Novel Library of Selenocompounds as Kinase Modulators


Abstract
:1. Introduction
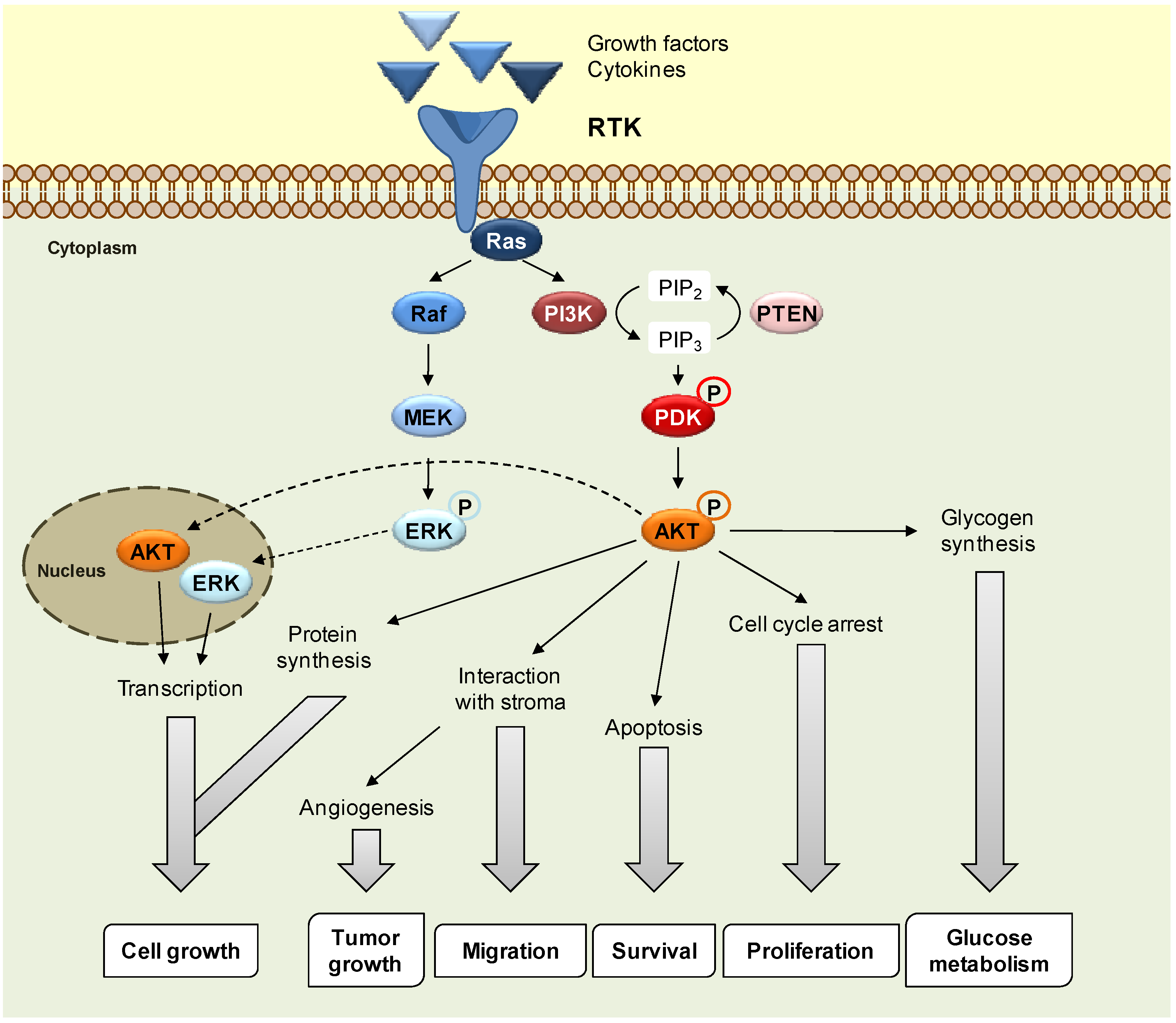
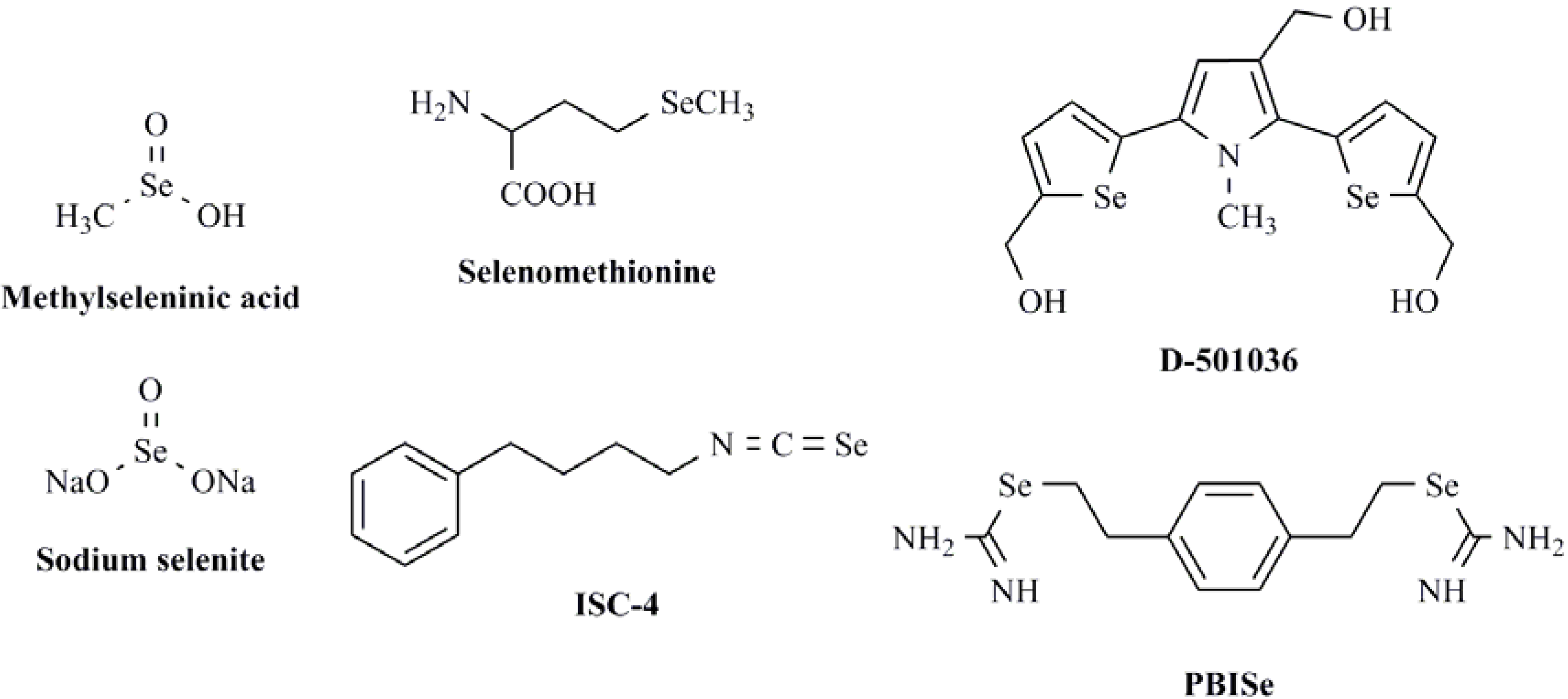
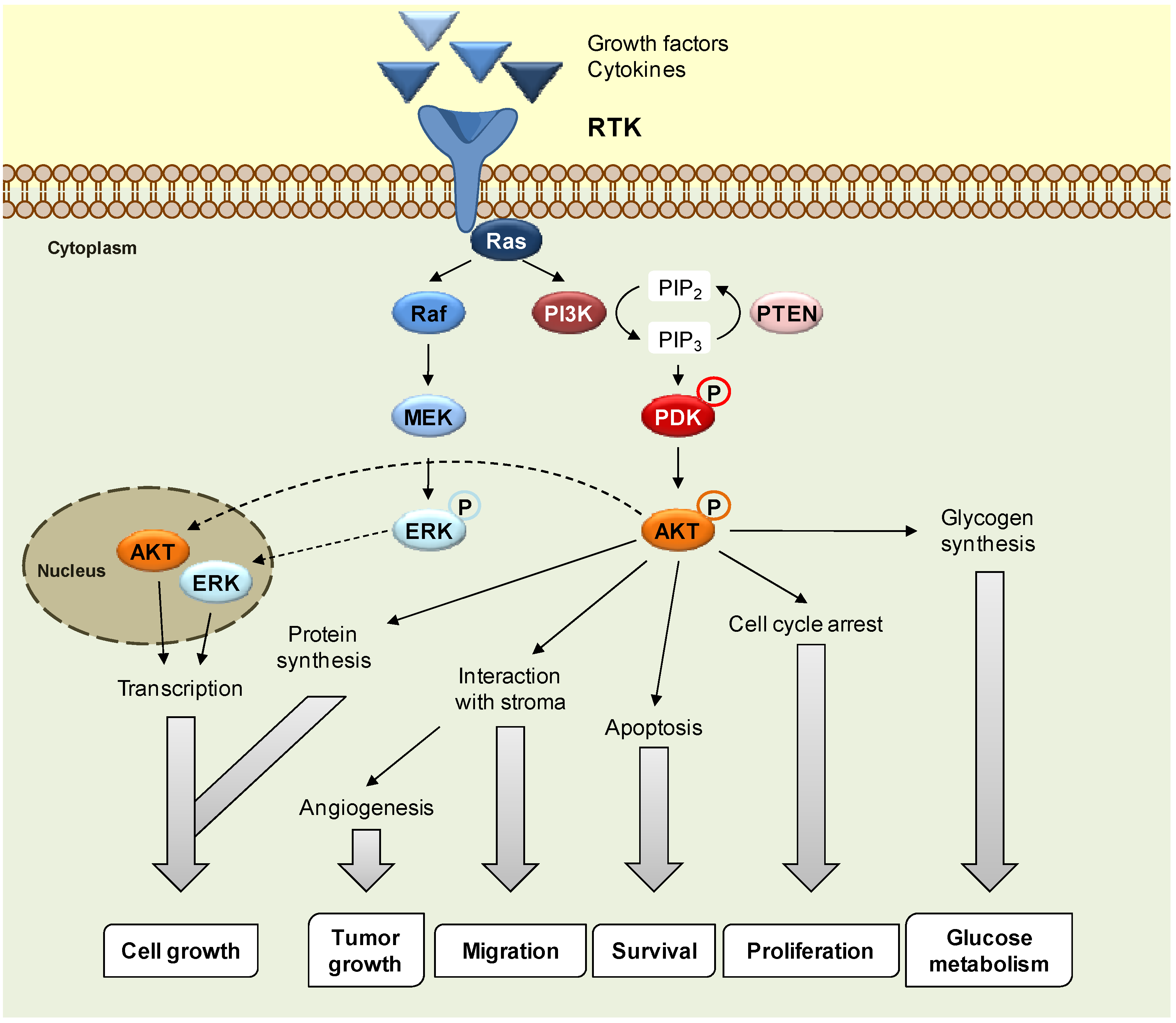
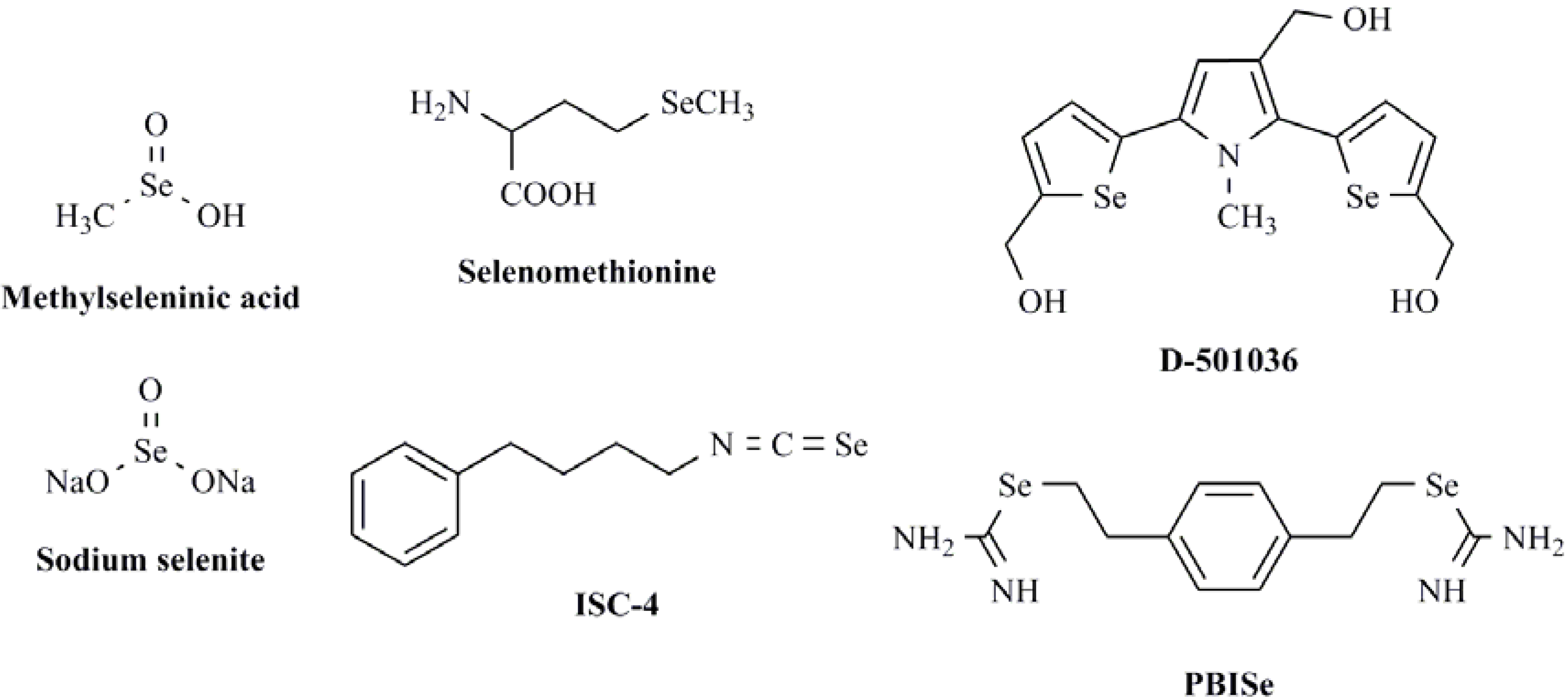
2. Results and Discussion
2.1. Chemistry

2.2. Biological Evaluation
2.2.1. Kinase screening

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinases | Compounds | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | A2 | A3 | A4 | A5 | B1 | B2 | C1 | C2 | C3 | D1 | D2 | E | |
