Peptide-Based Selective Inhibitors of Matrix Metalloproteinase-Mediated Activities

Abstract
:1. Introduction
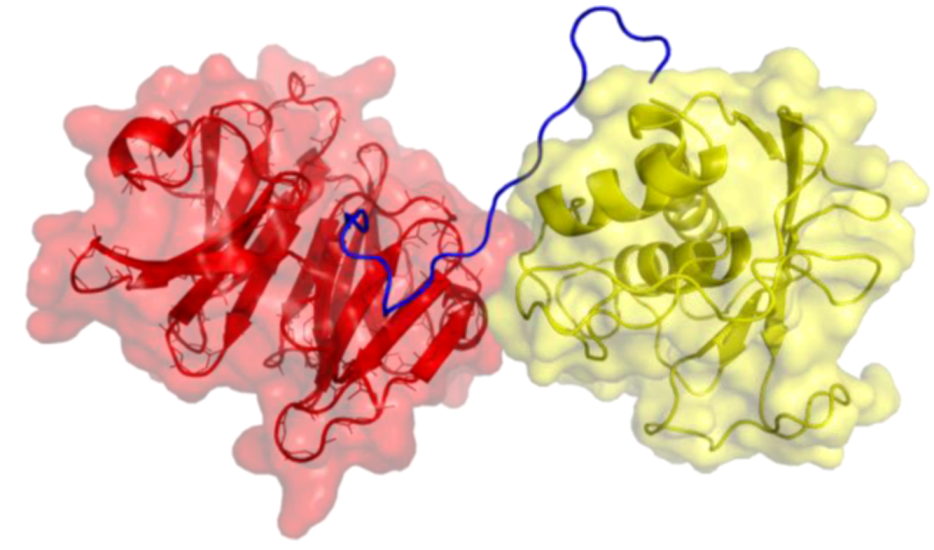
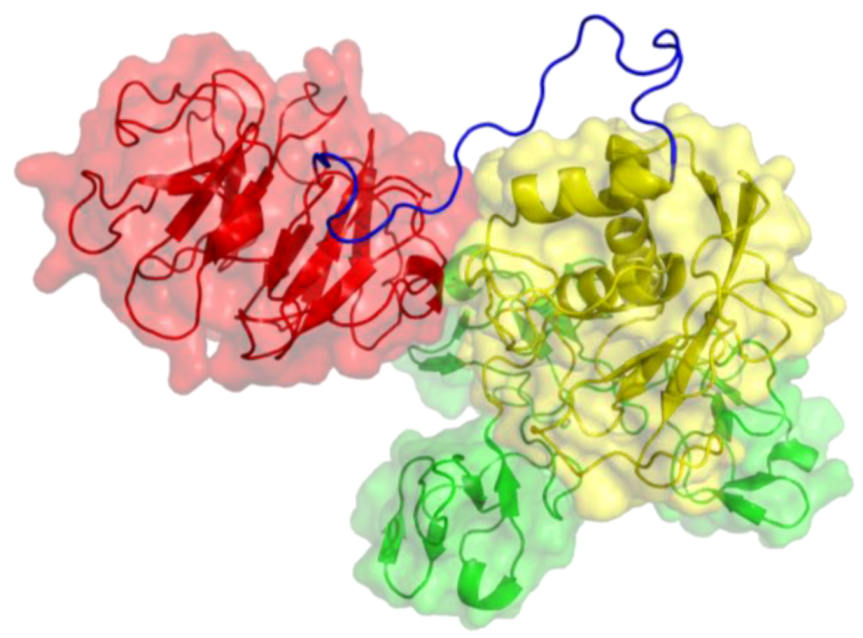
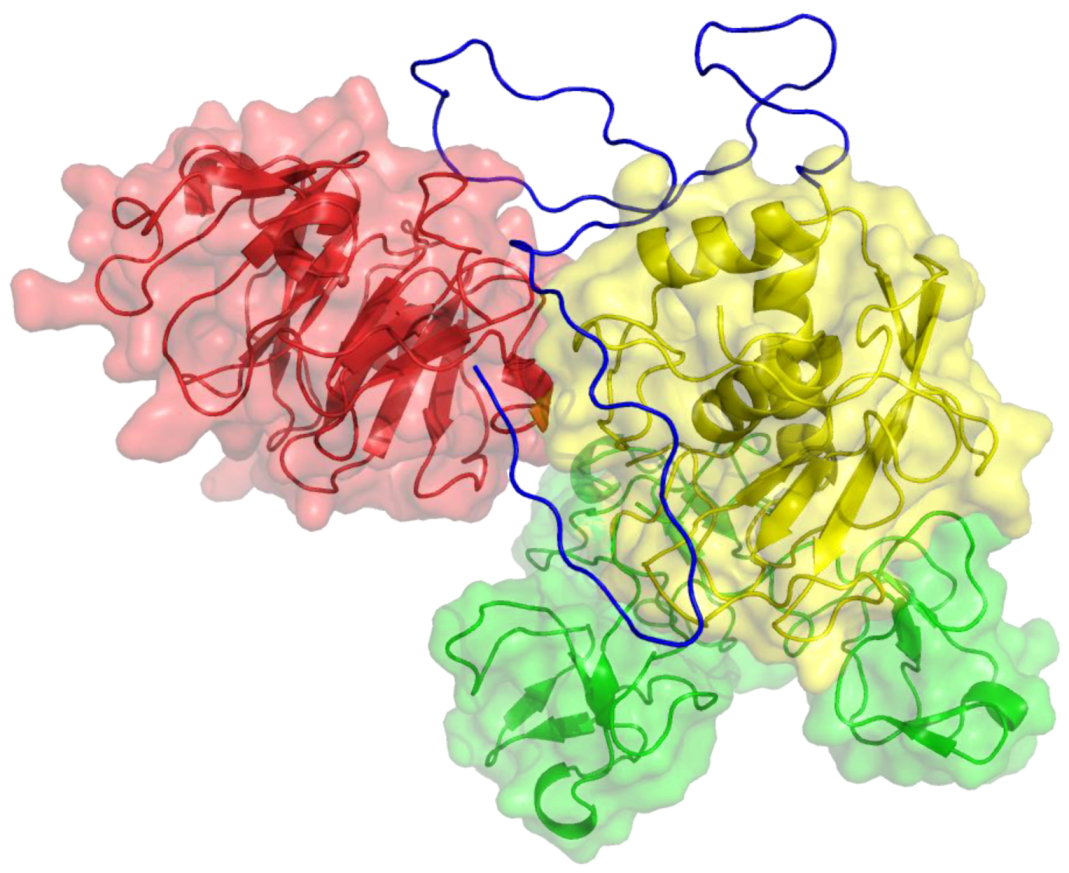
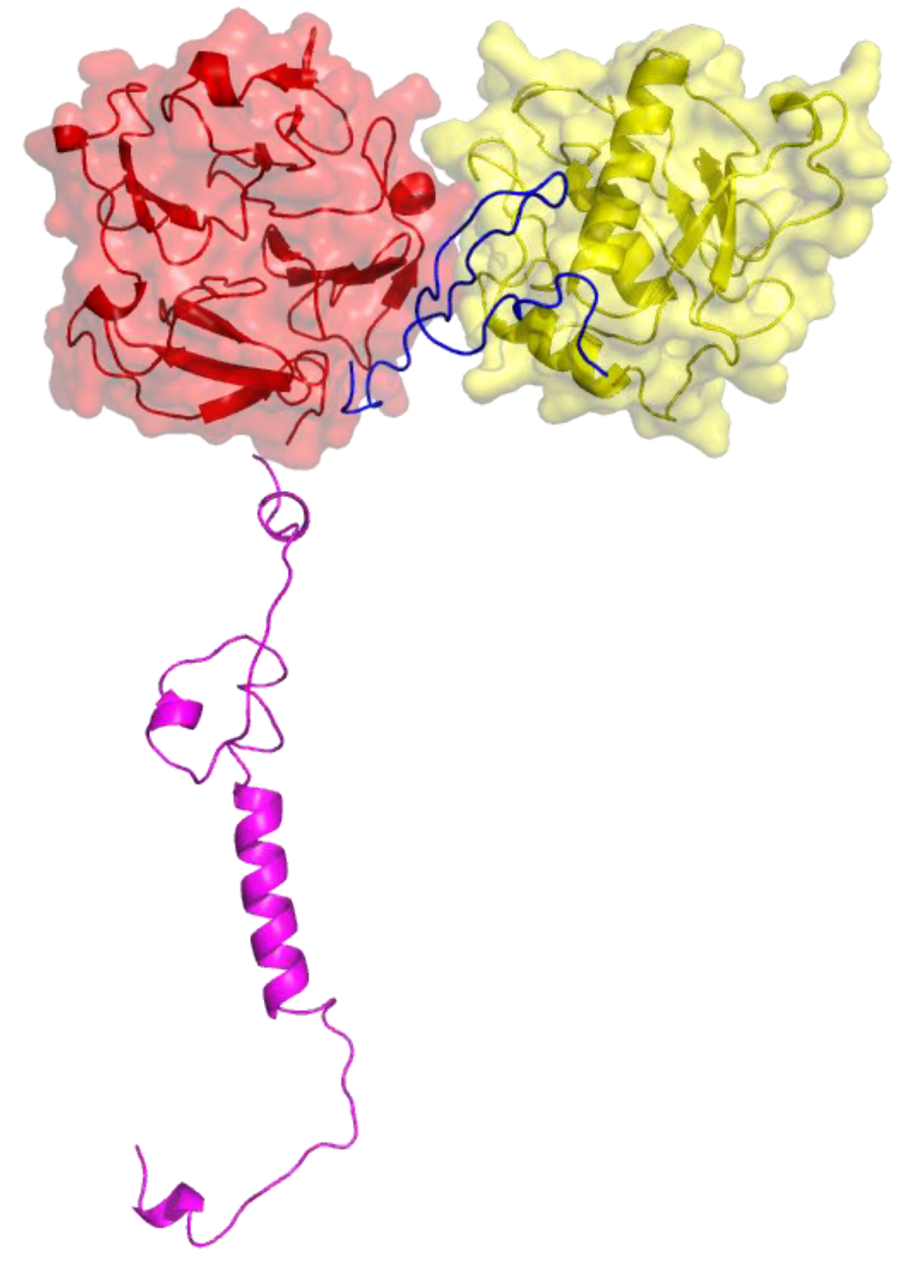
2. Peptides That Target Gelatinase Members of the MMP Family (MMP-2 and MMP-9)

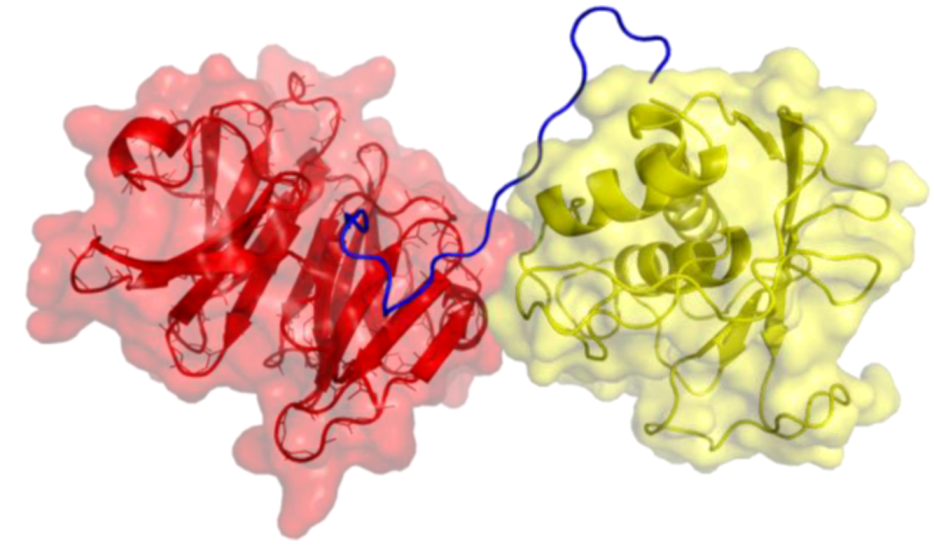{kind=link}
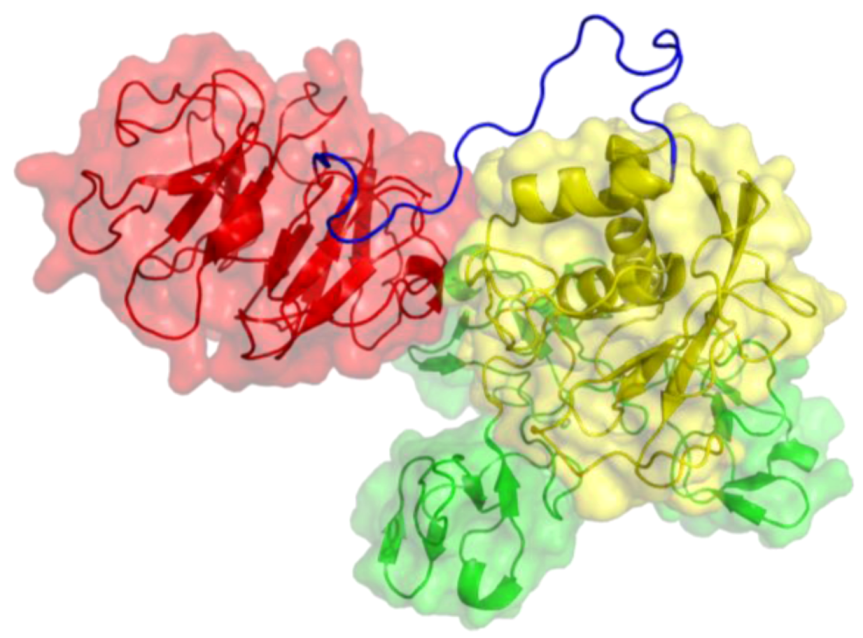{kind=link}
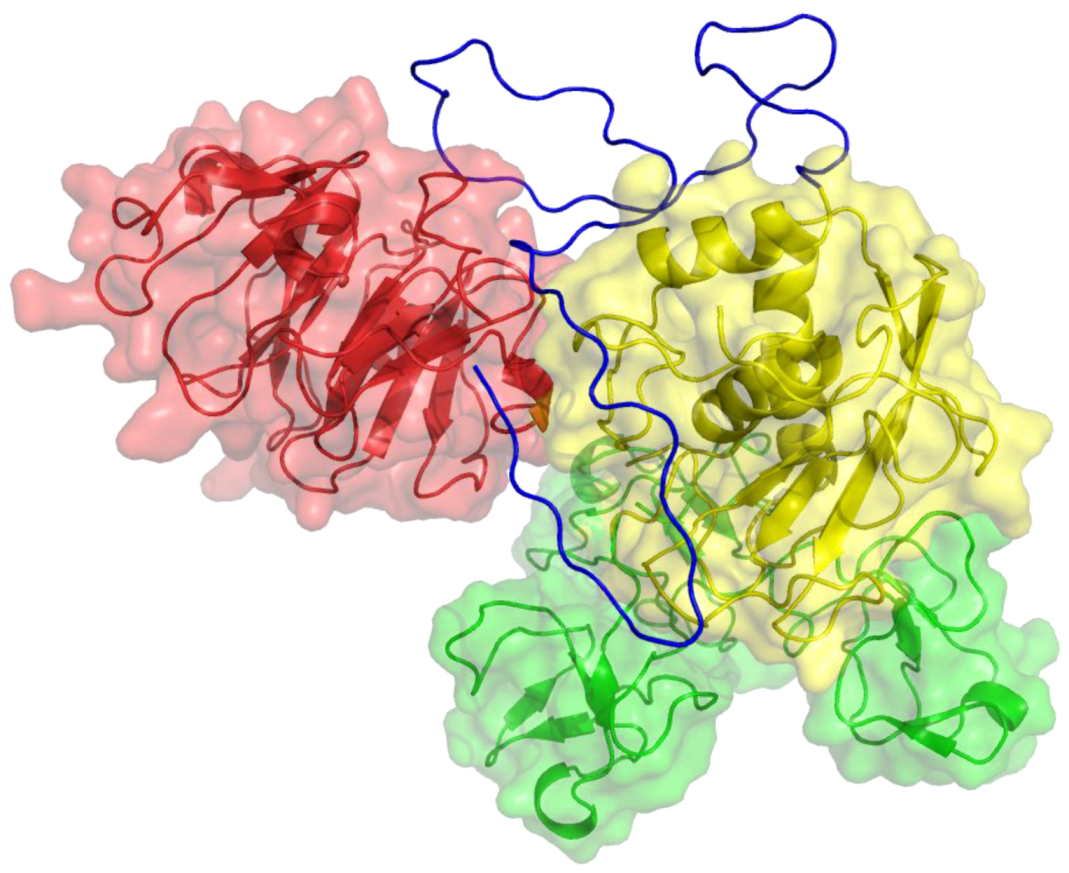{kind=link}
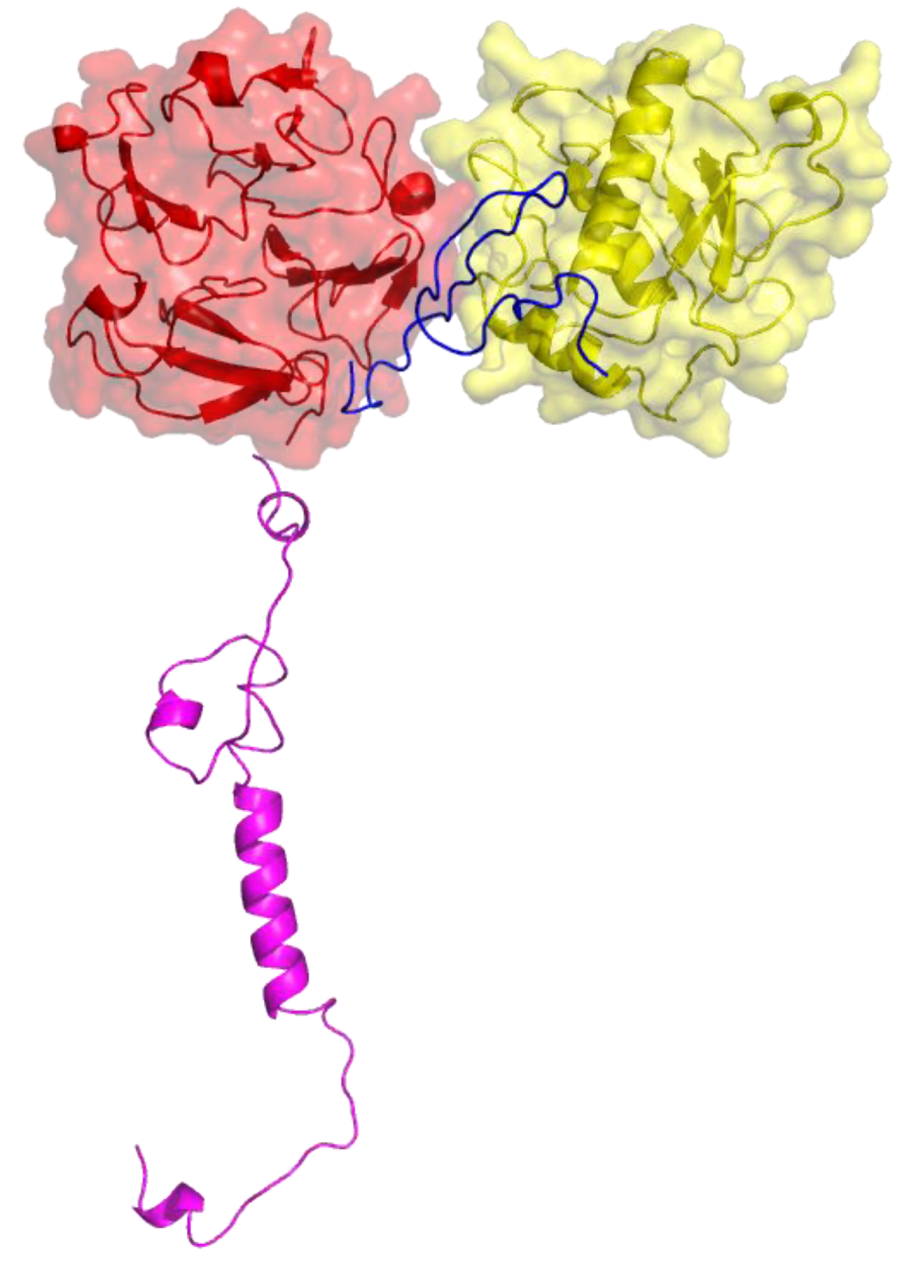{kind=link}
| Enzyme Affected | Peptide Sequence | IC50 (μM) | Reference |
|---|---|---|---|
| MMP-2 | CTTHWGFTLC (CTT) | 5–10 | [29] |
| “ | H-β3-Phe-β-Ala-β3-Trp-β3-His-OH | 225 | [31] |
| “ | HWWQWPSSLQLRGGGS (M204C4) | 78.0 | [32] |
| “ | HNWTRWLLHPDRGGGS (M205C4) | 38.8 | [32] |
| “ | ISYGNDALMP (APP-IP) | 0.030 | [33,34] |
| “ | MCMPCFTTDHQMARKCDDCCGGKGRGKCYGPQCLCR (Cltx) | 0.200 | [35] |
| “ | CGAOGAOGSQGA (P713) | 30 | [36] |
| MMP-9 | CRRHWGFEFC | ND | [29] |
| “ | GACLRSGRGCG (TCTP-1) | ND | [37] |
| “ | NQVDQVGY (IVS4) | 50 a | [38] |
| “ | SRPQGPFL (IS4) | 12 a | [38] |
| “ | FPGVPLDTHDVFQYREK (P3a) | 109–279 a | [39] |
| “ | CQVTGALRSGRGKMLLC-NH2 (cyclic LRSG) | ND | [40] |
| “ | CRVYGPYLLC | 200 b | [41] |
| PRCBCGE (Regasepin1) | ~1 | [42,43] | |
| “ | RC-[D-B]-[D-R] | 0.75 | [44] |
| “ | NENLLRFFVAPFPEVFG | 50 | [45] |
| “ | CSCSDMTDKECLYFCMSEMS (STX-S4-CT) | 1.0 d | [46] |
| MT1-MMP | ISYGNDALMP (APP-IP) | 2.0 | [33,34] |
| “ | GACFSIAHECGA (Peptide G) | 150 | [47] |
| “ | AHQLH | 165 c | [48] |
| “ | acetyl-VMDGYPMP-NH2 (IS4) | ND | [49] |
| “ | acetyl-GYPKSALR-NH2 (IVS4) | ND | [49] |
| “ | VFDEASLEP | 238 | Present study |
| MMP-1 | CSCSDMTDKECLYFCMSEMS (STX-S4-CT) | 4.5 d | [46] |
| Enzyme | Substrate | Ki(app) (μM) |
|---|---|---|
| MMP-2 | DQ gelatin | 52.26 ± 5.110 |
| “ | Knight SSP a | NI |
| “ | fTHP-15 b | NI |
| “ | α1(V)436-447 fTHP c | 143.5 ± 11.40 |
| MMP-9 | DQ gelatin | 54.42 ± 7.616 |
| “ | Knight SSP a | NI |
| “ | fTHP-15 b | NI |
| “ | α1(V)436-447 fTHP c | 122.7 ± 5.83 |
3. Inhibitors and Probes of the Membrane-Bound MMPs (MT1-MMP/MMP-14)
| Enzyme | Loop Peptide Sequence | IC50 (μM) b |
|---|---|---|
| MT1-MMP | LFW-[Nle]-PNG-NH2 (bIIIs1) c | 15,400 |
| “ | VFDEASLEP-NH2 (bIIs3-4) | 240 |
| “ | RKDGKFV-NH2 (bIIs2) | 3,000 |
| “ | VRNNQV-[Nle]-DGYP-[Nle]-P-NH2 (bIs4) c | 670 |
| “ | NNQKLKVEPGYPKSALRD-NH2 (bIVs4) | 5,400 |
| “ | acetyl-VMDGYPMP-NH2 (IS4) | 3,400 |
| MMP-1 | VFDEASLEP-NH2 (bIIs3-4) | NI |
| “ | VRNNQV-[Nle]-DGYP-[Nle]-P-NH2 (bIs4) c | NI |
4. Inhibitors of Matrix Metalloproteinase 1
5. Conclusions
Acknowledgments
References
- Barrett, A.J.; Rawlings, N.D.; Woessner, J.F. Handbook of Proteolytic Enzymes, 2nd ed; Elsevier/Academic Press: Amsterdam, The Netherlands, 2004; Volume 1. [Google Scholar]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Jackson, B.C.; Nebert, D.W.; Vasiliou, V. Update of human and mouse matrix metalloproteinase families. Hum. Genomics 2010, 4, 194–201. [Google Scholar]
- Murphy, G.; Nagase, H. Progress in matrix metalloproteinase research. Mol. Aspects Med. 2008, 29, 290–308. [Google Scholar] [CrossRef]
