielding 96% pure product. MP 130 °C; 1H-NMR δ (ppm): 9.6 (d, J = 6.05 Hz, 1H), 8.59 (q, J = 8.48 Hz, 2H), 8.31 (t, J = 8.55 Hz, 1H), 8.07 (q, J = 7.33 Hz, 2H), 4.98 (t, J = 7.39 Hz, 2H), 3.02 (s, 3H), 1.98 (m, 2H), 0.96 (t, J = 7.36 Hz 3H); 13C-NMR δ (ppm): 158.5 (C), 148.3 (C), 136.7 (C), 135.0 (CH), 129.5 (CH), 128.9 (CH), 127.1 (CH), 122.5 (CH), 119.3 (CH).
1-Butyl-4-methylquinolinium iodide (12): Lepidine (0.20 mL, 1.4 mmol) and iodobutane (0.32 mL, 0.28 mmol) were added to a microwave reaction vial. The microwave oven was set to 130 °C for 5 min with a ramp time of 2 min. After cooling, the product was filtered and washed with cold ether and acetone and dried in vacuo, yielding the product as yellow crystals in 81% yield. MP 125 °C; 1H-NMR δ (ppm): 9.6 (d, J = 6.05 Hz, 1H), 8.59 (q, J = 8.48 Hz, 2H), 8.3 (t, J = 8.55 Hz, 1H), 8.08 (q, J = 7.33 Hz, 2H), 5.0 (t, J = 7.49 Hz, 2H), 3.0 (s, 3H), 1.93 (m, 2H), 1.40 (m, 2H), 0.94 (t, J = 7.32 Hz, 3H); 13C-NMR δ (ppm): 158.5 (C), 148.3 (C), 136.6 (C), 135.0 (CH), 129.5 (CH), 128.9 (CH) 127.1 (CH), 122.6 (CH), 119.3 (CH), 56.7 (CH2), 19.7 (CH2), 19.1 (CH3), 13.4 (CH3).
1-(6-ethoxy-6-oxohexyl)-4-methylquinolinium iodide (13): Lepidine (0.23 mL, 1.754 mmol) and ethyl 6-iodohexanoate (0.35 mL, 1.929 mmol) were combined in a 5 mL Biotage microwave vial equipped with a stir bar. The vial was placed in the Biotage microwave with a ramp time of 4 min and held at 120 °C for 5 min. The vial with reaction mixture was allowed to sit for 5 min. and irradiated again with a ramp time of 4 min and held at 120 °C for 2 min. The resulting dark brown solid was dissolved in a mixture of ethyl acetate (5 mL) and methanol (0.5 mL). The dark brown mixture was purified by eluting on a coarse frit packed with silica gel. Ethyl acetate (400 mL) was used to wash the product and a mixture of dichloromethane and methanol (90:10) was used for subsequent washes. The dichloromethane and methanol washes were concentrated and dried to obtain brown oil in 56% yield. 1H-NMR δ (ppm): 9.45 (d, J = 6.05 Hz, 2H), 8.56 (m, 2H), 8.27 (m, 1H), 8.08 (m, 2H), 5.02 (t, J = 7.47 Hz, 2H), 4.02 (q, J = 7.11, Hz, 2H), 3.00 (s, 3H), 1.95 (m, 2H), 1.60 (m, 2H), 1.55 (m, 2H), 1.38 (m, 2H), 1.15 (t, J = 7.47 Hz, 3H). 13C-NMR δ (ppm): 172.7 (C), 158.5 (C), 148.4 (CH), 136.6 (CH), 135.0 (CH), 129.5 (CH), 128.9 (CH), 127.1 (CH), 122.6 (CH), 119.3 (CH), 59.7 (CH2), 56.6 (CH2), 33.1 (CH2), 29.0 (CH2), 25.1 (CH2), 23.8 (CH2), 19.7 (CH3), 14.1 (CH3).