| Abl a | -4 ± 3 | 61 ± 3 | 33 ± 6 | 82 ± 7 | 21 ± 8 | 4 ± 1 | 5 ± 10 | 10 ± 10 | 8 ± 5 | 9 ± 14 | 40 ± 4 | 25 ± 9 | 19 ± 14 |
| AKT1 a | 2 ± 2 | 71 ± 8 | 36 ± 9 | 89 ± 3 | 5 ± 1 | -12 ± 5 | -6 ± 5 | 12 ± 13 | 15 ± 12 | 13 ± 10 | 38 ± 12 | 9 ± 4 | 11 ± 2 |
| AurA a | 74 ± 3 | 5 ± 2 | -5 ± 2 | 13 ± 12 | 21 ± 7 | 17 ± 3 | 12 ± 6 | 3 ± 3 | 5 ± 1 | -7 ± 15 | 46 ± 4 | 42 ± 6 | 21 ± 7 |
| CDK2 a | -3 ± 5 | 64 ± 16 | 32 ± 11 | 73 ± 6 | 20 ± 9 | -2 ± 1 | -4 ± 2 | 26 ± 16 | 6 ± 1 | 3 ± 7 | 25 ± 5 | 8 ± 6 | 3 ± 6 |
| CDK9 a | 19 ± 2 | 45 ± 1 | 5 ± 6 | 24 ± 6 | 21 ± 3 | 17 ± 1 | 16 ± 4 | 6 ± 2 | 24 ± 4 | 17 ± 9 | 16 ± 2 | 14 ± 7 | 54 ± 2 |
| CK1A b | 18 ± 15 | 16 ± 11 | 21 ± 14 | 16 ± 19 | 13 ± 5 | 11 ± 1 | 15 ± 4 | 15 ± 6 | 17 ± 2 | 54 ± 1 | 12 ± 1 | 10 ± 1 | 12 ± 1 |
| CK2 b | 4 ± 1 | 19 ± 6 | 12 ± 3 | 6 ± 7 | 0 ± 1 | -1 ± 2 | 9 ± 5 | 18 ± 5 | 20 ± 3 | 15 ± 3 | 17 ± 3 | 9 ± 3 | 3 ± 3 |
