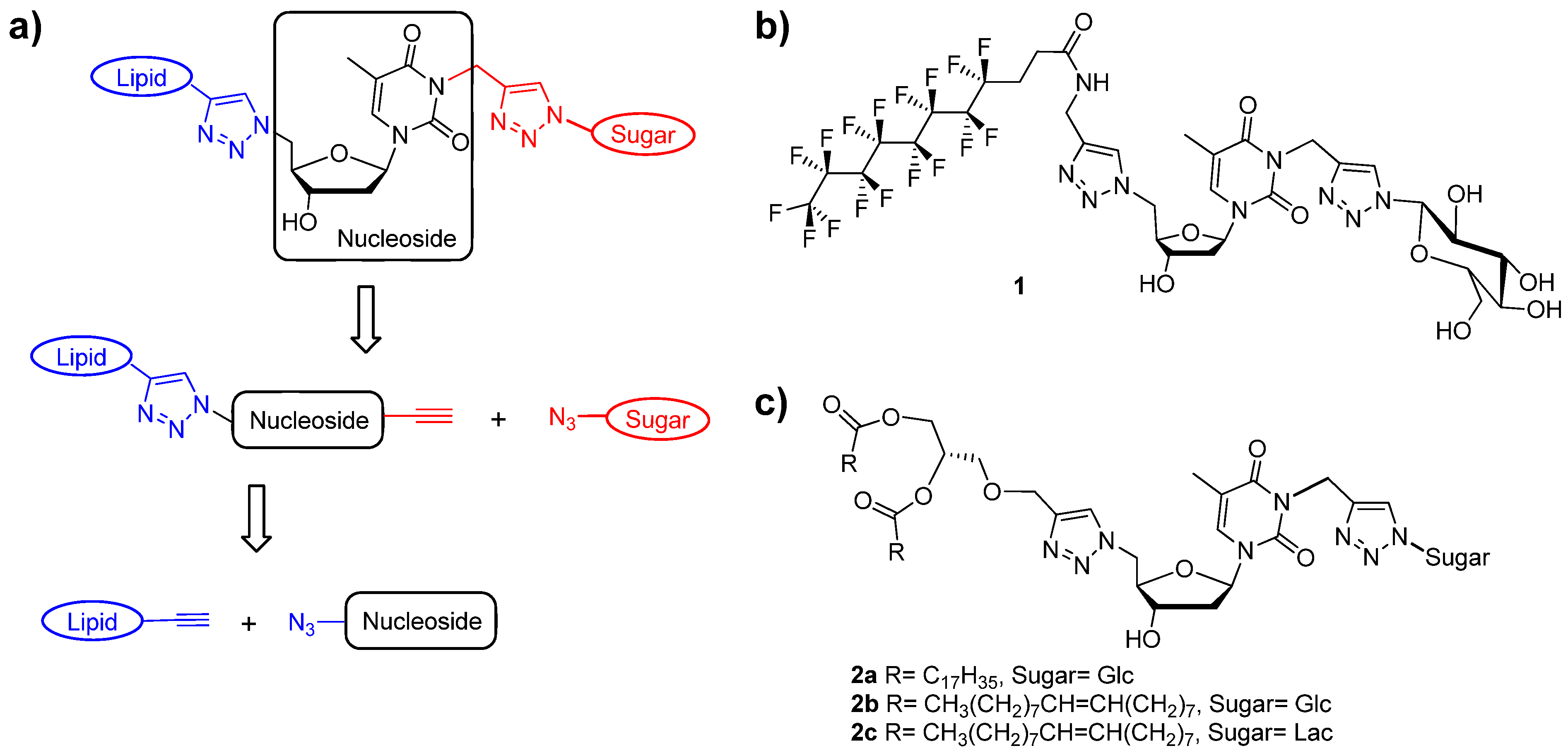
3.4. Syntheses
Dimethyl (S)-malate (
3b). Compound
3b was synthesized according to the literature procedure [
34]. The data agreed with the literature values.
1H-NMR (300 MHz, CDCl
3):
δ in ppm 2.73–2.89 (m, 2H, -C
H2-C=O), 3.32 (bs, 1H, -OH), 3.69 (s, 3H, -C
H3-O-C(=O)-CH
2-), 3.78 (s, 3H, -C
H3-O-C(=O)-CH-), 4.47–4.51 (m, 1H, -C
H-OH).
Methyl (4S)-(+)-2,2-dimethyl-1,3-dioxolane-4-acetate (
3). Compound
3 was synthesized according to the literature procedure [
34]. The data agreed with the literature values.
1H-NMR (300 MHz, CDCl
3):
δ in ppm 1.33 (s, 3H, -C-C
H3-), 1.39 (s, 3H, -C-C
H3-), 2.46–2.54 (m, 1H, -C
H'H≡-C=O), 2.66–2.73 (m, 1H, -CH'
H≡-C=O), 3.60–3.65 (m, 1H, -O- C
H'H≡-), 3.68 (s, 3H, -O-C
H3), 4.11–4.16 (m, 1H, -O- CH'
H≡-), 4.45 (quintet, 1H, -O-C
H-,
J = 6.45 Hz).
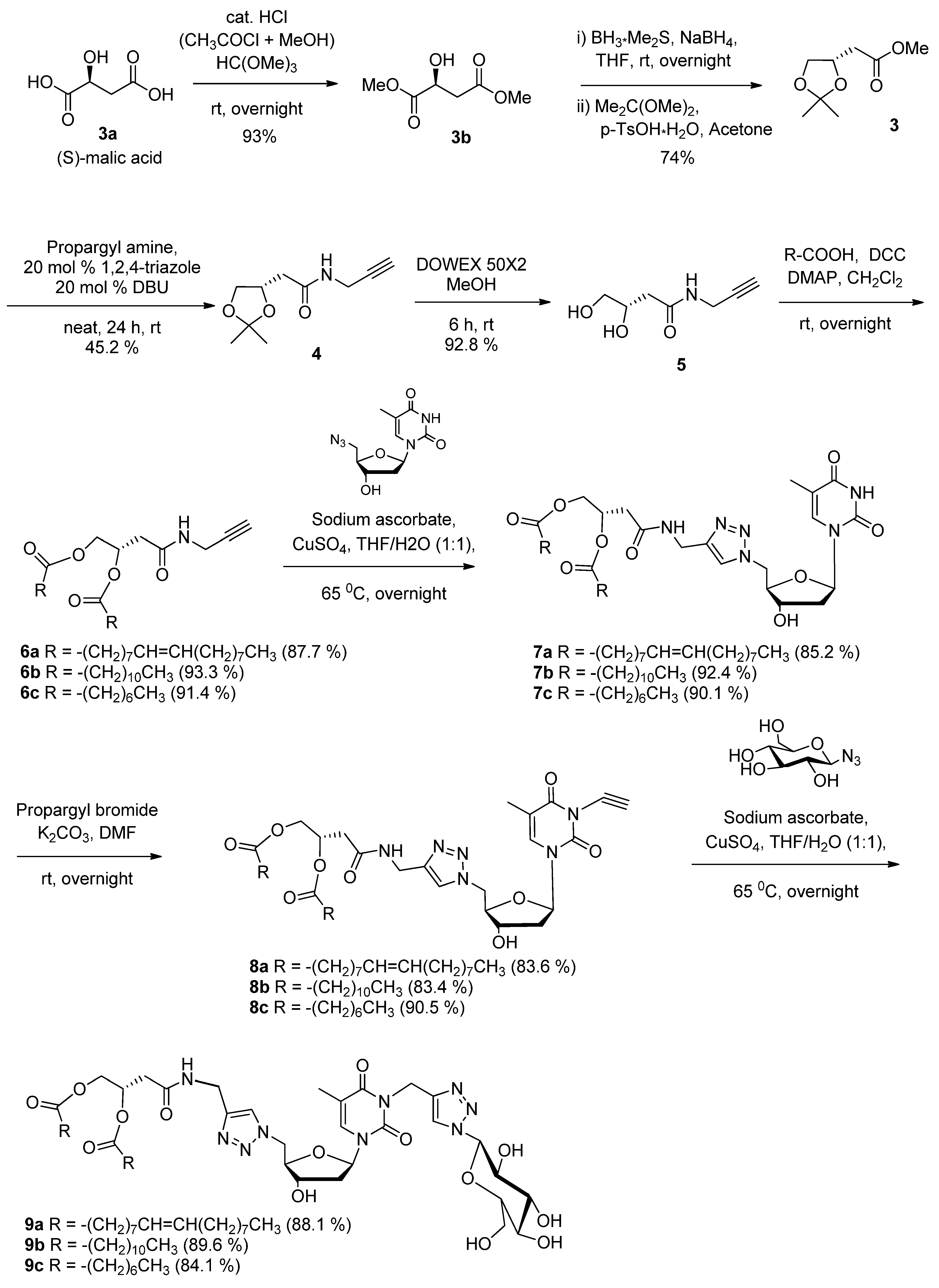
(S)-2-(2,2-Dimethyl-1,3-dioxolan-4-yl)-N-(prop-2-ynyl)acetamide (4). A reaction mixture consisting of propargyl amine (2.95 g, 3.5 mL, 53.63 mmol, 1.0 equiv), methyl (4S)-(+)-2,2-dimethyl-1,3-dioxolane-4-acetate (3, 11.2 g, 64.36 mmol, 1.2 equiv), 1,2,4-triazole (740 mg, 10.73 mmol, 0.20 equiv.) and DBU (1.63 g, 1.6 mL, 10.73 mmol, 0.20 equiv.) was stirred at room temperature for 48 h and then applied directly to a silica gel chromatographic column (diethyl ether-TEA 98:2). Product 4 was then isolated as a colourless oil. Yield: 4.77 g (45.2%). Rf: 0.5 (diethyl ether). 1H-NMR (300 MHz, CDCl3): δ in ppm 1.35 (s, 3H, CH3), 1.43 (s, 3H, CH3), 2.22 (t, 1H, -C≡CH, J = 2.5 Hz), 2.45–2.54 (m, 2H, -CH2-C=O), 3.59–3.64 (m, 1H, -O-CH′H″), 3.91–4.07 (m, 2H, -NH-CH2-C≡), 4.09–4.14 (m, 1H, -O-CH′H″), 4.36–4.45 (m, 1H, -CH-), 6.43 (bs, 1H, -NH-C=O-). 13C-NMR (75 MHz, CDCl3): δ in ppm 25.5 Η(-CH3), 26.8 (-CH3), 29.0 (-CH2-NH), 40.1 (-CH2-C=O), 68.9 (-O-CH2), 71.5 (≡CH), 72.2 (-CH), 79.4 (-C≡), 109.5 (O-C-O), 169.6 (-NH-C=O). High-resolution ESI-MS m/z calcd. for C10H15NO3Na 220.0944 [M+Na]+, found 220.0952.