- Li, J.; Brick, P.; O’Hare, M.C.; Skarzynski, T.; Lloyd, L.F.; Curry, V.A.; Clark, I.M.; Bigg, H.F.; Hazleman, B.L.; Cawston, T.E.; et al. Structure of full-length porcine synovial collagenase reveals a C-terminal domain containing a calcium-linked, four bladed β-propeller. Structure 1995, 15, 541–549. [Google Scholar]
- Iyer, S.; Visse, R.; Nagase, H.; Acharya, K.R. Crystal structure of an active form of human MMP-1. J. Mol. Biol. 2006, 362, 78–88. [Google Scholar] [CrossRef]
- Morgunova, E.; Tuuttila, A.; Bergmann, U.; Isupov, M.; Lindqvist, Y.; Schneider, G.; Tryggvason, K. Structure of human pro-matrix metalloproteinase-2: Activation mechanism revealed. Science 1999, 284, 1667–1670. [Google Scholar]
- Elkins, P.A.; Ho, S.H.; Smith, W.W.; Janson, C.A.; D'Alessio, K.J.; McQueney, M.S.; Cummings, M.D.; Romanic, A.M. Structure of the C-teminally truncated human proMMP9, a gelatin-binding matrix metalloproteinase. Acta Cryst. 2002, D58, 1182–1192. [Google Scholar]
- Cha, H.; Kopetzki, E.; Huber, R.; Lanzendorfer, M.; Brandstetter, H. Structural basis of the adaptive molecular recognition by MMP-9. J. Mol. Biol. 2002, 320, 1065–1079. [Google Scholar]
- Grossman, M.; Tworowski, D.; Dym, O.; Lee, M.H.; Levy, Y.; Murphy, G.; Sagi, I. The intrinsic protein flexibility of endogenous protease inhibitor TIMP-1 controls its binding interface and affects its function. Biochemistry 2010, 49, 6184–6192. [Google Scholar]
- Tochowicz, A.; Goettig, P.; Evans, R.; Visse, R.; Shitomi, Y.; Palmisano, R.; Ito, N.; Richter, K.; Maskos, K.; Franke, D.; et al. The dimer interface of the membrane type 1 matrix metalloproteinase hemopexin domain: Crystal structure and biological functions. J. Biol. Chem. 2011, 286, 7587–7600. [Google Scholar]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protocols 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008, 9, 40. [Google Scholar]
- Rosenberg, G.A. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet 2009, 8, 205–216. [Google Scholar]
- Hu, J.; Van den Steen, P.E.; Sang, Q.-X.A.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar]
- Troeberg, L.; Nagase, H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim. Biophys. Acta 2012, 1824, 133–145. [Google Scholar]
- Lindsey, M.L.; Zamilpa, R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc. Ther. 2012, 30, 31–41. [Google Scholar] [CrossRef]
- Newby, A.C. Matrix metalloproteinase inhibition therapy for vascular diseases. Vascul. Pharmacol. 2012, 56, 232–244. [Google Scholar] [CrossRef]
- Rivera, S.; Khrestchatisky, M.; Kaczmarek, L.; Rosenberg, G.A.; Jaworski, D.M. Metzincin proteases and their inhibitors: Foes or friends in nervous system physiology? J. Neurosci. 2010, 30, 15337–15357. [Google Scholar]
- Whittaker, M.; Floyd, C.D.; Brown, P.; Gearing, A.J.H. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999, 99, 2735–2776. [Google Scholar] [CrossRef]
- Jacobsen, J.A.; Jourden, J.L.M.; Miller, M.T.; Cohen, S.M. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim. Biophys. Acta 2010, 1803, 72–94. [Google Scholar]
- Saghatelian, A.; Jessani, N.; Joseph, A.; Humphrey, M.; Cravatt, B.F. Activity-based probes for the proteomic profiling of metalloproteases. Proc. Natl. Acad. Sci. USA 2004, 101, 10000–10005. [Google Scholar]
