3D-QSAR Studies of Dihydropyrazole and Dihydropyrrole Derivatives as Inhibitors of Human Mitotic Kinesin Eg5 Based on Molecular Docking

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Set
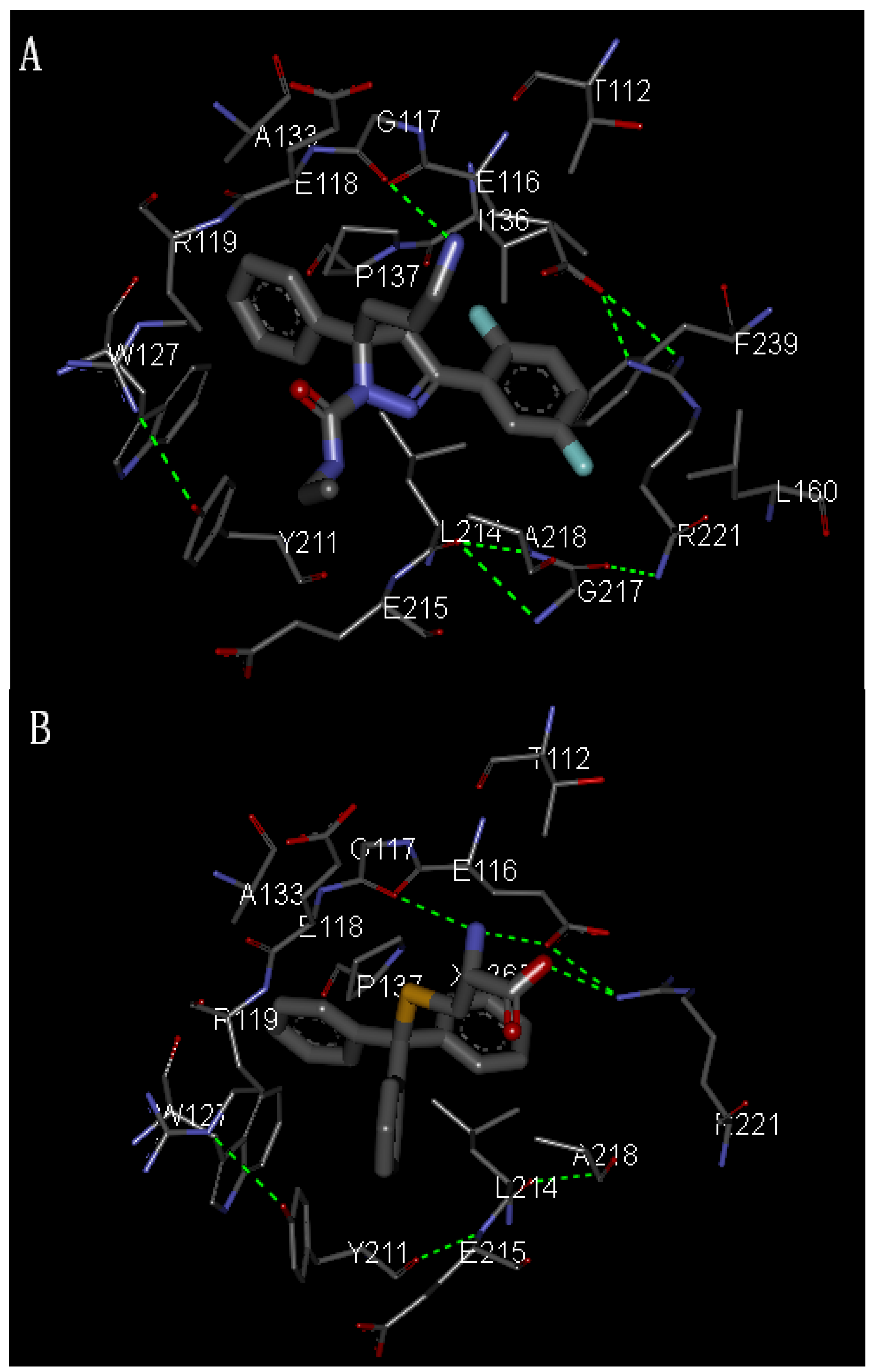
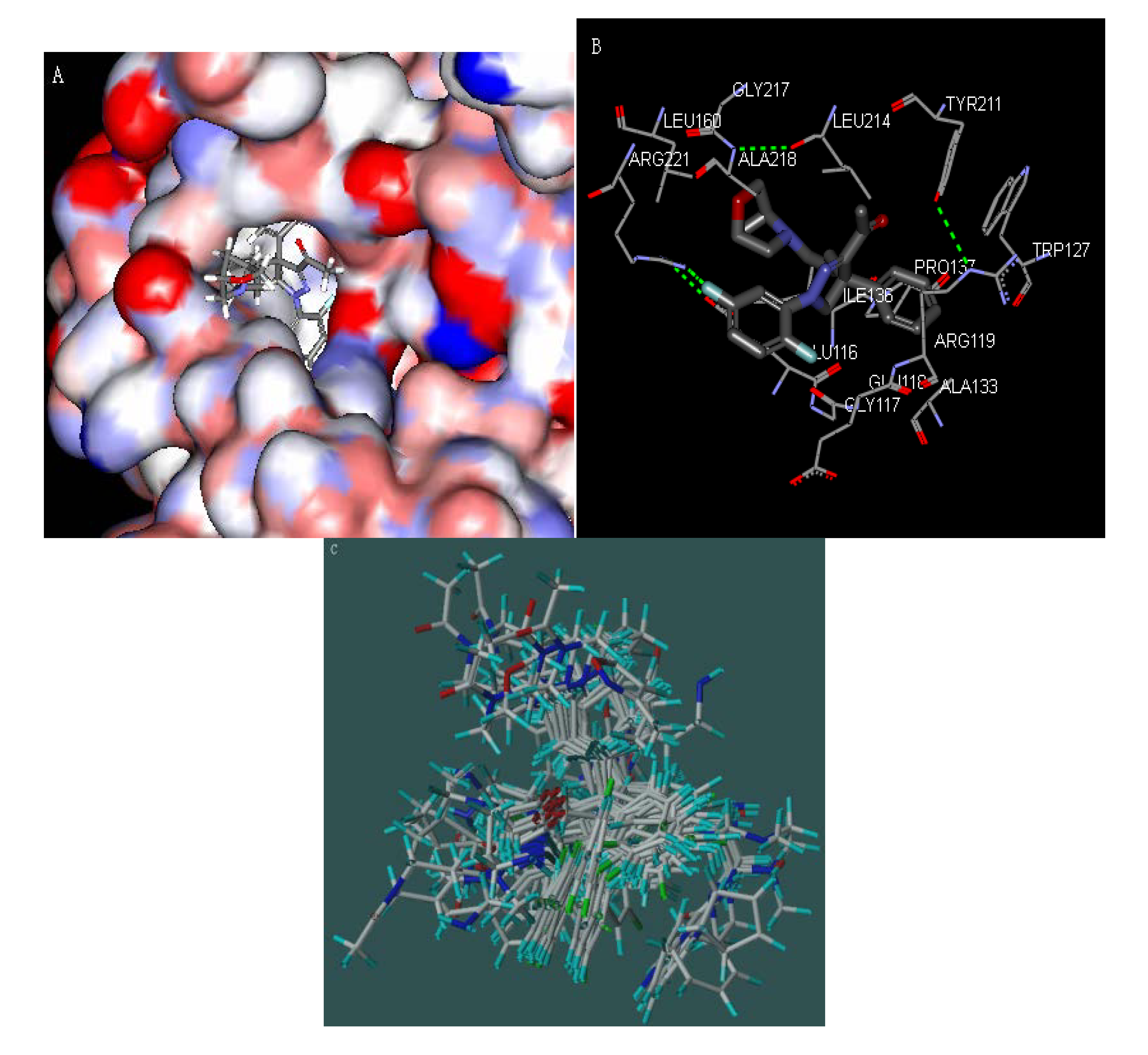
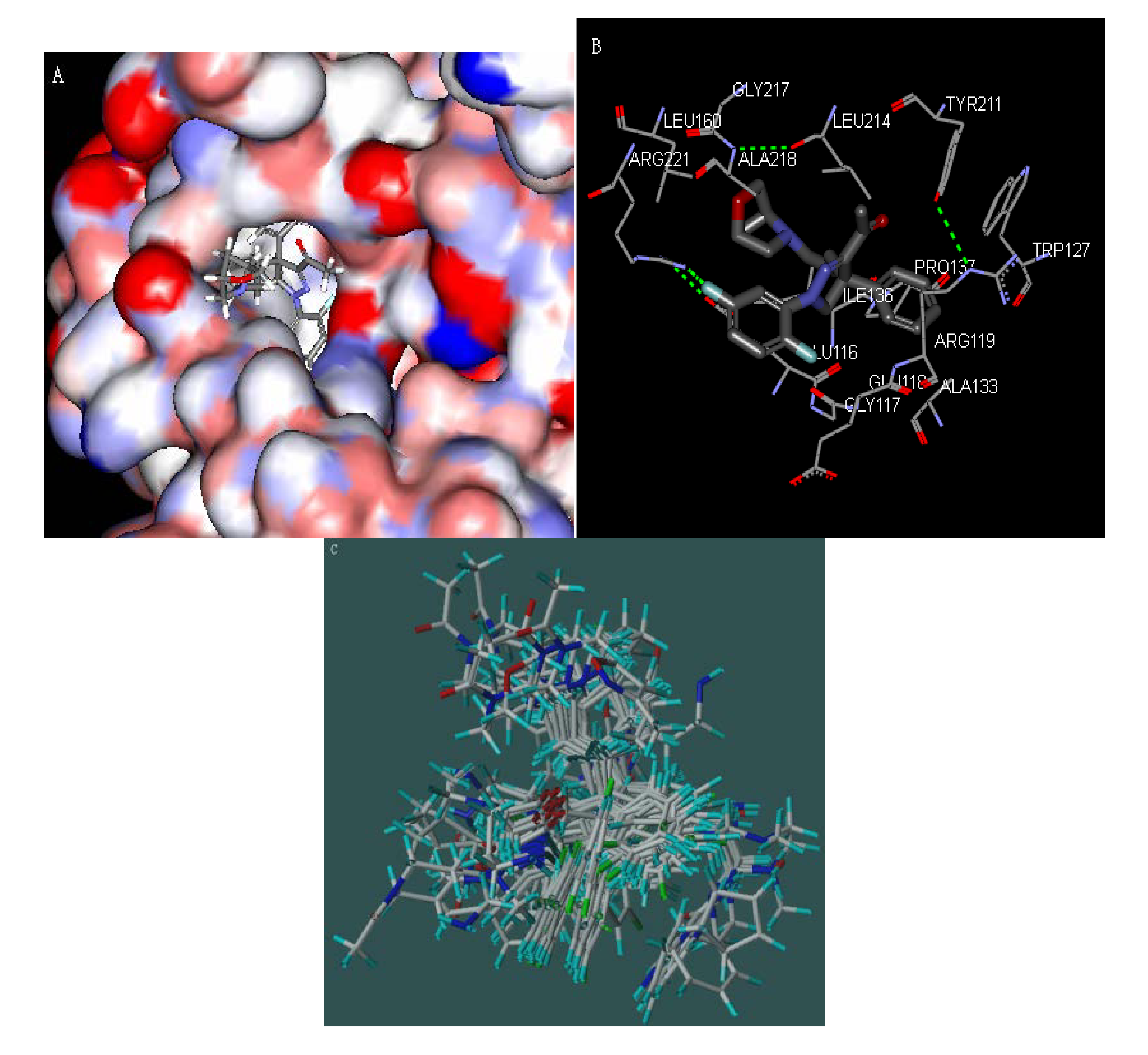
2.2. Molecular Docking


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO. | X | R\R1 | R2 | IC50 (nM) | pIC50 |
|---|---|---|---|---|---|
| A01 | F |  | - | 1.2 | 8.9208 |
| A02 * | CF3 |  | - | 2.0 | 8.699 |
| A03 | F |  | - | 2.1 | 8.6778 |
| A04 | F |  | - | 3.8 | 8.4202 |
| A05 * | F |  | - | 3.8 | 8.4202 |
| A06 | Cl |  | - | 3.9 | 8.4089 |
| A07 | F |  | - | 4.0 | 8.3979 |
| A08 | F |  | - | 4.2 | 8.3768 |
| A09 | Br |  | - | 4.7 | 8.3279 |
| A10 * | F |  | - | 5.2 | 8.284 |
| A11 | CF3 |  | - | 10.1 | 7.9957 |
| B01 | F | -(CH2)4- | 26.0 | 7.585027 | |
| B02 * | F | -(CH2)3- | 55.0 | 7.259637 | |
| B03 | F | -(CH2)5- | 85.0 | 7.070581 | |
| B04 | F | -(CH2)2O(CH2)2- | 122.0 | 6.91364 | |
| B05 | F | -NHBn | - | 100.0 | 7 |
| B06 | F | -NMe2 | - | 103.0 | 6.9872 |
| B07 | F |  | - | 119.0 | 6.9245 |
| B08 * | F |  | - | 391.0 | 6.4078 |
| B09 | F | -NMe2O | - | 585.0 | 6.2328 |
| B10 | F |  | - | 686.0 | 6.1637 |
| B11 | F |  | - | 829.0 | 6.0814 |
| B12 | F | CH2 CH3 | (CH2)3NH2 | 44.0 | 7.3565 |
| B13 | F | CH2 CH3 | (CH2)4NH2 | 67.0 | 7.1739 |
| B14 | F | CH3 | CH3 | 284.0 | 6.5467 |
| B15 | F | CH2 CH3 | (CH2)2NH2 | 390.0 | 6.4089 |
| B16 * | F | CH2 CH3 | (CH2)4OH | 697.0 | 6.1568 |
| B17 | F | CH2 CH3 | (CH2)3OH | 745.0 | 6.1278 |
| C01 | F |  | - | 5.2 | 8.284 |
| C02 | F |  | - | 7.4 | 8.1308 |
| C03 * | F |  | - | 11.0 | 7.9586 |
| C04 | F |  | - | 16.0 | 7.7959 |
