Antifungal Activity of Eugenol Analogues. Influence of Different Substituents and Studies on Mechanism of Action

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
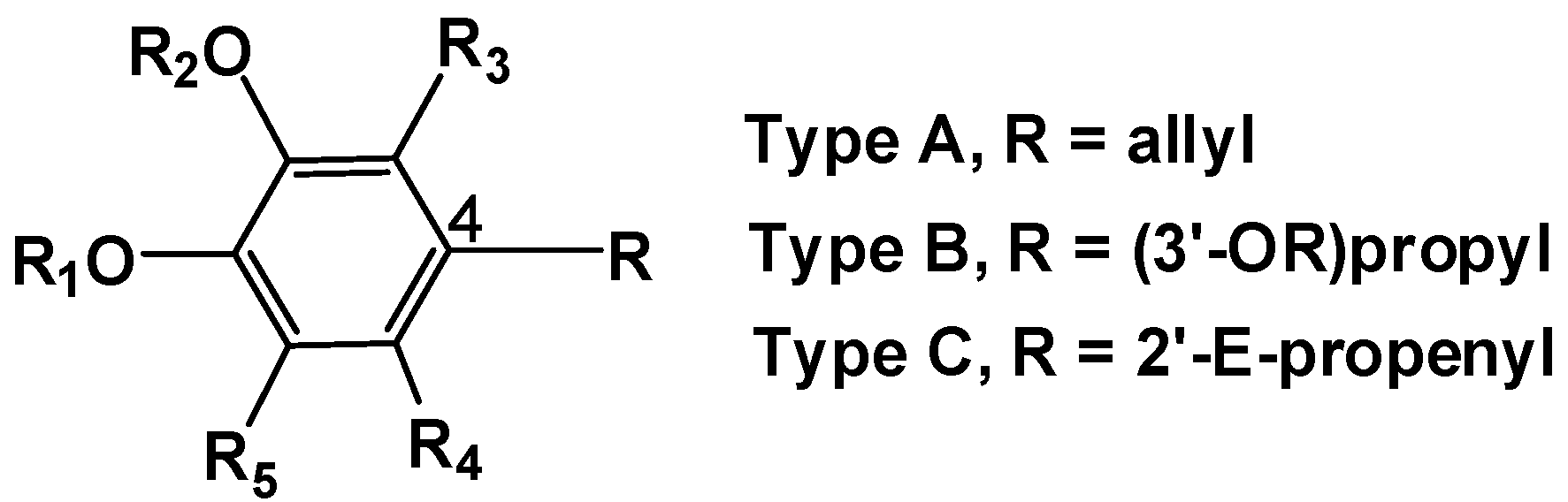
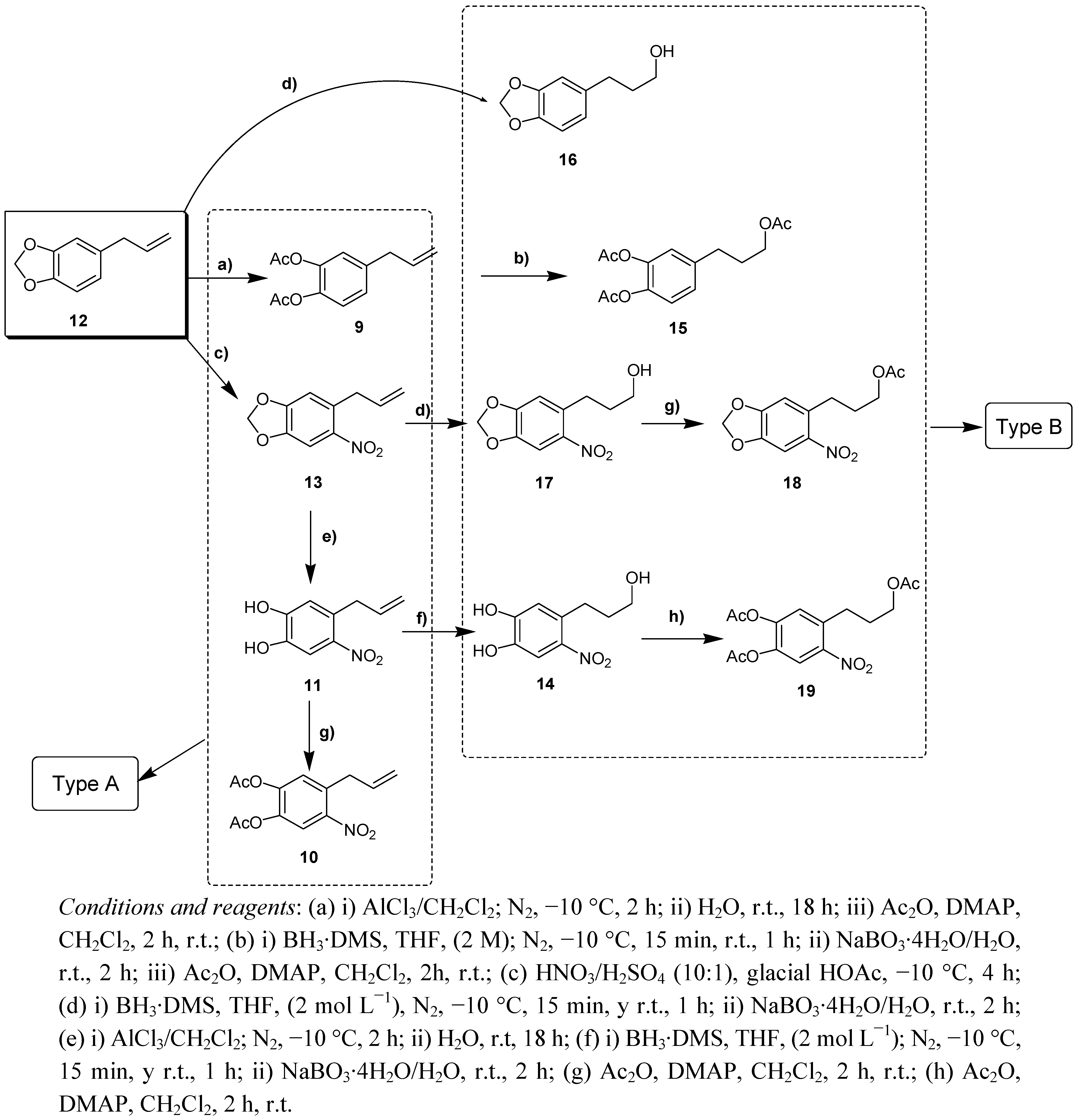
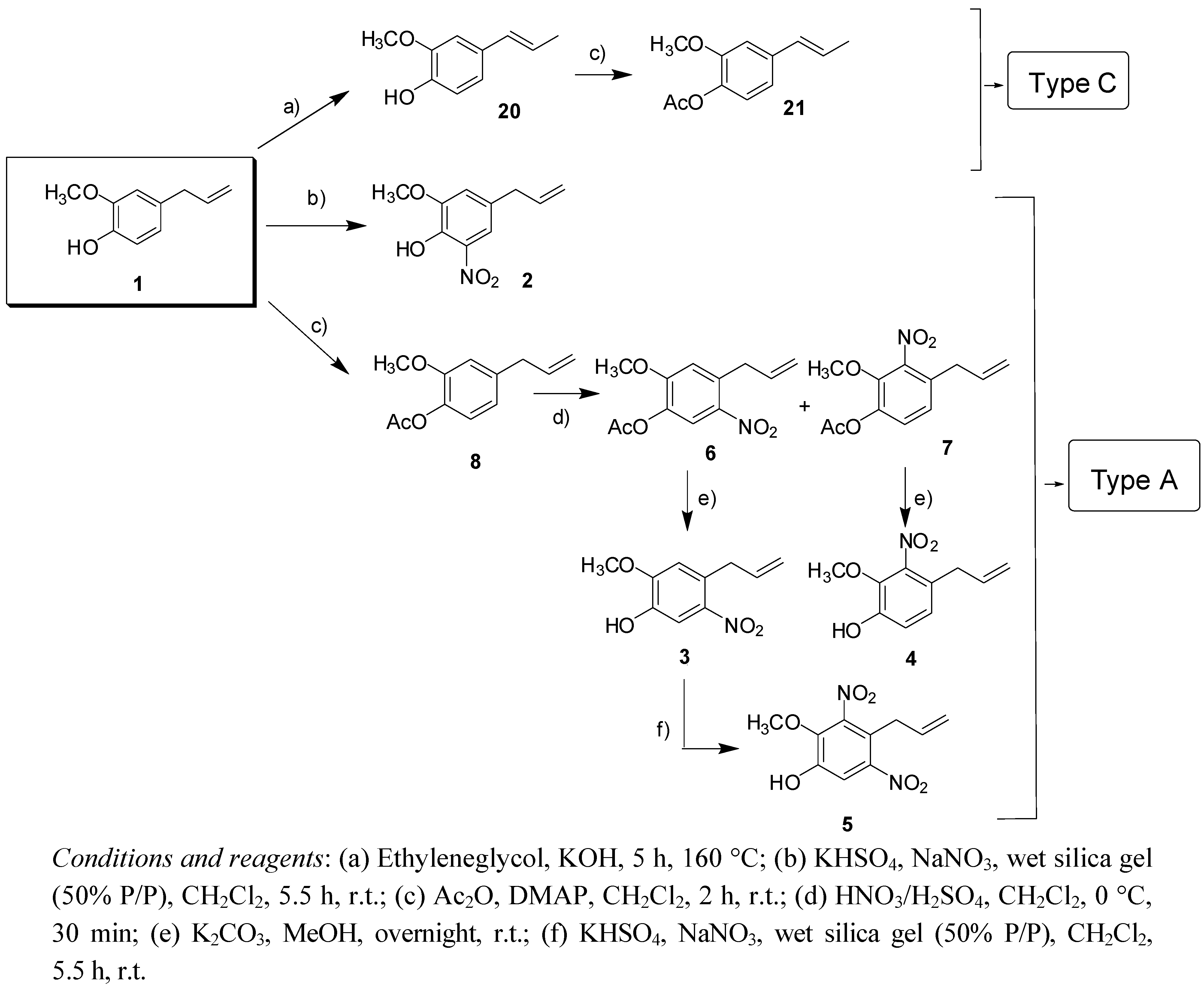
2.1. Chemistry

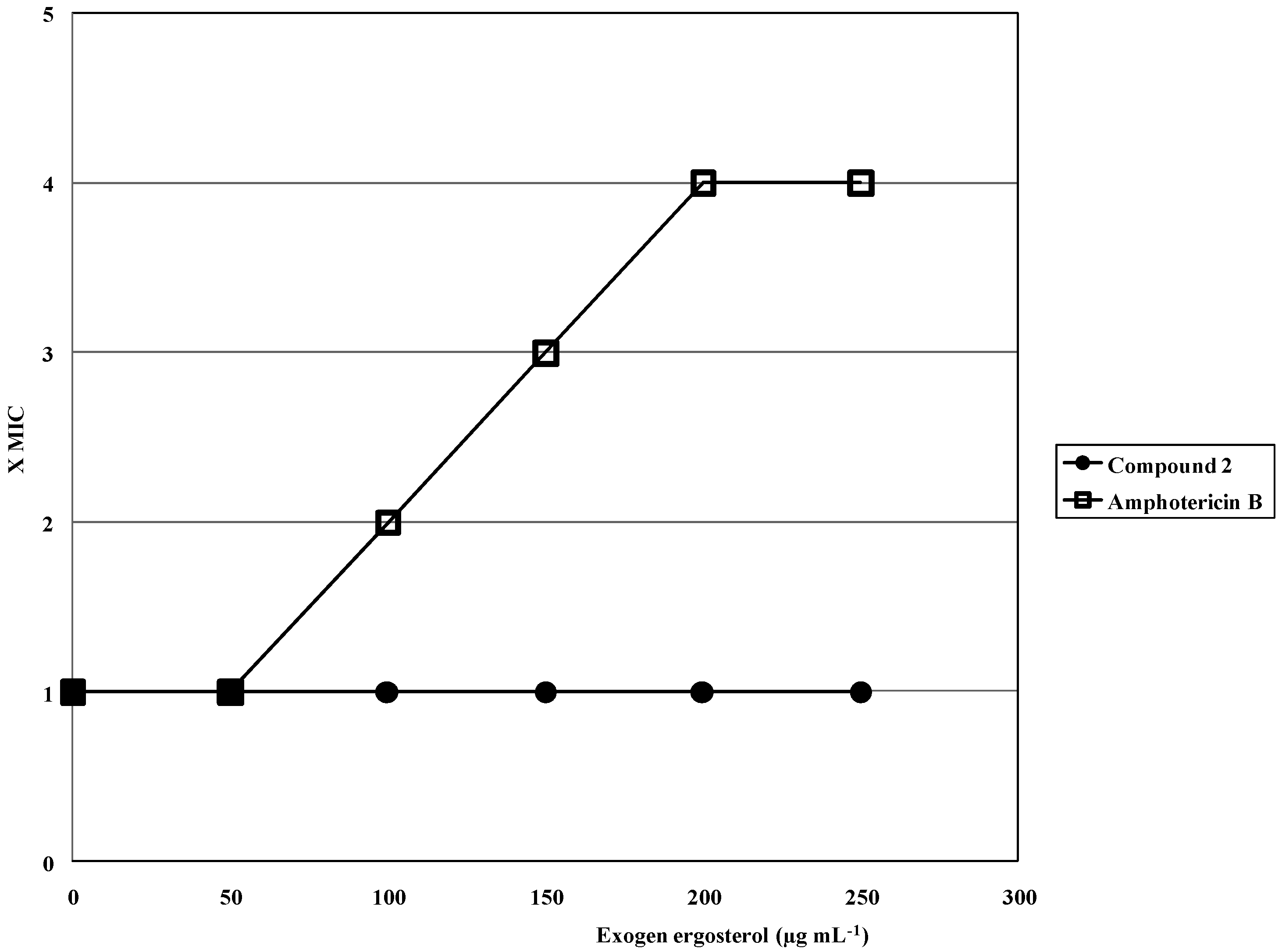
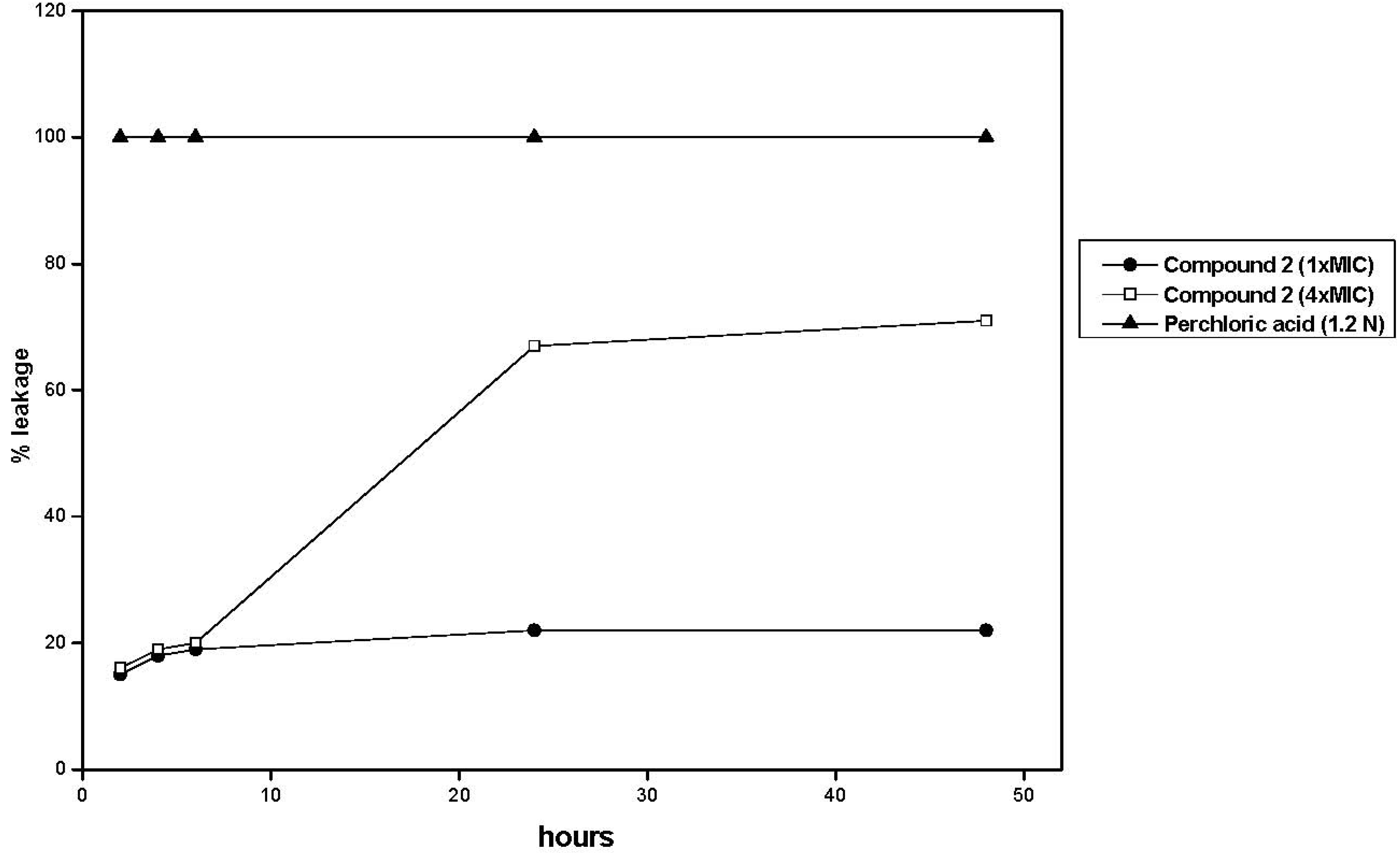
2.2. Antifungal Activity
2.2.1. First Order Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Type | R1 | R2 | R3 | R4 | R5 | R6 | Log P | Ca | Sc | Cn | Tr | Tm | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A | H | CH3 | H | H | H | - | 2.57 | i | i | 250 | 125 | 125 |
| 2 | A | H | CH3 | H | H | NO2 | - | 2.65 | 31 | 62 | 16 | 31 | 31 |
| 3 | A | H | CH3 | H | NO2 | H | - | 2.65 | 250 | 250 | 125 | 62 | 62 |
| 4 | A | H | CH3 | NO2 | H | H | - | 2.65 | 250 | 125 | 125 | 31 | 31 |
| 5 | A | H | CH3 | NO2 | NO2 | H | - | 2.61 | i | i | i | 250 | 250 |
| 6 | A | Ac | CH3 | H | NO2 | H | - | 2.77 | 125 | i | 250 | 62 | 62 |
| 7 | A | Ac | CH3 | NO2 | H | H | - | 2.77 | 250 | i | 250 | 62 | 62 |
| 8 | A | Ac | CH3 | H | H | H | - | 2.55 | i | i | i | 125 | 125 |
| 9 | A | Ac | Ac | H | H | H | - | 2.26 | 250 | i | 250 | 125 | 125 |
| 10 | A | Ac | Ac | H | NO2 | H | - | 2.38 | 250 | i | 125 | 125 | 125 |
| 11 | A | H | H | H | NO2 | H | - | 2.13 | 250 | i | 125 | 62 | 62 |
| 12 | A | -CH2- | H | H | H | - | 2.87 | i | i | i | i | i | |