(S)-3,4-Dihydroxy-N-(prop-2-ynyl)butanamide (5). A solution of compound 4 (4.2 g, 21.32 mmol) in methanol (100 mL) was treated with Dowex® 50X2 (10 g) at room temperature until completion of the reaction (≈4 to 5 h) as evidenced by TLC (methanol-ethyl acetate: 1:9). After removal of the solvent under reduced pressure, the crude deprotected product applied directly to a silica gel chromatographic column (methanol-ethyl acetate 1:9). Product 5 was isolated as a white solid. Yield: 3.1 g (92.8%). Rf: 0.33 (methanol-ethyl acetate 1:9). 1H-NMR (300 MHz, CD3OD): δ in ppm 2.26–2.45 (m, 2H,-CH2-C=O), 2.59 (t, 1H, -C≡CH, J = 2.5 Hz), 3.48 (d, 2H, J = 5.5, -CH2OH), 3.95–3.99 (m, 2H, NH-CH2-C≡), 4.00-4.09 (m, 1H, -CH-). 13C-NMR (75 MHz, CD3OD): δ in ppm 29.4 (-CH2-NH), 40.9 (-CH2-C=O), 66.8 (-CH2-OH), 70.4 (-CH-OH), 72.2 (≡CH), 80.5 (-C≡), 173.6 (-NH-C=O). High-resolution ESI-MS m/z calcd. for C7H11NO3Na 180.0631 [M+Na]+, found 180.0625.
General procedure for synthesis of coupling products 6a, 6b and 6c. To a solution of compound 5 (1 equiv.) in anhydrous dichloromethane was added DCC (3 equiv.), DMAP (3 equiv.) and the appropriate fatty acid 5a or 5b or 5c (3 equiv.), and the mixture was stirred overnight at room temperature. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel eluting with different ratios of ethylacetate/hexane to obtain pure product 5a, 5b and 5c.
(Z)-((S)-4-Oxo-4-(prop-2-ynylamino)butane-1,2-diyl) dioleate (6a). Anhydrous dichloromethane (15 mL), compound 5 (60 mg, 0.382 mmol, 1.0 equiv.), DCC (236 mg, 1.146 mmol, 3.0 equiv.), DMAP (140 mg, 1.146 mmol, 3.0 equiv.), oleic acid (5a, 324 mg, 364 μL, 1.146 mmol, 3.0 equiv.). Product 6a was isolated after purification on silica gel (ethyl acetate-hexane 1:3) as a white solid. Yield: 230 mg (87.7%). Rf: 0.32 (ethyl acetate-hexane 1:3). 1H-NMR (300 MHz, CDCl3): δ in ppm 0.87 (t, 6H, 2 CH3, J = 6.6 Hz), 1.26–1.29 (m, 40H, 20CH2 of the dioleyl chain), 1.51–1.62 (m, 4H, 2CH2-CH2-C=O), 1.88–2.09 (m, 8H, 2CH2-CH=CH-CH2), 2.23 (t, 1H, -C≡CH, J = 2.5 Hz), 2.27–2.33 (m, 4H, 2CH2-C=O of the dioleyl chain), 2.53 (d, 2H, CH2-C=O, J = 6.4 Hz), 4.02–4.05 (m, 2H, NH-H2-C≡), 4.12–4.18 (m, 1H, -O-CH′H″), 4.31-4.36 (m, 1H, -O-CH′H″), 5.28–5.35 (m, 4H, 2CH=CH of the dioleyl chain), 5.37–5.42 (m, 1H, -CH-), 5.93 (bs, 1H, -NH-C=O-). 13C-NMR (75 MHz, CDCl3): δ in ppm 14.1 (2CH3), 22.7 (2CH2-CH3), 24.8 (2CH2-CH2-C=O), 27.1 (CH2-CH=CH-CH2), 27.2 (CH2-CH=CH-CH2), 29.0–29.7 (CH2 of the dioleyl chain and -CH2-NH), 31.9 (2CH2-CH2-CH3), 34.0 (CH2-C=O of chain), 34.2 (CH2-C=O of chain), 37.9 (-CH2-C=O), 64.2 (-CH2-O-), 68.4 (≡CH), 71.8 (-CH-O-), 79.1 (-C≡), 129.7 (-CH=CH-), 130.0 (-CH=CH-), 168.2 (-NH-C=O), 172.9 (-O-C=O), 173.3 (-O-C=O). High-resolution ESI-MS m/z calcd. for C43H75NO5Na 708.5537 [M+Na]+, found 708.5532.
(S)-4-Oxo-4-(prop-2-ynylamino)butane-1,2-diyl didodecanoate (6b). Anhydrous dichloromethane (20 mL), compound 5 (150 mg, 0.95 mmol, 1.0 equiv.), DCC (590 mg, 2.86 mmol, 3.0 equiv.), DMAP (349 mg, 2.86 mmol, 3.0 equiv.), lauric acid (5b, 573 mg, 2.86 mmol, 3.0 equiv.). Product 6b was isolated after purification on silica gel (ethyl acetate-hexane 3:7) as a white solid. Yield: 465 mg (93.3%). Rf: 0.38 (ethyl acetate-hexane 3:7). 1H-NMR (300 MHz, CDCl3): δ in ppm 0.87 (t, 6H, 2 CH3, J = 6.6 Hz), 1.25 (s, 32H, 16CH2 of the dilauryl chain), 1.51–1.69 (m, 4H, 2CH2-CH2-C=O), 2.23 (t, 1H, -C≡CH, J = 2.54 Hz), 2.27–2.33 (m, 4H, 2CH2-C=O of the dilauryl chain), 2.53 (d, 2H, CH2-C=O, J = 6.4 Hz), 4.03–4.05 (m, 2H, NH-CH2-C≡), 4.12–4.18 (m, 1H, -O-CH′H″), 4.31–4.36 (m, 1H, -O-CH′H″), 5.35–5.44 (m, 1H,-CH-), 5.92 (bs, 1H, -NH-C=O-). 13C-NMR (75 MHz, CDCl3): δ in ppm 14.1 (2CH3), 22.6 (2CH2-CH3), 24.8 (2CH2-CH2-C=O), 29.0–29.6 (CH2 of the dilauryl chain and -CH2-NH), 31.9 (2CH2-CH2-CH3), 34.0 (CH2-C=O of chain), 34.2 (CH2-C=O of chain), 37.8 (-CH2-C=O), 64.1 (-CH2-O-), 68.4 (≡CH), 71.7 (-CH-O-), 79.1 (-C≡), 168.3 (-NH-C=O), 172.9 (-O-C=O), 173.4 (-O-C=O). High-resolution ESI-MS m/z calcd. for C31H55NO5Na 544.3972 [M+Na]+, found 544.3978.
(S)-4-Oxo-4-(prop-2-ynylamino)butane-1,2-diyl dioctanoate (6c). Anhydrous dichloromethane (20 mL), compound 5 (130 mg, 0.83 mmol, 1.0 equiv.), DCC (512 mg, 2.49 mmol, 3.0 equiv.), DMAP (303 mg, 2.49 mmol, 3.0 equiv.), octanoic acid (5c, 359 mg, 395 μL, 2.49 mmol, 3.0 equiv.). Product 6c was isolated after purification on silica gel (ethyl acetate-hexane 3:7) as a colourless oil (which may turn into a white solid upon standing). Yield: 310 mg (91.4%). Rf: 0.6 (ethyl acetate-hexane 2:3). 1H-NMR (300 MHz, CDCl3): δ in ppm 0.85 (t, 6H, 2 CH3, J = 6.6 Hz), 1.25 (s, 16H, 8CH2 of the dioctanoyl chain), 1.56–1.60 (m, 4H, 2CH2-CH2-C=O), 2.22 (t, 1H, -C≡CH, J = 2.54 Hz), 2.25–2.32 (m, 4H, 2CH2-C=O of the dioctanoyl chain), 2.53 (d, 2H, CH2-C=O, J = 6.4 Hz), 4.00–4.03 (m, 2H, NH-CH2-C≡), 4.11–4.17 (m, 1H, -O-CH′H″), 4.29–4.35 (m, 1H, -O-CH′H″), 5.34–5.42 (m, 1H, -CH-), 6.12 (bs, 1H, -NH-C=O-). 13C-NMR (75 MHz, CDCl3): δ in ppm 14.0 (2CH3), 22.5 (2CH2-CH3), 24.8 (2CH2-CH2-C=O), 28.9–29.2 (CH2 of the dioctanoyl chain and -CH2-NH), 31.6 (2CH2-CH2-CH3), 34.0 (CH2-C=O of chain), 34.2 (CH2-C=O of chain), 37.7 (-CH2-C=O), 64.1 (-CH2-O-), 68.3 (≡CH), 71.7 (-CH-O-), 79.2 (-C≡), 168.3 (-NH-C=O), 172.9 (-O-C=O), 173.4 (-O-C=O). High-resolution ESI-MS m/z calcd. for C23H39NO5Na 432.2720 [M+Na]+, found 432.2722.
General procedure for the synthesis of the first click products 7a,
7b and 7c. The above coupling products
6a or
6b or
6c, sodium ascorbate and copper sulfate were added to 5'-azido-5'-deoxythymidine [
37] in a water/THF (1:1) mixture. The reaction mixture was maintained at 65 °C under stirring for 10 h. After cooling to room temperature, the solvent was evaporated. The residual solid was dissolved in DCM and successively washed with water and brine. The organic layer was dried over Na
2SO
4 and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel eluted with different ratios of methanol/ethyl acetate to obtain purified compounds
7a,
7b or
7c.
(Z)-((S)-4-(1-(((2R,3S,5R)-3-Hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)yl) tetrahydrofuran -2-yl)methyl)-1H-1,2,3-triazol-4-ylamino)-4-oxobutane-1,2-diyl) dioleate (7a). Coupling product 6a (210 mg, 0.306 mmol, 1.0 equiv.), 5'-azido-5'-deoxythymidine (81.7 mg, 0.306 mmol, 1.0 equiv.), sodium ascorbate (31 mg, 0.153 mmol, 0.5 equiv.), copper sulfate (13 mg, 0.0765 mmol, 0.25 equiv.), water/THF (1:1) (20 mL). Product 7a was isolated after purification on silica gel (methanol-ethyl acetate:2:98) as a white solid. Yield: 248 mg (85.2%). Rf: 0.28 (methanol-ethyl acetate 2:98). 1H-NMR (300 MHz, CDCl3): δ in ppm 0.87 (t, 6H, 2 CH3 of the dioleyl chain, J = 6.7 Hz), 1.26–1.28 (m, 40H, 20CH2 of the dioleyl chain), 1.41–1.66 (m, 4H, 2CH2-CH2-C=O), 1.88 (s, 3H, CH3(base)), 1.96–2.09 (m, 8H, 2CH2-CH=CH-CH2), 2.23–2.42 (m, 6H, 2CH2-C=O of the dioleyl chain, 2H2′(sugar)), 2.47–2.60 (m, 2H, CH2-C=O), 3.38 (bs, 1H, -OH), 4.06–4.12 (m, 1H, NH-CH′H″-C=), 4.15–4.25 (m, 1H, -O-CH′H″), 4.28–4.36 (m, 1H, -O-CH′H″), 4.45–4.77 (m, 5H, NH-CH′H″-C=, H3′, H4′, 2H5′), 5.27–5.35 (m, 4H, 2CH=CH of the dioleyl chain), 5.37–5.46 (m, 1H, -CH-), 6.00 (t, 1H, J = 6.4 Hz, H1′), 6.98 (s, 1H, triazole), 7.17 (bs, 1H, -NH-C=O-), 7.77 (s, 1H, H-6(base)), 9.79 (s, 1H, -NH(base)). 13C-NMR (75 MHz, CDCl3): δ in ppm 12.4 (CH3(base)), 14.1 (2CH3 of the dioleyl chain), 22.6 (2CH2-CH3), 24.9 (2CH2-CH2-C=O), 27.2 (2CH2-CH=CH-CH2), 29.0–29.7 (CH2 of the dioleyl chain), 31.9 (2CH2-CH2-CH3), 34.0 (CH2-C=O of chain), 34.3 (CH2-C=O of chain), 34.7 (-CH2-NH), 37.6 (C2′), 38.6 (-CH2-C=O), 51.3 (C5′), 64.4 (-CH2-O-), 68.5 (C3′), 71.2 (-CH-O-), 83.8 (C4′), 87.1 (C1′), 111.3 (C5-thymine base), 124.5 (-CH triazole), 129.6 (-CH=CH-), 130.0 (-CH=CH-), 137.1 (C6-thymine base), 144.4 (=C- triazole), 150.3 (C=O(2) thymine base), 164.0 (C=O(4) thymine base), 169.3 (-NH-C=O), 173.2 (-O-C=O of the oleyl chain), 173.6 (-O-C=O of the oleyl chain). High-resolution ESI-MS m/z calcd. for C53H88N6O9Na 975.6505 [M+Na]+, found 975.6548.
