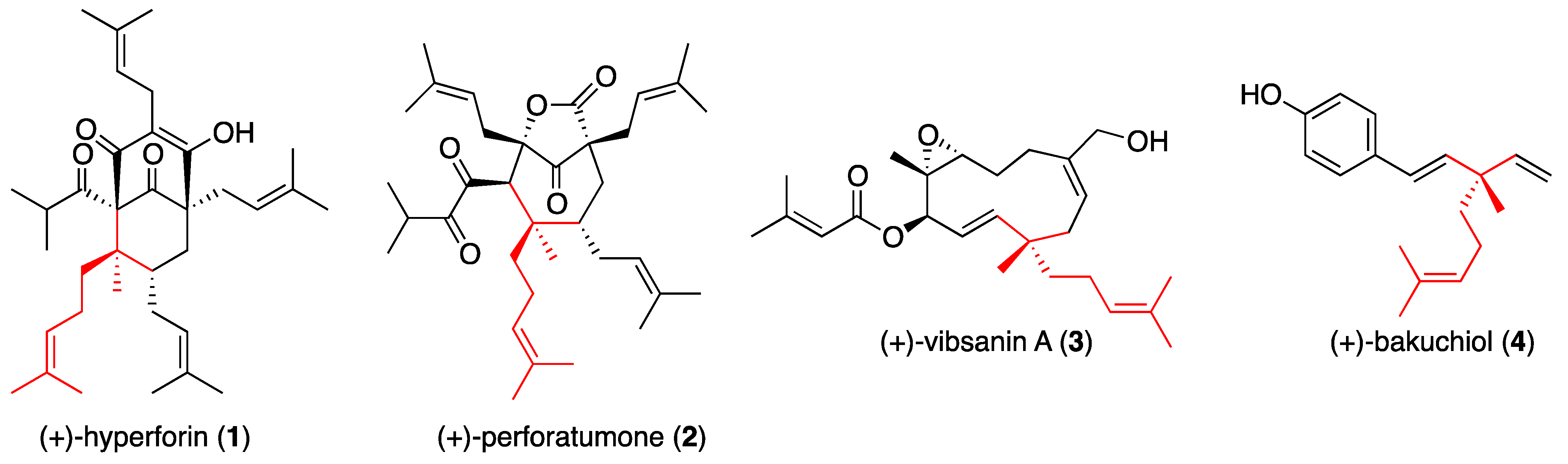
General
Melting points are uncorrected. Specific rotations were measured in a 100 mm cell. 1H-NMR spectra were recorded at 500 MHz with tetramethylsilane as an internal standard on a JEOL JNM-ECA500 spectrometer. 13C-NMR spectra were recorded at 125 MHz. All spectra were recorded in CDCl3. High-resolution mass spectra (HRMS) were measured in EI mode (70 eV) on a JEOL JMS-GCmate spectrometer. Thin-layer chromatography (TLC) was performed on Merck Kieselgel 60 F254 plates. The crude reaction mixtures and extracted materials were purified by column chromatography on Silica gel 60 (Merck) or Wakogel C-300 (Wako). Unless otherwise noted, reactions were carried out at room temperature. Combined organic extracts were dried over anhydrous Na2SO4. Solvents were removed from the reaction mixture and the combined organic extracts by concentration under reduced pressure using an evaporator with bath at 35–45 °C.
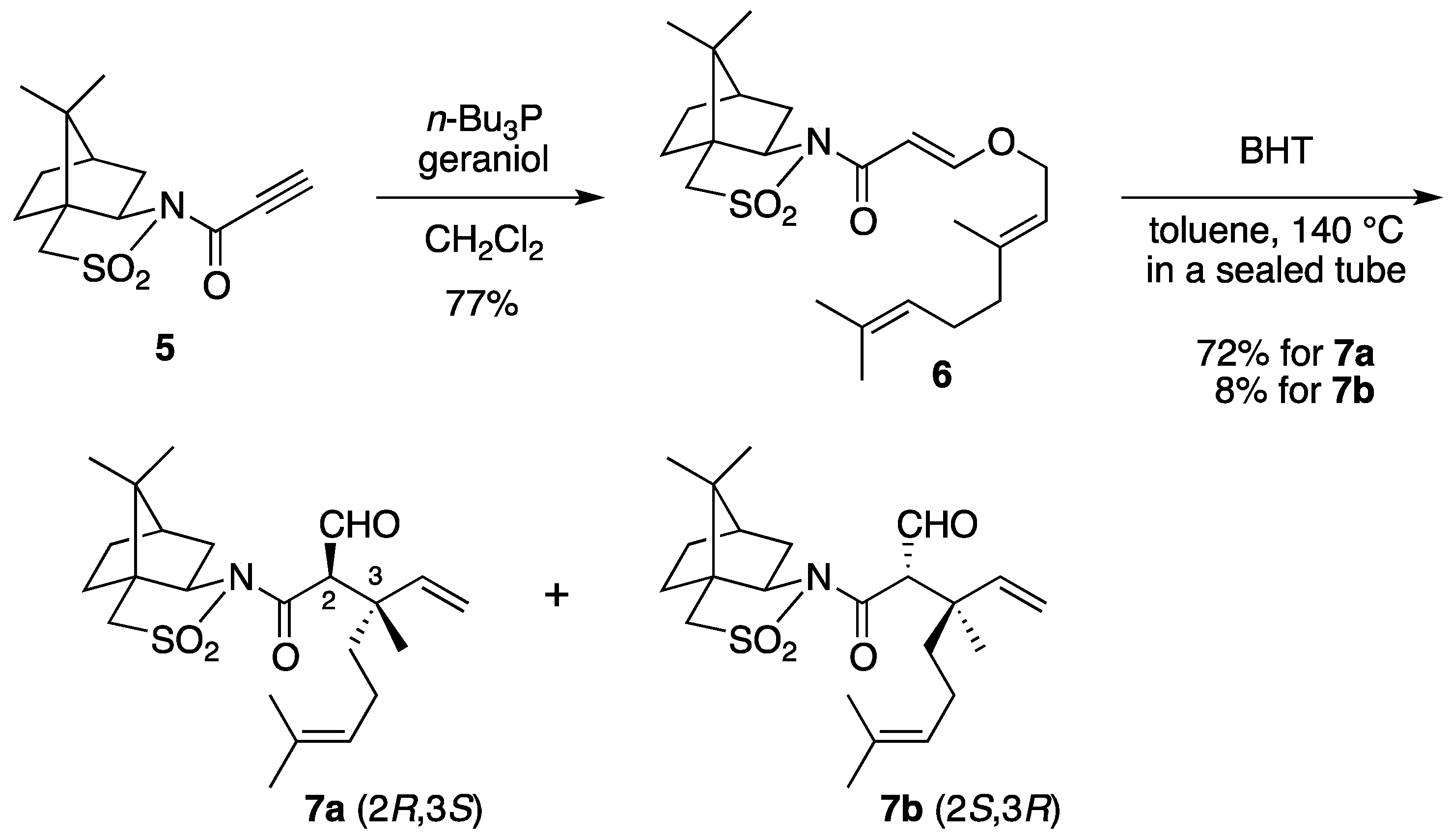
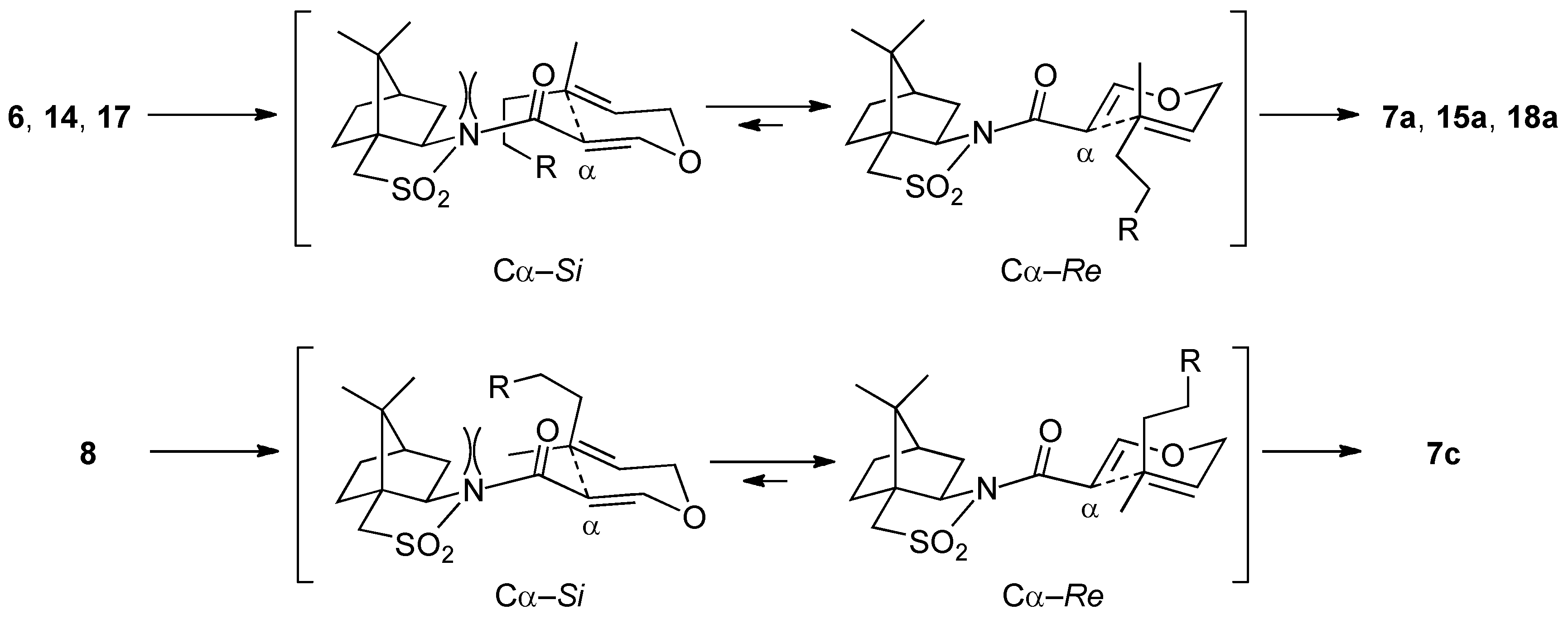
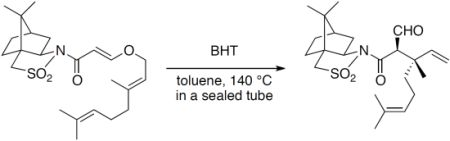
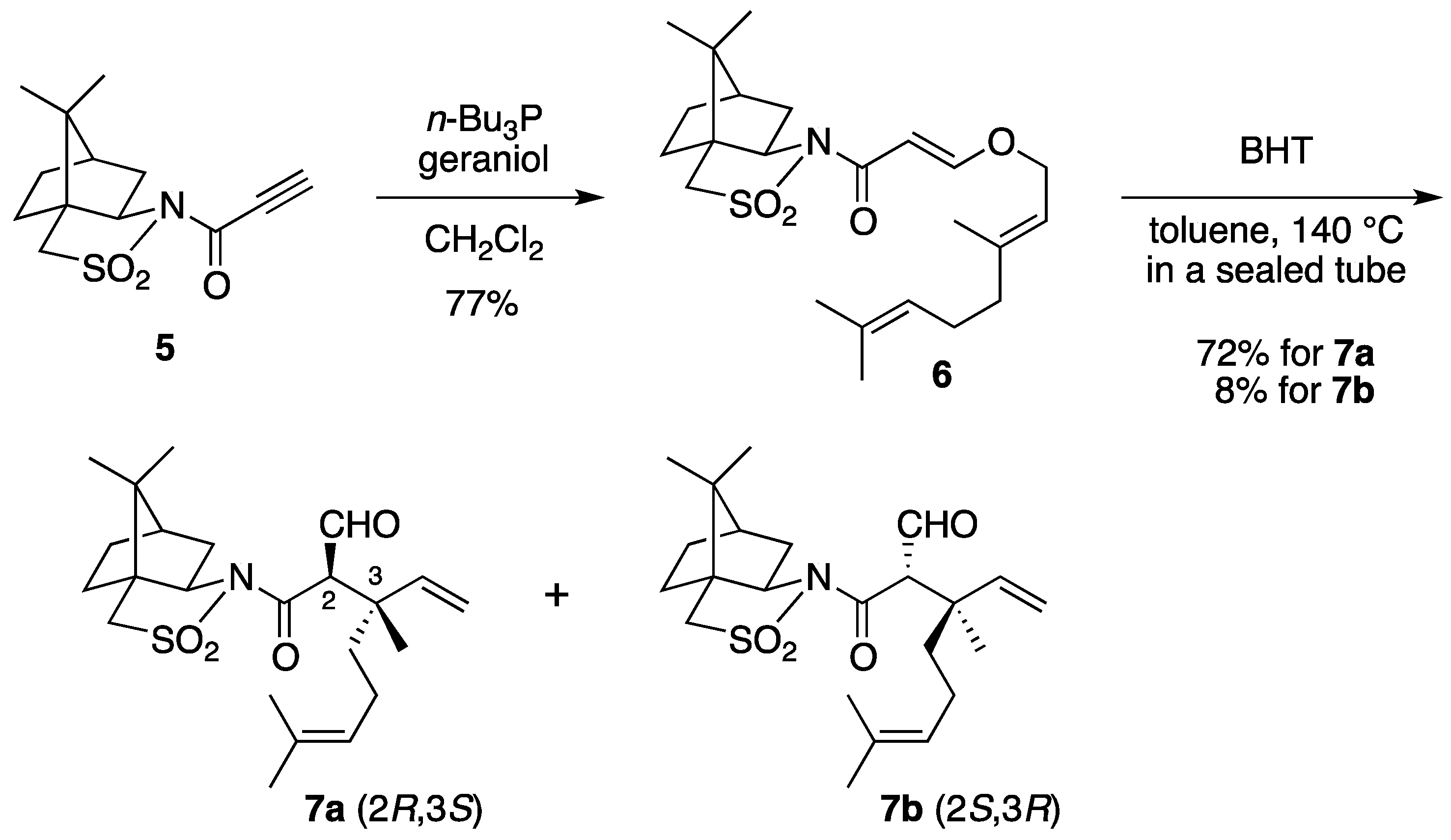
(2R)-N-{(E)-3-[((2E)-3,7-Dimethylocta-2,6-dien-1-yl)oxy]acryloyl}bornane-10,2-sultam (6). The following reaction was carried out under Ar. To a cooled (0 °C) stirred solution of 5 (302 mg, 1.13 mmol) in CH2Cl2 (11 mL) were added geraniol (218 μL, 1.24 mmol) and n-Bu3P (42 μL, 0.17 mmol). The mixture was stirred at 0 °C for 30 min, diluted with H2O (20 mL), and extracted with CH2Cl2 (10 mL × 3). The combined extracts were washed with saturated brine (20 mL), dried and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:30) to provide 368 mg (77%) of 6 as a colorless oil: TLC Rf 0.54 (EtOAc/hexane, 1:3); [α]D19–59.2 (c 1.19, CHCl3); IR (neat) 2962, 2885, 1678, 1608 cm−1; 1H-NMR (500 MHz): δ 0.97 (s, 3H), 1.18 (s, 3H), 1.34–1.45 (m, 2H), 1.60 (br s, 3H), 1.68 (br s, 3H), 1.71 (br s, 3H), 1.86–1.91 (m, 3H), 2.05–2.17 (m, 6H), 3.43 (d, 1H, J = 13.8 Hz), 3.48 (d, 1H, J = 13.8 Hz), 3.91 (dd, 1H, J = 4.9, 7.7 Hz), 4.45 (d, 2H, J = 6.9 Hz), 5.08 (m, 1H), 5.37 (qt, 1H, J = 1.0, 6.9 Hz), 5.97 (d, 1H, J = 12.1 Hz), 7.70 (d, 1H, J = 12.1 Hz); 13C-NMR (125 MHz) δ16.6, 17.6, 19.9, 20.7, 25.6, 26.1, 26.5, 32.7, 38.5, 39.4, 44.6, 47.7, 48.2, 53.0, 65.0, 68.1, 97.0, 117.5, 123.5, 131.9, 143.4, 163.3, 164.9; HRMS calcd for C23H35NO4S (M+) m/z 421.2287, found 421.2286.