| 13 | A | -CH2- | H | NO2 | H | - | 2.14 | 125 | i | 250 | i | i | |
| 14 | B | H | H | H | NO2 | H | H | 1.21 | i | i | i | i | i |
| 15 | B | Ac | Ac | H | H | H | Ac | 1.56 | i | i | i | i | i |
| 16 | B | H | H | H | H | H | H | 1.94 | i | i | i | i | i |
| 17 | B | -CH2- | H | NO2 | H | - | 1.22 | i | i | i | i | i | |
| 18 | B | H | H | H | NO2 | H | Ac | 1.81 | 250 | i | 250 | 125 | 125 |
| 19 | B | Ac | Ac | H | NO2 | H | Ac | 2.05 | i | i | i | i | i |
| 20 | C | H | CH3 | H | H | H | 2.52 | i | i | i | i | i | |
| 21 | C | Ac | CH3 | H | H | H | - | 2.50 | i | i | i | i | i |
| Amphotericin B | 0.78 | 0.50 | 0.25 | 0.075 | 0.075 | ||||||||
| Terbinafine | 1.56 | 3.12 | 0.39 | 0.01 | 0.025 | ||||||||
| Ketoconazole | 0.50 | 0.50 | 0.25 | 0.025 | 0.025 | ||||||||
2.2.2. Second Order Studies
| Strain | 2 | 6 | 13 | Amph. B | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Voucher specimen | MIC100 | MIC80 | MIC50 | MFC | MIC100 | MIC80 | MIC50 | MFC | MIC100 | MIC80 | MIC50 | MFC | MIC100 | |
| C. albicans | ATCC 10231 | 31 | 16 | 8 | 125 | 62 | 62 | 31 | >250 | 31 | 16 | 8 | 125 | 1.00 |
| C. albicans | C 126-2000 | 31 | 25 | 20 | 250 | i | 250 | 125 | >250 | 31 | 25 | 20 | 250 | 1.56 |
| C. albicans | C 127-2000 | 62 | 31 | 25 | 125 | i | i | i | >250 | 62 | 31 | 25 | 125 | 0.78 |
| C. albicans | C 128-2000 | 62 | 31 | 16 | 250 | 16 | 16 | 16 | >250 | 62 | 31 | 16 | 250 | 1.56 |
| C. albicans | C 129-2000 | 31 | 25 | 16 | 250 | i | 250 | 250 | >250 | 31 | 25 | 16 | 250 | 0.78 |
