3.1. General
Melting points were determined in open capillary tubes. Unless otherwise stated, NMR spectra were recorded at 25 °C in CDCl
3 solution:
1H-NMR (400 MHz),
13C-NMR (100.6 MHz),
19F (376.29 MHz). All chemical shifts (δ
H, δ
C and δ
F) are reported in parts per million (ppm) related to internal standards (CHCl
3 at δ
H = 7.26 ppm, δ
C = 77.0 ppm and CF
3COOH at δ
F −76.55 ppm). Assignments given for the
1H and
13C-NMR spectra are based on DEPT sequences,
1H/
1H COSY,
1H/
13C HETCOR (gHSQC and gHMBC sequences for one bond and long range correlations, respectively). In the case of triflate
3, previous assignment for the adduct of methyl 1-benzylcyclopenta-2,4-dienecarboxylate and triflate
1 were taken into account [
5]. Coupling constants
J are given in Hertz (Hz). Mass spectra were recorded on an LC/MSD-TOF instrunent (2006, Agilent Technologies), using electrospray (ESI-MS, positive mode, capillary: 3.5 kV, fragmentor: 215 V). Unless otherwise stated, IR spectra were recorded using the attenuated total reflection (ATR) technique and the absorption values are given as wavenumbers (cm
–1). Elemental analyses were done at the Microanalysis Service of the Institut de Química Avançada de Catalunya (IQAC, CSIC, Barcelona, Spain). Column chromatography was performed on silica gel 60 A C.C. (35–70 mesh). For the thin layer chromatography (TLC), aluminum-backed sheets with silica gel 60 F254 were used and spots were visualized with UV light and/or 1% aqueous KMnO
4.
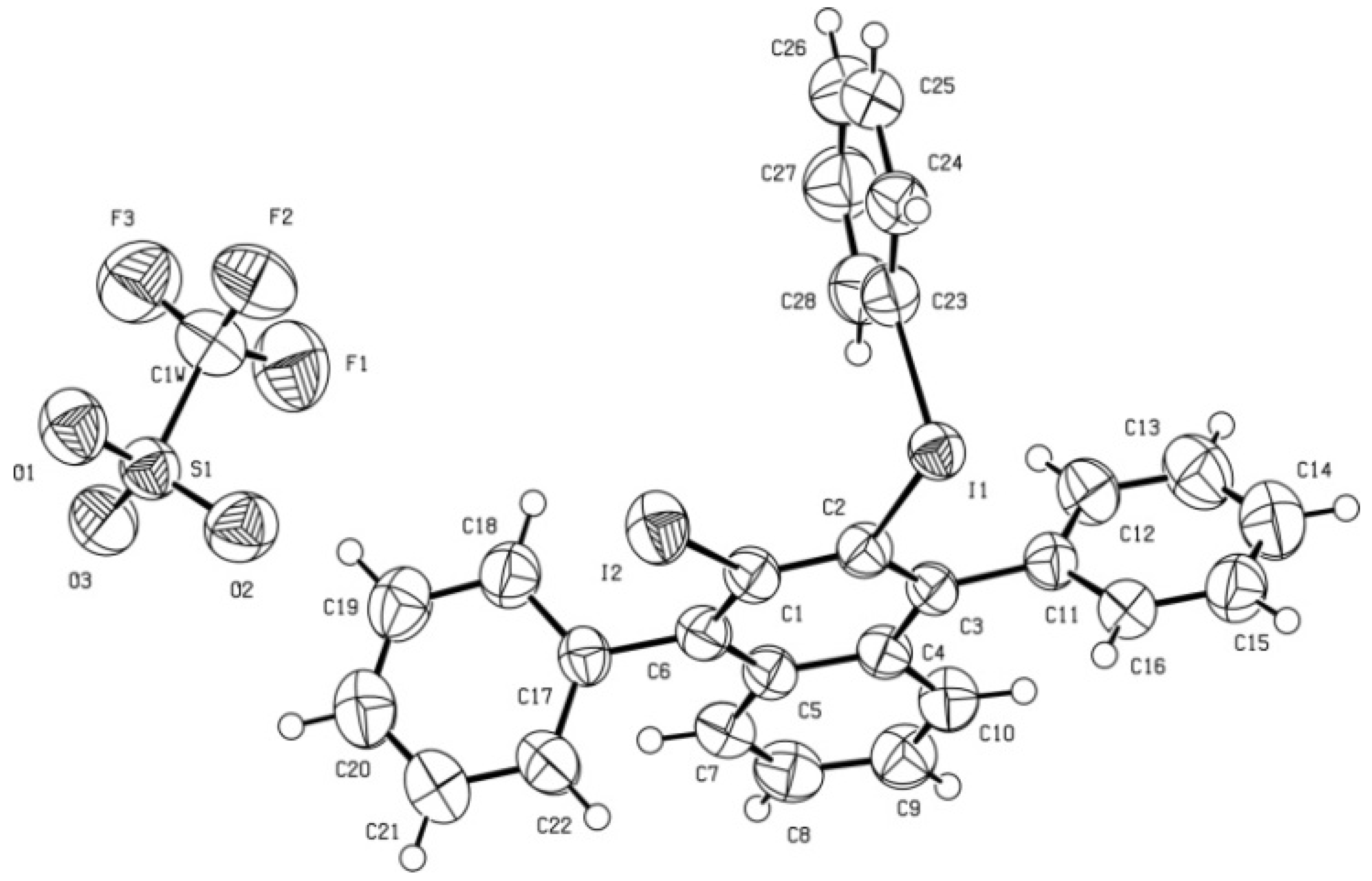
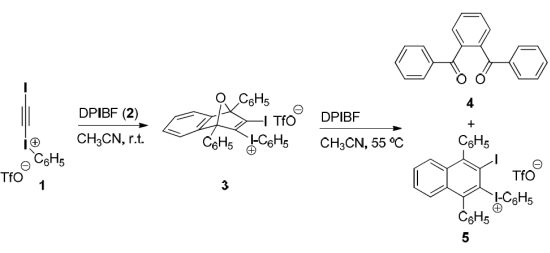
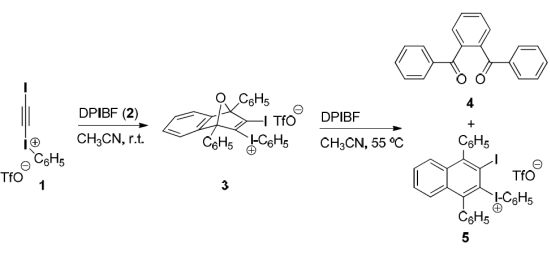
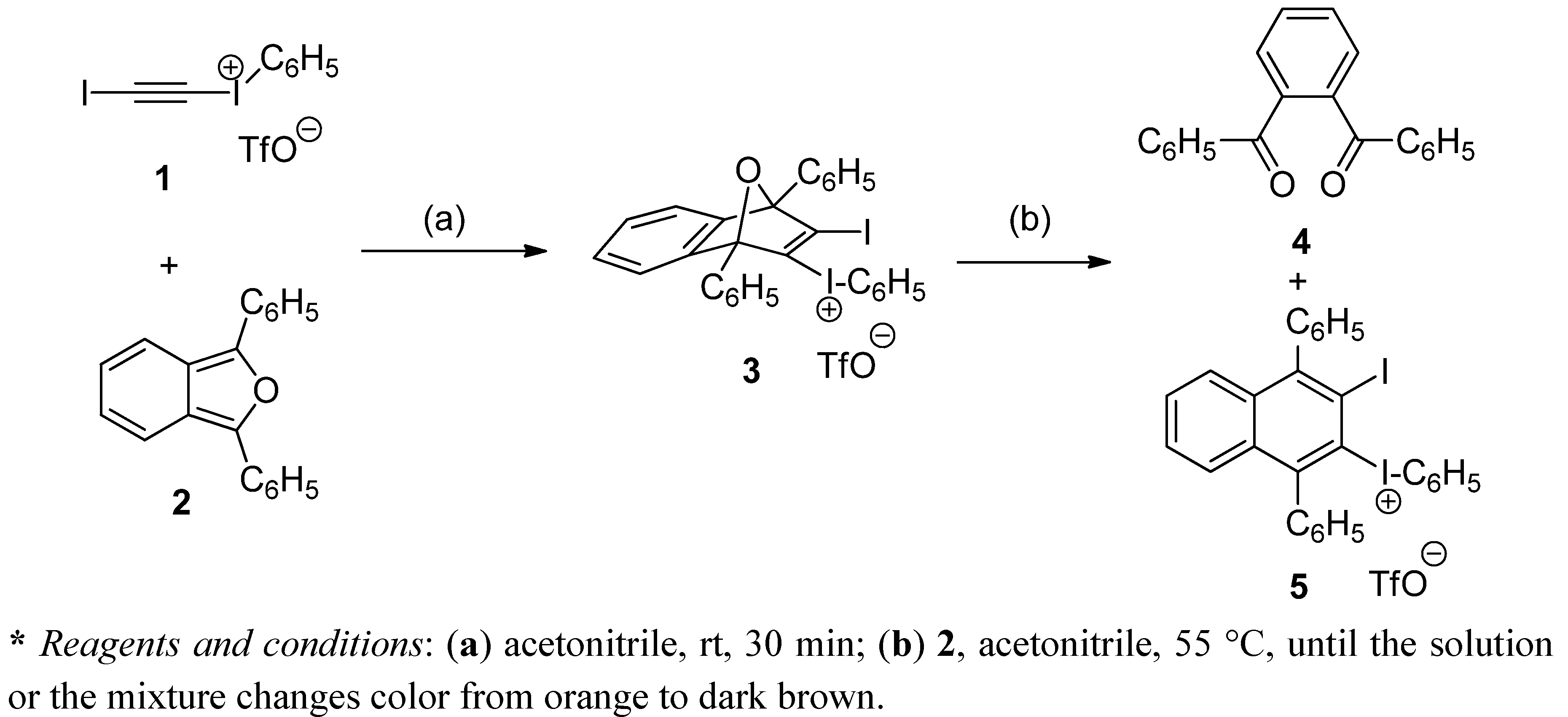
(3-Iodo-1,4-diphenyl-1,4-dihydro-1,4-epoxynaphthalen-2-yl)(phenyl)iodonium triflate (3). To a magnetically stirred suspension of DPIBF (2, 150 mg, 0.55 mmol) in anhydrous acetonitrile (2.5 mL), a solution of triflate 1 (233 mg, 0.46 mmol) in anhydrous acetonitrile (1 mL) was added dropwise under an Ar atmosphere and the mixture was vigorously stirred at room temperature for 20 h. The orange solution was concentrated in vacuo at room temperature and the orange solid residue (418 mg) was treated with CH2Cl2/Et2O (1:3, 2 mL). The solid triflate 3 (356 mg, quantitative yield) was isolated as a light yellow solid, m.p. 80–83 °C (dec.) by decantation and washing with Et2O (3 × 0.5 mL) and CH2Cl2 (3 × 0.5 mL). On drying in vacuo at room temperature this salt became black. The salt was stable for weeks at 4 °C, but decomposed after standing at room temperature for one day. The analytical sample was not fully