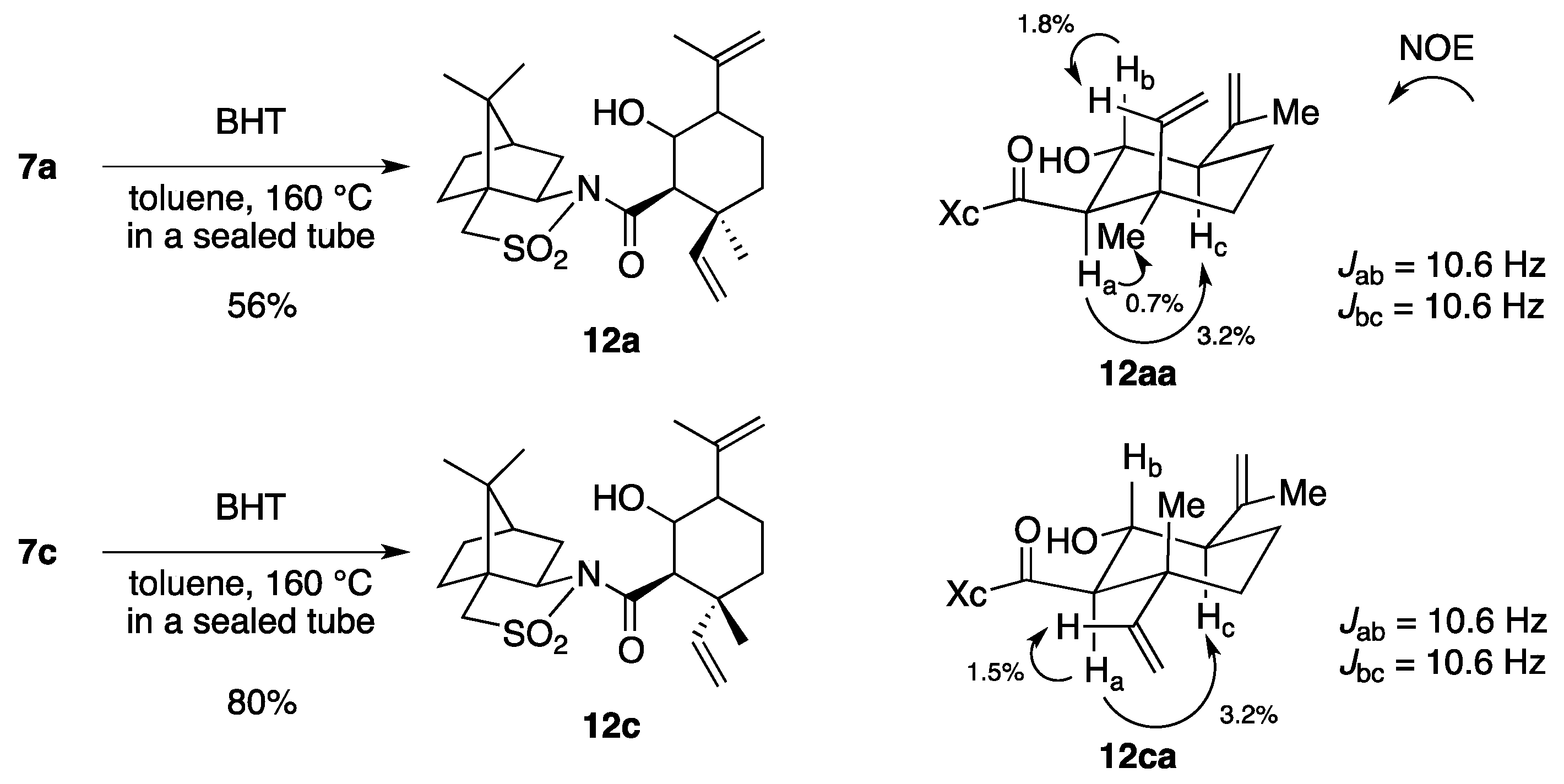
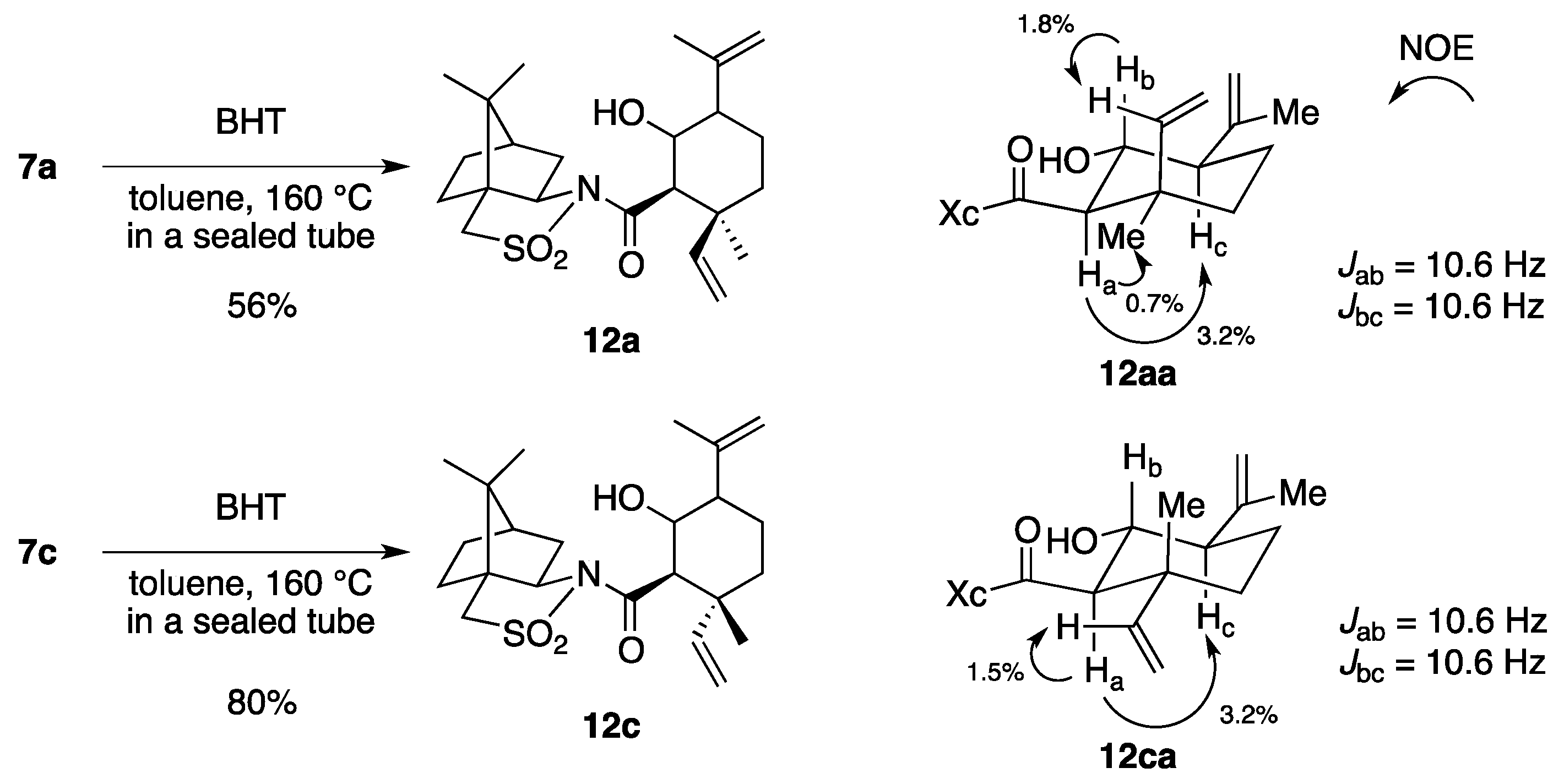
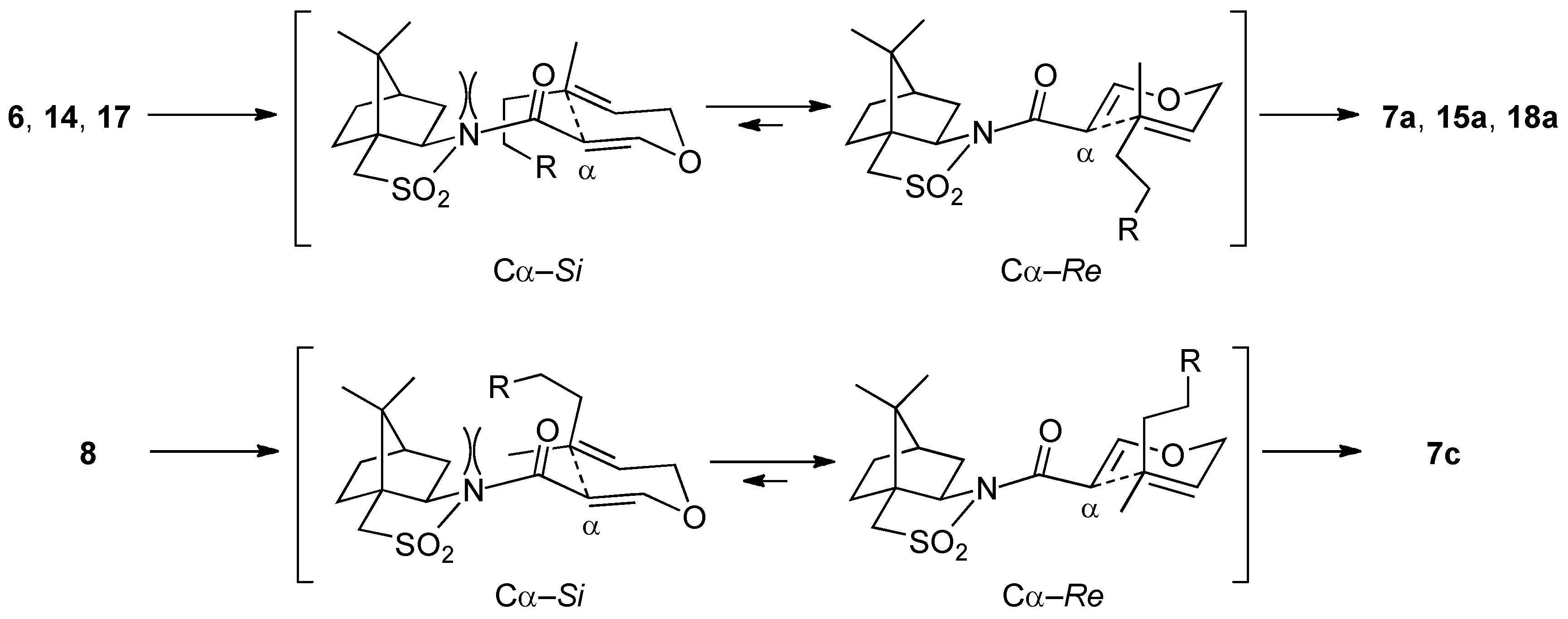
(2R)-N-[(2R,3S)-2-Formyl-3,7-dimethyl-3-vinyloct-6-enoyl]bornane-10,2-sultam (7a) and (2R)-N-[(2S,3R)]-isomer (7b). A solution of 6 (400 mg, 949 μmol) and BHT (10.5 mg, 47.5 μmol) in toluene (50 mL) was stirred at 140 °C for 65 h in a sealed tube and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:30) to provide 289 mg (72%) of 7a and 30.9 mg (8%) of 7b. Compound 7a was obtained as white crystals: mp 84–87 °C; TLC Rf 0.49 (EtOAc/hexane, 1:3); [α]D21–77.9 (c 2.55, CHCl3); IR (neat) 2960, 2925, 1730, 1680 cm−1; 1H-NMR (500 MHz) δ 0.98 (s, 3H), 1.15 (s, 3H), 1.26 (s, 3H), 1.37–1.46 (m, 3H), 1.56 (br s, 3H), 1.65 (br s, 3H), 1.67 (m, 1H), 1.87–1.96 (m, 5H), 2.06-2.15 (m, 2H), 3.44 (d, 1H, J = 13.8 Hz), 3.51 (d, 1H, J = 13.8 Hz), 3.96 (dd, 1H, J = 5.2, 7.4 Hz), 4.01 (d, 1H, J = 2.3 Hz), 5.02 (m, 1H), 5.07 (d, 1H, J = 17.4 Hz), 5.21 (d, 1H, J = 10.6 Hz), 5.92 (dd, 1H, J = 10.6, 17.4 Hz), 9.61 (d, 1H, J = 2.3 Hz); 13C-NMR (125 MHz) δ 17.6, 19.7, 19.9, 20.7, 22.2, 25.6, 26.4, 32.9, 38.5 (2C), 44.8, 45.5, 47.7, 48.1, 53.2, 65.4 (2C), 115.2, 123.7, 131.9, 142.3, 167.5, 197.3; HRMS calcd for C23H35NO4S (M+) m/z 421.2287, found 421.2283. Compound 7b was obtained as white crystals: mp 81–87 °C; TLC Rf 0.61 (EtOAc/hexane, 1:3); [α]D17+38.5 (c 0.965, CHCl3); IR (neat) 2960, 2925, 1730, 1700 cm−1; 1H-NMR (500 MHz) δ 0.95 (s, 3H), 1.10 (s, 3H), 1.31 (s, 3H), 1.34–1.43 (m, 2H), 1.54-1.68 (m, 2H), 1.56 (br s, 3H), 1.65 (br s, 3H), 1.88-1.93 (m, 5H), 2.08 (dd, 