| C. albicans | C 130-2000 | 62 | 31 | 25 | 250 | i | i | i | >250 | 62 | 31 | 25 | 250 | 0.39 |
| C. glabrata | C 115-2000 | 125 | 125 | 125 | 250 | i | i | i | >250 | 125 | 125 | 125 | 250 | 0.39 |
| C. parapsilopsis | C 124-2000 | 125 | 62 | 31 | >250 | i | 250 | 125 | >250 | 125 | 62 | 31 | >250 | 0.78 |
| C. lusitaniae | C 131-2000 | 62 | 50 | 25 | 250 | i | i | 250 | >250 | 62 | 50 | 25 | 250 | 0.39 |
| C. colliculosa | C 122-2000 | 62 | 31 | 25 | 250 | 31 | 31 | 16 | >250 | 62 | 31 | 25 | 250 | 0.36 |
| C. krusei | C 117-2000 | 125 | 100 | 50 | >250 | i | i | i | >250 | 125 | 100 | 50 | >250 | 0.39 |
| C. kefyr | C 123-2000 | 125 | 62 | 31 | >250 | i | i | i | >250 | 125 | 62 | 31 | >250 | 0.78 |
| C. tropicalis | C 131-1997 | 62 | 31 | 25 | >250 | i | i | i | >250 | 62 | 31 | 25 | >250 | 0.50 |
| 2 | 3 | 4 | 10 | 11 | Amp. B | Itz | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fungal sp. | Voucher specimen | MIC100 | MIC80 | MIC50 | MFC | MIC100 | MIC80 | MIC50 | MFC | MIC100 | MIC80 | MIC50 | MFC | MIC100 | MIC80 | MIC50 | MFC | MIC100 | MIC80 | MIC50 | MFC | CIM100 | ||
| Cn ATCC 32264 | 16 | 8 | 8 | 62 | 125 | 62 | 31 | 250 | 125 | 62 | 31 | 125 | 125 | 62 | 31 | 250 | 125 | 62 | 62 | >250 | 0.25 | 0.15 | ||
| Cn | IM 983040 | 31 | 16 | 8 | 250 | 125 | 62 | 62 | 250 | 125 | 31 | 16 | 125 | 250 | 250 | 125 | >250 | 250 | 125 | 16 | 250 | 0.13 | <0.015 | |
| Cn | IM 972724 | 31 | 16 | 8 | 250 | 125 | 125 | 62 | 250 | 125 | 31 | 31 | 125 | i | i | 250 | >250 | 250 | 125 | 16 | 250 | 0.06 | 0.25 | |