dried and contained traces of Et2O and CH2Cl2. IR (ATR) ν: 3061, 1548, 1494, 1472, 1446, 1287, 1228, 1220, 1180, 1166, 1156, 1021, 993, 916, 905, 851, 835, 757, 737, 703, 675, 653, 633, 591 cm−1. 1H-NMR δ: 7.10 (dt, J = 2.0 Hz, J′ = 9.0 Hz, 1H, 7-H), 7.15 (dt, J = 2.0 Hz, J′ = 9.0 Hz, 1H, 6-H), 7.19 (tm, J = 9.5 Hz, 2H, Ar-HmetaAr-I+), 7.30 (ddm, J = 8.8 Hz, J′ = 1.5 Hz, 2H, Ar-HorthoArI+), 7.38 (dd, J = 9.0 Hz, J′ = 1.5 Hz, 1H, 8-H), 7.48 (tm, J = 9.0 Hz, 1H, Ar-HparaArI+), 7.50–7.60 (complex signal, 7H, 5-H, C1-Ar-Hmeta, C4-Ar-Hmeta, C1-Ar-Hpara and C4-Ar-Hpara), 7.75 (dm, J = 8.0 Hz, 2H, C1-Ar-Hortho), 7.85 (dm, J = 7.5 Hz, 2H, C4-Ar-Hortho). 13C-NMR δ: 96.3 (C, C1), 97.6 (C, C4), 115.1 (C, C2-I+-Ar-Cipso), 120.0 (C, q, J = 320 Hz, CF3SO3), 121.8 (CH, C8), 122.4 (CH, C5), 126.4 (CH, C6), 126.5 (CH, C1-Ar-Cortho), 126.9 (CH, C7), 127.4 (CH, C4-Ar-Cortho and C, C1-Ar-Cipso), 128.5 (CH) and 129.3 (CH) (C1-Ar-Cmeta and C4-Ar-Cmeta), 129.6 (CH, C4-Ar-Cpara), 129.8 (CH, C1-Ar-Cpara), 132.0 (CH, C2-I+-Ar-Cmeta), 132.3 (CH, C2-I+-Ar-Cpara), 132.7 (C, C4-Ar-Cipso), 134.3 (CH, C2-I+-Ar-Cortho), 140.6 (C, C3), 142.2 (C, C2), 145.8 (C, C4a), 146.4 (C, C8a). 19F-NMR δ: −78.2 (CF3SO3). Accurate mass measurement: Calcd for C28H19I2O [M−CF3SO3]+: 624.9520. Found: 624.9515.