1H, J = 7.8, 13.9 Hz), 2.28 (m, 1H), 3.43 (d, 1H, J = 13.7 Hz), 3.48 (d, 1H, J = 13.7 Hz), 3.90 (dd, 1H, J = 4.9, 7.8 Hz), 4.21 (d, 1H, J = 0.9 Hz), 5.04 (m, 1H), 5.12 (dd, 1H, J = 0.6, 17.5 Hz), 5.26 (dd, 1H, J = 0.6, 10.8 Hz), 6.01 (dd, 1H, J = 10.8, 17.5 Hz), 9.60 (d, 1H, J = 0.9 Hz); 13C-NMR (125 MHz) δ 17.6, 19.3, 19.9, 20.4, 22.2, 25.7, 26.5, 32.7, 38.2, 38.9, 42.9, 44.5, 47.8, 48.2, 53.1, 65.1, 65.3, 115.1, 124.0, 131.7, 143.5, 166.3, 197.7; HRMS calcd for C23H35NO4S (M+) m/z 421.2287, found 421.2281.
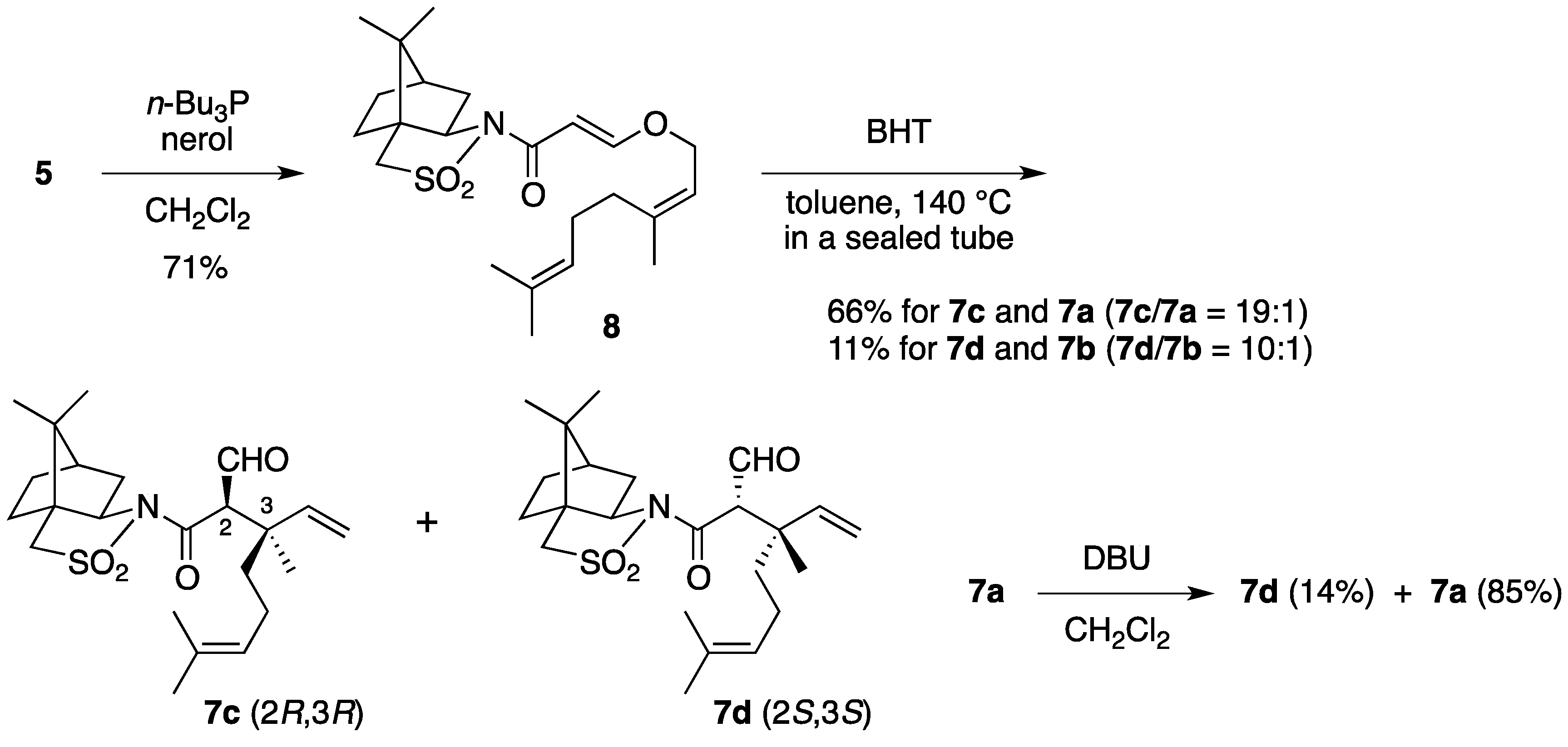
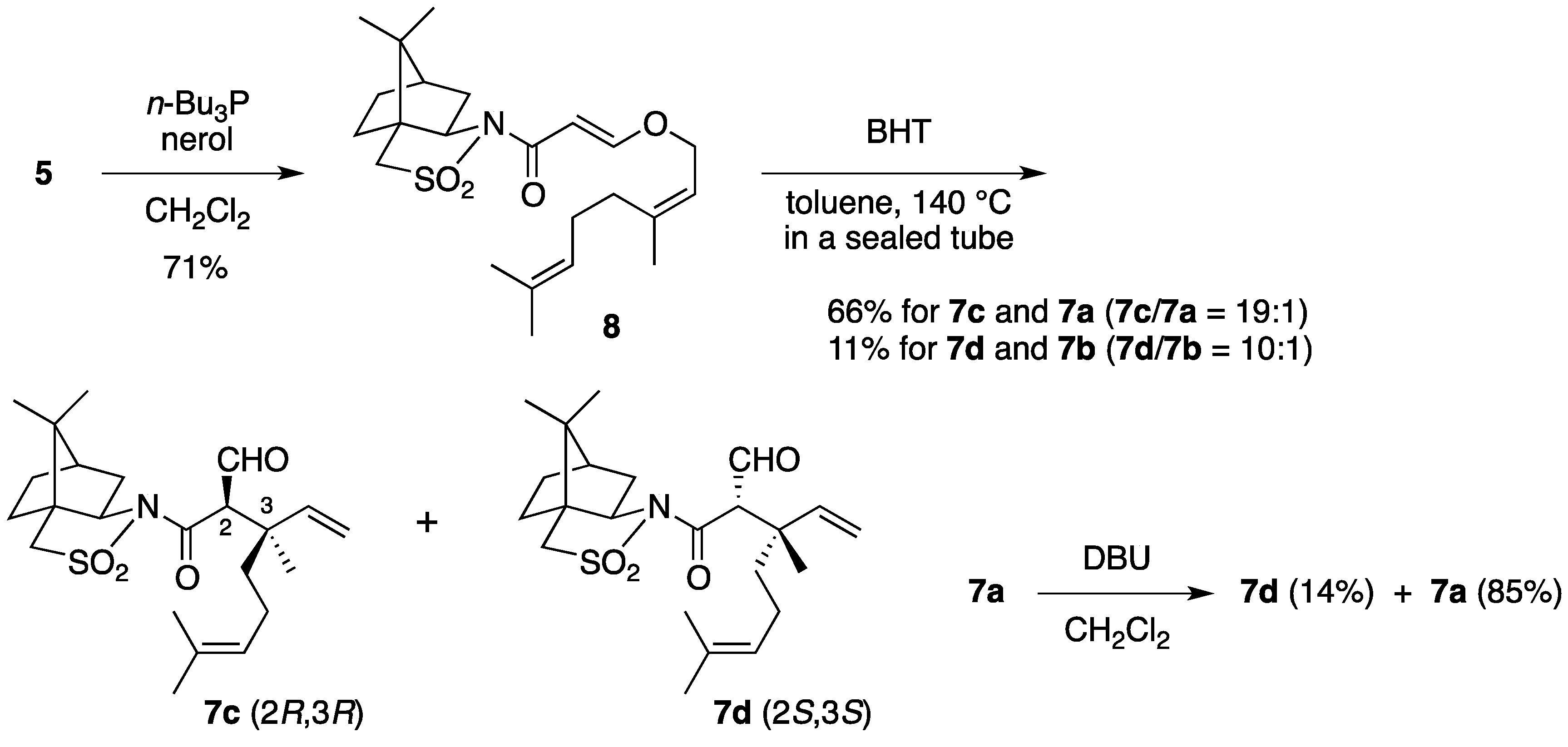
(2R)-N-{(E)-3-[((2Z)-3,7-Dimethylocta-2,6-dien-1-yl)oxy]acryloyl}bornane-10,2-sultam (8). As described for the preparation of 6, compound 5 (210 mg, 785 μmol) and nerol (155 μL, 882 μmol) were treated with n-Bu3P (32 μL, 0.12 mmol) in CH2Cl2 (8 mL) to provide 234 mg (71%) of 8 as white crystals: mp 62–64 °C; TLC Rf 0.52 (EtOAc/hexane, 1:3); [α]D26–71.0 (c 1.22, CHCl3); IR (neat) 2964, 2884, 1677, 1607 cm−1; 1H-NMR (500 MHz) δ 0.97 (s, 3H), 1.18 (s, 3H), 1.34–1.45 (m, 2H), 1.60 (br s, 3H), 1.69 (br s, 3H), 1.78 (br s, 3H), 1.87-1.91 (m, 3H), 2.05–2.17 (m, 6H), 3.43 (d, 1H, J = 13.7 Hz), 3.48 (d, 1H, J = 13.7 Hz), 3.91 (dd, 1H, J = 4.9, 7.8 Hz), 4.41 (d, 2H, J = 7.0 Hz), 5.08 (m, 1H), 5.39 (t, 1H, J = 7.0 Hz), 5.96 (d, 1H, J = 12.0 Hz), 7.69 (d, 1H, J = 12.0 Hz); 13C-NMR (125 MHz) δ 17.6, 19.9, 20.8, 23.5, 25.7, 26.5 (2C), 32.3, 32.8, 38.6, 44.7, 47.8, 48.2, 53.1, 65.0, 67.9, 97.0, 118.5, 123.3, 132.5, 143.8, 163.4, 165.0; HRMS calcd for C23H35NO4S (M+) m/z 421.2287, found 421.2287.