| Cn | IM 042074 | 31 | 16 | 8 | 250 | 125 | 125 | 62 | 250 | 125 | 62 | 31 | 125 | 250 | 250 | 31 | >250 | 250 | 125 | 62 | 250 | 0.25 | <0.015 | |
| Cn | IM 983036 | 31 | 16 | 16 | 250 | 125 | 62 | 31 | 250 | 125 | 62 | 62 | 125 | 250 | 250 | 31 | >250 | 250 | 125 | 31 | 250 | 0.13 | <0.015 | |
| Cn | IM 00319 | 31 | 16 | 8 | 250 | 125 | 31 | 31 | 250 | 125 | 62 | 15 | 125 | 250 | 125 | 62 | >250 | 250 | 125 | 62 | 250 | 0.25 | <0.015 | |
| Cn | IM 972751 | 31 | 16 | 8 | 250 | 125 | 62 | 31 | 250 | 125 | 62 | 31 | 250 | 250 | 250 | 62 | >250 | 125 | 62 | 62 | 250 | 0.25 | <0.015 | |
| Cn | IM 031631 | 31 | 16 | 4 | 250 | 250 | 125 | 31 | 250 | 125 | 62 | 31 | 250 | 250 | 250 | 125 | >250 | 125 | 125 | 16 | 250 | 0.13 | 0.25 | |
| Cn | IM 031706 | 62 | 31 | 16 | 125 | 125 | 62 | 15 | 250 | 125 | 62 | 15 | 250 | 250 | 125 | 31 | >250 | 250 | 125 | 31 | 250 | 0.25 | 0.50 | |
| Cn | IM 961951 | 31 | 16 | 8 | 250 | 250 | 125 | 62 | 250 | 125 | 62 | 15 | >250 | 250 | 125 | 31 | >250 | 250 | 62 | 31 | 250 | 0.06 | <0.015 | |
| Strain | Voucher specimen | 2 | 3 | 4 | 6 | 7 | 8 | 9 | 10 | 11 | 18 | Terb. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T. rubrum | C 110 | 16 | 16 | 62 | 62 | 31 | 125 | 62 | 31 | 31 | 125 | 0.006 |
| T. rubrum | C 135 | 31 | 31 | 62 | 62 | 62 | 125 | 125 | 31 | 31. | 125 | 0.006 |
| T. rubrum | C 136 | 31 | 31 | 62 | 125 | 62 | 125 | 125 | 62 | 62 | 125 | 0.006 |