| C05 | F | NMe2 | - | 38.0 | 7.4202 |
| C06 | F |  | - | 50.0 | 7.301 |
| C07 | F | NMe2 | - | 84.0 | 7.075721 |
| C08 | F | Me | - | 94.0 | 7.026872 |
| C09 | F | t-Bu | - | 113.0 | 6.946922 |
2.3. 3D-QSAR Modeling [25,26]
3. Results and Discussion
3.1. Molecular Docking

3.2. CoMFA and CoMSIA of 3D-QASR Models
| Model | q2 | n | r2 | SEE | F | S% | E% | H% | D% | A% |
|---|---|---|---|---|---|---|---|---|---|---|
| CoMFA | 0.798 | 4 | 0.980 | 0.127 | 304.977 | 34.9 | 65.1 | |||
| CoMSIA | 0.848 | 4 | 0.992 | 0.08 | 769.202 | 8.7 | 29.2 | 16.6 | 24.6 | 20.9 |
| No. | Actual pIC50 | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|
| Predicted | Residues | Predicted | Residues | ||
| A01 | 8.9208 | 8.999 | −0.0777 | 8.954 | −0.0337 |
| A02 * | 8.699 | 8.495 | 0.2036 | 8.17 | 0.5294 |
| A03 | 8.6778 | 8.679 | −0.0015 | 8.587 | 0.0909 |
| A04 | 8.4202 | 8.339 | 0.0816 | 8.459 | −0.0387 |
| A05 * | 8.4202 | 8.708 | −0.2881 | 8.603 | −0.1826 |
| A06 | 8.4089 | 8.445 | −0.0361 | 8.443 | −0.0342 |
| A07 | 8.3979 | 8.448 | −0.0499 | 8.424 | −0.0263 |
| A08 | 8.3768 | 8.399 | −0.0218 | 8.268 | 0.109 |
| A09 | 8.3279 | 8.334 | −0.0058 | 8.377 | −0.0494 |
| A10 * | 8.284 | 8.54 | −0.2558 | 8.454 | −0.1702 |
| A11 | 7.9957 | 7.977 | 0.0185 | 7.977 | 0.0188 |
| B01 | 7.585 | 7.747 | −0.1617 | 7.685 | −0.0997 |
| B02 * | 7.2596 | 7.372 | −0.1129 | 7.408 | −0.1481 |
| B03 | 7.0706 | 7.148 | −0.0774 | 7.009 | 0.0617 |
| B04 | 6.9136 | 6.761 | 0.1527 | 6.768 | 0.1461 |
| B05 | 7 | 6.884 | 0.1161 | 6.95 | 0.0499 |
| B06 | 6.9872 | 6.95 | 0.0368 | 7.014 | −0.0264 |
| B07 | 6.9245 | 6.978 | −0.0539 | 6.907 | 0.0174 |
| B08 * | 6.4078 | 6.183 | 0.2249 | 6.178 | 0.2301 |
| B09 | 6.2328 | 6.232 | 0.0007 | 6.18 | 0.0533 |
| B10 | 6.1637 | 6.031 | 0.1328 | 6.099 | 0.0648 |