- Morrison, C.J.; Butler, G.S.; Rodríguez, D.; Overall, C.M. Matrix metalloproteinase proteomics: Substrates, targets, and therapy. Curr. Opin. Cell Biol. 2009, 21, 645–653. [Google Scholar]
- Sela-Passwell, N.; Trahtenherts, A.; Krüger, A.; Sagi, I. New opportunities in drug design of metalloproteinase inhibitors: Combination between structure-function experimental approaches and systems biology. Expert Opin. Drug Discov. 2011, 6, 527–542. [Google Scholar] [CrossRef]
- Sela-Passwell, N.; Rosenblum, G.; Shoham, T.; Sagi, I. Structural and functional bases for allosteric control of MMP activities: Can it pave the path for selective inhibition? Biochim. Biophys. Acta 2010, 1803, 29–38. [Google Scholar]
- Bergers, G.; Brekken, R.A.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biol. 2000, 2, 737–744. [Google Scholar]
- Zamilpa, R.; Lopez, E.F.; Chiao, Y.A.; Dai, Q.; Escobar, G.P.; Hakala, K.; Weintraub, S.T.; Lindsey, M.L. Proteomic analysis identifies in vivo candidate matrix metalloproteinase-9 substrates in the left ventricle post-myocardial infarction. Proteomics 2010, 10, 2214–2223. [Google Scholar]
- Fernandes, K.S.; Brum, D.G.; Palei, A.C.; Sandrim, V.C.; Guerreiro, C.T.; Tanus-Santos, J.E.; Barreira, A.A. Functional MMP-9 polymorphisms modulate plasma MMP-9 levels in multiple sclerosis patients. J. Neuroimmunol. 2012, 249, 56–59. [Google Scholar] [CrossRef]
- Koivunen, E.; Arap, W.; Valtanen, H.; Rainisalo, A.; Medina, O.P.; Heikkila, P.; Kantor, C.; Gahmberg, C.G.; Salo, T.; Konttinen, Y.T.; et al. Tumor targeting with a selective gelatinase inhibitor. Nat. Biotech. 1999, 17, 768–774. [Google Scholar] [CrossRef]
- Medina, O.P.; Kairemo, K.; Valtanen, H.; Kangasniemi, A.; Kaukinen, S.; Ahonen, I.; Permi, P.; Annila, A.; Sneck, M.; Holopainen, J.M.; et al. Radionuclide imaging of tumor xenografts in mice using a gelatinase-targeting peptide. Anticancer Res. 2005, 25, 33–42. [Google Scholar]
- Mukai, T.; Suganuma, N.; Soejima, K.; Sasaki, J.; Yamamoto, F.; Maeda, M. Synthesis of a beta-tetrapeptide analog as a mother compound for the development of matrix metalloproteinase-2-imaging agents. Chem. Pharm. Bull. 2008, 56, 260–265. [Google Scholar]
- Lu, G.; Zheng, M.; Zhu, Y.; Sha, M.; Wu, Y.; Han, X. Selection of peptide inhibitor to matrix metalloproteinase-2 using phage display and its effects on pancreatic cancer cell lines PANC-1 and CFPAC-1. Int. J. Biol. Sci. 2012, 8, 650–662. [Google Scholar]
- Hashimoto, H.; Takeuchi, T.; Komatsu, K.; Miyazaki, K.; Sato, M.; Higashi, S. Structural basis for matrix metalloproteinase-2 (MMP-2)-selective inhibitory action of beta-amyloid precursor protein-derived inhibitor. J. Biol. Chem. 2011, 286, 33236–33243. [Google Scholar]
- Higashi, S.; Miyazaki, K. Identification of a region of β-amyloid precursor protein essential for its gelatinase A inhibitory activity. J. Biol. Chem. 2003, 278, 14020–14028. [Google Scholar] [CrossRef]
- Deshane, J.; Garner, C.C.; Sontheimer, H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J. Biol. Chem. 2003, 278, 4135–4144. [Google Scholar]