(2R)-N-[(2R,3R)-2-Formyl-3,7-dimethyl-3-vinyloct-6-enoyl]bornane-10,2-sultam (7c) and (2R)-N-[(2S,3S)]-isomer (7d). As described for the preparation of 7a and 7b from 6, a solution of 8 (223 mg, 529 μmol) and BHT (5.8 mg, 26 μmol) in toluene (27 mL) was heated at 140 °C for 26 h to provide 147 mg (66%) of a mixture of 7c and 7a (7c/7a = 19:1) and 25.0 mg (11%) of a mixture of 7d and 7b (7d/7b = 10:1), and 27.9 mg (13%) of 8 was recovered. A mixture of 7c and 7a (7c/7a = 19:1) was obtained as a colorless oil: TLC Rf 0.49 (EtOAc/hexane, 1:3); [α]D28–82.4 (c 1.26, CHCl3); IR (neat) 2965, 2930, 1727, 1684 cm−1; 1H-NMR (500 MHz) for 7c δ 0.97 (s, 3H), 1.16 (s, 3H), 1.26 (s, 3H), 1.34–1.49 (m, 3H), 1.55 (br s, 3H), 1.65 (br s, 3H), 1.68 (m, 1H), 1.84–1.93 (m, 5H), 2.03–2.09 (m, 2H), 3.43 (d, 1H, J = 13.8 Hz), 3.50 (d, 1H, J = 13.8 Hz), 3.89 (d, 1H, J = 3.5 Hz), 3.92 (dd, 1H, J = 5.5, 7.4 Hz), 5.02 (m, 1H), 5.02 (dd, 1H, J = 1.0, 17.4 Hz), 5.14 (dd, 1H, J = 1.0, 10.9 Hz), 6.02 (dd, 1H, J = 10.9, 17.4 Hz), 9.66 (d, 1H, J = 3.5 Hz); 13C-NMR (125 MHz) for 7c δ 17.6, 18.9, 19.9, 20.7, 22.2, 25.6, 26.4, 33.0, 38.2, 39.5, 44.7, 45.8, 47.7, 48.1, 53.3, 65.4, 65.5, 115.2, 123.7, 131.8, 141.7, 167.9, 197.8; HRMS calcd for C23H35NO4S (M+) m/z 421.2287, found 421.2289. A mixture of 7d and 7b (7d/7b = 10:1) was obtained as a colorless oil: TLC Rf 0.61 (EtOAc/hexane, 1:3); [α]D26+2.9 (c 1.25, CHCl3); IR (neat) 2964, 2924, 1728, 1697 cm−1; 1H-NMR (500 MHz) for 7d δ 0.95 (s, 3H), 1.09 (s, 3H), 1.31–1.42 (m, 2H), 1.34 (s, 3H), 1.57 (m, 1H), 1.57 (br s, 3H), 1.65 (br s, 3H), 1.76 (m, 1H), 1.87–1.95 (m, 5H), 2.07 (dd, 1H, J = 7.9, 14.0 Hz), 2.25 (m, 1H), 3.43 (d, 1H, J = 13.9 Hz), 3.49 (d, 1H, J = 13.9 Hz), 3.90 (dd, 1H, J = 4.9, 7.9 Hz), 4.19 (d, 1H, J = 1.1 Hz), 5.06 (m, 1H), 5.06 (d, 1H, J = 17.2 Hz), 5.17 (d, 1H, J = 10.9 Hz), 6.14 (dd, 1H, J = 10.9, 17.2 Hz), 9.68 (d, 1H, J = 1.1 Hz); 13C-NMR (125 MHz) for 7d δ 17.6, 19.9, 20.4, 21.2, 23.4, 25.6, 26.4, 32.7, 38.2, 39.1, 43.3, 44.5, 47.7, 48.2, 53.1, 65.3, 66.2, 114.7, 123.9, 131.7, 142.6, 166.3, 197.0; HRMS calcd for C23H35NO4S (M+) m/z 421.2287, found 421.2288.
Epimerization of 7a. To a stirred solution of 7a (7.9 mg, 19 μmol) in CH2Cl2 (1 mL) was added DBU (3.6 μL, 24 μmol). The mixture was stirred at room temperature for 45 min, diluted with 1 M aqueous HCl (1 mL), and extracted with CH2Cl2 (2 mL × 3). The combined extracts were washed with saturated brine (1 mL), dried and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:25) to provide 1.1 mg (14%) of 7d and 6.7 mg (85%) of 7a was recovered.
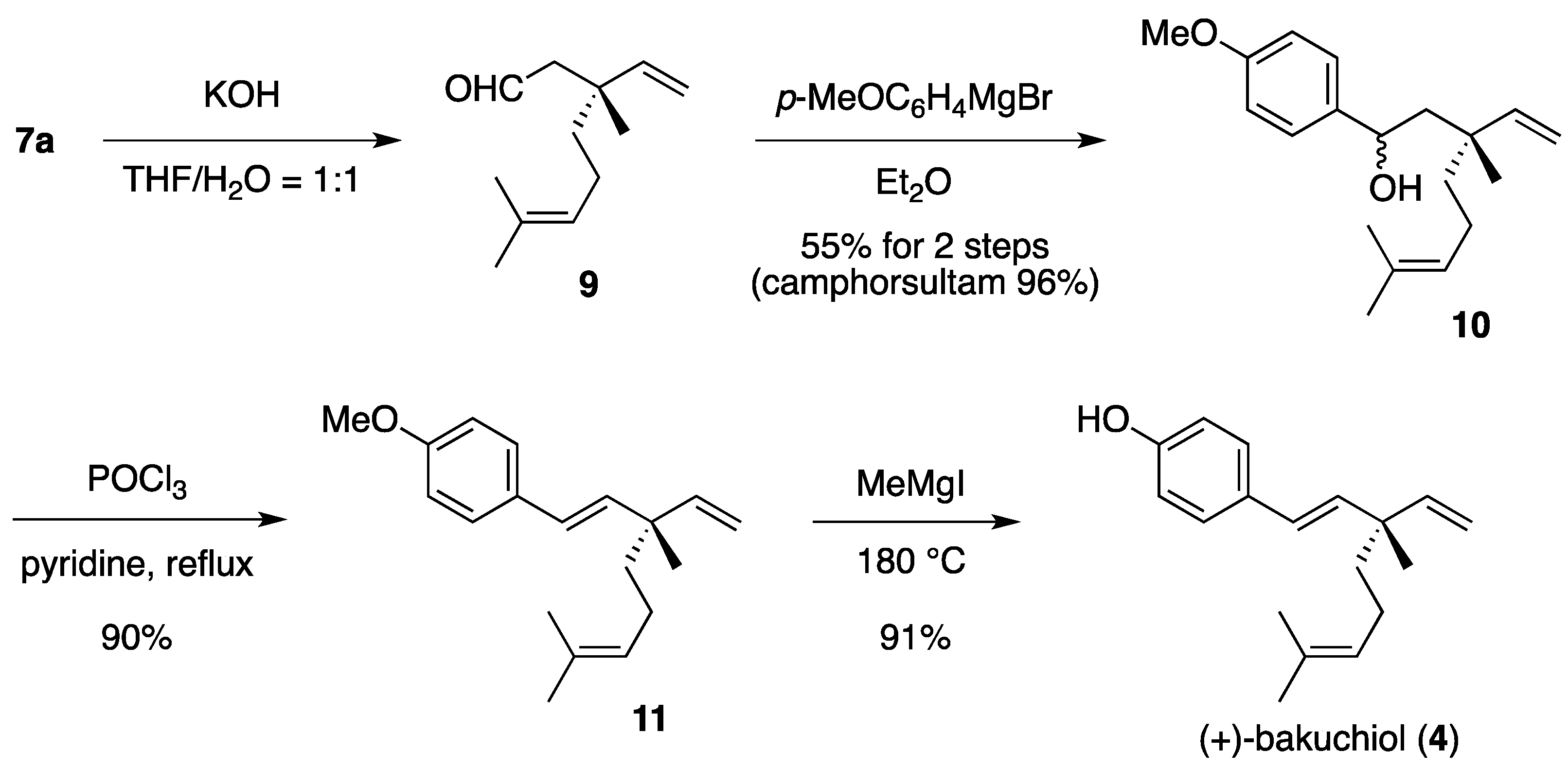
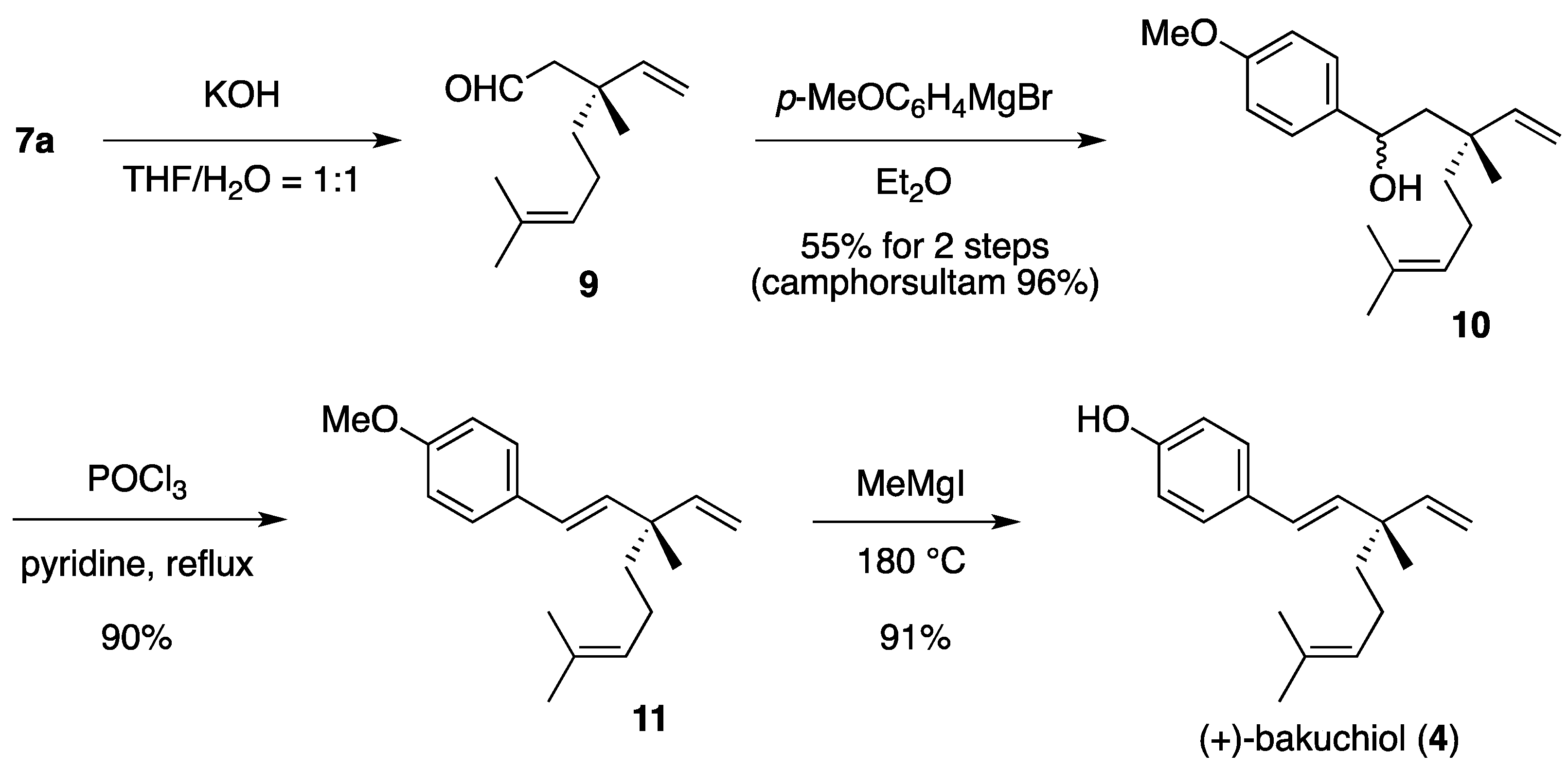
(1RS,3R)-1-(4-Methoxyphenyl)-3,7-dimethyl-3-vinyloct-6-enol (10). To a cooled (0 °C) stirred solution of 7a (233 mg, 553 μmol) in THF/H2O (1:1, 5 mL) was added 1.00 M aqueous KOH (1.11 mL, 1.11 mmol). The mixture was stirred at room temperature for 24 h, quenched with saturated aqueous NH4Cl (2 mL), diluted with H2O (2 mL), and extracted with Et2O (5 mL × 3). The combined extracts were washed with saturated brine (15 mL) and dried to provide a solution of aldehyde 9 in Et2O, which was used in the next step without further evaporation and purification.