| T. rubrum | C 137 | 16 | 31 | 31 | 62 | 31 | 125 | 125 | 31 | 62 | 125 | 0.006 |
| T. rubrum | C 139 | 16 | 16 | 31 | 62 | 31 | 125 | 62 | 62 | 62 | 125 | 0.012 |
| T. rubrum | C 140 | 16 | 62 | 16 | 62 | 31 | 125 | 62 | 62 | 31 | 125 | 0.003 |
| T. mentagrophytes | C 108 | 16 | 125 | 62 | 62 | 31 | 125 | 62 | 62 | 62 | 125 | 0.006 |
| T. mentagrophytes | C 364 | 16 | 62 | 31 | 62 | 31 | 250 | 62 | 62 | 62 | 125 | 0.006 |
| T. mentagrophytes | C 539 | 31 | 125 | 16 | 62 | 31 | 250 | 62 | 62 | 125 | 125 | 0.006 |
| T. mentagrophytes | C 738 | 16 | 62 | 31 | 62 | 31 | 125 | 125 | 62 | 31 | 125 | 0.006 |
| T. mentagrophytes | C 943 | 31 | 62 | 16 | 62 | 62 | 250 | 62 | 62 | 62 | 125 | 0.006 |
| T. mentagrophytes | C 944 | 31 | 31 | 31 | 62 | 62 | 125 | 62 | 62 | 31 | 125 | 0.006 |

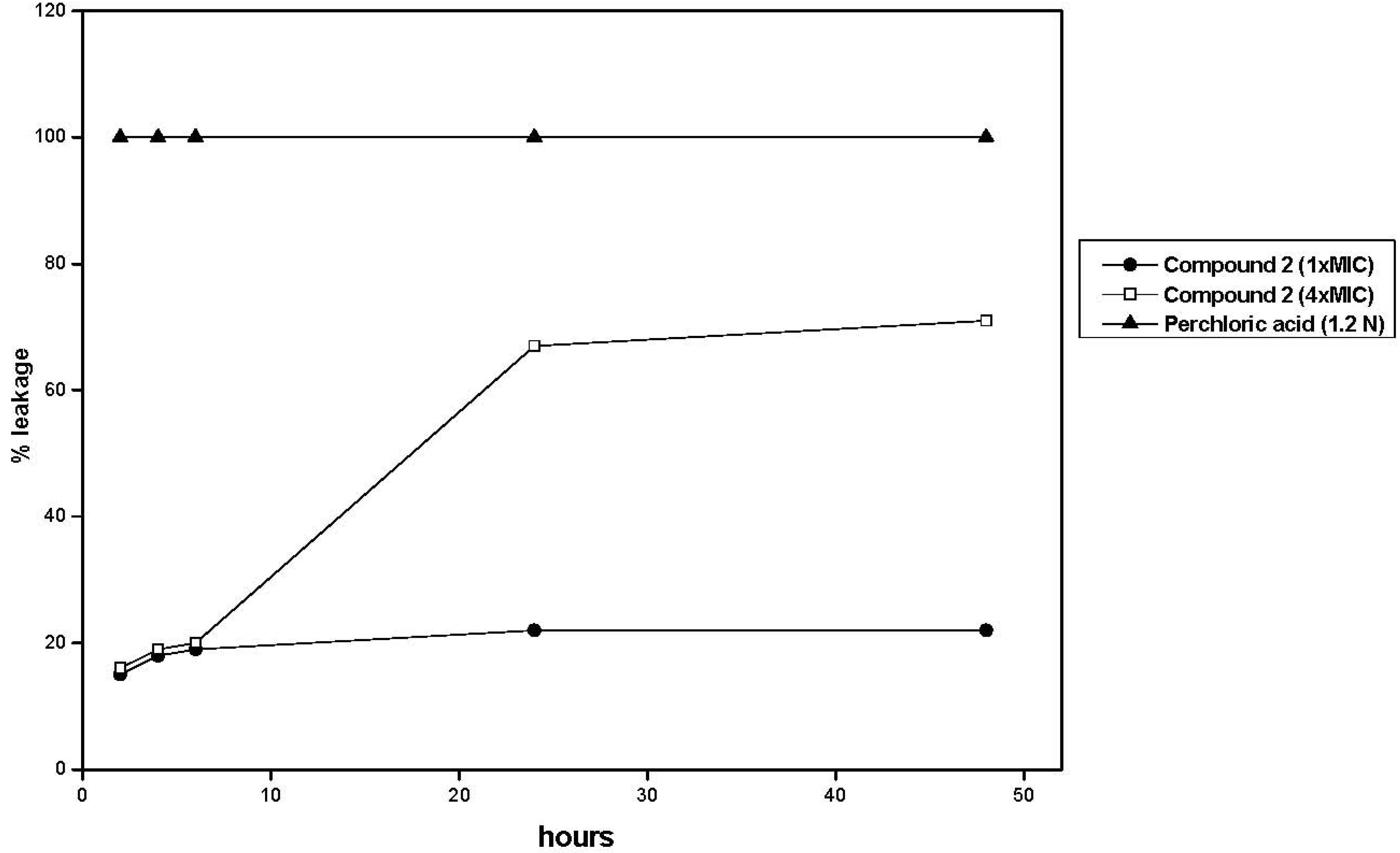
3. Experimental
3.1. General
3.2. Antifungal Susceptibility Testing
3.2.1. Microorganisms and Media
3.2.2. Determination of MICs and MFCs
3.2.3. Determination of MICs and MFCs
3.2.4. Statistical Analysis
4. Conclusions
Acknowledgements
References and Notes
- Chen, S.; Playford, E.; Sorrel, T. Antifungal therapy in invasive fungal infections. Curr. Opin. Pharmacol. 2010, 10, 522–530. [Google Scholar] [CrossRef]
- Monk, B.; Goffeau, A. Outwitting multidrug resistance to antifungals. Science 2008, 321, 367–369. [Google Scholar]
- Mathew, B.; Nath, M. Recent approaches to antifungal therapy for invasive mycoses. Chem. Med. Chem. 2009, 4, 310–323. [Google Scholar]