- Xu, X.; Chen, Z.; Wang, Y.; Bonewald, L.; Steffensen, B. Inhibition of MMP-2 gelatinolysis by targeting exodomain-substrate interactions. Biochem. J. 2007, 406, 147–155. [Google Scholar] [CrossRef]
- Ujula, T.; Huttunen, M.; Luoto, P.; Peräkylä, H.; Simpura, I.; Wilson, I.; Bergman, M.; Roivainen, A. Matrix metalloproteinase 9 targeting peptides: Syntheses, 68Ga-labeling, and preliminary evaluation in a rat melanoma xenograft model. Bioconjugate Chem. 2010, 21, 612–1621. [Google Scholar]
- Dufour, A.; Zucker, S.; Sampson, N.S.; Kuscu, C.; Cao, J. Role of matrix metalloproteinase-9 dimers in cell migration: design of inhibitory peptides. J. Biol. Chem. 2010, 285, 35944–35956. [Google Scholar]
- Ugarte-Berzal, E.; Bailón, E.; Amigo-Jiménez, I.; Vituri, C.L.; del Cerro, H.; Terol, M.J.; Albar, J.P.; Rivas, G.; García-Marco, J.A.; Garcío-Pardo, A. A 17-residue sequence from the matrix metalloproteinase-9 (MMP-9) hemopexin domain binds α4β1 integrin and inhibits MMP-9-induced functions in chronic lymphocytic leukemia B cells. J. Biol. Chem. 2012, 287, 27601–27613. [Google Scholar]
- Radjabi, A.R.; Sawada, K.; Jagadeeswaran, S.; Eichbichler, A.; Kenny, H.A.; Montag, A.; Bruno, K.; Lengyel, E. Thrombin induces tumor invasion through the induction and association of matrix metalloproteinase-9 and beta1-integrin on the cell surface. J. Biol. Chem. 2008, 283, 2822–2834. [Google Scholar]
- Björklund, M.; Heikkilä, P.; Koivunen, E. Peptide inhibition of catalytic and noncatalytic activities of matrix metalloproteinase-9 blocks tumor cell migration and invasion. J. Biol. Chem. 2004, 279, 29589–29597. [Google Scholar]
- Hu, J.; Van den Steen, P.E.; Dillen, C.; Opdenakker, G. Targeting neutrophil collagenase/matrix metalloproteinase-8 and gelatinase B/matrix metalloproteinase-9 with a peptidomimetic inhibitor protects against endotoxin shock. Biochem. Pharmacol. 2005, 70, 535–544. [Google Scholar]
- Hu, J.; Fiten, P.; Van den Steen, P.E.; Chaltin, P.; Opdenakker, G. Simulation of evolution-selected propeptide by high-throughput selection of a peptidomimetic inhibitor on a capillary DNA sequencer platform. Anal. Chem. 2005, 77, 2116–2124. [Google Scholar] [CrossRef]
- Qiu, Z.; Yan, M.; Li, Q.; Liu, D.; Van den Steen, P.E.; Wang, M.; Opdenakker, G.; Hu, J. Definition of peptide inhibitors from a synthetic peptide library by targeting gelatinase B/matrix metalloproteinase-9 (MMP-9) and TNF-alpha converting enzyme (TACE/ADAM-17). J. Enzym. Inhib. Med. Chem. 2012, 27, 533–540. [Google Scholar]
- Juillerat-Jeanneret, L.; Robert, M.C.; Juillerat, M.A. Peptides from Lactobacillus hydrolysates of bovine milk caseins inhibit prolyl-peptidases of human colon cells. J. Agric. Food Chem. 2011, 59, 370–377. [Google Scholar] [CrossRef]
- Lauer-Fields, J.L.; Marí, F.; Wei, S.; Fields, G.B.; Brew, K. Engineered Sarafotoxins as TIMP-like MMP Inhibitors. J. Biol. Chem. 2007, 282, 26948–26955. [Google Scholar]
- Suojanen, J.; Salo, T.; Koivunen, E.; Sorsa, T.; Pirila, E. A novel and selective membrane type-1 matrix metalloproteinase (MT1-MMP) inhibitor reduces cancer cell motility and tumor growth. Cancer Biol. Ther. 2009, 8, 2362–2370. [Google Scholar]