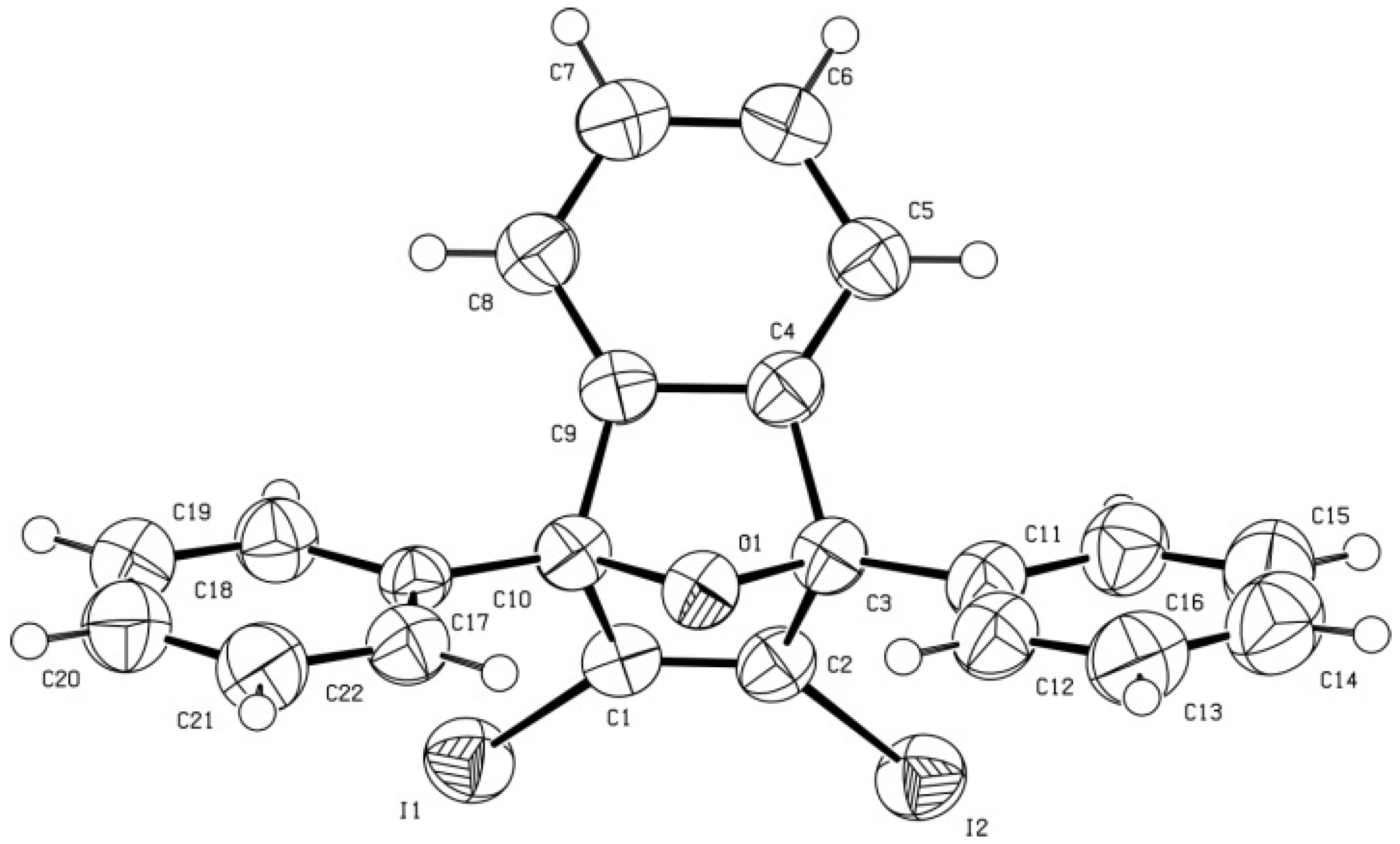
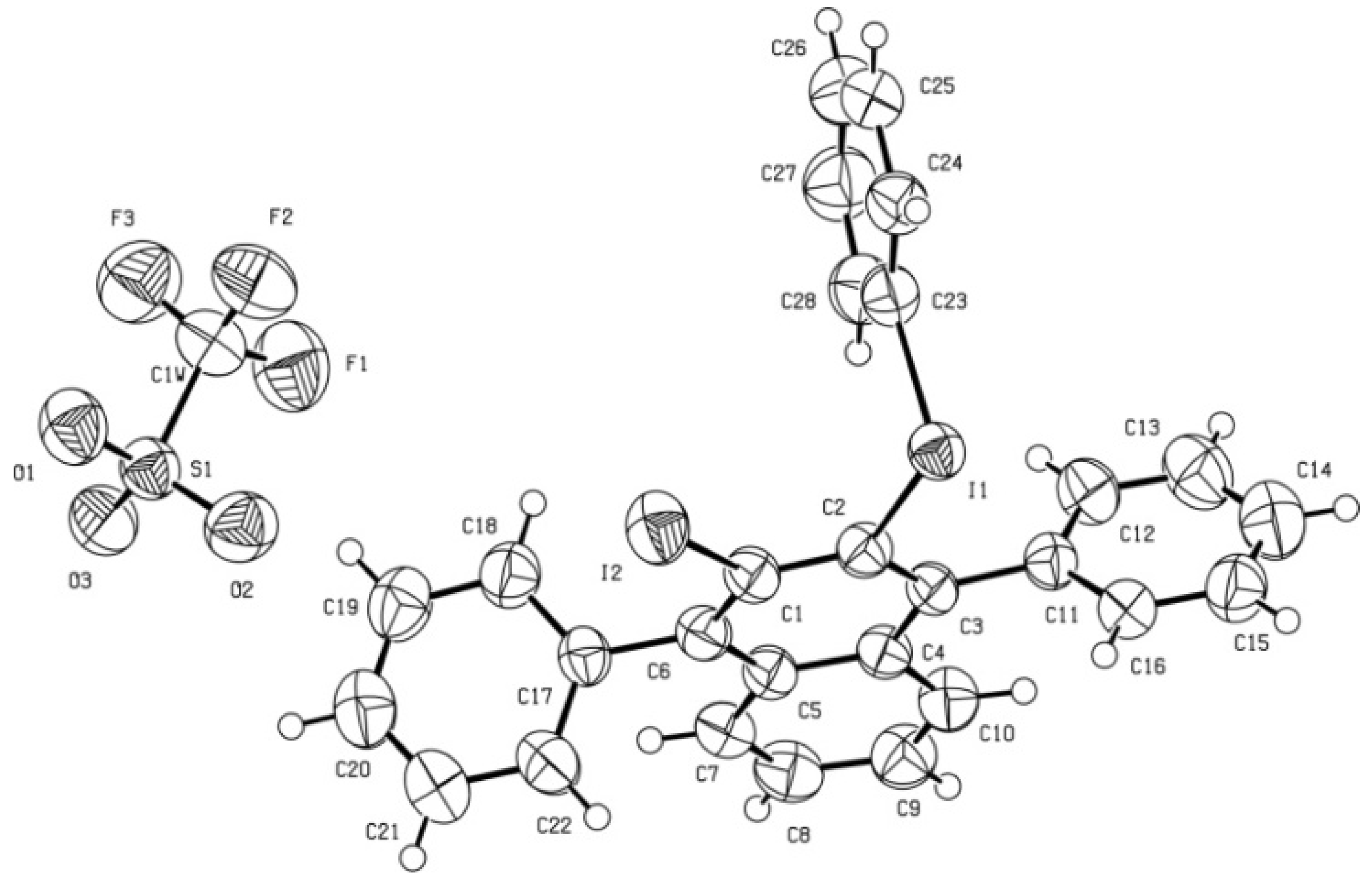
(3-Iodo-1,4-diphenylnaphthalen-2-yl)(phenyl)iodonium triflate (5). To a magnetically stirred suspension of DPIBF (2, 617 mg, 2.28 mmol) in anhydrous acetonitrile (5 mL), a solution of triflate 1 (575 mg, 1.14 mmol) in anhydrous acetonitrile (3 mL) was added dropwise under an Ar atmosphere and the mixture was vigorously stirred at room temperature for 20 h. The orange solution was concentrated in vacuo at 55 °C. In a certain moment the residue became dark brown. Crystallization of the above residue (1.31 g) from CH2Cl2/Et2O (3:10, 6.5 mL) gave a solid (1.02 g) mixture of triflate 5 and 1,2-phenylenebis(phenylmethanone) (4). After two crystallizations from CH2Cl2/toluene 3:10 (5.2 mL), triflate 5 (477 mg, 54% yield) was obtained as a beige solid, mp 207–208 °C (dec). IR (ATR) ν: 3058, 1490, 1470, 1443, 1282, 1266, 1222, 1153, 1023, 990, 835, 766, 722, 698, 666, 633, 599 cm–1. 1H-NMR (500 MHz) δ: 7.24–7.28 (complex signal, 4H, C1-Ar-Hortho and C4-Ar-Hortho), 7.37–7.42 (complex signal, 2H, Ar-Hmeta Ar-I+), 7.42–7.47 (complex signal, 4H, 5-H, 6-H, 7-H, 8-H), 7.51–7.62 (complex signal, 7H, C1-Ar-Hmeta, C4-Ar-Hmeta, C1-Ar-Hpara, C4-Ar-Hpara, and C2-I+-Ar-Hpara), 7.73 (dd, J = 8.5 Hz, J′ =1.0 Hz, 2H, Ar-Hortho ArI+). 