- Espinel-Ingroff, A. Novel antifungal agents, targets or therapeutic strategies for the treatment of invasive fungal diseases: A review of the literature (2005–2009). Rev. Iberoam. Micol. 2009, 26, 15–22. [Google Scholar] [CrossRef]
- Mukherjee, P.; Leidich, S.; Isham, N.; Leitner, I.; Ryder, N.; Ghannoum, M. Clinical Trichophyton rubrum strain exhibiting primary resistance to terbinafine. Antimicrob. Agents Chemother. 2003, 47, 82–86. [Google Scholar] [CrossRef]
- Vicente, M.F.; Basilio, A.; Cabello, A.; Peláez, F. Microbial natural products as a source of antifungals. Clin. Microbiol. Infect. 2003, 9, 15–32. [Google Scholar] [CrossRef]
- Zemek, J.; Košíková, B.; Augustin, J.; Joniak, D. Antibiotic properties of lignin components. Folia Microbiol. 1979, 24, 483–486. [Google Scholar] [CrossRef]
- Himejima, M.; Kubo, I. Antimicrobial agents from Licaria puchuri-major and their synergistic effect to polygodial. J. Nat. Prod. 1992, 55, 620–625. [Google Scholar] [CrossRef]
- Kubo, I.; Muroi, H.; Himejima, M. Combination effects of antifungal nagilactones against Candida albicans and two other fungi with phenylpropanoids. J. Nat. Prod. 1993, 56, 220–226. [Google Scholar] [CrossRef]
- Fujita, K.; Kubo, I. Potentiation of fungicidal activities of trans-anethole against Saccharomyces cerevisiae under hypoxic conditions. J. Biosci. Bioeng. 2004, 98, 490–492. [Google Scholar]