| B11 | 6.0814 | 6.201 | −0.1191 | 6.207 | −0.1256 |
| B12 | 7.3565 | 7.362 | −0.0054 | 7.42 | −0.063 |
| B13 | 7.1739 | 7.029 | 0.1451 | 7.165 | 0.0092 |
| B14 | 6.5467 | 6.568 | −0.0213 | 6.51 | 0.0362 |
| B15 | 6.4089 | 6.597 | −0.188 | 6.449 | −0.0404 |
| B16 * | 6.1568 | 6.365 | −0.2081 | 6.297 | −0.1406 |
| B17 | 6.1278 | 6.245 | −0.1176 | 6.286 | −0.1586 |
| C01 | 8.284 | 7.976 | 0.308 | 8.293 | −0.0092 |
| C02 | 8.1308 | 8.108 | 0.0225 | 8.09 | 0.0411 |
| C03 * | 7.9586 | 7.85 | 0.1088 | 7.682 | 0.2764 |
| C04 | 7.7959 | 7.71 | 0.0861 | 7.806 | −0.0103 |
| C05 | 7.4202 | 7.664 | −0.2433 | 7.519 | −0.0989 |
| C06 | 7.301 | 7.119 | 0.1821 | 7.177 | 0.1243 |
| C07 | 7.0757 | 7.135 | −0.0597 | 7.008 | 0.0677 |
| C08 | 7.0269 | 6.978 | 0.0491 | 7.01 | 0.0171 |
| C09 | 6.9469 | 7.039 | −0.0918 | 7.04 | -0.0929 |
3.3. Predictive Power of 3D-QSAR Analyses

3.4. Graphical Interpretation of the Fields

4. Conclusions
Acknowledgments
- Samples Availability: Not available.
References and Notes
- Blangy, A.; Lane, H.A.; d’Herin, P.; Harper, M.; Kress, M.; Nigg, E.A. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 1995, 83, 1159–1169. [Google Scholar] [CrossRef]
- Weil, D.; Garcon, L.; Harper, M.; Dumenil, D.; Dautry, F.; Kress, M. Targeting the kinesin Eg5 to monitor siRNA transfection in mammalian cells. Biotechniques 2002, 33, 1244–1248. [Google Scholar]
- Mayer, T.U.; Kapoor, T.M.; Haggarty, S.J.; King, R.W.; Schreiber, S.L.; Mitchison, T.J. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 1999, 286, 971–974. [Google Scholar] [CrossRef]
- Maliga, Z.; Kapoor, T.M.; Mitchison, T.J. Evidence that monastrol is an allosteric inhibitor of the mitotic kinesin Eg5. Chem. Biol. 2002, 9, 989–996. [Google Scholar] [CrossRef]