(S)-4-(1-(((2R,3S,5R)-3-Hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-ylamino)-4-oxobutane-1,2-diyl didodecanoate (7b). Coupling product 6b (350 mg, 0.67 mmol, 1.0 equiv.), 5'-azido-5'-deoxythymidine (180 mg, 0.67 mmol, 1.0 equiv.), sodium ascorbate (66 mg, 0.34 mmol, 0.5 equiv.), copper sulfate (27 mg, 0.17 mmol, 0.25 equiv.), water/THF (1:1) (15 mL). Product 7b was isolated after purification on silica gel (methanol-ethyl acetate 0:100 to 5:95) as a white solid. Yield: 490 mg (92.4%). Rf: 0.25 (methanol-ethyl acetate 2.5:97.5). 1H-NMR (300 MHz, CDCl3:CD3OD(9.5:0.5)): δ in ppm 0.75 (t, 6H, 2 CH3 of the dilauryl chain, J = 6.6 Hz), 1.13 (s, 32H, 16CH2 of the dilauryl chain), 1.35–1.54 (m, 4H, 2CH2-CH2-C=O), 1.78 (s, 3H, CH3(base)), 2.06–2.23 (m, 6H, 2CH2-C=O of the dilauryl chain, 2H2′(sugar)), 2.31–2.45 (m, 2H, CH2-C=O), 3.97–4.04 (m, 2H,-O-CH2), 4.15–4.21 (m, 2H, 2H5′(base)), 4.26 (s, 2H, NH-CH2-C=), 4.5–4.62 (m, 2H, H3′(sugar), H4′(sugar)), 5.22–5.34 (m, 1H, -CH-), 5.98 (t, 1H, J = 6.7 Hz, H1′(sugar)), 6.90 (s, 1H, triazole), 7.65 (s, 1H, H-6(base)). 13C-NMR (75 MHz, CDCl3:CD3OD(9.5:0.5)): δ in ppm 12.1 (CH3(base)), 13.9 (2CH3 of the dilauryl chain), 22.5 (2CH2-CH3), 24.7 (2CH2-CH2-C=O), 28.9–29.4 (CH2 of the dilauryl chain), 31.7 (2CH2-CH2-CH3), 33.9 (CH2-C=O of chain), 34.1 (CH2-C=O of chain), 34.2 (-CH2-NH), 37.2 (C2′), 38.5 (-CH2-C=O), 51.1 (C5′), 64.2 (-CH2-O-), 68.3 (C3′), 70.6 (-CH-O-), 83.5 (C4′), 86.0 (C1′), 111.1 (C5-thymine base), 124.5 (-CH triazole), 136.4 (C6-thymine base), 144.3 (=C- triazole), 150.3 (C=O(2) thymine base), 164.2 (C=O(4) thymine base), 169.5 (-NH-C=O), 173.2 (-O-C=O of the lauryl chain), 173.6 (-O-C=O of oleyl chain). High-resolution ESI-MS m/z calcd. for C41H68N6O9Na 811.4939 [M+Na]+, found 811.4972.
(S)-4-(1-(((2R,3S,5R)-3-Hydroxy-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-ylamino)-4-oxobutane-1,2-diyl dioctanoate (7c). Coupling product 6c (450 mg, 1.1 mmol, 1.0 equiv.), 5'-azido-5'-deoxythymidine (294 mg, 1.1 mmol, 1.0 equiv.), sodium ascorbate (44 mg, 0.22 mmol, 0.2 equiv.), copper sulfate (18 mg, 0.11 mmol, 0.1 equiv.), water/THF (1:1) (20 mL). Product 7c was isolated after purification on silica gel (methanol-ethyl acetate 1:9) as a white solid. Yield: 670 mg (90.1%). Rf: 0.33 (methanol-ethyl acetate 5:95). 1H-NMR (300 MHz, CD3OD): δ in ppm 0.90 (t, 6H, 2 CH3 of the dioctanoyl chain, J = 6.7 Hz), 1.29 (s, 16H, 8CH2 of the dioctanoyl chain), 1.48–1.67 (m, 4H, 2CH2-CH2-C=O), 1.90 (s, 3H, CH3(base)), 2.21–2.33 (m, 6H, 2CH2-C=O of the dioctanoyl chain, 2H2′ (sugar)), 2.43–2.60 (m, 2H, CH2-C=O), 4.03–4.18 (m, 2H, -O-CH2), 4.32–4.46 (m, 4H, 2H5′(base), NH-CH2-C=), 4.62–4.79 (m, 2H, H3′(sugar), H4′(sugar)), 5.37–5.49 (m, 1H, -CH-), 6.19 (t, 1H, J = 6.4 Hz, H1′), 7.32 (s, 1H, triazole), 7.88 (s, 1H, H-6(base)). 13C-NMR (75 MHz, CD3OD): δ in ppm 12.5 (CH3(base)), 14.5 (2CH3 of the dioctanoyl chain), 23.7 (2CH2-CH3), 26.0 (2CH2-CH2-C=O), 30.1-30.2 (CH2 of the dioctanoyl chain), 32.9 (2CH2-CH2-CH3), 34.9 (CH2-C=O of chain), 35.1 (CH2-C=O of chain), 35.6 (-CH2-NH), 38.2 (C2′), 39.6 (-CH2-C=O), 52.8 (C5′), 65.5 (-CH2-O-), 69.9 (C3′), 72.5 (-CH-O-), 85.6 (C4′), 87.0 (C1′), 111.9 (C5-thymine base), 125.4 (-CH triazole), 138.2 (C6-thymine base), 146.1 (=C- triazole), 152.1 (C=O(2) thymine base), 166.3 (C=O(4) thymine base), 171.7 (-NH-C=O), 174.4 (-O-C=O of the octanoyl chain), 174.9 (-O-C=O of octanoyl chain). High-resolution ESI-MS m/z calcd. for C33H52N6O9Na 699.3687 [M+Na]+, found 699.3683.
General procedure for the synthesis of the N-propargyl derivatives of click products 8a, 8b and 8c. Anhydrous DMF was added under nitrogen to the click products 7a, 7b or 7c, followed by K2CO3. Propargyl bromide was then added at room temperature, and the reaction mixture was stirred overnight. DMF was removed under reduced pressure, the residual solid was dissolved in ethyl acetate (or dichloromethane for 8a) and was washed twice with water and once with brine. The organic phase was dried over Na2SO4 and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel eluted with different ratios of methanol-ethyl acetate to afford the purified click products 8a, 8b or 8c.
(Z)-((S)-4-(1-(((2R,3S,5R)-3-Hydroxy-5-(5-methyl-2,4-dioxo-3-(prop-2-ynyl)-3,4-dihydropyrimidin-1(2H)-yl) tetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-ylamino)-4-oxobutane-1,2-diyl) dioleate (8a). Click product 7a (190 mg, 0.2 mmol, 1 equiv.), propargyl bromide (59.5 mg, 43.1 μL (80 wt.% solution in toluene), 0.4 mmol, 2 equiv.), K2CO3 (54 mg, 0.4 mmol, 2 equiv.), dry DMF (10 mL). Product 8a was isolated after purification on silica gel (methanol-ethyl acetate 0:100 to 2:98) as a colourless oil. Yield: 185 mg (83.6%). Rf: 0.41 (ethyl acetate). 1H-NMR (300 MHz, CDCl3): δ in ppm 0.87 (t, 6H, 2 CH3 of the dioleyl chain, J = 6.7 Hz), 1.26–1.28 (m, 40H, 20CH2 of the dioleyl chain), 1.44–1.69 (m, 4H, 2CH2-CH2-C=O), 1.95 (s, 3H, CH3(base)), 1.97–2.04 (m, 8H, 2CH2-CH=CH-CH2), 2.23–2.39 (m, 7H, 2CH2-C=O of the dioleyl chain, 2H2′(sugar), -C≡CH), 2.47–2.58 (m, 2H, CH2-C=O), 2.98 (bs, 1H, -OH), 4.05–4.12 (m, 1H, -O-CH′H″-C=), 4.21–4.31 (m, 2H, -O-CH′H″, NH-CH′H″-C=), 4.41–4.47 (m, 3H, NH-CH′H″-C=, CH′H″-C≡, H3′), 4.63–4.76 (m, 4H, H4′, 2H5′, -CH′H″-C≡), 5.27–5.35 (m, 4H, 2CH=CH of the dioleyl chain), 5.37–5.44 (m, 1H, -CH-), 6.14 (t, 1H, J = 6.7 Hz, H1′), 6.82 (bs, 1H, -NH-C=O-), 6.96 (s, 1H, triazole), 7.74 (s, 1H, H-6(base)). 13C-NMR (75 MHz, CDCl3): δ in ppm 13.1 (CH3(base)), 14.1 (2CH3 of the dioleyl chain), 22.6 (2CH2-CH3), 24.8 (2CH2-CH2-C=O), 27.2 (2CH2-CH=CH-CH2), 29.0–29.7 (CH2 of the dioleyl chain), 30.4 (-CH2-C≡), 31.9 (2CH2-CH2-CH3), 34.0 (CH2-C=O of chain), 34.2 (CH2-C=O of chain), 34.7 (-CH2-NH), 37.8 (C2′), 38.6 (-CH2-C=O), 51.4 (C5′), 64.3 (-CH2-O-), 68.4 (C3′), 70.9 (≡CH), 71.5 (-CH-O-), 78.1 (-C≡), 83.9 (C4′), 87.5 (C1′), 110.7 (C5-thymine base), 124.4 (-CH triazole), 129.6 (-CH=CH-), 130.0 (-CH=CH-), 135.0 (C6-thymine base), 144.3 (=C- triazole), 150.0 (C=O(2) thymine base), 162.2 (C=O(4) thymine base), 169.1 (-NH-C=O), 173.2 (-O-C=O of the oleyl chain), 173.6 (-O-C=O of oleyl chain). High-resolution ESI-MS m/z calcd. for C56H90N6O9Na 1013.6661 [M+Na]+, found 1013.6711.