3.4. Synthesis of Sulfoindolenium Salts
1,2,3,3-Tetramethyl-5-sulfo-3H-indolium (14): Iodomethane (0.11 mL, 2.2 mmol) and potassium 2,3,3-trimethyl-3H-indole-5-sulfonate (0.10 g, 0.36 mmol) in 1 mL of acetonitrile were added to a microwave reaction vial. The CEM microwave oven was set to 130 °C for 30 min with a ramp time of 1 min. After cooling, the product was boiled in acetone for 5 min and was recrystallized upon cooling. The solid was filtered and washed with acetone and cold ether and dried in vacuo, yielding a red solid in 84% yield. MP 253 °C; 1H-NMR δ (ppm): 7.96 (s, 1H) 7.84 (d, J = 8.34 Hz, 2H), 7.76 (d, J = 1.5 Hz, 2H), 3.95 (s, 3H), 2.77 (s, 3H), 2.09 (s, 6H); 13C-NMR δ (ppm): 195.5 (C), 148.0 (CH), 140.7 (CH), 139.9 (CH), 124.9 (C), 119.3 (C), 113.2 (C), 52.7 (C), 33.5 (CH3), 20.3 (CH3), 13.0 (CH3).
1-Ethyl-2,3,3-Trimethyl-5-sulfo-3H-indolium (15): Iodoethane (0.11 mL, 1.4 mmol) and potassium 2,3,3-trimethyl-3H-indole-5-sulfonate (0.10 g, 0.36 mmol) in 1 mL of acetonitrile were added to a microwave reaction vial. The CEM microwave oven was set to 130 °C for 30 min with a ramp time of 1 min. After cooling, the red solid was filtered and washed with acetone and cold ether and dried in vacuo, yielding a red solid in 67% pure product. MP 224–229 °C; 1H-NMR δ (ppm): 8.02 (s, 1H), 7.92 (d, J = 8.35 Hz, 1H), 7.82 (d, J = 1.5 Hz, 1H), 4.49 (q, J = 7.34 Hz, 2H), 2.84 (s, 3H), 2.09 (s, 6H), 1.44 (t, J = 7.33 Hz, 3H); 13C-NMR δ (ppm): 195.6 (C), 148.1 (CH), 140.3 (CH), 139.3 (CH), 125.1 (C), 119.5 (C), 113.5 (C), 52.9 (C), 41.9 (CH2), 20.5 (CH3), 12.7 (CH3), 11.3 (CH3).
1-Propyl-2,3,3-trimethyl-5-sulfo-3H-indolium (16): Iodopropane (0.20 mL, 2.1 mmol) and potassium 2,3,3-trimethyl-3H-indole-5-sulfonate (0.10 g, 0.36 mmol) in 1 mL of acetonitrile were added to a microwave reaction vial. The CEM microwave oven was set to 150 °C for 30 min with a ramp time of 1 min. After cooling, the product was concentrated under reduced pressure. The resulting red solid was stirred in acetone then filtered and washed with acetone and cold ether. The light pink solid was dried in vacuo, yielding 63% pure product. MP 208–211 °C; 1H-NMR δ (ppm): 8.03 (s, 1H), 7.95 (d, J = 8.38 Hz, 2H), 7.81 (d, J = 1.49 Hz, 2H), 4.44 (t, J = 7.69 Hz, 2H), 2.86 (s, 3H), 1.86 (m, 2H), 1.55 (s, 6H), 0.99 (t, J = 7.40 Hz, 3H); 13C-NMR δ (ppm): 195.7 (C), 149.3 (CH), 141.5 (CH), 140.9 (CH), 126.3 (C), 120.7 (C), 114.9 (C), 54.2 (C), 48.9 (CH2), 21.9 (CH2), 20.7 (CH3), 14.1 (CH3), 10.7 (CH3).