- Turner, J.; Anderson, R.; Guo, J.; Beraud, C.; Fletterick, R.; Sakowicz, R. Crystal structure of the mitotic spindle kinesin Eg5 reveals a novel conformation of the neck-linker. J. Biol. Chem. 2001, 276, 25496–25502. [Google Scholar]
- DeBonis, S.; Skoufias, D.A.; Lebeau, L.; Lopez, R.; Robin, G.; Margolis, R.L.; Wade, R.H.; Kozielski, F. In vitro screening for inhibitors of the human mitotic kinesin Eg5 with antimitotic and antitumor activities. Mol. Cancer Ther. 2004, 3, 1079–1090. [Google Scholar]
- Skoufias, D.A.; DeBonis, S.; Saoudi, Y.; Lebeau, L.; Crevel, I.; Cross, R.; Wade, R.H.; Hackney, D.; Kozielski, F. S-trityl-L-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. J. Biol. Chem. 2006, 281, 17559–17569. [Google Scholar]
- Gartner, M.; Sunder-Plassmann, N.; Seiler, J.; Utz, M.; Vernos, I.; Surrey, T.; Giannis, A. Development and biological evaluation of potent and specific inhibitors of mitotic Kinesin Eg5. ChemBioChem 2005, 6, 1173–1177. [Google Scholar] [CrossRef]
- Kozielski, F.; DeBonis, S.; Skoufias, D.A. Screening for inhibitors of microtubule-associated motor proteins. Methods Mol. Med. 2007, 137, 189–207. [Google Scholar] [CrossRef]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef]
- Kaan, H.Y.; Weiss, J.; Menger, D.; Ulaganathan, V.; Tkocz, K.; Laggner, C.; Popowycz, F.; Joseph, B.; Kozielski, F. Structure-activity relationship and multidrug resistance study of new S-trityl-L-cysteine derivatives as inhibitors of Eg5. J. Med. Chem. 2011, 54, 1576–1586. [Google Scholar] [CrossRef]
- Barsanti, P.A.; Wang, W.; Ni, Z.-J.; Duhl, D.; Brammeier, N.; Martin, E.; Bussiere, D.; Walter, A.O. The discovery of tetrahydro-β-carbolines as inhibitors of the kinesin Eg5. Bioorg. Med. Chem. Lett. 2010, 20, 157–160. [Google Scholar]