(S)-4-(1-(((2R,3S,5R)-3-Hydroxy-5-(5-methyl-2,4-dioxo-3-(prop-2-ynyl)-3,4-dihydropyrimidin-1(2H)-yl) tetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-ylamino)-4-oxobutane-1,2-diyl didodecanoate (8b). Click product 7b (440 mg, 0.56 mmol, 1 equiv.), propargyl bromide (116 mg, 120 μL (80 wt.% solution in toluene), 1.12 mmol, 2 equiv.), K2CO3 (154 mg, 1.12 mmol, 2 equiv.), dry DMF (15 mL). Product 8b was isolated after purification on silica gel (methanol-ethyl acetate 0:100 to 2:98) as a colourless oil. Yield: 385 mg (83.4%). Rf: 0.30 (methanol-ethyl acetate 2:98). 1H-NMR (300 MHz, CDCl3): δ in ppm 0.86 (t, 6H, 2CH3 of the dilauryl chain, J = 6.6 Hz), 1.23 (s, 32H, 16CH2 of the dilauryl chain), 1.46–1.65 (m, 4H, 2CH2-CH2-C=O), 1.93 (s, 3H, CH3(base)), 2.11–2.42 (m, 7H, 2CH2-C=O of the dilauryl chain, 2H2′(sugar), -C≡CH), 2.47–2.56 (m, 2H, CH2-C=O), 4.05–4.11 (m, 1H, -O-CH′H″-C=), 4.19–4.30 (m, 2H, -O-CH′H″, NH-CH′H″-C=), 4.37–4.49 (m, 3H, NH-CH′H″-C=, CH′H″-C≡, H3′), 4.58–4.80 (m, 4H, H4′, 2H5′, -CH′H″-C≡), 5.35–5.40 (m, 1H, -CH-), 6.15 (t, 1H, J = 6.7 Hz, H1′), 6.96 (bs, 2H, -NH-C=O, triazole), 7.73 (s, 1H, H-6(base)). 13C-NMR (75 MHz, CDCl3): δ in ppm 13.1 (CH3(base)), 14.1 (2CH3 of the dilauryl chain), 22.6 (2CH2-CH3), 24.8 (2CH2-CH2-C=O), 29.0–29.6 (CH2 of the dilauryl chain), 30.4 (-CH2-C≡), 31.8 (2CH2-CH2-CH3), 34.0 (CH2-C=O of chain), 34.2 (CH2-C=O of chain), 34.7 (-CH2-NH), 37.7 (C2′), 38.5 (-CH2-C=O), 51.2 (C5′), 64.3 (-CH2-O-), 68.4 (C3′), 70.9 (≡CH), 71.3 (-CH-O-), 78.1 (-C≡), 83.9 (C4′), 87.2 (C1′), 110.7 (C5-thymine base), 124.3 (-CH triazole), 135.0 (C6-thymine base), 144.4 (=C- triazole), 150.0 (C=O(2) thymine base), 162.2 (C=O(4) thymine base), 169.1 (-NH-C=O), 173.2 (-O-C=O of the lauryl chain), 173.6 (-O-C=O of oleyl chain). High-resolution ESI-MS m/z calcd. for C44H70N6O9Na 849.5096 [M+Na]+, found 849.5111.
(S)-4-(1-(((2R,3S,5R)-3-Hydroxy-5-(5-methyl-2,4-dioxo-3-(prop-2-ynyl)-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-ylamino)-4-oxobutane-1,2-diyl dioctanoate (8c). Click product 7c (565 mg, 0.84 mmol, 1 equiv), propargyl bromide (248 mg, 180 μL (80 wt.% solution in toluene), 1.67 mmol, 2 equiv.), K2CO3 (231 mg, 1.67 mmol, 2 equiv.), dry DMF (20 mL). Product 8c was isolated after purification on silica gel (methanol-ethyl acetate 5:95) as a colourless oil. Yield: 540 mg (90.5%). Rf: 0.45 (methanol-ethyl acetate 5:95). 1H-NMR (300 MHz, DMSO-d6): δ in ppm 0.84 (t, 6H, 2CH3 of dioctanoyl chain, J = 6.2 Hz), 1.23 (s, 16H, 16CH2 of the dioctanoyl chain), 1.37–1.57 (m, 4H, 2CH2-CH2-C=O), 1.87 (s, 3H, CH3(base)), 2.15–2.27 (m, 6H, 2CH2-C=O of the dioctanoyl chain, 2H2′(sugar)), 2.44 (d, 2H, J = 6.6 Hz, CH2-C=O), 3.10 (t, 1H, J = 2.26 Hz, -C≡CH), 3.99–4.11 (m, 2H, -O-CH2), 4.22–4.30 (m, 4H, 2H5′(base), NH-CH2-C=), 4.52 (s, 2H, -CH2-C≡), 4.56–4.73 (m, 2H, H3′(sugar), H4′(sugar)), 5.26–5.34 (m, 1H, -CH-), 5.56 (d, 1H, J = 4.14 Hz, -NH-C=O, ), 6.21 (t, 1H, J = 6.9 Hz, H1′), 7.49 (s, 1H, triazole), 8.49 (t, 1H, J = 5.47 Hz, H-6(base)). 13C-NMR (75 MHz, DMSO-d6): δ in ppm 12.7 (CH3(base)), 14.0 (2CH3 of the dioctanoyl chain), 22.1 (2CH2-CH3), 24.4–24.5 (2CH2-CH2-C=O), 28.4–28.5 (CH2 of the dioctanoyl chain), 30.1 (-CH2-C≡), 31.2 (2CH2-CH2-CH3), 33.4 (CH2-C=O of chain), 33.6 (CH2-C=O of chain), 34.2 (-CH2-NH), 36.4 (C2′), 38.0 (-CH2-C=O), 51.2 (C5′), 64.1 (-CH2-O-), 68.3 (C3′), 70.8 (≡CH) 73.1 (-CH-O-), 79.1 (-C≡), 84.3 (C4′), 85.3 (C1′), 109.1 (C5-thymine base), 123.6 (-CH triazole), 135.3 (C6-thymine base), 144.7 (=C- triazole), 149.8 (C=O(2) thymine base), 161.8 (C=O(4) thymine base), 168.3 (-NH-C=O), 172.1 (-O-C=O of the octanoyl chain), 172.6 (-O-C=O of the octanoyl chain). High-resolution ESI-MS m/z calcd. for C36H54N6O9Na 737.3844 [M+Na]+, found 737.3823.
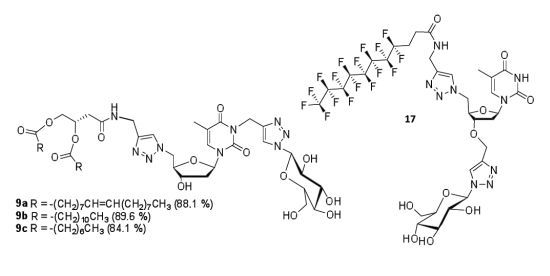
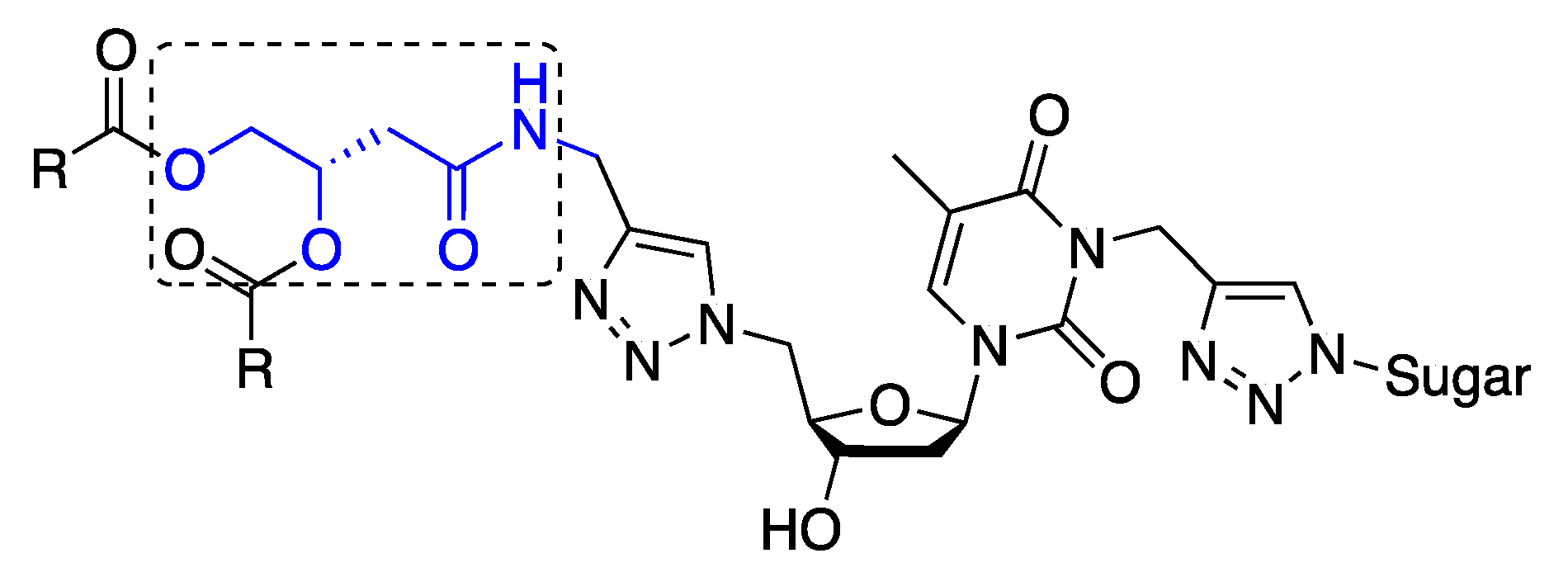
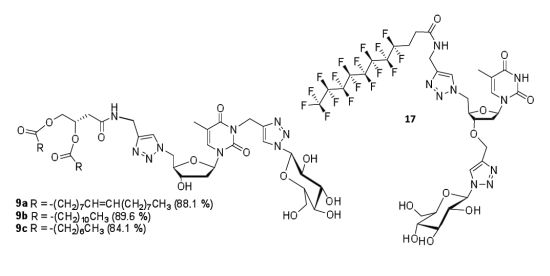
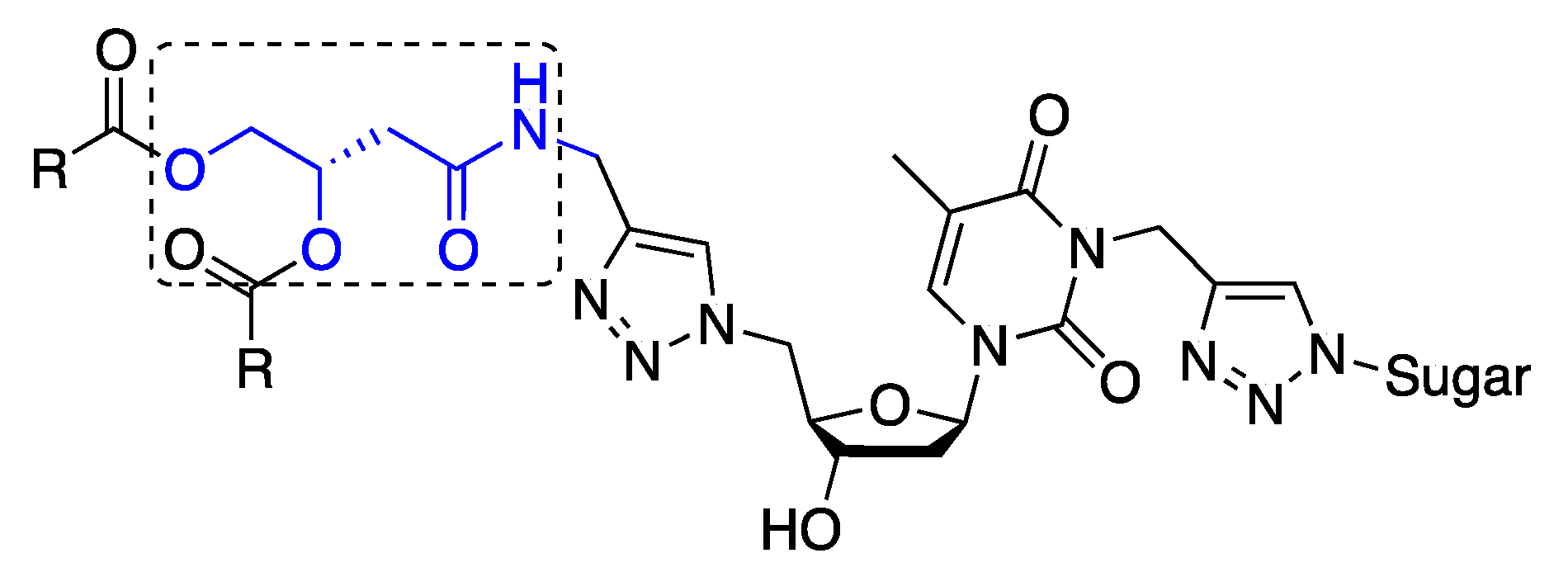
General procedure for the synthesis of GNLs 9a, 9b and 9c. The above N-propargyl derivative 8a, 8b or 8c, sodium ascorbate and copper sulfate were added to 1-azido-β-d-glucopyranoside in water/THF (1:1) mixture. The reaction mixture was maintained at 65 °C under stirring for 10 h, then cooled to room temperature and the solvent was evaporated. The residual solid was dissolved in DCM and successively washed by water and brine. The organic layer was dried over Na2SO4 and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel eluted with different ratios of methanol/dichloromethane to obtain pure click product 9a, 9b or 9c.