- Liang, Z.J.; Huang, J.S.; Cui, J.; Xiong, L.L.; Mao, C.Q. Selection and molecular simulation of binding peptides dual-targeting MMP-14 and metal ions. Chin. J. Biochem. Mol. Biol. 2011, 27, 341–349. [Google Scholar]
- Zarrabi, K.; Dufour, A.; Li, J.; Kuscu, C.; Pulkoski-Gross, A.; Zhi, J.; Hu, Y.; Sampson, N.S.; Zucker, S.; Cao, J. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J. Biol. Chem. 2011, 286, 33167–33177. [Google Scholar]
- Kuhnast, B.; Bodenstein, C.; Haubner, R.; Wester, H.J.; Senekowitsch-Schmidtke, R.; Schwaiger, M.; Weber, W.A. Targeting of gelatinase activity with a radiolabeled cyclic HWGF peptide. Nucl. Med. Biol. 2004, 31, 337–344. [Google Scholar] [CrossRef]
- Sprague, J.E.; Li, W.P.; Liang, K.; Achilefu, S.; Anderson, C.J. In vitro and in vivo investigation of matrix metalloproteinase expression in metastatic tumor models. Nucl. Med. Biol. 2006, 33, 227–237. [Google Scholar]
- Hanaoka, H.; Mukai, T.; Habashita, S.; Asano, D.; Ogawa, K.; Kuroda, Y.; Akizawa, H.; Iida, Y.; Endo, K.; Saga, T.; et al. Chemical design of a radiolabeled gelatinase inhibitor peptide for the imaging of gelatinase activity in tumors. Nucl. Med. Biol. 2007, 34, 503–510. [Google Scholar]
- Suojanen, J.; Vilen, S.-T.; Nyberg, P.; Heikkilä, P.; Penate-Medina, O.; Saris, P.E.J.; Hagström, J.; Ranta, T.-M.; Salo, T.; Sorsa, T.; et al. Selective gelatinase inhibitor peptide is effective in targeting tongue carcinoma cell tumors in vivo. Anticancer Res. 2011, 31, 3659–3664. [Google Scholar]
- Cheng, R.P.; Gellman, S.H.; DeGrado, W.F. β-Peptides: From structure to function. Chem. Rev. 2001, 101, 3219–3232. [Google Scholar]
- Gademann, K.; Kimmerlin, T.; Hoyer, D.; Seebach, D. Peptide folding induces high and selective affinity of a linear and small beta-peptide to the human somatostatin receptor 4. J. Med. Chem. 2001, 44, 2460–2468. [Google Scholar] [CrossRef]
- Seebach, D.; Beck, A.K.; Bierbaum, D.J. The world of β- and γ-peptides comprised of homologated proteinogenic amino acids and other components. Chem. Biodiver. 2004, 1, 1111–1239. [Google Scholar] [CrossRef]
- Steer, D.L.; Lew, R.A.; Perlmutter, P.; Smith, A.I.; Aguilar, M.I. β-amino acids: Versatile peptidomimetics. Curr. Med. Chem. 2002, 9, 811–822. [Google Scholar] [CrossRef]
- Sisodia, S.S.; Koo, E.H.; Beyreuther, K.; Unterbeck, A.; Price, D.L. Evidence that β-amyloid protein in Alzheimer’s disease is not derived by normal processing. Science 1990, 248, 492–495. [Google Scholar]
- Esch, F.S.; Keim, P.S.; Beattie, E.C.; Blacher, R.W.; Culwell, A.R.; Oltersdorf, T.; McClure, D.; Ward, P.J. Cleavage of amyloid β peptide during constitutive processing of its precursor. Science 1990, 248, 1122–1124. [Google Scholar]
- Miyazaki, K.; Hasegawa, M.; Funahashi, K.; Umeda, M. A metalloproteinase inhibitor domain in Alzheimer amyloid protein precursor. Nature 1993, 362, 839–841. [Google Scholar]
- Higashi, S.; Miyazaki, K. Novel processing of β-amyloid precursor protein catalyzed by membrane type 1 matrix metalloproteinase releases a fragment lacking the inhibitor domain against gelatinase A. Biochemistry 2003, 42, 6514–6526. [Google Scholar] [CrossRef]
- Higashi, S.; Miyazaki, K. Identification of amino acid residues of the matrix metalloproteinase-2 essential for its selective inhibition by β-amyloid precursor protein-derived inhibitor. J. Biol. Chem. 2008, 283, 10068–10078. [Google Scholar] [CrossRef]
