A Convenient Route to 4-Carboxy-4-Anilidopiperidine Esters and Acids

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
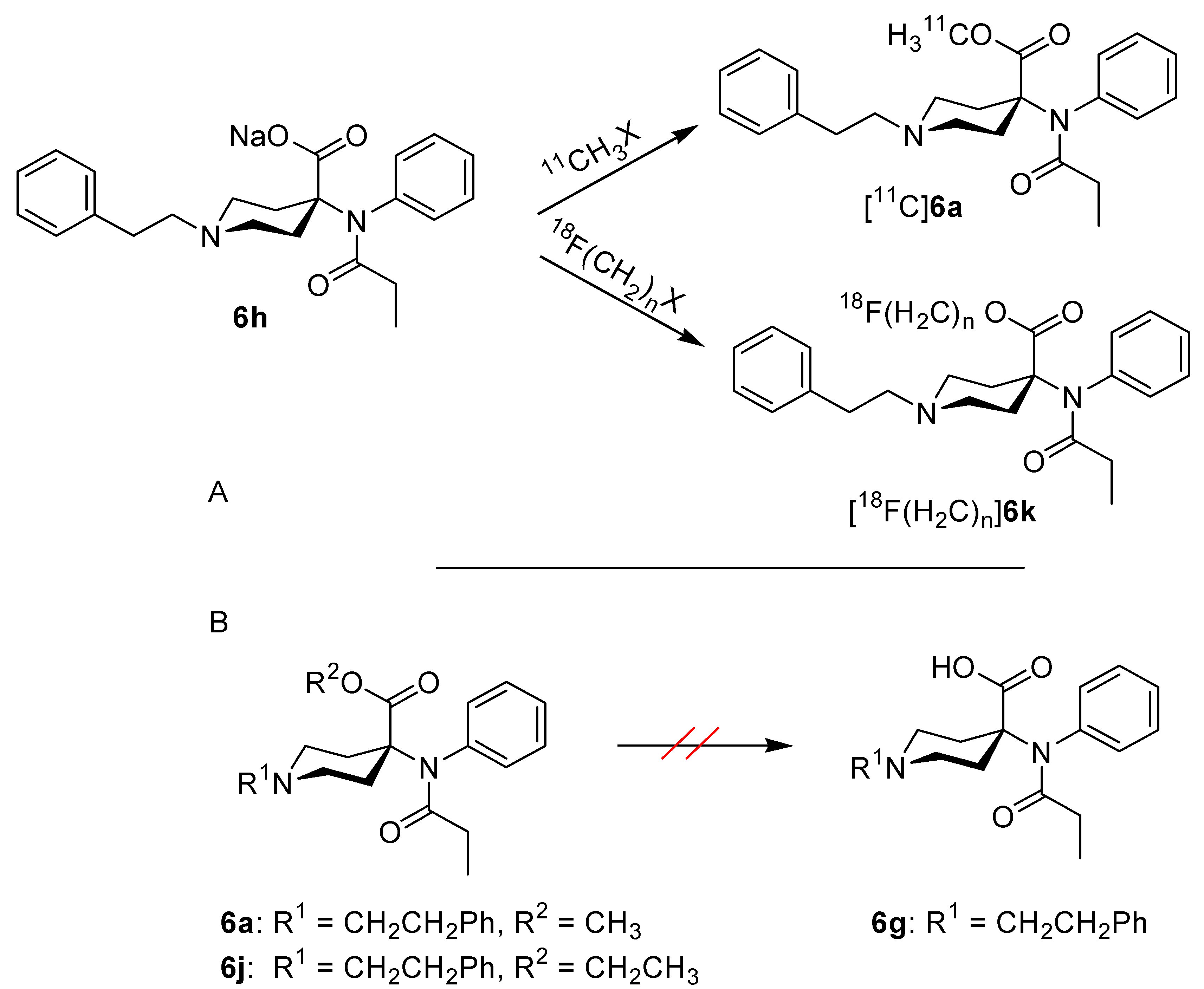
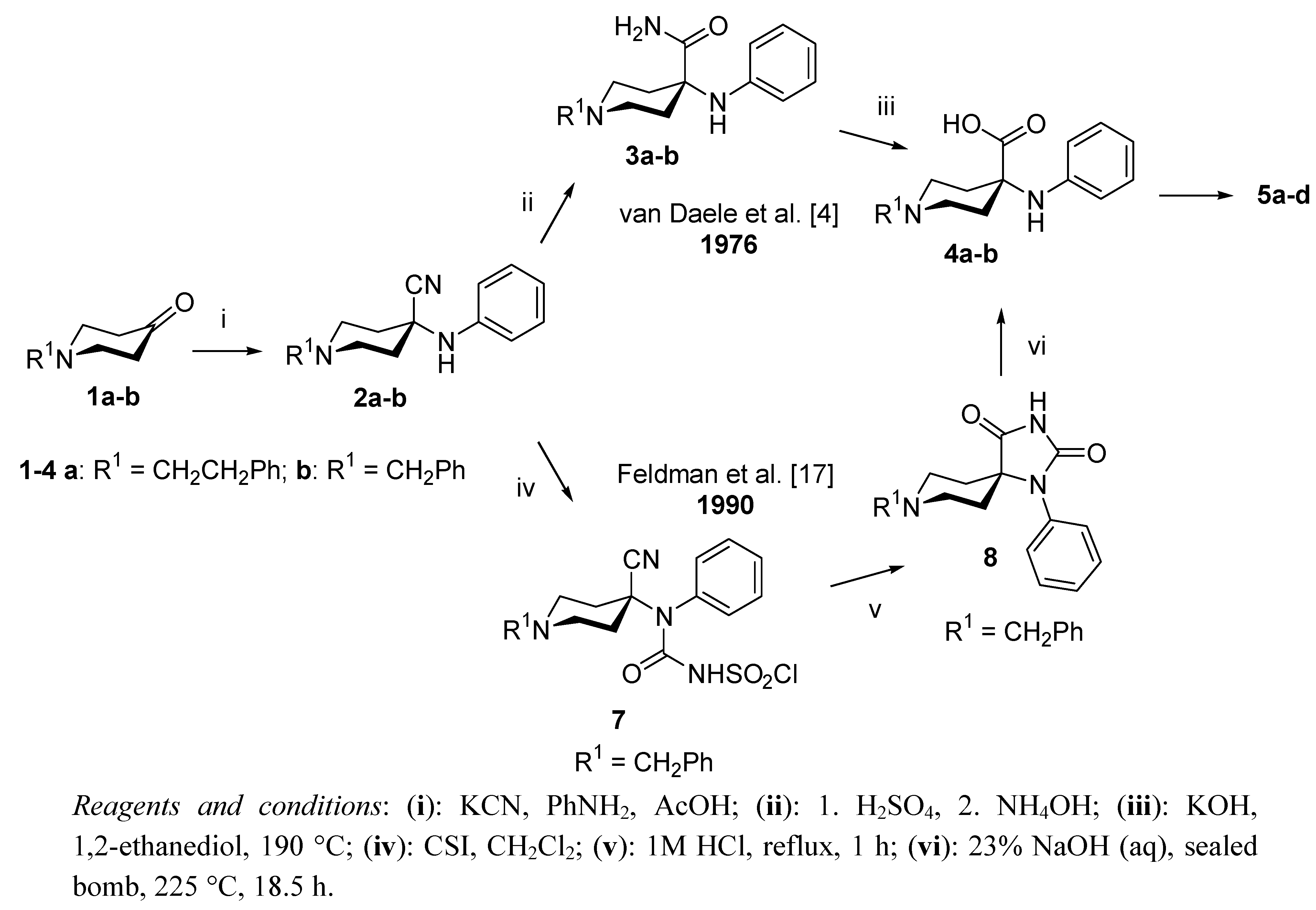
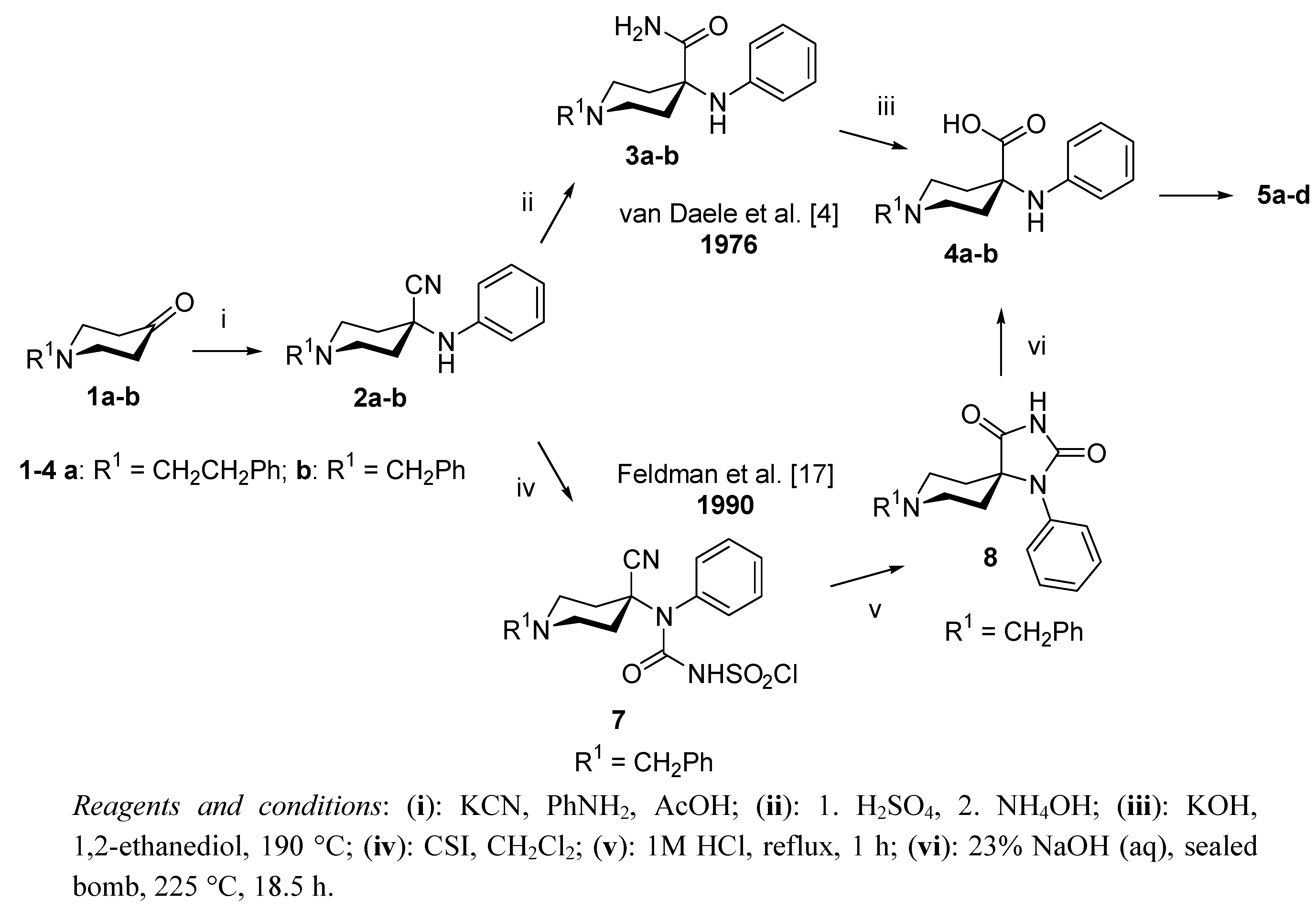
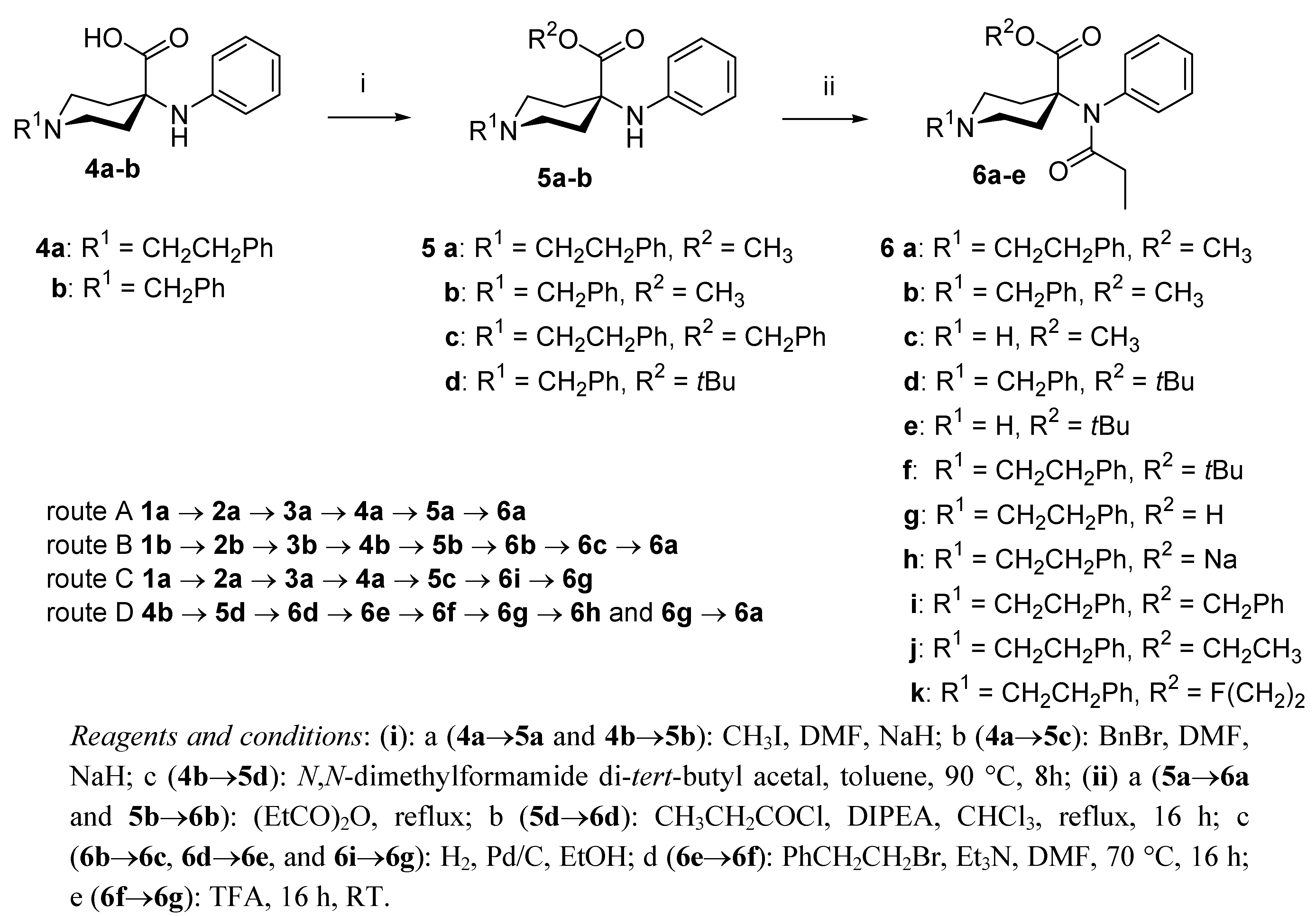
2. Results and Discussion
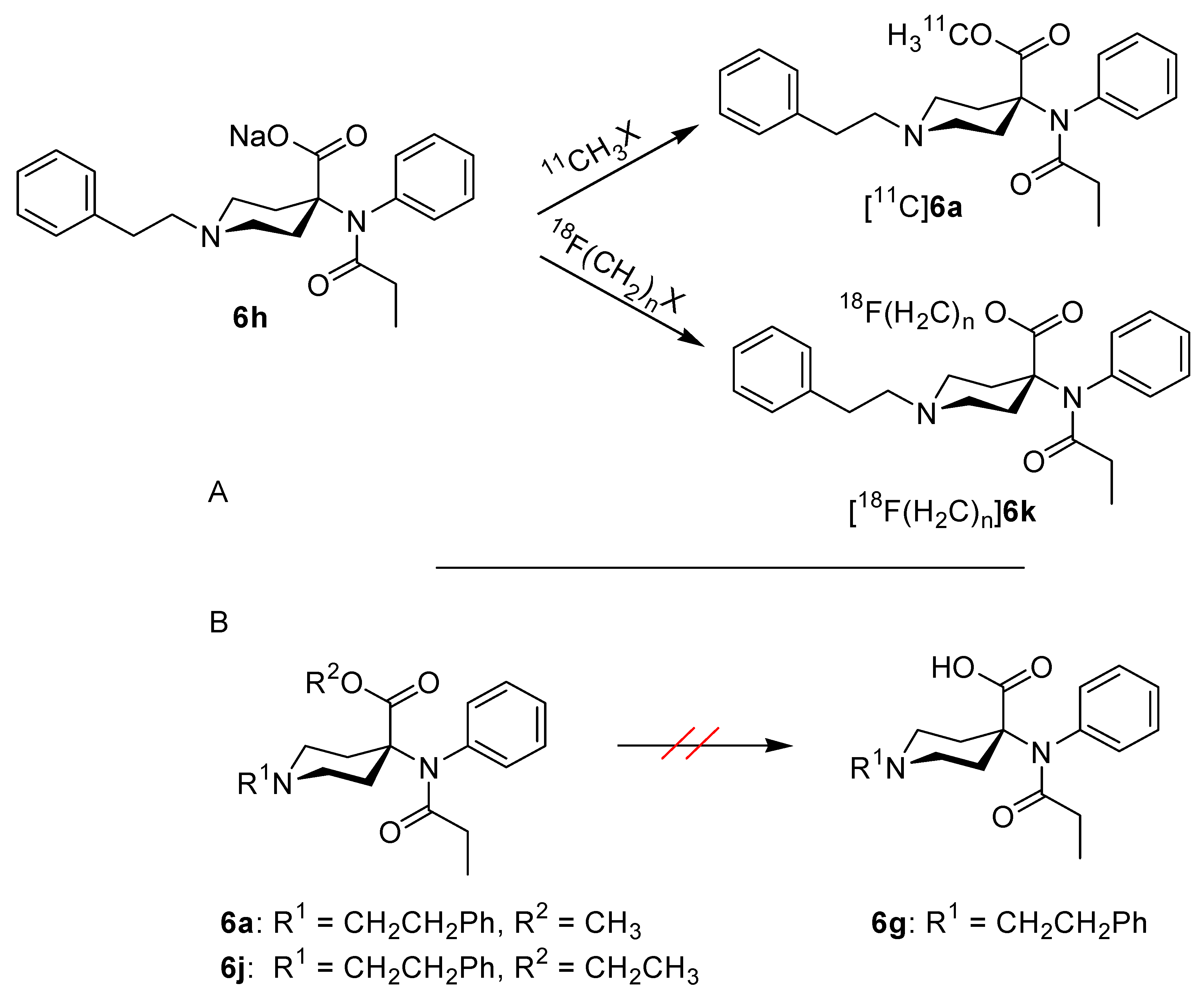
3. Experimental
3.1. General
3.2. Chemistry
4. Conclusions
Supplementary Materials
References and Notes
- Subramanian, G.; Paterlini, M.G.; Portoghese, P.S.; Ferguson, D.M. Molecular docking reveals a novel binding site model for fentanyl at the mu-opioid receptor. J. Med. Chem. 2000, 43, 381–391. [Google Scholar] [CrossRef]
- Lee, Y.S.; Nyberg, J.; Moye, S.; Agnes, R.S.; Davis, P.; Ma, S.-W.; Lai, J.; Porreca, F.; Vardanyan, R.; Hruby, V.J. Understanding the structural requirements of 4-anilidopiperidine analogues for biological activities at mu and delta opioid receptors. Bioorg. Med. Chem. Lett. 2007, 17, 2161–2165. [Google Scholar] [CrossRef]