13C-NMR (125.8 MHz) δ: 105.4 (C, C3), 116.3 (C, C2-I+-Ar-Cipso), 120.3 (C, q, J = 320 Hz, CF3SO3), 128.26 (CH), 128.30 (CH), 128.8 (CH), 129.3 (CH), 130.0 (CH) and 130.1 (CH) (C5, C6, C7, C8, C1-Ar-Cpara and C4-Ar-Cpara), 128.9 (CH), 129.1 (CH), 129.2 (CH) and 129.9 (CH) (C1-Ar-Cortho, C4-Ar-Cortho, C1-Ar-Cmeta and C4-Ar-Cmeta), 129.7 (C, C2), 131.9 (CH, Ar-Cmeta Ar-I+), 132.4 (CH, C2-I+-Ar-Cpara), 132.5 (C) and 134.6 (C) (C4a and C8a), 134.4 (CH, C2-I+-Ar-Cortho), 141.7 (C, C1-Ar-Cipso), 145.2 (C, C4-Ar-Cipso), 149.3 (C) and 149.8 (C) (C1 and C4). 19F-NMR δ: −78.2 (CF3SO3). Accurate mass measurement: Calcd for C28H19I2 [M–CF3SO3]+: 608.9571. Found: 608.9567. Anal. Calcd for C29H19F3I2O3S·0.1CH2Cl2: C 45.58; H 2.52; I 33.10; F 7.43. Found: C 45.35; H 2.50; I 32.60; F 7.16.
X-Ray diffraction data for triflate
5. A prismatic crystal (0.1 × 0.1 × 0.2 mm) was selected and mounted on a MAR345 diffractometer with an image plate detector. Unit-cell parameters were determined from 177 reflections (3 < θ < 31°) and refined by least-squares method. Intensities were collected with graphite monochromatized MoKα radiation; 23,699 reflections were measured in the range 1.91 ≤ θ ≤ 32.36. 8,297 of which were non-equivalent by symmetry [Rint (on I) = 0.058]. Reflections (5,839) were assumed as observed by applying the condition I >2σ(I). Lorentz-polarization but no absorption corrections were made. The structure was solved by direct methods, using SHELXS computer program and refined by full-matrix least-squares method with SHELX-97 computer program [
8], using 23,699 reflections (very negative intensities were not assumed). The minimized function was Σw||Fo|
2 – |Fc|
2|
2, where w = [σ
2(I) + (0.0339P)
2 + 1.5431P]
−1, and P = (|Fo|
2+ 2|Fc|
2)/3, f, f', and f" were taken from International Tables of X-ray Crystallography [
9]. All H atoms were computed and refined, using a riding model, with an isotropic temperature factor equal to 1.2 times the equivalent temperature factor of the atom to which it is linked. The final R (on F) factor was 0.050, wR(on |F|
2) = 0.108 and goodness of fit = 1.138 for all observed reflections. Number of refined parameters was 343. Max. shift/esd = 0.00, mean shift/esd = 0.00. Max. and min. peaks in final difference synthesis were 0.970 and −0.582 eÅ
−3, respectively [
10].