- DeBin, J.A.; Strichartz, G.R. Chloride channel inhibition by the venom of the scorpion Leiurus quinquestriatus. Toxicon 1991, 29, 1403–1408. [Google Scholar] [CrossRef]
- Soroceanu, L.; Gillespie, Y.; Khazaeli, M.B.; Sontheimer, H. Use of chlorotoxin for targeting of primary brain tumors. Cancer Res. 1998, 58, 4871–4879. [Google Scholar]
- Lyons, S.A.; O’Neal, J.; Sontheimer, H. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia 2002, 39, 162–173. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Ratnikov, B.; Monosov, E.; Postnova, T.I.; DiScipio, R.; Smith, J.W.; Strongin, A.Y. MT1-MMP initiates activation of pro-MMP-2 and integrin avb3 promotes maturation of MMP-2 in breast carcinoma cells. Exp. Cell Res. 2001, 263, 209–223. [Google Scholar] [CrossRef]
- Veiseh, M.; Gabikian, P.; Bahrami, S.-B.; Veiseh, O.; Zhang, M.; Hackman, R.C.; Ravanpay, A.C.; Stroud, M.R.; Kusuma, Y.; Hansen, S.J.; et al. Tumor paint: A chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res. 2007, 67, 6882–6888. [Google Scholar]
- Banyai, L.; Patthy, L. Evidence for the involvement of type II domains in collagen binding by 72 kDa type IV procollagenase. FEBS Lett. 1991, 282, 23–25. [Google Scholar] [CrossRef]
- Senior, R.M.; Griffin, G.L.; Fliszar, C.J.; Shapiro, S.D.; Goldberg, G.I.; Welgus, H.G. Human 92- and 72-kilodalton type IV collagenases are elastases. J. Biol. Chem. 1991, 266, 7870–7875. [Google Scholar]
- Shipley, J.M.; Doyle, G.A.; Fliszar, C.J.; Ye, Q.Z.; Johnson, L.L.; Shapiro, S.D.; Welgus, H.G.; Senior, R.M. The structural basis for the elastolytic activity of the 92-kDa and 72-kDa gelatinases. Role of the fibronectin type II-like repeats. J. Biol. Chem. 1996, 271, 4335–4341. [Google Scholar]
- Steffensen, B.; Bigg, H.F.; Overall, C.M. The involvement of the fibronectin type II-like modules of human gelatinase A in cell surface localization and activation. J. Biol. Chem. 1998, 273, 20622–20628. [Google Scholar] [CrossRef]
- Steffensen, B.; Wallon, U.M.; Overall, C.M. Extracellular matrix binding properties of recombinant fibronectin type II-like modules of human 72-kDa gelatinase/type IV collagenase. High affinity binding to native type I collagen but not native type IV collagen. J. Biol. Chem. 1995, 270, 11555–11566. [Google Scholar] [CrossRef]
- Murphy, G.; Nguyen, Q.; Cockett, M.I.; Atkinson, S.J.; Allan, J.A.; Knight, C.G.; Willenbrock, F.; Docherty, A.J. Assessment of the role of the fibronectin-like domain of gelatinase A by analysis of a deletion mutant. J. Biol. Chem. 1994, 269, 6632–6636. [Google Scholar]
- Lauer-Fields, J.L.; Whitehead, J.K.; Li, S.; Hammer, R.P.; Brew, K.; Fields, G.B. Selective modulation of matrix metalloproteinase 9 (MMP-9) functions via exosite inhibition. J. Biol. Chem. 2008, 283, 20087–20095. [Google Scholar]
- Tauzin, J.; Miclo, L.; Gaillard, J.L. Angiotensin-I-converting enzyme inhibitory peptides from tryptic hydrolysate of bovine αS2-casein. FEBS Lett. 2002, 531, 369–374. [Google Scholar]