The following reaction was carried out under Ar. To a cooled (0 °C) stirred solution of aldehyde 9 in Et2O obtained above was added 4-methoxyphenylmagnesium bromide (1.50 M solution in Et2O, total 6.27 mL, total 9.41 mmol) in ten times over a period of 2 h. The mixture was quenched with saturated aqueous NH4Cl (30 mL), diluted H2O (10 mL), and extracted with CH2Cl2 (40 mL × 3). The combined extracts were dried and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:40) to provide 86.7 mg (55%) of 10 and 114 mg (96%) of camphorsultam. Compound 10 (dr = 1:1) was obtained as a colorless oil: TLC Rf 0.61 (EtOAc/hexane, 1:3); IR (neat) 3442, 2965, 2924, 1612, 1512 cm−1; 1H-NMR (500 MHz) δ 1.10 (s, 3H × 1/2), 1.11 (s, 3H × 1/2), 1.34 (t, 2H × 1/2, J = 8.5 Hz), 1.40–1.43 (m, 2H × 1/2), 1.57 (br s, 3H × 1/2), 1.59 (br s, 3H × 1/2), 1.66 (br s, 3H × 1/2), 1.67 (br s, 3H × 1/2), 1.80–1.93 (m, 4H), 3.79 (s, 3H), 4.74 (dd, 1H × 1/2, J = 2.6, 8.6 Hz), 4.79 (dd, 1H × 1/2, J = 2.6, 9.3 Hz), 5.02 (dd, 1H × 1/2, J = 1.1, 17.7 Hz), 5.07 (dd, 1H × 1/2, J = 1.1, 10.8 Hz), 5.07 (m, 1H), 5.07 (dd, 1H × 1/2, J = 0.9, 17.7 Hz), 5.14 (dd, 1H × 1/2, J = 0.9, 10.8 Hz), 5.83 (dd, 1H × 1/2, J = 10.8, 17.7 Hz), 5.97 (dd, 1H × 1/2, J = 10.8, 17.7 Hz), 6.86 (d, 2H × 1/2, J = 8.8 Hz), 6.86 (d, 2H × 1/2, J = 8.6 Hz), 7.24 (d, 2H × 1/2, J = 8.8 Hz), 7.25 (d, 2H × 1/2, J = 8.6 Hz); 13C-NMR (125 MHz) δ 17.6, 21.3 (1/2C), 22.5 (1/2C), 22.7 (1/2C), 23.5 (1/2C), 25.7, 39.5, 40.5 (1/2C), 42.5 (1/2C), 50.3 (1/2C), 51.1 (1/2C), 55.3, 71.4 (1/2C), 71.5 (1/2C), 112.2 (1/2C), 112.9 (1/2C), 113.7, 113.8, 124.6 (1/2C), 124.8 (1/2C), 126.9 (2C), 131.2 (1/2C), 131.3 (1/2C), 137.7 (1/2C), 138.3 (1/2C), 147.4 (1/2C), 147.7 (1/2C), 158.8; HRMS calcd for C19H28O2 (M+) m/z 288.2089, found 288.2090.
(1E,3S)-1-(4-Methoxyphenyl)-3,7-dimethyl-3-vinylocta-1,6-diene (11). The following reaction was carried out under Ar. To a stirred solution of 10 (22.5 mg, 78.0 μmol) in pyridine (1 mL) was added POCl3 (8.6 μL, 95 μmol). The mixture was refluxed for 4 h, diluted with EtOAc (15 mL), and washed with H2O (10 mL) and saturated brine (10 mL). The organic layer was dried and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:100) to provide 18.9 mg (90%) of 11 as a colorless oil: TLC Rf 0.80 (EtOAc/hexane, 1:3); [α]D25+28.4 (c 0.855, CHCl3); IR (neat) 2966, 2916, 1609, 1511 cm−1; 1H-NMR (500 MHz) δ 1.20 (s, 3H), 1.48–1.51 (m, 2H), 1.58 (br s, 3H), 1.67 (br s, 3H), 1.93–1.98 (m, 2H), 3.80 (s, 3H), 5.01 (dd, 1H, J = 1.4, 17.5 Hz), 5.03 (dd, 1H, J = 1.4, 10.7 Hz), 5.11 (m, 1H), 5.88 (dd, 1H, J = 10.7, 17.5 Hz), 6.06 (d, 1H, J = 16.4 Hz), 6.26 (d, 1H, J = 16.4 Hz), 6.83 (d, 2H, J = 8.7 Hz), 7.29 (d, 2H, J = 8.7 Hz); 13C-NMR (125 MHz) δ 17.6, 23.2, 23.4, 25.7, 41.3, 42.5, 55.3, 111.8, 113.9 (2C), 124.8, 126.5, 127.1 (2C), 130.7, 131.3, 135.8, 146.0, 158.7; HRMS calcd for C19H26O (M+) m/z 270.1984, found 270.1983.
(+)-Bakuchiol (4). The following reaction was carried out under Ar. To a cooled (0 °C) solution of 11 (30.2 mg, 112 μmol) in Et2O (1 mL) was added MeMgI (0.500 M solution in Et2O, 1.57 mL, 785 μmol). The solvent was removed under reduced pressure. The residue was heated at 180 °C for 15 min and cooled to room temperature. The mixture was quenched with 1 M aqueous HCl (2 mL), diluted with H2O (2 mL), and extracted with CH2Cl2 (5 mL × 3). The combined extracts were dried and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:60) to provide 26.1 mg (91%) of 4 as a pale yellow oil: TLC Rf 0.63 (EtOAc/hexane, 1:3); [α]D29 + 25.6 (c 0.795, CHCl3); IR (neat) 3359, 2967, 2924, 1610, 1513 cm−1; 1H-NMR (500 MHz) δ 1.19 (s, 3H), 1.47-1.51 (m, 2H), 1.58 (br s, 3H), 1.67 (br s, 3H), 1.93–1.97 (m, 2H), 4.85 (br, 1H, OH), 5.01 (dd, 1H, J = 1.5, 17.4 Hz), 5.03 (dd, 1H, J = 1.5, 10.8 Hz), 5.11 (m, 1H), 5.88 (dd, 1H, J = 10.8, 17.4 Hz), 6.05 (d, 1H, J = 16.2 Hz), 6.25 (d, 1H, J = 16.2 Hz), 6.76 (d, 2H, J = 8.6 Hz), 7.24 (d, 2H, J = 8.6 Hz); 13C-NMR (125 MHz) δ 17.6, 23.2, 23.3, 25.7, 41.3, 42.5, 111.9, 115.3 (2C), 124.8, 126.4, 127.4 (2C), 130.9, 131.3, 135.9, 145.9, 154.6; HRMS calcd for C18H24O (M+) m/z 256.1827, found 256.1829.
(2R)-N-[(1R,2S,5R,6R)-6-Hydroxy-5-isopropenyl-2-methyl-2-vinylcyclohexanecarbonyl]bornane-10,2-sultam (12aa) and its diastereomers. A solution of 7a (22.8 mg, 54.1 μmol) and BHT (a crystal) in toluene (6 mL) was stirred at 160 °C for 50 h in a sealed tube and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:15) to provide 4.9 mg (21%) of 12aa, 3.2 mg (14%) of 12ab, 3.3 mg (14%) of 12ac, and 1.7 mg (7%) of 12ad. Compound 12aa was obtained as white crystals: mp 198–200 °C; TLC Rf 0.43 (EtOAc/hexane, 1:2); [α]D21–10.5 (c 0.27, CHCl3); IR (neat) 3520, 2960, 1695 cm−1; 1H-NMR (500 MHz) δ 0.97 (s, 3H), 1.16 (s, 3H), 1.17 (s, 3H), 1.36–1.44 (m, 2H), 1.51–1.56 (m, 2H), 1.67-1.79 (m, 2H), 1.73 (br s, 3H), 1.86–1.92 (m, 3H), 2.02 (d, 1H, J = 8.3 Hz, OH), 2.08–2.15 (m, 3H), 2.99 (d, 1H, J = 10.6 Hz), 3.47 (d, 1H, J = 13.9 Hz), 3.53 (d, 1H, J = 13.9 Hz), 3.94 (dt, 1H, J = 8.3, 10.6 Hz), 4.00 (dd, 1H, J = 5.1, 7.7 Hz), 4.83 (s, 1H), 4.85 (s, 1H), 4.98 (dd, 1H, J = 1.2, 17.5 Hz), 5.12 (dd, 1H, J = 1.2, 11.2 Hz), 6.43 (dd, 1H, J = 11.2, 17.5 Hz); 13C-NMR (125 MHz) δ 19.2, 20.0, 20.9, 26.4, 26.5, 27.8, 33.0, 38.7, 39.0, 42.5, 44.8, 47.6, 47.7, 53.6, 54.1, 60.7, 65.9, 70.3, 112.5, 113.1, 141.5, 146.2, 171.9; HRMS calcd for C23H35NO4S (M+) m/z 421.2287, found 421.2291.
(2R)-N-[(1R,2R,5R,6R)-6-Hydroxy-5-isopropenyl-2-methyl-2-vinylcyclohexanecarbonyl]bornane-10,2-sultam (12ca) and its diastereomers. As described for the preparation of 12aa and its diastereomers from 7a, a solution of 7c (23.5 mg, 55.7 μmol) and BHT (a crystal) in toluene (6 mL) was heated at 160 °C for 40 h to provide 8.7 mg (37%) of 12ca, 9.2 mg (39%) of a mixture of 12cb and 12cc, and 0.9 mg (4%) of 12cd. Compound 12ca was obtained as white crystals: TLC Rf 0.32 (EtOAc/hexane, 1:2); 1H-NMR (500 MHz) δ 0.95 (s, 3H), 1.15 (s, 3H), 1.19 (s, 3H), 1.23–1.41 (m, 3H), 1.50–1.70 (m, 3H), 1.78 (br s, 3H), 1.81–1.96 (m, 3H), 2.02–2.06 (m, 3H), 2.10 (dt, 1H, J = 5.0, 10.6 Hz), 3.00 (d, 1H, J = 10.5 Hz), 3.45 (d, 1H, J = 13.9 Hz), 3.50 (d, 1H, J = 13.9 Hz), 3.95 (dd, 1H, J = 5.2, 7.8 Hz), 4.00 (q, 1H, J = 10.5 Hz), 4.85 (s, 1H), 4.86 (s, 1H), 4.90 (d, 1H, J = 10.6 Hz), 4.98 (d, 1H, J = 17.5 Hz), 6.00 (dd, 1H, J = 10.6, 17.5 Hz).