- Janssen, P.A.J.; van Daele, G.H.P. N-(4-piperidinyl)-N-phenylamides and -carbamates. U.S. Patent 3,998,834, 1976. [Google Scholar]
- van Daele, P.G.H.; de Bruyn, M.F.L.; Boey, J.M.; Sanczuk, S.; Agten, J.T.M.; Janssen, P.A.J. Synthetic analgesics: N-(1-[2-arylethyl]-4-substituted 4-piperidinyl) N-arylalkanamides. Arzneimittel-Forsch. 1976, 26, 1521–1531. [Google Scholar]
- Greenwald, M.K.; Steinmiller, C.L. Imaging Human Brain Opioid Receptors: Applications to Substance Use Disorders. In Opiate Receptors and Antagonists, From Bench to Clinic; Dean, R.L., Bilsky, E.J., Negus, S.S., Eds.; Humana Press: New York, NY, USA, 2009; pp. 45–65, Chapter 3. [Google Scholar]
- Henriksen, G.; Willoch, F. Imaging of opioid receptors in the central nervous system. Brain 2008, 131, 1171–1196. [Google Scholar] [CrossRef]
- Henriksen, G.; Herz, M.; Schwaiger, M.; Wester, H.-J. Synthesis of [18F]fluoroalkyl esters of carfentanil. J. Labelled Comp. Radiopharm. 2005, 48, 771–779. [Google Scholar] [CrossRef]
- Wadsak, W.; Key Mien, L.; Ettlinger, D.E.; Feitscher, S.; Lanzenberger, R.; Marton, J.; Dudczak, R.; Kletter, K.; Mitterhauser, M. Preparation and radiosynthesis of [18F]FE@CFN (2-[18F]fluoroethyl 4-[N-(1-oxopropyl)-N-phenylamino]-1-(2-phenylethyl)-4-piperidinecarboxylate): a potential mu-opioid receptor imaging agent. Radiochim. Acta 2007, 95, 33–38. [Google Scholar] [CrossRef]
- Dannals, R.F.; Ravert, H.T.; Frost, J.J.; Wilson, A.A.; Burns, H.D.; Wagner, H.N., Jr. Radiosynthesis of an opiate receptor binding radiotracer: [11C]carfentanil. Int. J. Appl. Radiat. Isot. 1985, 36, 303–306. [Google Scholar] [CrossRef]