- Robert, M.C.; Razaname, A.; Mutter, M.; Juillerat, M.A. Identification of angiotensin-I-converting enzyme inhibitory peptides derived from sodium caseinate hydrolysates produced by Lactobacillus helveticus NCC 2765. J. Agric. Food Chem. 2004, 52, 6923–6931. [Google Scholar] [CrossRef]
- Mook, O.R.; Frederiks, W.M.; Van Noorden, C.J. The role of gelatinases in colorectal cancer progression and metastasis. Biochim. Biophys. Acta 2004, 1705, 69–89. [Google Scholar]
- Basset, P.; Okada, A.; Chenard, M.P.; Kannan, R.; Stoll, I.; Anglard, P.; Bellocq, J.P.; Rio, M.C. Matrix metalloproteinases as stromal effectors of human carcinoma progression: therapeutic implications. Matrix Biol. 1997, 15, 535–541. [Google Scholar] [CrossRef]
- Redondo-Munoz, J.; Ugarte-Berzal, E.; Garcia-Marco, J.A.; del Cerro, M.H.; Van den Steen, P.E.; Opdenakker, G.; Terol, M.J.; Garcia-Pardo, A. α4β1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood 2008, 112, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Dufour, A.; Sampson, N.S.; Zucker, S.; Cao, J. Role of the hemopexin domain of matrix metalloproteinases in cell migration. J. Cell. Physiol. 2008, 217, 643–651. [Google Scholar]
- Stefanidakis, M.; Karjalainen, K.; Jaalouk, D.E.; Gahmberg, C.G.; O’Brien, S.; Pasqualini, R.; Arap, W.; Koivunen, E. Role of leukemia cell invadosome in extramedullary infiltration. Blood 2009, 114, 3008–3017. [Google Scholar] [CrossRef]
- Sabeh, F.; Li, X.Y.; Saunders, T.L.; Rowe, R.G.; Weiss, S.J. Secreted versus membrane-anchored collagenases: Relative roles in fibroblast-dependent collagenolysis and invasion. J. Biol. Chem. 2009, 284, 23001–23011. [Google Scholar] [CrossRef]
- Van de Vijver, M.J.; He, Y.D.; van’t Veer, L.J.; Dai, H.; Hart, A.A.; Voskuil, D.W.; Schreiber, G.J.; Peterse, J.L.; Roberts, C.; Marton, M.J.; et al. A gene-expression signature as a predictor of survival in breast cancer. New Eng. J. Med. 2002, 347, 1999–2009. [Google Scholar]
- Wang, Y.; Klijn, J.G.; Zhang, Y.; Sieuwerts, A.M.; Look, M.P.; Yang, F.; Talantov, D.; Timmermans, M.; Meijer-van Gelder, M.E.; Yu, J.; et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005, 365, 671–679. [Google Scholar]
- Pawitan, Y.; Bjohle, J.; Amler, L.; Borg, A.L.; Egyhazi, S.; Hall, P.; Han, X.; Holmberg, L.; Huang, F.; Klaar, S.; et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005, 7, R953–R964. [Google Scholar]
- Seftor, R.E.; Seftor, E.A.; Kirschmann, D.A.; Hendrix, M.J. Targeting the tumor microenvironment with chemically modified tetracyclines: inhibition of laminin 5 γ2 chain promigratory fragments and vasculogenic mimicry. Mol. Cancer Ther. 2002, 1, 1173–1179. [Google Scholar]
- Oku, N.; Sasabe, E.; Ueta, E.; Yamamoto, T.; Osaki, T. Tight junction protein claudin-1 enhances the invasive activity of oral squamous cell carcinoma cells by promoting cleavage of laminin-5 γ2 chain via matrix metalloproteinase (MMP)-2 and membrane-type MMP-1. Cancer Res. 2006, 66, 5251–5257. [Google Scholar] [CrossRef]
- Liang, Z.; Huang, J.; Huang, T.; Cui, J.; Zeng, L.; Xiong, L.; Wu, F.; Mao, C. Selection and finding of lead peptides dual-targeting MMP-14 and metal ions by subtractive cell surface panning and molecular docking. Int. J. Pept. Res. Ther. 2012, 18, 31–40. [Google Scholar] [CrossRef]