- Liu, M.; Yu, H.; Huo, L.; Liu, J.; Li, M.; Zhou, J. Validating the mitotic kinesin Eg5 as a therapeutic target in pancreatic cancer cells and tumor xenografts using a specific inhibitor. Biochem. Pharmacol. 2008, 76, 169–178. [Google Scholar]
- Xiao, S.; Shi, X.-X. The first highly stereoselective approach to the mitotic kinesin Eg5 inhibitor HR22C16 and its analogues. Tetrahedron: Asymmetry 2010, 21, 226–231. [Google Scholar]
- Cox, C.D.; Torrent, M.; Breslin, M.J.; Mariano, B.J.; Whitman, D.B.; Coleman, P.J.; Buser, C.A.; Walsh, E.S.; Hamilton, K.; Schaber, M.D. . Kinesin spindle protein (KSP) inhibitors. Part 4:1 Structure-based design of 5-alkylamino-3,5-diaryl-4,5-dihydropyrazoles as potent, water-soluble inhibitors of the mitotic kinesin KSP. Bioorg. Med. Chem. Lett. 2006, 16, 3175–3179. [Google Scholar]
- Fraley, M.E.; Garbaccio, R.M.; Arrington, K.L.; Hoffman, W.F.; Tasber, E.S.; Coleman, P.J.; Buser, C.A.; Walsh, E.S.; Hamilton, K.; Fernandes, C. Kinesin spindle protein (KSP) inhibitors. Part 2: The design, synthesis, and characterization of 2,4-diaryl-2,5-dihydropyrrole inhibitors of the mitotic kinesin KSP. Bioorg. Med. Chem. Lett. 2006, 16, 1775–1779. [Google Scholar]
- Roecker, A.J.; Coleman, P.J.; Mercer, S.P.; Schreier, J.D.; Buser, C.A.; Walsh, E.S.; Hamilton, K.; Lobell, R.B.; Tao, W.; Diehl, R.E. Kinesin spindle protein (KSP) inhibitors. Part 8: Design and synthesis of 1,4-diaryl-4,5-dihydropyrazoles as potent inhibitors of the mitotic kinesin KSP. Bioorg. Med. Chem. Lett. 2007, 17, 5677–5682. [Google Scholar]
- Brier, S.; Lemaire, D.; Debonis, S.; Forest, E.; Kozielski, F. Identification of the protein binding region of S-trityl-L-cysteine, a new potent inhibitor of the mitotic kinesin Eg5. Biochemistry 2004, 43, 13072–13082. [Google Scholar] [CrossRef]