(Z)-4-(1-(((2R,3S,5R)-3-Hydroxy-5-(5-methyl-2,4-dioxo-3-((1-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)-1H-1,2,3-triazol-4-yl)methyl)-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-ylamino)-4-oxobutane-1,2-diyl dioleate (9a). N-propargyl derivative 8a (160 mg, 0.16 mmol, 1.0 equiv), 1-azido-β-d-glucopyranoside (33 mg, 0.16 mmol, 1.0 equiv.), sodium ascorbate (16 mg, 0.2 mmol, 0.5 equiv.), copper sulfate (6.5 mg, 0.1 mmol, 0.25 equiv.), water-THF (1:1) (15 mL). Product 9a was isolated after purification on silica gel (methanol-dichloromethane 15:85) as a white solid. Yield: 170 mg (88.1%). Rf: 0.4 (methanol-dichloromethane 15:85). 1H-NMR (300 MHz, DMSO-d6): δ in ppm 0.84 (t, 6H, 2CH3 of the dioleyl chain, J = 6.0 Hz), 1.23 (s, 40H, 20CH2 of the dioleyl chain), 1.38–1.57 (m, 4H, 2CH2-CH2-C=O), 1.88 (s, 3H, CH3(base)), 1.90–2.07 (m, 8H, 2CH2-CH=CH-CH2), 2.15–2.30 (m, 6H, 2CH2-C=O of the dioleyl chain, 2H2′(sugar)), 2.44 (d, 2H, J = 6.2 Hz, CH2-C=O), 3.17–3.23 (m, 1H, -O-CH- (glucose-C5)), 3.32–3.45 (m, 4H, -CH2-OH(glucose-C6), -CH(glucose-C4), -OH(glucose (C6)), 3.65–3.77 (m, 4H, -CH(glucose-C3), H3′, -OH(C3′-sugar), -OH(glucose-C3)), 4.00-4.14 (m, 3H, -O-CH2-, -OH(glucose-C4)), 4.26–4.28 (m, 5H, -OH(glucose-C2), 2H5′, -CH2-NH-C=O), 4.56–4.72 (m, 2H, H4′, -CH(glucose-C2)), 5.05 (s, 2H, N-CH2-triazole), 5.25–5.35 (m, 5H, -CH-, 2CH=CH of the dioleyl chain 5.48 (d, 1H, -N-CH-O (glucose-C1), J = 9.2 Hz), 6.22 (t, 1H, H1′, J = 6.7 Hz), 7.49 (s, 1H, triazole-between nucleoside and glucose), 7.92 (s, 1H, triazole-between alkyl chain and nucleoside), 8.11 (s, 1H, H-6(base), 8.49 (bs, 1H, -NH-C=O-). 13C-NMR (75 MHz, DMSO-d6): δ in ppm 12.8 (CH3(base)), 14.0 (2CH3 of the dioleyl chain), 22.2 (2CH2-CH3), 24.4 (CH2-CH2-C=O), 24.5 (CH2-CH2-C=O), 26.6 (CH2-CH=CH-CH2), 26.7 (CH2-CH=CH-CH2), 28.5–29.2 (CH2 of the dioleyl chain), 31.4 (2CH2-CH2-CH3), 33.4 (CH2-C=O of chain), 33.6 (CH2-C=O of chain), 34.2 (-CH2-NH), 36.2 (N-CH2-triazole), 36.4 (C2′), 38.1 (-CH2-C=O), 51.2 (C5′), 60.7 (-CH2-OH (glucose-C6)), 64.2 (-CH2-O-), 68.3 (C3′), 69.6 (-CH(glucose-C2)), 70.8 (-CH(glucose-C4)), 71.9 (-CH-O-), 77.0 (-CH(glucose-C3)), 80.0 (-CH(glucose-C5)), 84.2 (-CH(glucose-C1)), 85.2 (C4′), 87.4 (C1′), 109.2 (C5-thymine base), 122.5 (-CH triazole between thymidine and alkyl chain), 123.6 (-CH triazole between glucose and thymidine), 129.6 (2-CH=CH-), 135.1 (C6-thymine base), 142.6 (=C- triazole triazole between glucose and thymidine), 144.7 (=C- triazole between thymidine and alkyl chain), 150.3 (C=O(2) thymine base), 162.3 (C=O(4) thymine base), 168.3 (-NH-C=O), 172.0 (-O-C=O of the oleyl chain), 172.5 (-O-C=O of oleyl chain). High-resolution FD-MS m/z calcd. for C62H102N9O14 1196.7546 [M]+, found 1196.7449.
4-(1-(((2R,3S,5R)-3-Hydroxy-5-(5-methyl-2,4-dioxo-3-((1-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)-1H-1,2,3-triazol-4-yl)methyl)-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-ylamino)-4-oxobutane-1,2-diyl didodecanoate (9b). N-propargyl derivative 8b (335 mg, 0.4 mmol, 1.0 equiv), 1-azido-β-d-glucopyranoside (83 mg, 0.4 mmol, 1.0 equiv.), sodium ascorbate (40 mg, 0.2 mmol, 0.5 equiv.), copper sulfate (16.2 mg, 0.1 mmol, 0.25 equiv.), water/THF (1:1) (20 mL). Product 9b was isolated after purification on silica gel (methanol-dichloromethane 15:85) as a white solid. Yield: 374 mg (89.6%). Rf: 0.52 (methanol-dichloromethane 15:85). 1H-NMR (300 MHz, DMSO-d6): δ in ppm 0.85 (t, 6H, 2CH3 of the dilauryl chain, J = 6.2 Hz), 1.23 (s, 32H, 16CH2 of the dilauryl chain), 1.46–1.49 (m, 4H, 2CH2-CH2-C=O), 1.88 (s, 3H, CH3(base)), 2.07–2.27 (m, 6H, 2CH2-C=O of the dilauryl chain, 2H2′(sugar)), 2.45 (d, 2H, J = 6.6 Hz, CH2-C=O), 3.18–3.24 (m, 1H, -O-CH- (glucose-C5)), 3.33–3.46 (m, 4H, -CH2-OH(glucose-C6), -CH(glucose-C4), -OH(glucose (C6)), 3.65–3.77 (m, 3H, -CH(glucose-C3), H3′,-OH(C3′-sugar)), 4.00–4.11 (m, 3H, -O-CH2-, -OH (glucose-C3)), 4.26–4.28 (m, 4H, -OH(glucose-C4), H5′, -CH2-NH-C=O), 4.57–4.73 (m, 3H, H4′, H5″, -CH(glucose-C2)), 5.05 (s, 2H, N-CH2-triazole), 5.29–5.36 (m, 2H, -CH-, -OH (glucose-C2)), 5.48 (d, 1H, -N-CH-O (glucose-C1), J = 9.2 Hz), 6.23 (t, 1H, H1′, J = 6.8 Hz), 7.49 (s, 1H, triazole-between nucleoside and glucose), 7.92 (s, 1H, triazole-between alkyl chain and nucleoside), 8.10 (s, 1H, H-6(base), 8.48 (t,1H, -NH-C=O-,J = 5.4 Hz). 13C-NMR (75 MHz, DMSO-d6): δ in ppm 12.7 (CH3(base)), 13.9 (2CH3 of the dilauryl chain), 22.1 (2CH2-CH3), 24.4 (CH2-CH2-C=O), 24.5 (CH2-CH2-C=O), 28.4-29.1 (CH2 of the dilauryl chain), 31.3 (2CH2-CH2-CH3), 33.4 (CH2-C=O of chain), 33.6 (CH2-C=O of chain), 34.1 (-CH2-NH), 36.1 (N-CH2-triazole), 36.4 (C2′), 38.1 (-CH2-C=O), 51.1 (C5′), 60.7 (-CH2-OH (glucose-C6)), 64.1 (-CH2-O-), 68.3 (C3′), 69.5 (-CH(glucose-C2)), 70.7 (-CH(glucose-C4)), 71.9 (-CH-O-), 77.0 (-CH(glucose-C3)), 80.0 (-CH(glucose-C5)), 84.2 (-CH(glucose-C1)), 85.1 (C4′), 87.4 (C1′), 109.1 (C5-thymine base), 122.5 (-CH triazole between thymidine and alkyl chain), 123.5 (-CH triazole between glucose and thymidine), 135.1 (C6-thymine base), 142.6 (=C- triazole triazole between glucose and thymidine), 144.6 (=C- triazole between thymidine and alkyl chain), 150.3 (C=O(2) thymine base), 162.3 (C=O(4) thymine base), 168.3 (-NH-C=O), 172.0 (-O-C=O of the lauryl chain), 172.5 (-O-C=O of the lauryl chain). High-resolution ESI-MS m/z calcd. for C50H81N9O14Na 1054.5795 [M+Na]+, found 1054.5854.