- Mori, H.; Tomari, T.; Koshikawa, N.; Kajita, M.; Itoh, Y.; Sato, H.; Tojo, H.; Yana, I.; Seiki, M. CD44 directs membrane-type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin-like domain. EMBO J. 2002, 21, 3949–3959. [Google Scholar] [CrossRef]
- Lichte, A.; Kolkenbrock, H.; Tschesche, H. The recombinant catalytic domain of membrane-type matrix metalloproteinase-1 (MT1-MMP) induces activation of progelatinase A and progelatinase A complexed with TIMP-2. FEBS Lett. 1996, 397, 277–282. [Google Scholar]
- Cao, J.; Kozarekar, P.; Pavlaki, M.; Chiarelli, C.; Bahou, W.F.; Zucker, S. Distinct roles for the catalytic and hemopexin domains of membrane type 1-matrix metalloproteinase in substrate degradation and cell migration. J. Biol. Chem. 2004, 279, 14129–14139. [Google Scholar]
- Zhu, L.; Wang, H.; Wang, L.; Wang, Y.; Jiang, K.; Li, C.; Ma, Q.; Gao, S.; Wang, L.; Li, W.; et al. High-affinity peptide against MT1-MMP for in vivo tumor imaging. J. Control. Release 2011, 150, 248–255. [Google Scholar] [CrossRef]
- Overall, C.M.; Kleifeld, O. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [Google Scholar] [CrossRef]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases (TIMPs): evolution, structure and function. Biochem. Biophys. Acta 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta 2010, 2010, 55–71. [Google Scholar] [CrossRef]
- Nagase, H.; Brew, K. Engineering of tissue inhibitor of metalloproteinases mutants as potential therapeutics. Arthritis Res. 2002, 4 (Suppl. 3), S51–S61. [Google Scholar]
- Wei, S.; Chen, Y.; Chung, L.; Nagase, H.; Brew, K. Protein engineering of the tissue inhibitor of metalloproteinase 1 (TIMP-1) inhibitory domain. J. Biol. Chem. 2003, 278, 9831–9834. [Google Scholar]
- Hamze, A.B.; Wei, S.; Bahudhanapati, H.; Kota, S.; Acharya, K.R.; Brew, K. Constraining specificity in the N-domain of tissue inhibitor of metalloproteinases-1; gelatinase-selective inhibitors. Protein Sci. 2007, 16, 1905–1913. [Google Scholar]
- Bahudhanapati, H.; Zhang, Y.; Sidhu, S.S.; Brew, K. Phage display of tissue inhibitor of metalloproteinases-2 (TIMP-2). J. Biol. Chem. 2011, 286, 31761–31770. [Google Scholar]
- Schlippe, Y.V.G.; Hartman, M.C.T.; Josephson, K.; Szostak, J.W. In vitro selection of highly modified cyclic peptides that act as tight binding inhibitors. J. Am. Chem. Soc. 2012, 134, 10469–10477. [Google Scholar]
- Sample Availability: Contact the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ndinguri, M.W.; Bhowmick, M.; Tokmina-Roszyk, D.; Robichaud, T.K.; Fields, G.B. Peptide-Based Selective Inhibitors of Matrix Metalloproteinase-Mediated Activities. Molecules 2012, 17, 14230-14248. https://doi.org/10.3390/molecules171214230
Ndinguri MW, Bhowmick M, Tokmina-Roszyk D, Robichaud TK, Fields GB. Peptide-Based Selective Inhibitors of Matrix Metalloproteinase-Mediated Activities. Molecules. 2012; 17(12):14230-14248. https://doi.org/10.3390/molecules171214230
Chicago/Turabian StyleNdinguri, Margaret W., Manishabrata Bhowmick, Dorota Tokmina-Roszyk, Trista K. Robichaud, and Gregg B. Fields. 2012. "Peptide-Based Selective Inhibitors of Matrix Metalloproteinase-Mediated Activities" Molecules 17, no. 12: 14230-14248. https://doi.org/10.3390/molecules171214230