- Yi Kristal Kaan, H.; Ulaganathan, V.; Hackney, D.D.; Kozielski, F. An allosteric transition trapped in an intermediate state of a new kinesin-inhibitor complex. Biochem. J. 2010, 425, 55–60. [Google Scholar] [CrossRef]
- Debonis, S.; Skoufias, D.A.; Indorato, R.L.; Liger, F.; Marquet, B.; Laggner, C.; Joseph, B.; Kozielski, F. Structure-activity relationship of S-trityl-L-cysteine analogues as inhibitors of the human mitotic kinesin Eg5. J. Med. Chem. 2008, 51, 1115–1125. [Google Scholar] [CrossRef]
- Kozielski, F.; Skoufias, D.A.; Indorato, R.L.; Saoudi, Y.; Jungblut, P.R.; Hustoft, H.K.; Strozynski, M.; Thiede, B. Proteome analysis of apoptosis signaling by S-trityl-L-cysteine, a potent reversible inhibitor of human mitotic kinesin Eg5. Proteomics 2008, 8, 289–300. [Google Scholar]
- Prokopcova, H.; Dallinger, D.; Uray, G.; Kaan, H.Y.; Ulaganathan, V.; Kozielski, F.; Laggner, C.; Kappe, C.O. Structure-activity relationships and molecular docking of novel dihydropyrimidine-based mitotic Eg5 inhibitors. ChemMedChem 2010, 5, 1760–1769. [Google Scholar] [CrossRef]
- Kubinyi, H. QSAR and 3D QSAR in drug design Part 1: Methodology. Drug Discov. Today 1997, 2, 457–467. [Google Scholar] [CrossRef]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef]
- Puntambekar, D.; Giridhar, R.; Yadav, M.R. 3D-QSAR studies of farnesyltransferase inhibitors: A comparative molecular field analysis approach. Bioorg. Med. Chem. Lett. 2006, 16, 1821–1827. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, C.; Li, H. CoMFA and CoMSIA Studies of nAChRs Ligands: Epibatidine Analogues. QSAR Comb. Sci. 2004, 23, 80–88. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, C.; Min, J.; Wang, Y.; He, J.; Yu, Z. Rational questing for potential novel inhibitors of FabK from Streptococcus pneumoniae by combining FMO calculation, CoMFA 3D-QSAR modeling and virtual screening. J. Mol. Model. 2010, 17, 1483–1492. [Google Scholar]
- AbdulHameed, M.D.; Hamza, A.; Liu, J.; Huang, X.; Zhan, C.G. Human microsomal prostaglandin E synthase-1 (mPGES-1) binding with inhibitors and the quantitative structure-activity correlation. J. Chem. Inf. Model. 2008, 48, 179–185. [Google Scholar] [CrossRef]
- Venkatachalam, C.M.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
- Sippl, W. Application of Structure-Based Alignment Methods for 3D QSAR Analyses. In Pharmacophores and Pharmacophore Searches; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 223–249. [Google Scholar]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Luo, X.; Shu, M.; Wang, Y.; Liu, J.; Yang, W.; Lin, Z. 3D-QSAR Studies of Dihydropyrazole and Dihydropyrrole Derivatives as Inhibitors of Human Mitotic Kinesin Eg5 Based on Molecular Docking. Molecules 2012, 17, 2015-2029. https://doi.org/10.3390/molecules17022015
Luo X, Shu M, Wang Y, Liu J, Yang W, Lin Z. 3D-QSAR Studies of Dihydropyrazole and Dihydropyrrole Derivatives as Inhibitors of Human Mitotic Kinesin Eg5 Based on Molecular Docking. Molecules. 2012; 17(2):2015-2029. https://doi.org/10.3390/molecules17022015
Chicago/Turabian StyleLuo, Xingyan, Mao Shu, Yuanqiang Wang, Jin Liu, Wenjuan Yang, and Zhihua Lin. 2012. "3D-QSAR Studies of Dihydropyrazole and Dihydropyrrole Derivatives as Inhibitors of Human Mitotic Kinesin Eg5 Based on Molecular Docking" Molecules 17, no. 2: 2015-2029. https://doi.org/10.3390/molecules17022015