4-(1-(((2R,3S,5R)-3-Hydroxy-5-(5-methyl-2,4-dioxo-3-((1-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)-1H-1,2,3-triazol-4-yl)methyl)-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-ylamino)-4-oxobutane-1,2-diyl dioctanoate (9c). N-propargyl derivative 8c (490 mg, 0.7 mmol, 1.0 equiv.), 1-azido-β-d-glucopyranoside (141 mg, 0.7 mmol, 1.0 equiv.), sodium ascorbate (28 mg, 0.14 mmol, 0.2 equiv.), copper sulfate (11 mg, 0.07 mmol, 0.1 equiv.), water/THF (1:1) (20 mL). Product 9c was isolated after purification on silica gel (methanol-dichloromethane 15:85) as a white solid. Yield: 530 mg (84.1%). Rf: 0.46 (methanol-dichloromethane 15:85). 1H-NMR (300 MHz, DMSO-d6): δ in ppm 0.85 (t, 6H, 2CH3 of the dioctanoyl chain, J = 6.2 Hz), 1.23 (s, 32H, 16CH2 of the dioctanoyl chain), 1.36–1.60 (m, 4H, 2CH2-CH2-C=O), 1.88 (s, 3H, CH3(base)), 2.16–2.28 (m, 6H, 2CH2-C=O of the dioctanoyl chain, 2H2′(sugar)), 2.45 (d, 2H, J = 6.8 Hz, CH2-C=O), 3.17–3.25 (m, 1H, -O-CH- (glucose-C5)), 3.32–3.47 (m, 3H, -CH2-OH(glucose-C6), -CH(glucose-C4), 3.64–3.78 (m, 2H, -CH(glucose-C3, H3′), 4.01–4.12 (m, 2H, -O-CH2-), 4.23–4.28 (m, 4H, -OH(glucose-C4), H5′, -CH2-NH-C=O,), 4.57–4.73 (m, 3H, H4′, H5″,-CH(glucose-C2)), 5.05 (s, 2H, N-CH2-triazole), 5.14 (d, 1H, -OH(glucose (C6), J = 5.3 Hz), 5.24 (d, 1H, -OH (glucose-C3), J = 4.7 Hz), 5.29–5.31 (m, 1H, -CH-), 5.34 (d, 1H, -OH (glucose-C2), J = 6.0 Hz) 5.48 (d, 1H, -N-CH-O (glucose-C1), J = 9.2 Hz), 5.53 (d, 1H, -OH(C3′-sugar), J = 4.2 Hz) 6.23 (t, 1H, H1′, J = 6.8 Hz), 7.49 (s, 1H, triazole-between nucleoside and glucose), 7.91 (s, 1H, triazole-between alkyl chain and nucleoside), 8.10 (s, 1H, H-6(base), 8.48 (t, 1H, -NH-C=O-, J = 5.4 Hz). 13C-NMR (75 MHz, DMSO-d6): δ in ppm 12.7 (CH3(base)), 13.9 (2CH3 of the dioctanoyl chain), 22.1 (2CH2-CH3), 24.4 (CH2-CH2-C=O), 24.5 (CH2-CH2-C=O), 28.3–28.4 (CH2 of the dioctanoyl chain), 31.1 (2CH2-CH2-CH3), 33.4 (CH2-C=O of chain), 33.6 (CH2-C=O of chain), 34.1 (-CH2-NH), 36.1 (N-CH2-triazole), 36.4 (C2′), 38.1 (-CH2-C=O), 51.1 (C5′), 60.7 (-CH2-OH (glucose-C6)), 64.1 (-CH2-O-), 68.3 (C3′), 69.5 (-CH(glucose-C2)), 70.7 (-CH(glucose-C4)), 71.9 (-CH-O-), 77.0 (-CH(glucose-C3)), 80.0 (-CH(glucose-C5)), 84.2 (-CH(glucose-C1)), 85.2 (C4′), 87.4 (C1′), 109.1 (C5-thymine base), 122.5 (-CH triazole between glucose and thymidine), 123.6 (-CH triazole between thymidine and alkyl chain), 135.1 (C6-thymine base), 142.6 (=C- triazole triazole between glucose and thymidine), 144.7 (=C- triazole between thymidine and alkyl chain), 150.3 (C=O(2) thymine base), 162.3 (C=O(4) thymine base), 168.3 (-NH-C=O), 172.1 (-O-C=O of the dioctanoyl chain), 172.6 (-O-C=O of the dioctanoyl chain). High-resolution ESI-MS m/z calcd. for C42H65N9O14Na 942.4543 [M+Na]+, found 942.4562.
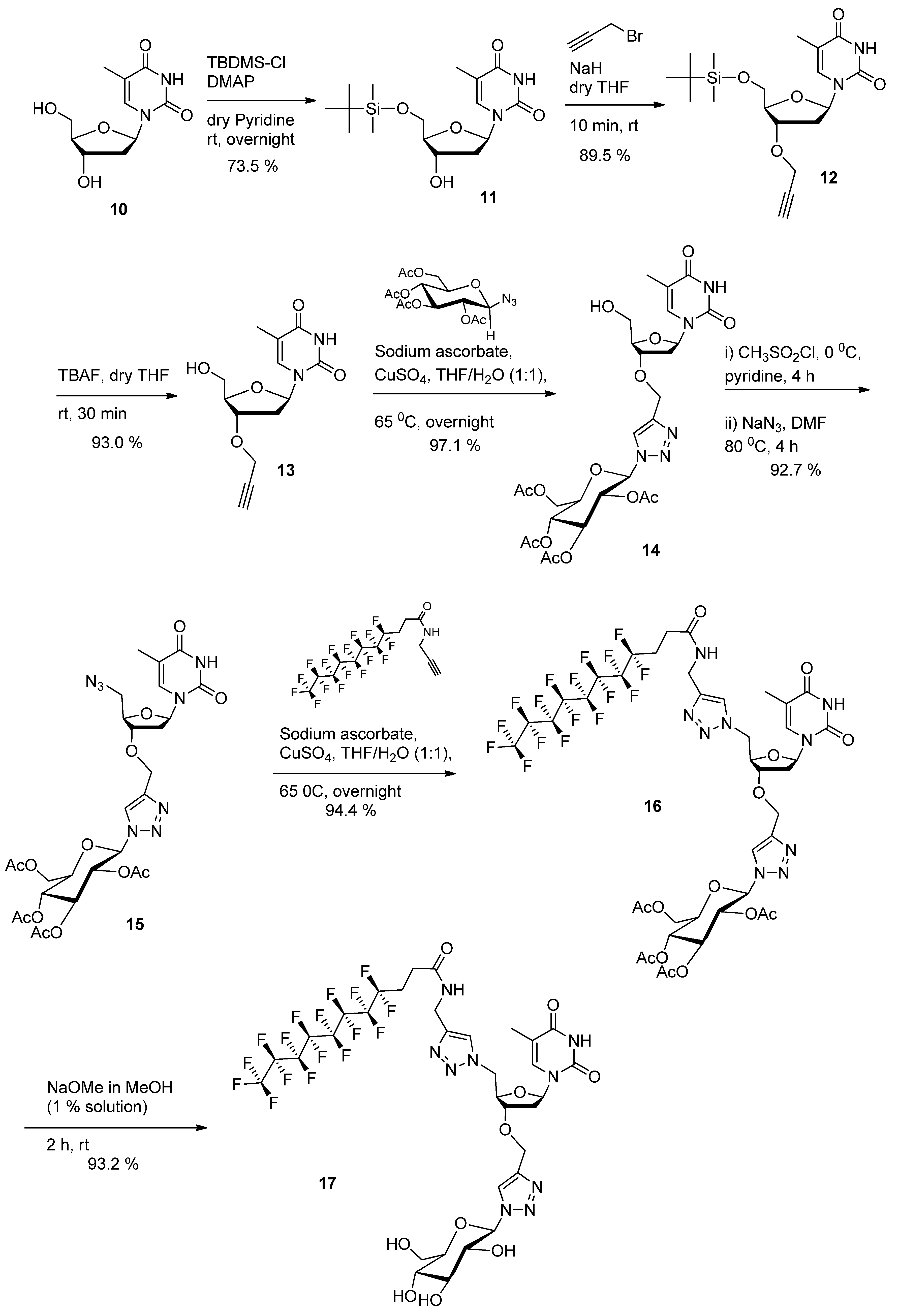
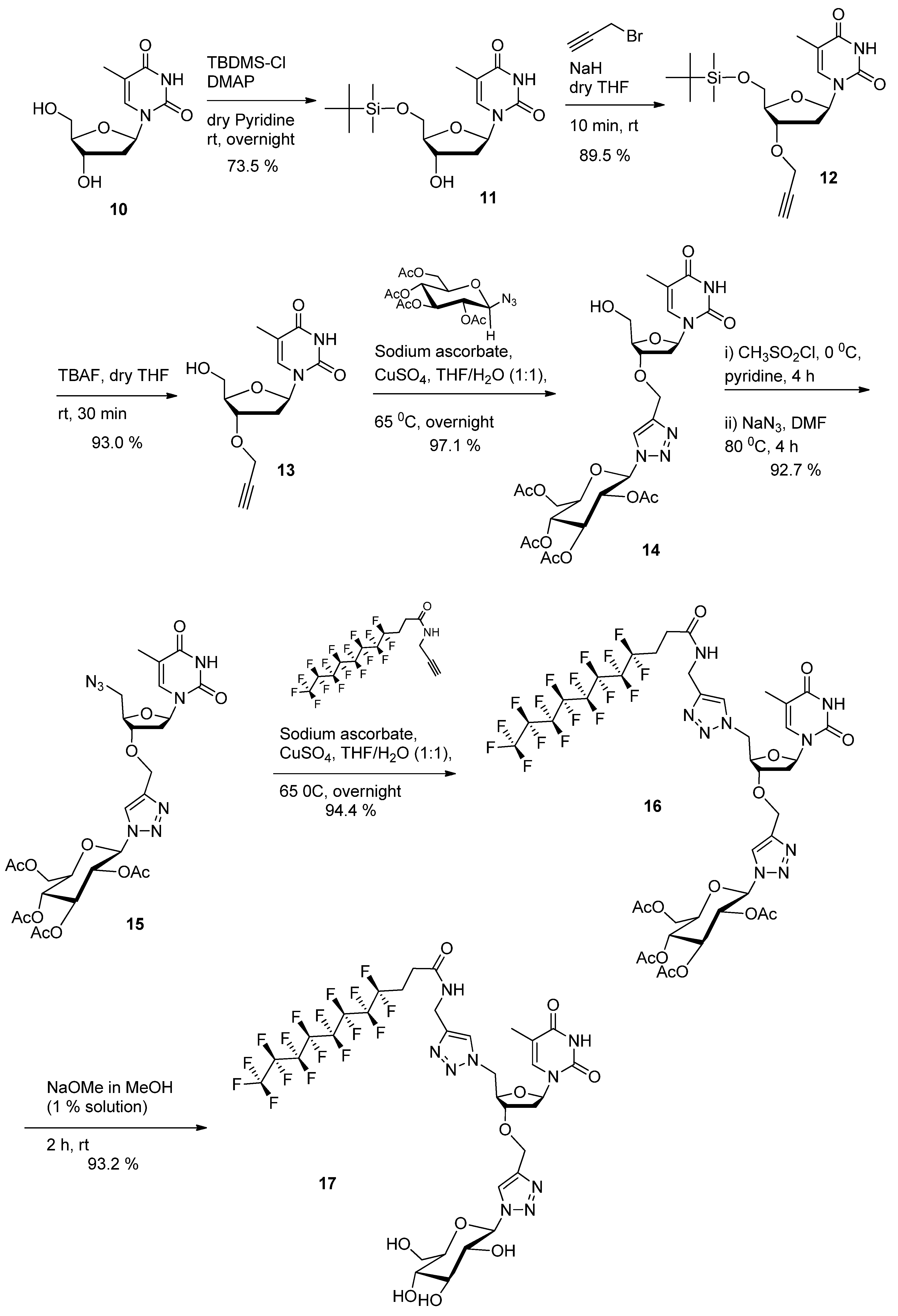
1-((2R,4S,5R)-5-((tert-Butyldimethylsilyloxy)methyl)-4-hydroxytetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (
11). To a solution of thymidine
10 (5 g, 20.6 mmol, 1.0 equiv) in pyridine (125 mL) was added DMAP (0.126 g, 1.03 mmol, 0.05 equiv.) and TBDMS-Cl (3.48 g, 23.1 mmol, 1.1 equiv.) sequentially. The reaction mixture was stirred overnight at room temperature. After removal of the solvent under reduced pressure, the crude reaction mixture was dissolved in DCM and successively washed with water, aqueous NaHCO
3 solution (5%) and brine. The organic layer was dried over Na
2SO
4 and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel (ethyl acetate-hexane-TEA (79:20:1)) to give pure
11 as a white solid. Yield: 5.4 g (73.5%). R
f: 0.45 (ethyl acetate-hexane-TEA 79:20:1). The data agreed with the literature values [
41,
42].