- Frost, J.J.; Wagner, H.N., Jr.; Dannals, R.F.; Ravert, H.T.; Links, J.M.; Wilson, A.A.; Burns, H.D.; Wong, D.F.; McPherson, R.W.; Rosenbaum, A.E.; Kuhar, M.J.; Snyder, S.H. Imaging opiate receptors in the human brain by positron tomography. J. Comput. Assist. Tomogr. 1985, 9, 231–236. [Google Scholar] [CrossRef]
- Saji, H.; Tsutsumi, D.; Magata, Y.; Iida, Y.; Konishi, J.; Yokoyama, A. Preparation and biodistribution in mice of [11C]carfentanil: A radiopharmaceutical for studying brain mu-opioid receptors by positron emission tomography. Ann. Nucl. Med. 1992, 6, 63–67. [Google Scholar] [CrossRef]
- Jewett, D.M. A simple synthesis of [11C]carfentanil using an extraction disk instead of HPLC. Nucl. Med. Biol. 2001, 28, 733–734. [Google Scholar] [CrossRef]
- Studenov, A.R.; Jivan, S.; Buckley, K.R.; Adam, M.J. Efficient in-loop synthesis of high specific radioactivity [11C]carfentanil. J. Labelled Comp. Radiopharm. 2003, 46, 837–842. [Google Scholar] [CrossRef]