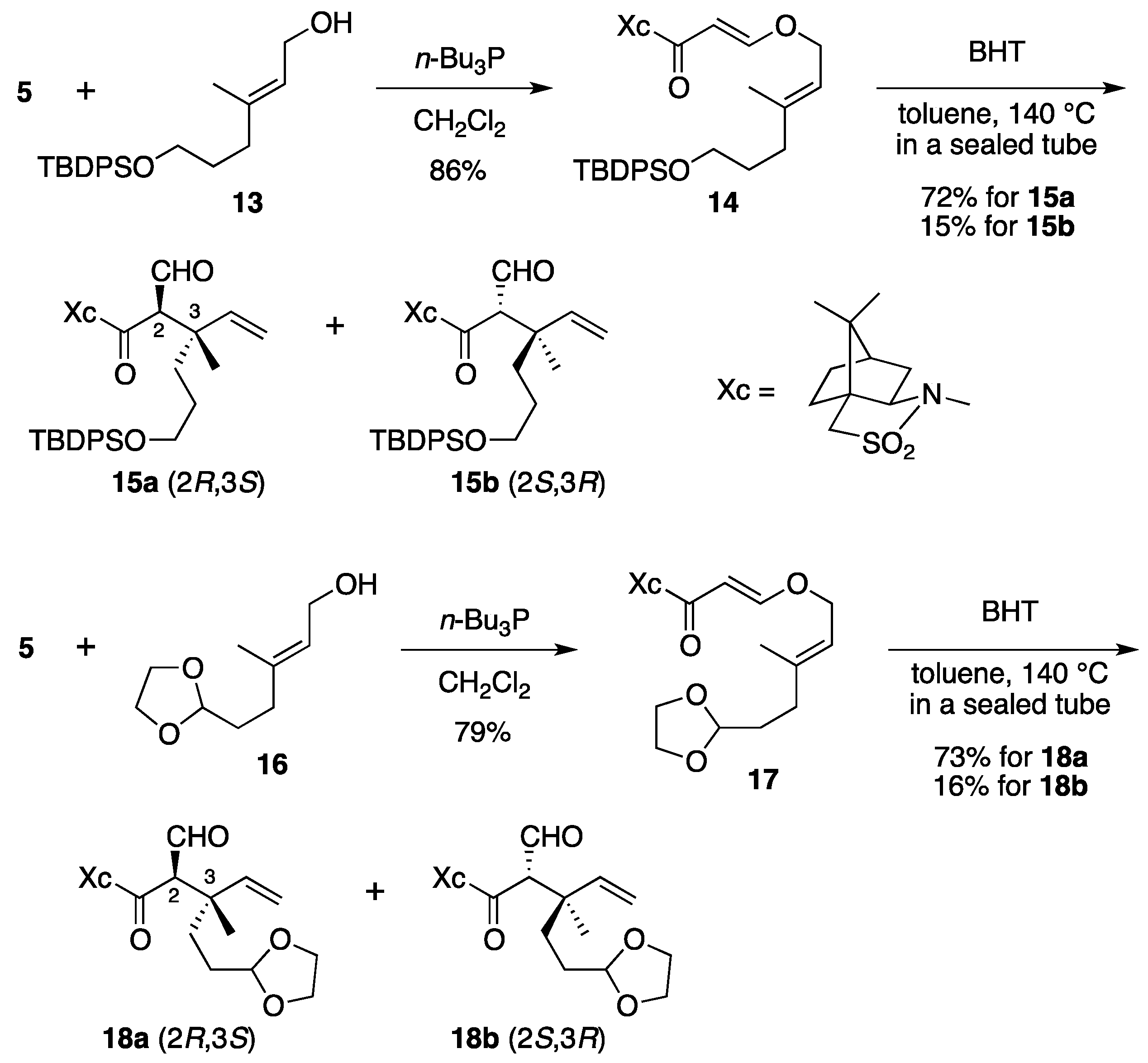
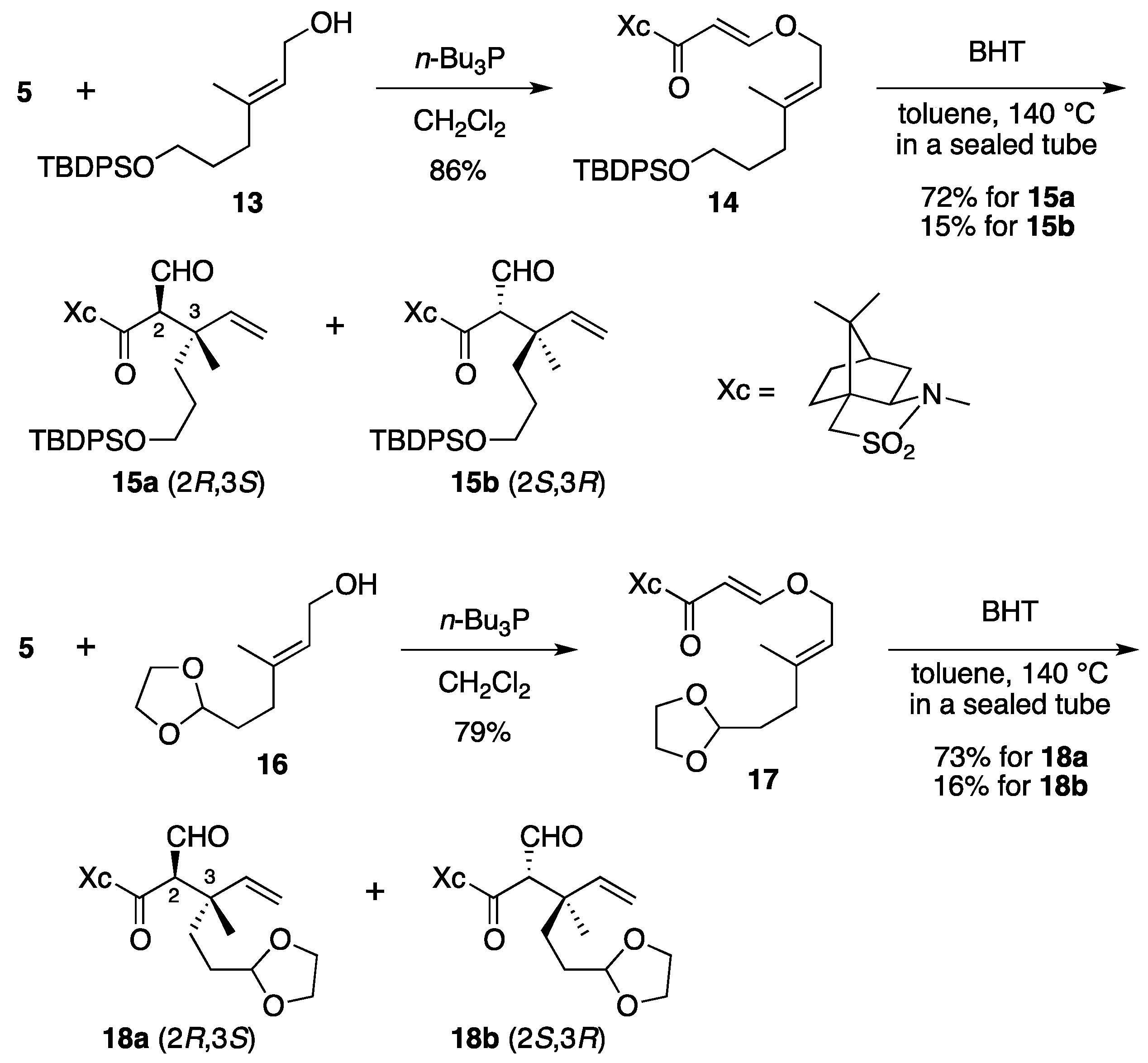
(2R)-N-{(E)-3-[((2E)-6-(tert-Butyldiphenysilyloxy)-3-methylhex-2-en-1-yl)oxy]acryloyl}bornane-10,2-sultam (14). As described for the preparation of 6, compound 5 (109 mg, 408 μmol) and 13 (165 mg, 448 μmol) were treated with n-Bu3P (15 μL, 61 μmol) in CH2Cl2 (4 mL) to provide 223 mg (86%) of 14 as white crystals: mp 74–77 °C; TLC Rf 0.78 (EtOAc/toluene, 1:4); [α]D26 – 45.6 (c 1.02, CHCl3); IR (neat) 2958, 2858, 1678, 1608 cm−1; 1H-NMR (500 MHz) δ 0.97 (s, 3H), 1.05 (s, 9H), 1.17 (s, 3H), 1.36–1.45 (m, 2H), 1.65–1.70 (m, 2H), 1.67 (s, 3H), 1.86–1.91 (m, 3H), 2.08 (dd, 1H, J = 7.8, 13.8 Hz), 2.13 (t, 2H, J = 7.8 Hz), 2.15 (m, 1H), 3.42 (d, 1H, J = 13.8 Hz), 3.48 (d, 1H, J = 13.8 Hz), 3.64 (t, 2H, J = 6.3 Hz), 3.91 (dd, 1H, J = 4.9, 7.8 Hz), 4.41 (d, 2H, J = 7.1 Hz), 5.35 (t, 1H, J = 7.1 Hz), 5.96 (d, 1H, J = 12.0 Hz), 7.36–7.44 (m, 6H), 7.65–7.67 (m, 4H), 7.70 (d, 1H, J = 12.0 Hz); 13C-NMR (125 MHz) δ 16.6, 19.2, 19.9, 20.8, 26.5, 26.9 (3C), 30.5, 32.8, 35.7, 38.6, 44.7, 47.8, 48.2, 53.1, 63.3, 65.0, 68.1, 97.0, 117.6, 127.6 (4C), 129.5 (2C), 134.0 (2C), 135.6 (4C), 143.4, 163.4, 165.0; HRMS calcd for C32H40NO5SSi (M+–t-C4H9) m/z 578.2396, found 578.2398.
(2R)-N-[(2R,3S)-6-(tert-Butyldiphenysilyloxy)-2-formyl-3-methyl-3-vinylhexanoyl]bornane-10,2-sultam (15a) and (2R)-N-[(2S,3R)]-Isomer (15b). As described for the preparation of 7a and 7b from 6, a solution of 14 (209 mg, 329 μmol) and BHT (3.6 mg, 16 μmol) in toluene (17 mL) was heated at 140 °C for 71 h to provide 150 mg (72%) of 15a and 32.1 mg (15%) of 15b. Compound 15a was obtained as a colorless oil: TLC Rf 0.59 (EtOAc/toluene, 1:5); [α]D23–88.2 (c 1.46, CHCl3); IR (neat) 2961, 2859, 1731, 1686 cm−1; 1H-NMR (500 MHz) δ 0.95 (s, 3H), 1.14 (s, 9H), 1.11 (s, 3H), 1.23 (s, 3H), 1.35–1.40 (m, 2H), 1.48-1.54 (m, 2H), 1.74 (m, 1H), 1.83–1.91 (m, 4H), 2.07–2.13 (m, 2H), 3.43 (d, 1H, J = 13.8 Hz), 3.50 (d, 1H, J = 13.8 Hz), 3.60 (t, 2H, J = 6.3 Hz), 3.95 (dd, 1H, J = 5.4, 7.5 Hz), 4.01 (d, 1H, J = 2.5 Hz), 5.05 (d, 1H, J = 17.5 Hz), 5.19 (d, 1H, J = 10.9 Hz), 5.88 (dd, 1H, J = 10.9, 17.5 Hz), 7.35–7.43 (m, 6H), 7.63–7.65 (m, 4H), 9.60 (d, 1H, J = 2.5 Hz); 13C-NMR (125 MHz) δ 19.2, 19.8, 19.9, 20.8, 26.4, 26.7, 26.8 (3C), 32.9, 34.5, 38.5, 44.7, 45.3, 47.7, 48.1, 53.2, 63.9, 65.4, 65.5, 115.3, 127.6 (4C), 129.5 (2C), 134.0 (2C), 135.6 (4C), 142.3, 167.4, 197.2; HRMS calcd for C32H40NO5SSi (M+–t-C4H9) m/z 578.2396, found 578.2401. Compound 15b was obtained as a colorless oil: TLC Rf 0.69 (EtOAc/toluene, 1:5); [α]D24+6.7 (c 1.50, CHCl3); IR (neat) 2961, 2859, 1728, 1696 cm−1; 1H-NMR (500 MHz) δ 0.94 (s, 3H), 1.04 (s, 9H), 1.10 (s, 3H), 1.27 (s, 3H), 1.33–1.39 (m, 2H), 1.51 (m, 1H), 1.62 (m, 1H), 1.70 (m, 1H), 1.87–1.91 (m, 4H), 2.06 (dd, 1H, J = 7.8, 14.0 Hz), 2.26 (m, 1H), 3.41 (d, 1H, J = 13.7 Hz), 3.48 (d, 1H, J = 13.7 Hz), 3.61 (t, 2H, J = 6.5 Hz), 3.87 (dd, 1H, J = 4.9, 7.8 Hz), 4.20 (s, 1H), 5.09 (d, 1H, J = 17.5 Hz), 5.23 (d, 1H, J = 10.7 Hz), 5.96 (dd, 1H, J = 10.7, 17.5 Hz), 7.36–7.43 (m, 6H), 7.64–7.66 (m, 4H), 9.59 (s, 1H); 13C-NMR (125 MHz) δ 19.2, 19.5, 19.9, 20.4, 26.4, 26.7, 26.9 (3C), 32.8, 34.9, 38.2, 42.7, 44.5, 47.7, 48.2, 53.1, 64.0, 65.2, 65.3, 115.1, 127.6 (4C), 129.5 (2C), 134.0 (2C), 135.6 (4C), 143.4, 166.3, 197.7; HRMS calcd for C32H40NO5SSi (M+–t-C4H9) m/z 578.2396, found 578.2389.
(2R)-N-{(E)-3-[((2E)-5-(1,3-Dioxolan-2-yl)-3-methylpent-2-en-1-yl)oxy]acryloyl}bornane-10,2-sultam (17). As described for the preparation of 6, compound 5 (171 mg, 640 μmol) and 16 (121 mg, 703 μmol) were treated with n-Bu3P (24 μL, 97 μmol) in CH2Cl2 (6 mL) to provide 222 mg (79%) of 17 as a colorless oil: TLC Rf 0.67 (EtOAc/toluene, 1:3); [α]D25–59.7 (c 1.16, CHCl3); IR (neat) 2958, 2885, 1677, 1609 cm−1; 1H-NMR (500 MHz) δ 0.97 (s, 3H), 1.18 (s, 3H), 1.34–1.45 (m, 2H), 1.72 (s, 3H), 1.77–1.81 (m, 2H), 1.87–1.91 (m, 3H), 2.07 (dd, 1H, J = 7.8, 13.9 Hz), 2.14 (m, 1H), 2.18 (t, 2H, J = 8.1 Hz), 3.43 (d, 1H, J = 13.8 Hz), 3.48 (d, 1H, J = 13.8 Hz), 3.84–3.86 (m, 2H), 3.91 (dd, 1H, J = 5.0, 7.8 Hz), 3.95–3.98 (m, 2H), 4.45 (d, 2H, J = 6.9 Hz), 4.86 (t, 1H, J = 4.7 Hz), 5.41 (t, 1H, J = 6.9 Hz), 5.96 (d, 1H, J = 12.1 Hz), 7.69 (d, 1H, J = 12.1 Hz); 13C-NMR (125 MHz) δ 16.7, 19.9, 20.8, 26.5, 31.8, 32.7, 33.6, 38.5, 44.6, 47.7, 48.2, 53.0, 64.9 (2C), 65.0, 68.0, 97.0, 104.0, 117.8, 142.7, 163.3, 164.9; HRMS calcd for C22H33NO6S (M+) m/z 439.2029, found 439.2035.