1-Butyl-2,3,3-trimethyl-5-sulfo-3H-indolium (17): Iodobutane (0.20 mL, 1.8 mmol) and potassium 2,3,3-trimethyl-3H-indole-5-sulfonate (0.10 g, 0.36 mmol) in 1 mL of acetonitrile were added to a microwave reaction vial. The CEM microwave oven was set to 155 °C for 35 min with a ramp time of 1 min. After cooling, the product was concentrated under reduced pressure then stirred in acetone until residue formed. The solution was decanted and the residue was suspended in acetone until solid product had further formed. The solid was filtered and washed with acetone and cold ether and dried in vacuo, yielding a red solid in 58% yield. MP 145–149 °C; 1H-NMR δ (ppm): 8.02 (s, 1H), 7.95 (d, J = 8.38 Hz, 1H), 7.81 (d, J = 1.34 Hz, 1H), 4.46 (t, J = 7.63 Hz, 2H), 2.87 (s, 3H), 1.82 (q, J = 7.08 Hz, 2H), 1.55 (s, 6H), 1.42 (q, J = 7.78 Hz, 2H), 0.93 (t, J = 7.36 Hz, 3H); 13C-NMR δ (ppm): 197.1 (C), 149.1 (CH), 141.4 (CH), 140.8 (CH), 126.2 (C), 120.5 (C), 114.9 (C), 54.1 (C), 47.5 (CH2), 30.6 (CH2), 29.0 (CH2), 21.8 (CH2), 19.2 (CH3), 14.0 (CH3), 13.4 (CH3).
1-(2-Hydroxyethyl)-2,3,3-trimethyl-5-sulfo-3H-indolium (18): 2-bromoethanol (0.20 mL, 2.8 mmol) and potassium 2,3,3-trimethyl-3H-indole-5-sulfonate (0.10 g, 0.36 mmol) were added to a microwave reaction vial. The CEM microwave oven was set to 125 °C for 5 min with a ramp time of 1 min. After cooling, the product was stirred in 1:1:3 of chloroform/acetone/ether. The light grey solid was filtered and washed with cold ether and dried in vacuo, yielding 69% pure product. MP 255–260 °C; 1H-NMR δ (ppm): 8.03 (s, 1H), 7.94 (d, J = 8.38 Hz, 2H), 7.80 (d, J = 1.40 Hz, 2H), 4.61 (t, J = 4.84 Hz, 2H), 3.87 (t, J = 4.86 Hz, 2H), 2.85 (s, 3H), 1.56 (s, 6H); 13C-NMR δ (ppm): 198.1 (C), 149.1 (CH), 141.4 (CH), 141.0 (CH), 126.1 (C), 115.0 (C), 57.7 (C), 54.4 (C), 50.4 (CH2), 21.9 (CH2), 14.6 (CH3).
1-(5-Carboxypentyl)-2,3,3-trimethyl-5-sulfo-3H-indolium (19): 6-Bromohexanioc acid (0.23 g, 1.2 mmol) and potassium 2,3,3-trimethyl-3H-indole-5-sulfonate (0.10 g, 0.36 mmol) in 1 mL of acetonitrile were added to a microwave reaction vial. The CEM microwave oven was set to 150 °C for 30 min with a ramp time of 1 min. After cooling for one hour, the vial was reintroduced to the microwave oven which was set to 150 °C for 15 min. After sitting and cooling for one hour, the precipitate was induced by agitation. The light pink solid was filtered and washed with 1:1 2-propanol/ether and dried in vacuo, yielding 72% pure product. MP 169–172 °C; 1H-NMR δ (ppm): 8.02 (s, 1H), 7.94 (d, J = 8.37 Hz, 1H), 7.81 (d, J = 1.31 Hz,1H), 4.46 (t, J = 7.41 Hz, 2H), 2.87 (s, 3H), 2.53 (q, J = 1.75 Hz, 2H), 2.22 (t, J = 7.20 Hz, 2H), 1.84 (q, J = 7.02 Hz, 2H), 1.55 (s, 6H), 1.41 (q, J = 7.03 Hz, 2H); 13C-NMR δ (ppm): 196.1 (C), 173.1 (C), 148.1 (CH), 140.3 (CH), 139.7 (CH), 125.1 (C), 119.5 (C), 113.7 (C), 53.0 (C), 46.3 (CH2), 32.1 (CH2), 25.6 (CH2), 24.1 (CH2), 22.8 (CH2), 20.6 (CH3), 12.9 (CH3).