2,3-Diiodo-1,4-diphenyl-1,4-dihydro-1,4-epoxynaphthalene (6). To a cold (−35 to −40 °C), magnetically stirred solution of NaI (23 mg, 0.154 mmol) and CuI (29 mg, 0.154 mmol) in anhydrous acetonitrile (2.3 mL) under an Ar atmosphere, triflate 3 (120 mg, 0.154 mmol) was added. The mixture was allowed to warm to room temperature and was stirred overnight at this temperature. The solvent and volatile products were eliminated under reduced pressure and the residue was extracted with CH2Cl2 (4 × 2 mL). The combined organic extracts were filtered through a PTFE filter and were concentrated under reduced pressure to give diodide 6 as a light yellow solid (74 mg, 87% yield). The analytical sample of 6 was obtained as a light yellow solid (58 mg, 69% yield of crystallized product) by crystallization from CH2Cl2/MeOH 5:2 (0.7 mL), m.p. 158–160 °C (dec.). IR ν 3053, 1599, 1544, 1492, 1449, 1342, 1327, 1304, 1132, 1025, 998, 943, 917, 905, 854, 761, 755, 738, 705, 698, 674, 652, 590 cm−1. 1H-NMR δ 7.10–7.12 [m, 4H, 6(7)-H], 7.46–7.50 [complex signal, 4H, 5(8)-H and phenyl-Hpara], 7.52–7.56 (m, 4H, phenyl-Hmeta), 7.89–7.92 (dm, J= 8.0 Hz, 4H, phenyl-Hortho). 13C-NMR δ 96.7 [(C, C1(4)], 121.2 [CH, C5(8)], 125.7 [CH, C6(7)], 126.4 [(C, C2(3)], 128.0 (CH, phenyl-Cortho), 128.2 (CH, phenyl-Cmeta), 128.9 (CH, phenyl-Cpara), 134.1 (C, phenyl-Cipso), 148.3 [C, C4a(8a)]. Accurate mass measurement: Calcd for C22H15I2O [M+H]+: 548.9207. Found: 548.9188; Calcd for [M+H−I]+: 422.0162. Found: 422.0158. Anal. Calcd for C22H14I2O: C 48.20, H 2.57, I 46.30. Found: C 48.25; H 2.64; I 46.22.
X-ray diffraction data for diiodide
6. A prismatic crystal (0.1 × 0.1 × 0.2 mm) was selected and mounted on a MAR345 diffractometer with an image plate detector. Unit-cell parameters were determined from 289 reflections (3 < θ < 31°) and refined by least-squares method. Intensities were collected with graphite monochromatized MoKα radiation; 10,713 reflections were measured in the range 1.92 ° θ ° 32.03. 4,303 of which were non-equivalent by symmetry (Rint (on I) = 0.045). 3,705 reflections were assumed as observed by applying the condition I >2σ(I). Lorentz-polarization but no absorption corrections were made. The structure was solved by direct methods, using SHELXS computer program and refined by full-matrix least-squares method with SHELX-97 computer program [
8], using 10,713 reflections (very negative intensities were not assumed). The minimized function was Σw||Fo|
2–|Fc|
2|
2, where w = [σ
2(I) + (0.0258P)
2 + 1.0315P]
–1, and P = (|Fo|
2 + 2|Fc|
2)/3, f, f', and f" were taken from International Tables of X-ray Crystallography [
9]. All H atoms were computed and refined, using a riding model, with an isotropic temperature factor equal to 1.2 times the equivalent temperature factor of the atom to which it is linked. The final R (on F) factor was 0.031, wR(on |F|
2) = 0.072 and goodness of fit = 1.171 for all observed reflections. Number of refined parameters was 226. Max. shift/esd = 0.00, mean shift/esd = 0.00. Max. and min. peaks in final difference synthesis were 0.395 and −0.620 eÅ
−3, respectively [
10].