(2R)-N-[(2R,3S)-5-(1,3-Dioxolan-2-yl)-2-formyl-3-methyl-3-vinylpentanoyl]bornane-10,2-sultam (18a) and (2R)-N-[(2S,3R)]-Isomer (18b). As described for the preparation of 7a and 7b from 6, a solution of 17 (219 mg, 498 μmol) and BHT (5.5 mg, 25 μmol) in toluene (25 mL) was heated at 140 °C for 116 h to provide 159 mg (73%) of 18a and 34.0 mg (16%) of 18b. Compound 18a was obtained as white crystals: mp 116–118 °C; TLC Rf 0.67 (EtOAc/toluene, 1:2); [α]D21–119 (c 1.34, CHCl3); IR (neat) 2964, 2886, 1731, 1684 cm−1; 1H-NMR (500 MHz) δ 0.98 (s, 3H), 1.16 (s, 3H), 1.24 (s, 3H), 1.34–1.43 (m, 2H), 1.53–1.63 (m, 2H), 1.81 (m, 1H), 1.88–1.96 (m, 4H), 2.11–2.12 (m, 2H), 3.44 (d, 1H, J = 13.7 Hz), 3.51 (d, 1H, J = 13.7 Hz), 3.80–3.83 (m, 2H), 3.91–3.94 (m, 2H), 3.96 (t, 1H, J = 6.6 Hz), 4.01 (d, 1H, J = 2.3 Hz), 4.81 (t, 1H, J = 4.2 Hz), 5.08 (d, 1H, J = 17.5 Hz), 5.22 (d, 1H, J = 10.6 Hz), 5.89 (dd, 1H, J = 10.6, 17.5 Hz), 9.62 (d, 1H, J = 2.3 Hz); 13C-NMR (125 MHz) δ 19.7, 19.9, 20.8, 26.4, 28.0, 32.0, 33.0, 38.5, 44.8, 45.0, 47.7, 48.1, 53.2, 64.9 (2C), 65.4 (2C), 104.3, 115.6, 141.9, 167.4, 197.1; HRMS calcd for C22H33NO6S (M+) m/z 439.2029, found 439.2036. Compound 18b was obtained as a colorless oil: TLC Rf 0.75 (EtOAc/toluene, 1:2); [α]D22+10.4 (c 1.67, CHCl3); IR (neat) 2962, 2885, 1728, 1697 cm−1; 1H-NMR (500 MHz) δ 0.94 (s, 3H), 1.10 (s, 3H), 1.29 (s, 3H), 1.32–1.42 (m, 2H), 1.58–1.65 (m, 2H), 1.74 (m, 1H), 1.87–1.94 (m, 4H), 2.07 (dd, 1H, J = 7.8, 13.8 Hz), 2.26 (m, 1H), 3.43 (d, 1H, J = 13.9 Hz), 3.48 (d, 1H, J = 13.9 Hz), 3.81–3.85 (m, 2H), 3.90–3.95 (m, 3H), 4.21 (s, 1H), 4.82 (t, 1H, J = 4.4 Hz), 5.12 (d, 1H, J = 17.5 Hz), 5.26 (d, 1H, J = 10.9 Hz), 5.98 (dd, 1H, J = 10.9, 17.5 Hz), 9.61 (s, 1H); 13C-NMR (125 MHz) δ 19.2, 19.9, 20.4, 26.4, 28.1, 32.6, 32.7, 38.1, 42.5, 44.5, 47.7, 48.2, 53.0, 64.9 (2C), 65.2 (2C), 104.4, 115.5, 143.0, 166.2, 197.5; HRMS calcd for C22H33NO6S (M+) m/z 439.2029, found 439.2032.
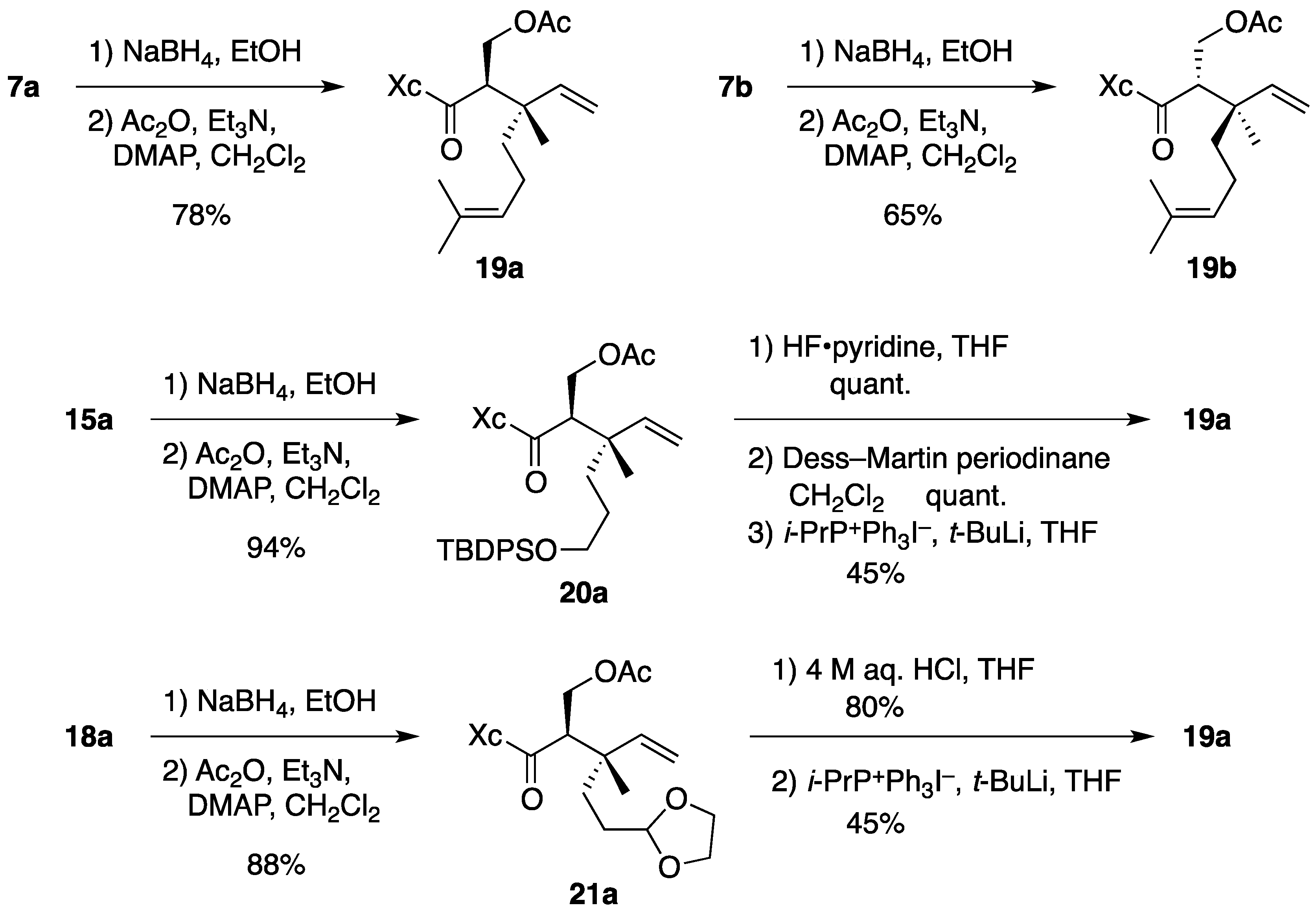
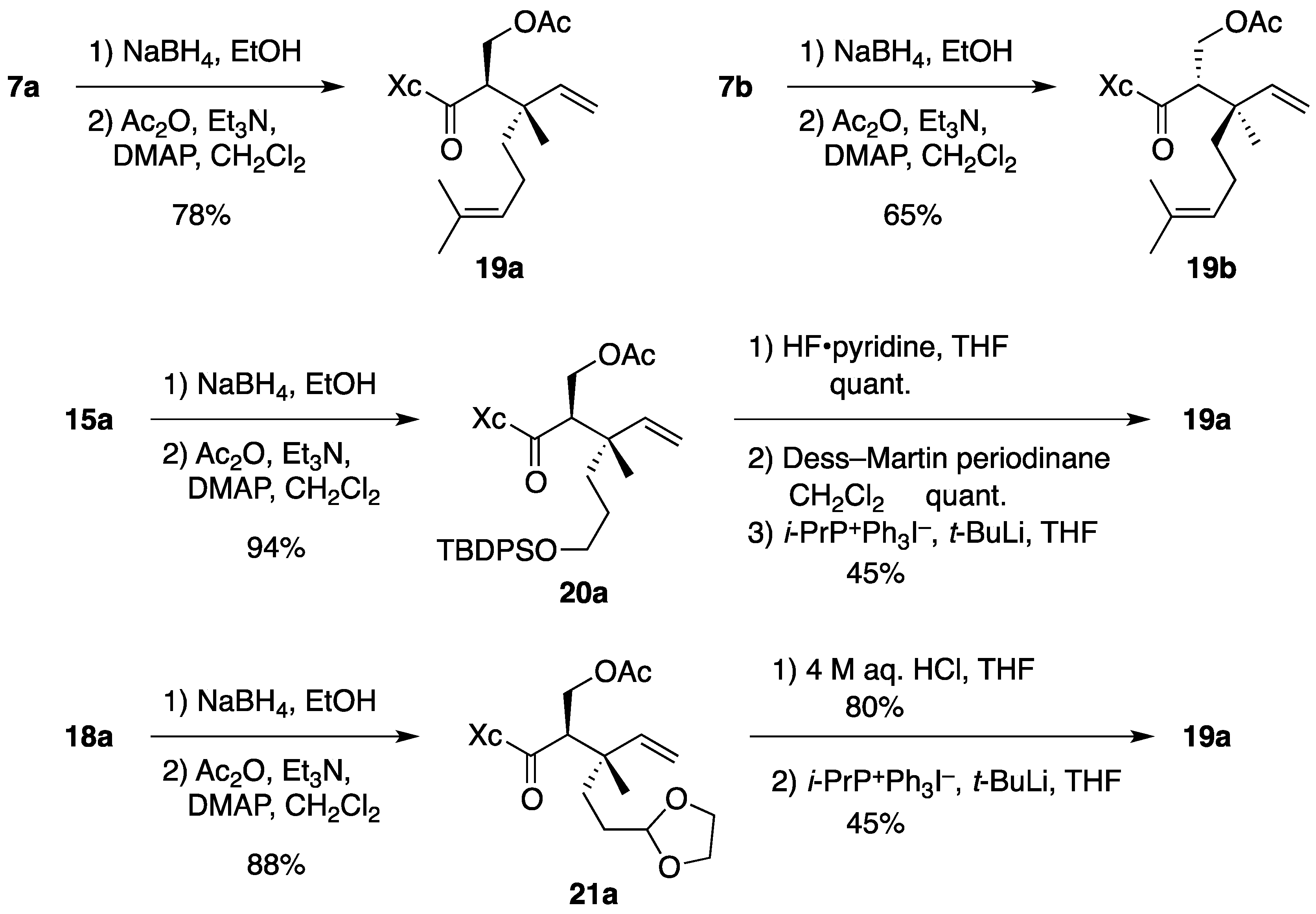
(2R)-N-[(2R,3S)-2-(Acetoxymethyl)-3,7-dimethyl-3-vinyloct-6-enoyl]bornane-10,2-sultam (19a). To a cooled (0 °C) stirred solution of 7a (158 mg, 375 μmol) in EtOH (4 mL) was added NaBH4 (14.2 mg, 375 μmol). The mixture was stirred at 0 °C for 4 h, quenched with saturated aqueous NH4Cl (1 mL), diluted with H2O (20 mL), and extracted with CH2Cl2 (10 mL × 3). The combined extracts were dried and concentrated under reduced pressure to provide crude alcohol (152 mg), which was used in the next step without further purification.