- Zacchino, S.; López, S.; Pezzenati, G.; Furlán, R.; Santecchia, C.; Muñoz, L.; Giannini, F.; Rodríguez, A.; Enriz, R. In vitro evaluation of antifungal properties of phenylpropanoids and related compounds acting against dermatophytes. J. Nat. Prod. 1999, 62, 1353–1357. [Google Scholar] [CrossRef]
- Faria, T.; Ferreira, R.; Yassumoto, L.; Pinto de Souza, J.; Ishikawa, N.; de Melo Barbosa, A. Antifungal activity of essential oil isolated from Ocimum gratissimum L. (eugenol chemotype) against phytopathogenic fungi. Braz. Arch. Biol. Technol. 2006, 49, 867–871. [Google Scholar]
- Wang, S.; Chen, P.P.; Chang, S. Antifungal activities of essential oils from indigenous cinnamon (Cinnamomum osmophloeum) leaves. Bioresour. Technol. 2005, 96, 813–818. [Google Scholar] [CrossRef]
- Campaniello, D.; Sinigaglia, M. Antifungal activity of eugenol against Penicillium, Aspergillus, and Fusarium specie. J. Food Prot. 2010, 73, 1124–1128. [Google Scholar]
- Cos, P.; Vlietinck, J.; Vanden Berghe, D.; Maes, L. Antiinfective potntial of natural products: How to develop a steronger in vitro proor of concept. J. Ethnopharmacol. 2006, 106, 290–302. [Google Scholar] [CrossRef]
- Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts. Document M27-A3, Approved Standard, 3th ed; Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2008; Volume 28, N° 14, p. 25.
- Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts. Document M38-A2, Approved Standard, 2nd ed; Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2008; Volume 28, N° 16, p. 35.