1H-NMR (300 MHz, CDCl
3):
δ in ppm 0.1 (s, 6H, -Si(CH
3)
2), 0.91 (s, 9H, -Si-C(CH
3)
2), 1.90 (s, 3H, CH
3(base)), 2.04-2.12 (m, 1H, H2'(sugar)), 2.35-2.42 (m, 1H, H2ʺ(sugar)), 3.12 (bs, 1H,-OH(sugar)), 3.80–3.92 (m, 2H, 2H5'(sugar)), 4.02–4.11 (m, 1H, H3'(sugar)), 4.44 (bs, 1H, H4'(sugar)), 6.37–6.42 (m, 1H, 1H'(sugar)), 7.53 (s, 1H, H-6(base)), 9.39 (s, 1H, -NH(base)).
1-((2R,4S,5R)-5-((tert-Butyldimethylsilyloxy)methyl)-4-(prop-2-ynyloxy)tetrahydrofuran-2-yl)-5-methylpyrimidine- 2,4(1H,3H)-dione (
12). Anhydrous THF (100 mL) was added under nitrogen to TBDMS protected thymidine
11 (5 g, 14.03 mmol, 1 equiv.). The mixture was cooled at 0 °C and NaH (1.4 g (60% in oil), 35.06 mmol, 2.5 equiv.) was added by small portion. Propargyl bromide (4.69 g, 3.4 mL (80 wt.% solution in toluene), 31.55 mmol, 2.25 equiv) was then added at room temperature. The reaction mixture was maintained at 50 °C under stirring for 24 h. The reaction was stopped by addition of methanol (20 mL). The solvent was removed under reduced pressure. The residual mixture was dissolved in dichloromethane and successively washed by water and brine. The organic layer then dried over Na
2SO
4 and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel (ethyl acetate-hexane-TEA 39:60:1 to 49:50:1). Product
12 was isolated as a colourless oil. Yield: 4.95 g (89.5%). R
f: 0.44 (ethyl acetate-hexane-TEA (49:50:1)). The data agreed with the literature values [
39].
1H-NMR (300 MHz, CDCl
3):
δ in ppm 0.09 (s, 6H, -Si(CH
3)
2), 0.89 (s, 9H, -Si-C(CH
3)
2), 1.88 (s, 3H, CH
3(base)), 1.92–2.01 (m, 1H, H2'(sugar)), 2.40–2.47 (m, 2H, H2ʺ(sugar), -C≡C
H), 3.75–3.90 (m, 2H, 2H5'), 4.07–4.24 (m, 3H, H3'(sugar), O-C
H2-C≡), 4.31–4.33 (m, 1H, H4'(sugar)), 6.25–6.29 (m, 1H, 1H'(sugar)), 7.48 (s, 1H, H-6(base)), 9.76 (s, 1H, -NH(base)).
13C-NMR (75 MHz, CDCl
3):
δ in ppm −5.56 (-Si-
CH
3), −5.5 (-Si-
CH
3), 12.4 (CH
3(base)), 18.2 (-
C-(CH
3)
3), 25.8 (-C-(
CH
3)
3), 37.5 (C2′), 56.2 (-O-
CH
2-C≡), 63.4 (C5'), 75.0 (≡
CH), 78.4 (C3'), 79.0 (-C≡), 84.7 (C4'), 84.8 (C1'), 110.6 (C5-thymine base), 135.2 (C6-thymine base), 150.5 (C=O(2) thymine base), 164.1 (C=O(4) thymine base). High-resolution ESI-MS
m/z calcd. for C
19H
30N
2O
5NaSi 417.1816 [M+Na]
+, found. 417.1834.
1-((2R,4S,5R)-5-(Hydroxymethyl)-4-(prop-2-ynyloxy)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (13). To a mixture of compound 12 (4.85 g, 12.3 mmol, 1.0 equiv.) in THF (40 mL) was added 1M solution of TBAF (14.7 mL, 14.75 mmol, 1.2 equiv.) in THF and the resulting mixture was stirred at room temperature for 1h. After removal of the solvent under reduced pressure, the crude deprotected product was applied directly to a silica gel chromatographic column (methanol-ethyl acetate 0:100 to 5:95). Product 13 was isolated as a white solid. Yield: 3.2 g (93.0%). Rf: 0.55 (ethyl acetate). 1H-NMR (300 MHz, DMSO-d6): δ in ppm 1.77 (s, 3H, CH3(base)), 2.04–2.33 (m, 2H, H2'(sugar)), 3.47 (s, -C≡CH), 3.58 (bs 2H, 2H5'), 3.93 (bs, 1H, H3'(sugar)), 4.21 (bs, 3H, - OH, O-CH2-C≡), 5.15(s, 1H, H4'(sugar)), 6.10 (bs, 1H, 1H'(sugar)), 7.70 (s, 1H, H-6(base)), 11.34 (s, 1H, -NH(base)). 13C-NMR (75 MHz, DMSO-d6): δ in ppm 12.4 (CH3(base)), 36.1 (C2′), 55.9 (-O-CH2-C≡), 61.6 (C5'), 77.4 (≡CH), 78.8 (C3'), 80.3 (-C≡), 83.9 (C4'), 84.5 (C1'), 109.7 (C5-thymine base), 136.1 (C6-thymine base), 150.6 (C=O(2) thymine base), 163.9 (C=O(4) thymine base). High-resolution ESI-MS m/z calcd. for C13H16N2O5Na 303.0951 [M+Na]+, found. 303.0945.
(2R,3R,4S,5R,6R)-2-(Acetoxymethyl)-6-(4-(((2R,3S,5R)-2-(hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yloxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (14). 1-Azido-2,3,4,6-tetra-O-acetyl-β-d-glucopyranoside (2.5 g, 6.69 mmol, 1 equiv.), sodium ascorbate (265.5 mg, 1.34 mmol, 0.2 equiv.) and copper sulfate (106.8 mg, 0.66 mmol, 0.1 equiv.) were added to compound 13 (1.87 g, 6.69 mmol, 1 equiv.) in water/THF (1:1) mixture (100 mL). The reaction mixture was maintained at 65 °C under stirring for 10 h. The mixture was cooled down to room temperature. After removal of the solvent under reduced pressure, the crude product was applied directly to a silica gel chromatographic column (methanol-ethyl acetate 0:100 to 5:95). Product 14 was isolated as a white solid (foam). Yield: 4.24 g (97.1%). Rf: 0.25 (ethyl acetate). 1H-NMR (300 MHz, CDCl3): δ in ppm 1.88 (s, 3H, CH3(base)), 1.89–2.08 (m, 12H, 4CH3-C=O(glucose)), 2.24–2.33 (m, 1H, H2'(sugar)), 2.37–2.45 (m, 1H, H2ʺ(sugar), 3.31 (bs, 1H, -OH), 3.73–4.17 (m, 5H, 2H5'(sugar), H3'(sugar), O-CH2-triazole), 4.25–4.34 (m, 2H, H4'(sugar), -O-CH(glucose-C5)), 4.68 (q, 2H, -CH2-OAc(glucose), J = 12.7 Hz), 5.25 (t, 1H, -CH(glucose-C4), J = 9.5 Hz), 5.34-5.48 (m, 2H, 2-CH(glucose-C2, C3)), 5.88 (d, 1H, -N-CH-O-(glucose), J = 8.85 Hz), 6.15 (t, 1H, 1H'(sugar), J = 6.8 Hz), 7.46 (s, 1H, triazole), 7.86 (s, 1H, H-6(base)), 9.10 (s, 1H, -NH(base)). 13C-NMR (75 MHz, CDCl3): δ in ppm 12.4 (CH3(base)), 20.1–20.6 (4 -CH3 (acetyl)), 36.1 (C2′), 61.4 (-O-CH2-triazole), 62.0 (C5'), 62.2 (-CH2-OAc), 67.5 (-CH (glucose-C3)), 70.5 (-CH (glucose-C2)), 72.3 (-CH (glucose-C4)), 74.9 (-CH (glucose-C5)), 78.0 (-CH (glucose-C1)), 84.9 (C3'), 85.5 (C4'), 86.0 (C1'), 110.8 (C5-thymine base), 121.4 (-CH triazole), 136.7 (C6-thymine base), 145.2 (=C- triazole), 150.5 (C=O(2) thymine base), 164.1 (C=O(4) thymine base), 169.3–169.9 (3 C=O(acetyl at C2, C3, C4)), 170.5 (-CH2-C=O(acetyl at C6)). High-resolution ESI-MS m/z calcd. for C27H35N5O14Na 676.2072 [M+Na]+, found. 676.2089.
(2R,3R,4S,5R,6R)-2-(Acetoxymethyl)-6-(4-(((2R,3S,5R)-2-(azidomethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yloxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (15). Anhydrous pyridine (80 mL) was added to click product 14 (4 g,6.12 mmol, 1 equiv.). The mixture was cooled to 0 °C and methanesulfonyl chloride (0.92 g, 0.62 mL, 7.95 mmol, 1.3 equiv.) was added dropwise. The mixture was stirred at room temperature for 4 h. The solvent was evaporated under reduced pressure and the residual compound was used directly in the following step without further purification. To the above crude product, DMF (80 mL) and sodium azide (1.99 g, 30.6 mmol, 5 equiv) were added. The reaction mixture was maintained at 80 °C under stirring for 4 h. DMF was removed under reduced pressure. The residual solid was dissolved in ethyl acetate and washed successively twice with aqueous 5% NaHCO3 solution and once with brine. The organic phase was dried on Na2SO4 and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel (ethyl acetate). Product 15 was isolated as a white solid (foam). Yield: 3.85 g (92.7%). Rf: 0.45 (ethyl acetate). 1H NMR (300 MHz, CDCl3): δ in ppm 1.88 (s, 3H, CH3(base)), 1.92–2.08 (m, 12H, 4CH3-C=O(glucose)), 2.11–2.15 (m, 1H, H2'(sugar)), 2.38–2.46 (m, 1H, H2ʺ(sugar), 3.54–3.73 (m, 2H, 2H5'(sugar), 3.99–4.19 (m, 4H, H3'(sugar), O-CH2-triazol, -O-CH(glucose-C5)), 4.28–4.34 (m, 1H, H4'(sugar), 4.66 (q, 2H, -CH2-OAc(glucose), J = 12.2 Hz), 5.24 (t, 1H, -CH(glucose-C4), J = 9.5 Hz), 5.35–5.48 (m, 2H, 2-CH(glucose-C2, C3)), 5.88 (d, 1H, -N-CH-O-(glucose), J = 8.86 Hz), 6.23–6.28 (m, 1H, 1H'(sugar)), 7.32 (s, 1H, triazole), 7.83 (s, 1H, H-6(base)), 8.95 (s, 1H, -NH(base)). 13C-NMR (75 MHz, CDCl3): δ in ppm 12.5 (CH3(base)), 20.0-20.6 (4 -CH3 (acetyl)), 36.9 (C2′), 52.3 (C5'), 61.4 (-O-CH2-triazole), 62.5 (-CH2-OAc), 67.5 (-CH (glucose-C3)), 70.4 (-CH (glucose-C2)), 72.2 (-CH (glucose-C4)), 74.9 (-CH (glucose-C5)), 78.5 (-CH (glucose-C1)), 82.3 (C3'), 84.8 (C4'), 85.6 (C1'), 111.3 (C5-thymine base), 121.2 (-CH triazole), 135.3 (C6-thymine base), 145.0 (=C- triazole), 150.3 (C=O(2) thymine base), 163.8 (C=O(4) thymine base), 168.9–169.8 (3 C=O(acetyl at C2, C3, C4)), 170.4 (-CH2-C=O(acetyl at C6)). High-resolution ESI-MS m/z calcd. for C27H34N8O13Na 701.2137 [M+Na]+, found. 701.2139.