To a cooled (0 °C) stirred solution of crude alcohol in CH2Cl2 (4 mL) were added Ac2O (85 μL, 0.90 mmol), Et3N (150 μL, 1.08 mmol), and DMAP (4.4 mg, 36 μmol). The mixture was stirred at room temperature for 2.5 h, diluted with CH2Cl2 (20 mL), and washed with H2O (10 mL × 2). The combined aqueous layers were extracted with CH2Cl2 (30 mL). The combined organic layer and extract were dried and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:15) to provide 137 mg (78% for 2 steps) of 19a as a colorless oil: TLC Rf 0.61 (EtOAc/hexane, 1:2); [α]D21–38.4 (c 1.46, CHCl3); IR (neat) 2964, 2884, 1745, 1690 cm−1; 1H-NMR (500 MHz) δ 0.96 (s, 3H), 1.10 (s, 3H), 1.13 (s, 3H), 1.27–1.47 (m, 3H), 1.55 (br s, 3H), 1.64 (br s, 3H), 1.65 (m, 1H), 1.81–1.92 (m, 5H), 1.99 (s, 3H), 2.09-2.13 (m, 2H), 3.35 (m, 1H), 3.43 (d, 1H, J = 13.7 Hz), 3.50 (d, 1H, J = 13.7 Hz), 3.94 (t, 1H, J = 6.4 Hz), 4.22 (t, 1H, J = 10.6 Hz), 4.37 (dd, 1H, J = 3.4, 10.6 Hz), 5.01 (m, 1H), 5.01 (d, 1H, J = 17.5 Hz), 5.16 (d, 1H, J = 10.9 Hz), 5.79 (dd, 1H, J = 10.9, 17.5 Hz); 13C-NMR (125 MHz) δ 17.5, 17.8, 20.0, 20.4, 20.9, 22.3, 25.6, 26.5, 32.9, 38.6, 38.7, 43.3, 44.5, 47.7 (2C), 52.2, 53.3, 63.0, 65.6, 114.5, 124.1, 131.5, 143.3, 171.0, 172.7; HRMS calcd for C25H39NO5S (M+) m/z 465.2549, found 465.2556.
(2R)-N-[(2S,3R)-2-(Acetoxymethyl)-3,7-dimethyl-3-vinyloct-6-enoyl]bornane-10,2-sultam (19b). As described for the preparation of 19a from 7a, compound 7b (33.7 mg, 79.9 μmol) was treated with NaBH4 (1.5 mg, 40 μmol) in EtOH (1 mL) to provide crude alcohol (37.0 mg), which was then treated with Ac2O (19 μL, 0.20 mmol), Et3N (33 μL, 0.24 mmol), and DMAP (1.1 mg, 9.0 μmol) in CH2Cl2 (1 mL) to provide 24.2 mg (65% for 2 steps) of 19b as a colorless oil: TLC Rf 0.68 (EtOAc/hexane, 1:2); [α]D20–54.7 (c 1.06, CHCl3); IR (neat) 2966, 2886, 1742, 1687 cm−1; 1H-NMR (500 MHz) δ 0.97 (s, 3H), 1.18 (s, 3H), 1.19 (s, 3H), 1.35–1.48 (m, 3H), 1.57 (br s, 3H), 1.61 (m, 1H), 1.65 (br s, 3H), 1.86–1.93 (m, 5H), 1.95 (s, 3H), 2.10 (dd, 1H, J = 7.8, 13.8 Hz), 2.19 (m, 1H), 3.26 (dd, 1H, J = 3.7, 10.6 Hz), 3.47 (d, 1H, J = 13.8 Hz), 3.52 (d, 1H, J = 13.8 Hz), 3.94 (dd, 1H, J = 5.2, 7.8 Hz), 4.06 (t, 1H, J = 10.6 Hz), 4.56 (dd, 1H, J = 3.7, 10.6 Hz), 5.02 (d, 1H, J = 17.4 Hz), 5.06 (m, 1H), 5.16 (d, 1H, J = 11.3 Hz), 5.84 (dd, 1H, J = 11.3, 17.4 Hz); 13C-NMR (125 MHz) δ 17.6, 18.6, 19.9, 20.8, 21.1, 22.5, 25.7, 26.3, 33.0, 37.9, 38.6, 42.4, 44.6, 47.7, 47.8, 52.0, 53.3, 64.6, 65.8, 114.2, 124.5, 131.2, 143.9, 170.6, 172.6; HRMS calcd for C25H39NO5S (M+) m/z 465.2549, found 465.2558.
(2R)-N-[(2R,3S)-2-(Acetoxymethyl)-6-(tert-butyldiphenysilyloxy)-3-methyl-3-vinylhexanoyl]bornane-10,2-sultam (20a). As described for the preparation of 19a from 7a, compound 15a (150 mg, 236 μmol) was treated with NaBH4 (4.4 mg, 0.12 mmol) in EtOH (3 mL) to provide crude alcohol (152 mg), which was then treated with Ac2O (56 μL, 0.59 mmol), Et3N (99 μL, 0.71 mmol), and DMAP (3.0 mg, 25 μmol) in CH2Cl2 (3 mL) to provide 151 mg (94% for 2 steps) of 20a as a colorless oil: TLC Rf 0.66 (EtOAc/toluene, 1:5); [α]D23–28.6 (c 2.01, CHCl3); IR (neat) 2960, 2859, 1744, 1691 cm−1; 1H-NMR (500 MHz) δ 0.92 (s, 3H), 1.03 (s, 9H), 1.03 (s, 3H), 1.07 (s, 3H), 1.32–1.46 (m, 5H), 1.72 (m, 1H), 1.80 (m, 1H), 1.88–1.90 (m, 2H), 1.98 (s, 3H), 2.06–2.09 (m, 2H), 3.34 (m, 1H), 3.41 (d, 1H, J = 13.8 Hz), 3.47 (d, 1H, J = 13.8 Hz), 3.54–3.60 (m, 2H), 3.92 (t, 1H, J = 6.3 Hz), 4.22 (t, 1H, J = 10.6 Hz), 4.36 (dd, 1H, J = 3.4, 10.6 Hz), 4.99 (d, 1H, J = 17.5 Hz), 5.13 (d, 1H, J = 11.0 Hz), 5.75 (dd, 1H, J = 11.0, 17.5 Hz), 7.35–7.43 (m, 6H), 7.63-7.64 (m, 4H); 13C-NMR (125 MHz) δ 18.2, 19.1, 19.9, 20.5, 20.9, 26.5, 26.8 (3C), 27.0, 32.9, 34.4, 38.6, 43.0, 44.4, 47.6 (2C), 52.2, 53.3, 63.0, 64.1, 65.6, 114.6, 127.6 (4C), 129.5 (2C), 133.9, 134.0, 135.5 (2C), 135.6 (2C), 143.2, 171.0, 172.6; HRMS calcd for C34H44NO6SSi (M+–t-C4H9) m/z 622.2659, found 622.2677.
Synthesis of 19a from 20a. To a cooled (0 °C) stirred solution of 20a (12.2 mg, 17.9 μmol) in THF (3 mL) was added HF·pyridine (0.2 mL). The mixture was stirred at room temperature for 5 h and quenched with saturated aqueous NaHCO3 (1 mL). This was diluted with H2O (15 mL) and extracted with CH2Cl2 (10 mL × 4). The combined extracts were dried and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:2) to provide 8.0 mg (quant.) of alcohol as white crystals: mp 113-115 °C; TLC Rf 0.24 (EtOAc/hexane, 1:2); [α]D20 –42.7 (c 1.02, CHCl3); IR (neat) 3529, 2961, 2882, 1741, 1690 cm−1; 1H-NMR (500 MHz) δ 0.97 (s, 3H), 1.09 (s, 3H), 1.16 (s, 3H), 1.35–1.58 (m, 5H), 1.67 (m, 1H), 1.88-1.92 (m, 3H), 1.99 (s, 3H), 2.09–2.15 (m, 2H), 3.37 (m, 1H), 3.44 (d, 1H, J = 13.9 Hz), 3.50 (d, 1H, J = 13.9 Hz), 3.54 (m, 1H), 3.61 (m, 1H), 3.95 (t, 1H, J = 6.5 Hz), 4.20 (t, 1H, J = 10.7 Hz), 4.40 (dd, 1H, J = 3.4, 10.7 Hz), 5.02 (d, 1H, J = 17.5 Hz), 5.15 (d, 1H, J = 11.3 Hz), 5.80 (dd, 1H, J = 11.3, 17.5 Hz); 13C-NMR (125 MHz) δ 18.7, 19.9, 20.5, 20.9, 26.5, 27.1, 32.9, 34.4, 38.6, 42.9, 44.5, 47.7 (2C), 51.8, 53.3, 63.0, 63.1, 65.7, 114.5, 143.1, 171.2, 172.6; HRMS calcd for C22H35NO6S (M+) m/z 441.2185, found 441.2192.
To a cooled (0 °C) stirred solution of alcohol (20.9 mg, 47.3 μmol) in CH2Cl2 (1 mL) was added Dess–Martin periodinane (30.3 mg, 71.4 μmol). The mixture was stirred at room temperature for 2 h and Dess–Martin periodinane (31.1 mg, 73.3 μmol) was added. After being stirred at room temperature for 2.5 h, the mixture was quenched with saturated aqueous Na2S2O3 (3 mL) and saturated aqueous NaHCO3 (3 mL), diluted with H2O (4 mL), and extracted with CH2Cl2 (15 mL × 3). The combined extracts were washed with saturated brine (20 mL), dried and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:7) to provide 20.8 mg (quant.) of aldehyde as a colorless oil, which was immediately used in the next step: TLC Rf 0.33 (EtOAc/hexane, 1:2); 1H-NMR (300 MHz) δ 0.97 (s, 3H), 1.07 (s, 3H), 1.17 (s, 3H), 1.33–1.48 (m, 4H), 1.68 (m, 1H), 1.89–2.04 (m, 2H), 1.99 (s, 3H), 2.11–2.13 (m, 2H), 2.41 (t, 2H, J = 7.8 Hz), 3.38 (m, 1H), 3.44 (d, 1H, J = 13.9 Hz), 3.52 (d, 1H, J = 13.9 Hz), 3.95 (t, 1H, J = 6.5 Hz), 4.21 (t, 1H, J = 10.7 Hz), 4.37 (dd, 1H, J = 3.6, 10.7 Hz), 5.05 (d, 1H, J = 17.5 Hz), 5.20 (d, 1H, J = 10.7 Hz), 5.78 (dd, 1H, J = 10.7, 17.5 Hz), 9.73 (s, 1H).
The following reaction was carried out under Ar. To a cooled (0 °C) stirred suspension of i-PrP+Ph3I− (21.9 mg, 49.1 μmol) in THF (1 mL) was added t-BuLi (1.61 M solution in pentane, 29 μL, 47 μmol). The mixture was stirred at 0 °C for 30 min and a solution of aldehyde (6.9 mg, 16 μmol) in THF (1 mL) was added. After being stirred at 0 °C for 20 min, the mixture was diluted with H2O (10 mL) and extracted with CH2Cl2 (15 mL × 3). The combined extracts were dried and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:15) to provide 3.3 mg (45%) of 19a.
(2R)-N-[(2R,3S)-2-(Acetoxymethyl)-5-(1,3-dioxolan-2-yl)-3-methyl-3-vinylpentanoyl]bornane-10,2-sultam (21a). As described for the preparation of 19a from 7a, compound 18a (154 mg, 350 μmol) was treated with NaBH4 (6.5 mg, 0.17 mmol) in EtOH (3 mL) to provide crude alcohol (158 mg), which was then treated with Ac2O (83 μL, 0.88 mmol), Et3N (146 μL, 1.05 mmol), and DMAP (4.4 mg, 36 μmol) in CH2Cl2 (4 mL) to provide 150 mg (88% for 2 steps) of 21a as a colorless oil: TLC Rf 0.65 (EtOAc/toluene, 1:2); [α]D21–39.5 (c 1.02, CHCl3); IR (neat) 2962, 2884, 1743, 1690 cm−1; 1H-NMR (500 MHz) δ 0.97 (s, 3H), 1.08 (s, 3H), 1.16 (s, 3H), 1.36 (m, 1H), 1.43–1.50 (m, 2H), 1.55–1.60 (m, 2H), 1.78 (m, 1H), 1.87–1.91 (m, 3H), 1.99 (s, 3H), 2.11–2.18 (m, 2H), 3.35 (m, 1H), 3.43 (d, 1H, J = 13.7 Hz), 3.50 (d, 1H, J = 13.7 Hz), 3.78–3.81 (m, 2H), 3.91–3.95 (m, 3H), 4.22 (t, 1H, J = 10.7 Hz), 4.38 (dd, 1H, J = 3.5, 10.7 Hz), 4.76 (t, 1H, J = 4.6 Hz), 5.01 (d, 1H, J = 17.5 Hz), 5.17 (d, 1H, J = 10.7 Hz), 5.77 (dd, 1H, J = 10.7, 17.5 Hz); 13C-NMR (125 MHz) δ 18.1, 20.0, 20.5, 20.9, 26.5, 28.3, 32.3, 32.9, 38.6, 42.8, 44.5, 47.7 (2C), 52.2, 53.3, 63.0, 64.7, 64.8, 65.6, 104.5, 114.9, 142.9, 171.0, 172.5; HRMS calcd for C24H37NO7S (M+) m/z 483.2291, found 483.2291.
Synthesis of 19a from 21a. A solution of 21a (80.5 mg, 166 μmol) in THF (12 mL) and 4 M aqueous HCl (12 mL) was stirred at 0 °C for 15 h, diluted with saturated aqueous NaHCO3 (50 mL), and extracted with CH2Cl2 (60 mL × 3). The combined extracts were dried and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:7) to provide 58.2 mg (80%) of aldehyde, which was identical with the aldehyde derived from 20a and converted into 19a as described above.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}