1-Butyl-2,3,3-trimethylindolium iodide (20): 2,3,3-Trimethylindolenine (0.52 mL, 3.2 mmol) and iodobutane (0.76 mL, 6.6 mmol) were added to a microwave reaction vial. The microwave oven was set to 130 °C for 20 min with a ramp time of 2 min. After cooling, the product was stirred in acetone and ether. The yellow solid was filtered, washed with cold ether and dried in vacuo, yielding 70% pure product. MP 105-110 °C; 1H-NMR δ (ppm): 0.95 (t, J = 7.29 Hz, 3H), 1.44 (m, 2H), 1.55 (s, 6H), 1.83 (m, 2H), 2.87 (s, 3H), 4.47 (t, J = 7.72 Hz, 2H), 7.64 (m, 1H), 7.8 (m, 1H), 8 (m, 1H); 13C-NMR δ (ppm): 196.3 (C), 141.8 (CH), 141.0 (CH), 129.3 (C), 128.9 (C), 123.5 (C), 115.4 (CH), 54.1 (C), 47.4 (CH2), 29.2 (CH2), 22.0 (CH2), 19.3 (CH3), 14.1 (CH3), 13.6 (CH3).

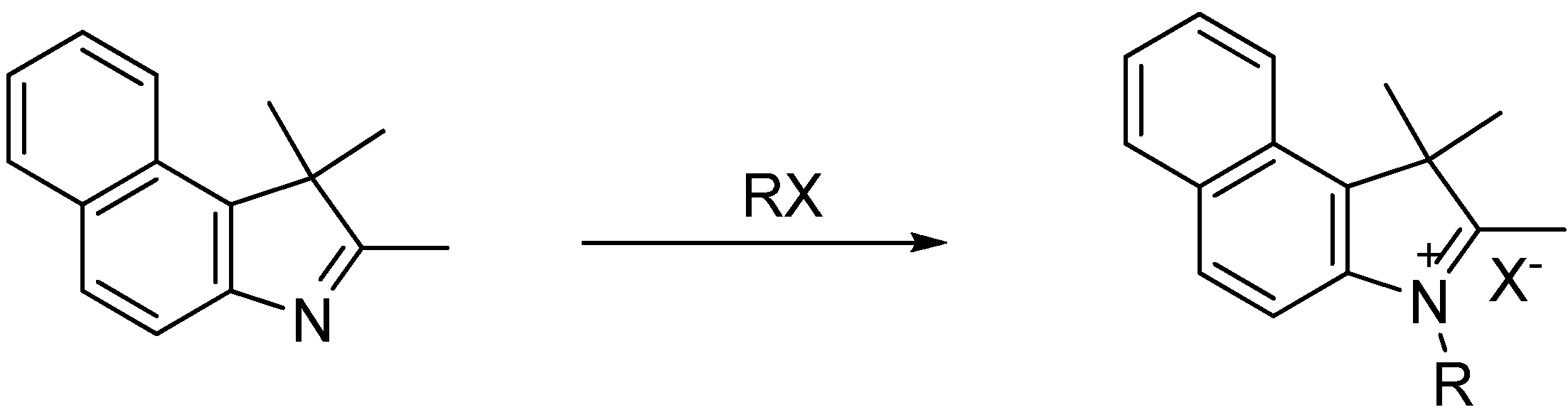

{kind=link}
{kind=link}
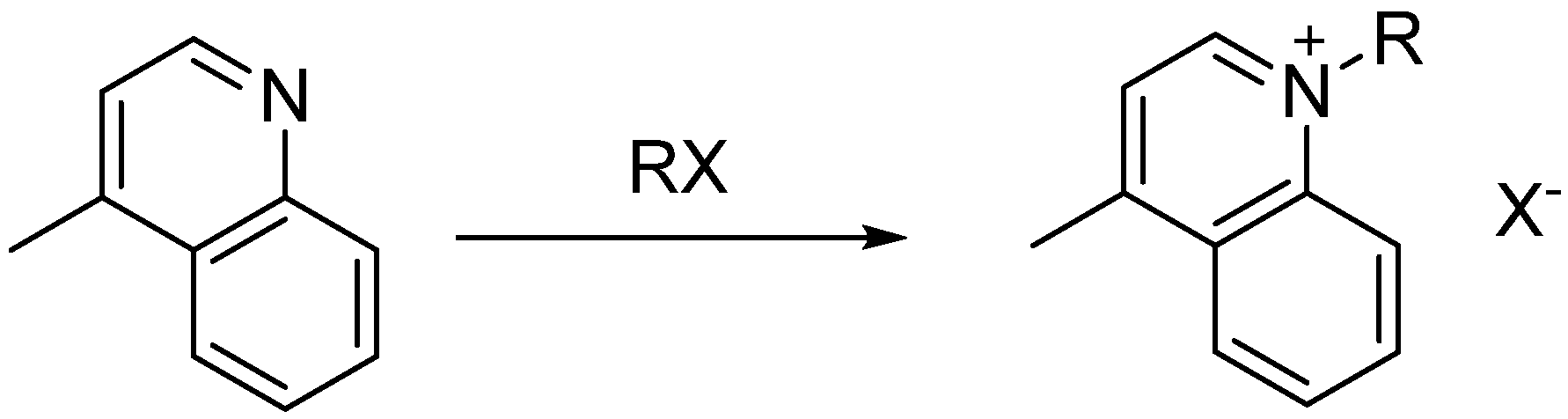{kind=link}
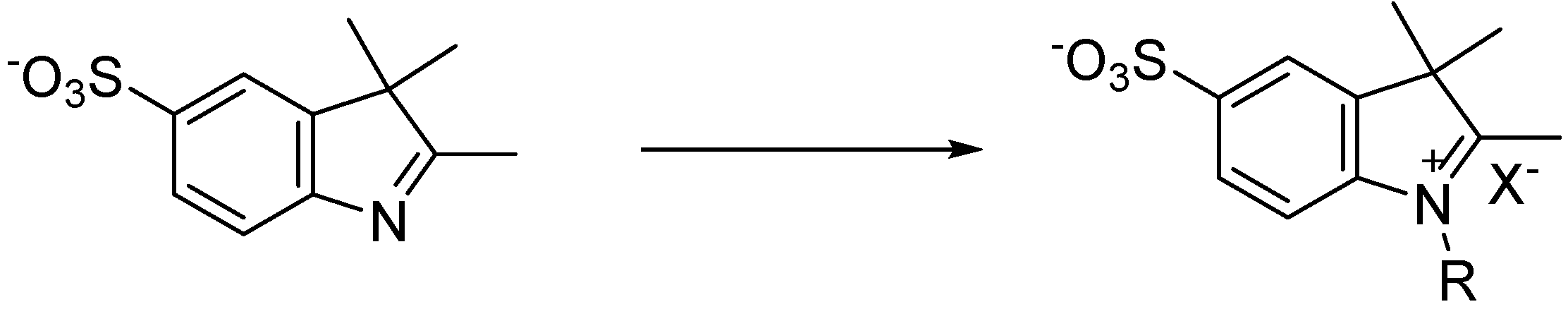{kind=link}