2,3-Diiodo-1,4-diphenylnaphthalene (7). To a cold (−35 to −40 °C), magnetically stirred solution of NaI (92 mg, 0.61 mmol) and CuI (117 mg, 0.61 mmol) in anhydrous acetonitrile (10 mL) under an Ar atmosphere, triflate 5 (455 mg, 0.60 mmol) was added. The mixture was allowed to warm to room temperature and was stirred overnight at this temperature. The solution was separated from the inorganic salts by decantation. The salts were washed with CH2Cl2 (2 × 2 mL). The combined organic phases were concentrated under reduced pressure to give a solid residue (758 mg) that was subjected to column chromatography (silica gel, 30 g, hexane/EtOAc mixtures) to give the volatile iodobenzene (12 mg, 10%) on elution with hexane and product 7 as a white solid, m.p. 269–271 °C (328 mg, quantitative yield) on elution with hexane/EtOAc 99:1. IR ν 3058, 2921, 2844, 1490, 1480, 1439, 1364, 1270, 1260, 1107, 1073, 1027, 840, 764, 719, 697, 667, 599 cm−1. 1H-NMR δ 7.23–7.27 (dm, J = 6.8 Hz, 4H, Ar-Hortho phenyl), 7.28–7.32 [m, 2H, 6(7)-H], 7.30–7.35 [m, 2H, 5(8)-H], 7.48 (tm, J = 8.5 Hz, 2H, Ar-Hpara phenyl), 7.55 (m, 4H, Ar-Hmeta phenyl). 13C-NMR δ 112.7 [C, C2(3)], 126.9 [CH, C6(7)], 127.8 [CH, C5(8)], 128.0 (CH, Ar-Cpara phenyl), 128.6 (CH, Ar-Cmeta phenyl), 129.6 (CH, Ar-Cortho phenyl), 132.4 [C, C4a(8a)], 146.4 (C, Ar-Cipso phenyl), 146.7 [C, C1(4)]. Anal. Calcd for C22H14I2: C 49.65, H 2.65, I 47.69. Found: C 49.81; H 2.71; I 47.55.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
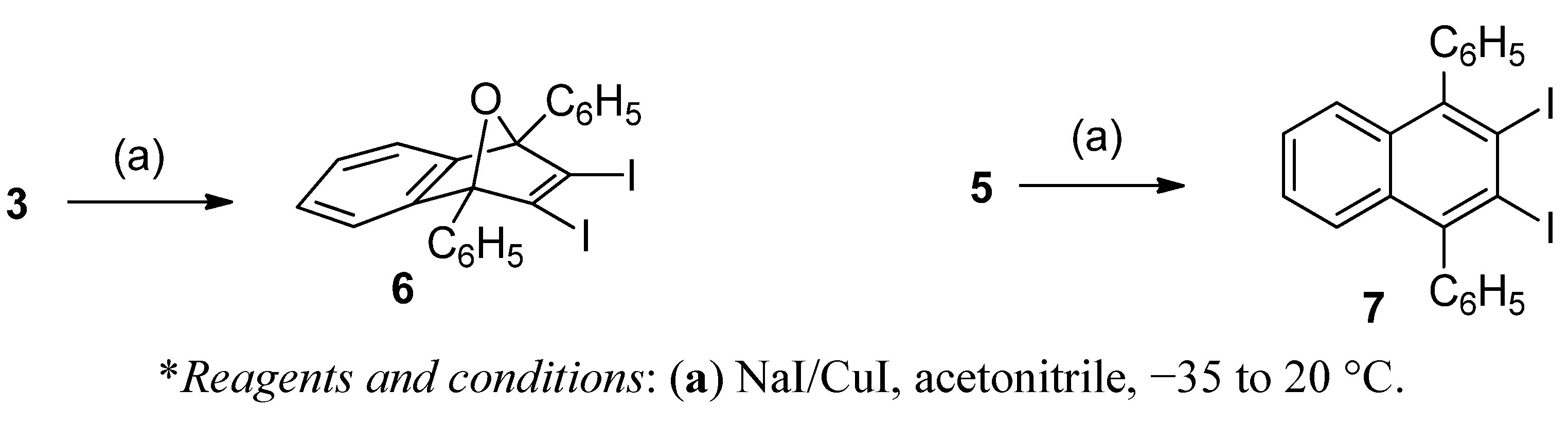{kind=link}
{kind=link}