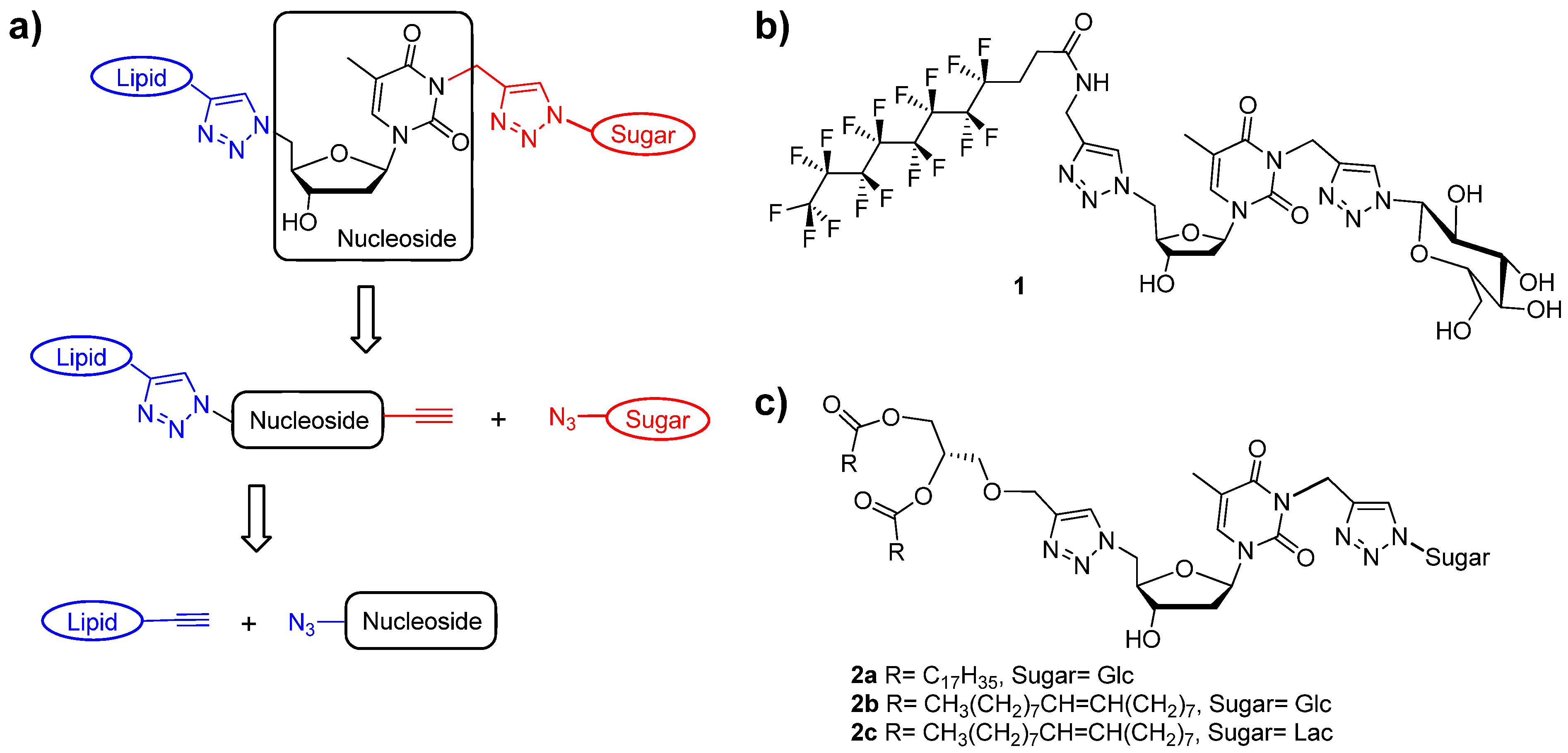
(2R,3R,4S,5R,6R)-2-(Acetoxymethyl)-6-(4-(((2R,3S,5R)-2-((4-((4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecanamido)methyl)-1H-1,2,3-triazol-1-yl)methyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yloxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydro-2H-pyran-3,4,5-triyl triacetate (
16).
N-propargyl-1
H,1
H,2
H,2
H-perfluoroundecanoyl amide [
29] (480 mg, 0.91 mmol, 1 equiv.), sodium ascorbate (36 mg, 0.18 mmol, 0.2 equiv.) and copper sulfate (14.5 mg, 0.091 mmol, 0.1 equiv.) were added to azido derivative
15 (616 mg, 0.91 mmol, 1 equiv.) in water/THF (1:1) mixture (50 mL). The reaction mixture was maintained at 65 °C under stirring for 10 h. The mixture was cooled down to room temperature. After removal of the solvent under reduced pressure, the crude product applied directly to a silica gel chromatographic column (methanol-ethyl acetate 0:100 to 5:95). Product
16 was isolated as a white sticky foam. Yield: 1.03 g (94.4%). R
f: 0.2 (ethyl acetate).
1H-NMR (300 MHz, CDCl
3):
δ in ppm 1.69 (s, 3H, CH
3(base)), 1.80, 2.00 and 2.11 (s, 12H, 4CH
3-C=O(glucose)), 2.13–2.73 (m, 6H, 2H2'(sugar), CF
2-C
H2-C
H2-C=O), 4.07–4.45 (m, 6H, H5≡(sugar), -C
H2-OAc(glucose-C6), -O-C
H(glucose-C5), triazole-CH'
H≡-NH-C=O, H3'(sugar)), 4.61–4.70 (m, 3H, O-C
H2-triazole, H5'(sugar)), 4.80–4.89 (m, 2H, H4'(sugar), triazole-C
H'Hʺ-NH-C=O), 5.40–5.51 (m, 2H, 2-CH(glucose-C3, C4)), 5.69–5.78 (m, 1H, -CH(glucose-C2)), 5.82–5.90 (m, 1H-N-C
H-O-(glucose-C1)), 6.22–6.27 (m, 1H, 1H'(sugar)), 6.54 (s, 1H, triazole-between nucleoside and glucose), 7.35 (bs, 1H, -N
H-C=O-), 7.64 (s, 1H, triazole-between F-chain and nucleoside) 8.35 (s, 1H, H-6(base)), 9.27 (s, 1H, -NH(base)).
13C-NMR (75 MHz, CDCl
3):
δ in ppm 11.9 (CH
3(base)), 20.0–20.6 (4 -CH
3 (acetyl)), 26.5 (-CH
2-CH
2-chain) 34.2 (triazole-
CH
2-NH) 35.2 (C2′), 49.8 (C5'), 61.5 (-O-
CH
2-triazole), 63.2 (-
CH
2-OAc), 67.8 (-CH (glucose-C3)), 70.8 (-CH (glucose-C2)), 72.2 (-CH (glucose-C4)), 74.2 (-CH (glucose-C5)), 75.1 (-CH (glucose-C1)), 81.0 (C3'), 84.6 (C4'), 85.9 (C1'), 111.2 (C5-thymine base), 121.9 (-CH triazole-between F-chain and nucleoside), 124.3 (-CH triazole-between nucleoside and glucose) 136.3 (C6-thymine base), 144.4 (=C- triazole-between nucleoside and glucose), 144.5 (=C- triazole-between F-chain and nucleoside), 150.3 (C=O(2) thymine base), 163.7 (C=O(4) thymine base), 169.1–170.2 (3 C=O(acetyl at C2, C3, C4)), 170.3 (-CH
2-
C=O(acetyl at C6)), 170.6 (-NH-
C=O). High-resolution FD-MS
m/z calcd. for C
41H
42F
17N
9O
14 1207.2580, found. 1207.2582.
4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-Heptadecafluoro-N-((1-(((2R,3S,5R)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-((1-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)-1H-1,2,3-triazol-4-yl)methoxy)tetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl) undecanamide (17). To a solution of double click product 16 (950 mg, 0.79 mmol, 1.0 equiv.) in methanol (40 mL) was added sodium methoxide powder (850 mg, 15.74 mmol, 20 equiv.) and the resulting mixture was stirred at room temperature for 2 h. After removal of the solvent under reduced pressure, the crude deprotected product was applied directly to a silica gel chromatographic column (methanol-dichloromethane 1:4). Product 17 was isolated as a white solid. Yield: 762 m g (93.2%). Rf: 0.35 (methanol-dichloromethane 1:4). 1H-NMR (300 MHz, DMSO-d6): δ in ppm 1.80 (s, 3H, CH3(base)), 2.20–2.33 (m, 2H, 2H2'(sugar)), 2.43–2.56 (m, 4H, CF2-CH2-CH2-C=O), 3.15–3.26 (m, 1H, -O-CH- (glucose-C5)), 3.40–3.45 (m, 3H, -CH2-OH(glucose-C6), -CH(glucose-C4), 3.63–3.80 (m, 2H, -CH(glucose-C3), H3′), 4.20–4.40 (m, 4H, -OH (glucose-C4), H5′, -CH2-NH-C=O,), 4.58 (s, 2H, -O-CH2-triazole), 4.63–4.76 (m, 3H, H4′, H5″, -CH(glucose-C2)), 5.19 (d, 1H, -OH(glucose (C6), J = 5.5 Hz), 5.33 (d, 1H, -OH (glucose-C3), J = 4.7 Hz), 5.44 (d, 1H, -OH (glucose-C2), J = 6.0 Hz), 5.53 (d, 1H, -N-CH-O (glucose-C1), J = 9.4 Hz), 6.12 (t, 1H, 1H'(sugar), J = 7.3 Hz), 7.45 (s, 1H, triazole-between nucleoside and glucose), 7.98 (s, 1H, triazole-between F-chain and nucleoside) 8.33 (s, 1H, H-6(base)), 8.57 (t, 1H, -NH-C=O-, J = 5.46 Hz), 11.37 (s, 1H, -NH(base)). 13C-NMR (75 MHz, DMSO-d6): δ in ppm 12.0 (CH3(base)), 25.7–25.8 (-CH2-CH2-chain) 34.4 (triazole-CH2-NH) 35.1 (C2′), 51.4 (C5'), 60.8 (-O-CH2-triazole), 62.1 (-CH2-OH (glucose-C6)), 69.6 (-CH(glucose-C2)), 72.1 (-CH(glucose-C4)), 77.0 (-CH(glucose-C3)), 79.1 (C3′), 80.0 (-CH(glucose-C5)), 81.6 (-CH(glucose-C1)), 84.5 (C4′), 87.5 (C1′), 109.9 (C5-thymine base), 123.3 (-CH triazole between thymidine and alkyl chain), 123.6 (-CH triazole between glucose and thymidine), 136.1 (C6-thymine base), 143.5 (=C- triazole triazole between glucose and thymidine), 144.7 (=C- triazole between thymidine and alkyl chain), 150.4 (C=O(2) thymine base), 163.7 (C=O(4) thymine base), 169.3 (-NH-C=O). MS m/z calcd. for C33H34F17N9O10Na 1062.6, found. 1062.3.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