- Colapret, J.A.; Diamantidis, G.; Spencer, K.H.; Spaulding, T.C.; Rudo, F.G. Synthesis and Pharmacological Evaluation of 4,4-Disubstituted Piperidines. J. Med. Chem. 1989, 32, 968–974. [Google Scholar] [CrossRef]
- Henriksen, G.; Platzer, S.; Marton, J.; Hauser, A.; Berthele, A.; Schwaiger, M.; Marinelli, L.; Lavecchia, A.; Novellino, E.; Wester, H.-J. Syntheses, biological evaluation, and molecular modeling of 18F-labeled 4-anilidopiperidines as mu-opioid receptor imaging agents. J. Med. Chem. 2005, 48, 7720–7732. [Google Scholar]
- Kiessling, A.J.; McClure, C.K. The Conversion of Amides to Esters with Meerwein’s Reagent. Application to the Synthesis of a Carfentanil Precursor. Synth. Commun. 1997, 27, 923–937. [Google Scholar] [CrossRef]
- Feldman, P.L.; Brackeen, M.F. A novel route to the 4-anilido-4-(methoxycarbonyl)piperidine class of analgetics. J. Org. Chem. 1990, 55, 4207–4209. [Google Scholar] [CrossRef]
- Meusel, M.; Guetschow, M. Recent developments in hydantoin chemistry. A review. Org. Prep. Proced. Int. 2004, 36, 391–443. [Google Scholar] [CrossRef]
- Abdulla, R.F.; Brinkmeyer, R.S. The chemistry of formamide acetals. Tetrahedron 1979, 35, 1675–1735. [Google Scholar] [CrossRef]
- Armstrong, A.; Brackenridge, I.; Jackson, R.F.W.; Kirk, J.M. A new method for the preparation of tertiary butyl ethers and esters. Tetrahedron Lett. 1988, 29, 2483–2486. [Google Scholar] [CrossRef]
- Galzi, J.-L.; Mejean, A.; Ilien, B.; Mollereau, C.; Meunier, J.-C.; Goeldner, M.; Hirth, C. Photoactivatable opiate Derivatives as Irreversible Probes of the mu-Opioid Receptor. J. Med. Chem. 1990, 33, 2456–2464. [Google Scholar] [CrossRef]
- Feldman, P.L.; James, M.K.; Brackeen, M.F.; Bilotta, J.M.; Schuster, S.V.; Lahey, A.P.; Lutz, M.W.; Johnson, M.R.; Leighton, H.J. Design, synthesis, and pharmacological evaluation of ultrashort- to long-acting opioid analgetics. J. Med. Chem. 1991, 34, 2202–2208. [Google Scholar]
- Sample Availability: Samples of the compounds 3b, 4b, 5d, 6d–g are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Marton, J.; Glaenzel, B.; Roessler, J.; Golaszewski, D.; Henriksen, G. A Convenient Route to 4-Carboxy-4-Anilidopiperidine Esters and Acids. Molecules 2012, 17, 2823-2832. https://doi.org/10.3390/molecules17032823
Marton J, Glaenzel B, Roessler J, Golaszewski D, Henriksen G. A Convenient Route to 4-Carboxy-4-Anilidopiperidine Esters and Acids. Molecules. 2012; 17(3):2823-2832. https://doi.org/10.3390/molecules17032823
Chicago/Turabian StyleMarton, János, Brita Glaenzel, Julia Roessler, Daniela Golaszewski, and Gjermund Henriksen. 2012. "A Convenient Route to 4-Carboxy-4-Anilidopiperidine Esters and Acids" Molecules 17, no. 3: 2823-2832. https://doi.org/10.3390/molecules17032823