- Chami, N.; Bennis, S.; Chami, F.; Aboussekhra, A.; Remmal, A. Study of anticandidal activity of carvacrol and eugenol in vitro and in vivo. Oral Microbiol. Immunol. 2005, 20, 106–111. [Google Scholar] [CrossRef]
- Sikemma, J.; de Bont, J.; Poolman, B. Mechanisms of membrane toxicity of hydrocarbons. Microbiol. Rev. 1995, 59, 201–222. [Google Scholar]
- Gill, A.; Holley, R. Disruption of Escherichia coli, Listeria monocytogenes and Lactobacillus sakei cellular membranes by plant oil aromatics. Int. J. Food Microbiol. 2006, 108, 1–9. [Google Scholar] [CrossRef]
- Voda, K.; Boh, B.; Vrtačnik, M. A quantitative structure-antifungal activity relationship study of oxygenated aromatic essential oil compounds using data structuring and PLS regression analysis. J. Mol. Model. 2004, 10, 76–84. [Google Scholar] [CrossRef]
- Ntamila, M.S.; Hassanali, A. Isolation of oil of clove and separation of eugenol and acetyl eugenol. J. Chem. Educ. 1976, 53, 263. [Google Scholar] [CrossRef]
- Carrasco Altamirano, H.; Espinoza, L.; Gallardo, C.; Cardona, W.; Catalán, K.; Russo, A.; Cardile, V.; Lombardo, L.; Cuellar, M. Eugenol and its synthetic analogues inhibit cell growth of human cancer cells (Part I). J. Braz. Chem. Soc. 2008, 19, 543–548. [Google Scholar] [CrossRef]
- Hidalgo, M.E.; de la Rosa, C.; Carrasco, H.; Cardona, W.; Gallardo, C.; Espinoza, L. Antioxidant capacity of eugenol derivatives. Quim. Nova 2009, 32, 1467–1470. [Google Scholar] [CrossRef]
- Ernst, E.; Roling, E.; Petzold, R.; Keele, D.; Klepser, M. In vitro activityof micafungin (FK-463) against Candida spp.: microdilution, time-kill, and postantifungal-effect studies. Antimicrob. Agents Chemother. 2002, 46, 3846–3853. [Google Scholar] [CrossRef]
- Klepser, M.; Ernst, E.; Ernst, M.; Messer, S.; Pfaller, M. Evaluation of endpoints for antifungal susceptibility determinations with LY303366. Antimicrob. Agents Chemother. 1998, 42, 1387–1391. [Google Scholar]
- Pfaller, M.A.; Diekema, D. Epidemiology of invasive candidiasis: A persistent public health problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar] [CrossRef]
- Kontoyannis, D.; Mantadakis, E.; Samonis, G. Systemic mycoses in the immunocompromised host: An update in antifungal therapy. J. Hosp. Infect. 2003, 53, 243–258. [Google Scholar] [CrossRef]
- Singh, N. Treatment of opportunistic mycoses: How long is long enough? Lancet Infect. Dis. 2003, 3, 703–708. [Google Scholar] [CrossRef]
- Weitzman, R.; Summerbell, I. The dermatophytes. Clin. Microb. Rev. 1995, 8, 240–259. [Google Scholar]
- Frost, D.; Brandt, K.; Cugier, D.; Goldman, R. A whole-cell Candida albicans assay for the detection of inhibitors towards fungal cell wall synthesis and assembly. J. Antibiot. 1995, 48, 306–310. [Google Scholar] [CrossRef]
- Escalante, A.; Gattuso, M.; Pérez, P.; Zacchino, S. Evidence for the mechanism of action of the antifungal phytolaccoside B isolated from Phytolacca tetramera Hauman. J. Nat. Prod. 2008, 71, 1720–1725. [Google Scholar] [CrossRef]
- Lunde, C.; Kubo, I. Effect of polygodial on the mitochondrial ATPase of Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 2000, 44, 1943–1953. [Google Scholar] [CrossRef]
- Matsumori, N.; Sawada, Y.; Murata, M. Mycosamine orientation of amphotericin B controlling interaction with ergosterol: Sterol-dependent activity of conformation-restricted derivatives with an amino-carbonyl bridge. J. Am. Chem. Soc. 2005, 127, 10667–10675. [Google Scholar] [CrossRef]
- Gruda, I.; Nadeau, P.; Brajtburg, J. Application of differential spectra in the ultraviolet-visible region to study the formation of amphotericin B-sterol complexes. Biochim. Biophys. Acta 1980, 602, 260–268. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–21 are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Carrasco, H.; Raimondi, M.; Svetaz, L.; Liberto, M.D.; Rodriguez, M.V.; Espinoza, L.; Madrid, A.; Zacchino, S. Antifungal Activity of Eugenol Analogues. Influence of Different Substituents and Studies on Mechanism of Action. Molecules 2012, 17, 1002-1024. https://doi.org/10.3390/molecules17011002
Carrasco H, Raimondi M, Svetaz L, Liberto MD, Rodriguez MV, Espinoza L, Madrid A, Zacchino S. Antifungal Activity of Eugenol Analogues. Influence of Different Substituents and Studies on Mechanism of Action. Molecules. 2012; 17(1):1002-1024. https://doi.org/10.3390/molecules17011002
Chicago/Turabian StyleCarrasco, Héctor, Marcela Raimondi, Laura Svetaz, Melina Di Liberto, María V. Rodriguez, Luis Espinoza, Alejandro Madrid, and Susana Zacchino. 2012. "Antifungal Activity of Eugenol Analogues. Influence of Different Substituents and Studies on Mechanism of Action" Molecules 17, no. 1: 1002-1024. https://doi.org/10.3390/molecules17011002