| CKIT a | -15 ± 3 | 64 ± 15 | -1 ± 11 | 58 ± 8 | 3 ± 10 | 1 ± 14 | -1 ± 3 | 13 ± 13 | -15 ± 8 | -10 ± 5 | 3 ± 8 | 18 ± 15 | -5 ± 4 |
| CRAF a | -8 ± 2 | -6 ± 1 | -6 ± 1 | -6 ± 3 | -7 ± 1 | 1 ± 3 | 0 ± 2 | 1 ± 4 | 2 ± 1 | -1 ± 1 | 2 ± 1 | 15 ± 9 | -10 ± 4 |
| EGFR a | -4 ± 9 | 7 ± 6 | 0 ± 3 | 11 ± 7 | 1 ± 1 | -4 ± 5 | 1 ± 2 | −2 ± 5 | -4 ± 2 | -4 ± 5 | 3 ± 5 | 2 ± 2 | -2 ± 4 |
| ErbB4 a | 24 ± 3 | 24 ± 12 | 43 ± 9 | 15 ± 10 | 29 ± 4 | 30 ± 1 | 29 ± 1 | 34 ± 5 | 32 ± 6 | 22 ± 12 | 65 ± 4 | 70 ± 2 | 35 ± 2 |
| FAK2 a | 4 ± 1 | 0 ± 4 | 1 ± 2 | 5 ± 2 | -1 ± 3 | 5 ± 1 | 5 ± 1 | 2 ± 1 | 4 ± 2 | 6 ± 2 | 8 ± 4 | 5 ± 1 | 3 ± 1 |
| FGFR1 a | -6 ± 2 | 1 ± 1 | -9 ± 6 | 3 ± 3 | 6 ± 2 | 7 ± 2 | -2 ± 1 | -2 ± 2 | 1 ± 1 | -3 ± 10 | 2 ± 3 | 3 ± 2 | -2 ± 2 |
| GSK3a b | 8 ± 2 | 50 ± 7 | 41 ± 3 | 42 ± 10 | 22 ± 9 | 23 ± 8 | 28 ± 4 | 37 ± 2 | 31 ± 8 | 44 ± 4 | 34 ± 6 | 37 ± 10 | 46 ± 13 |
| IGF1R a | -2 ± 4 | -2 ± 4 | 3 ± 2 | -18 ± 4 | -4 ± 4 | -9 ± 2 | -2 ± 7 | -98 ± 9 | 2 ± 4 | -5 ± 4 | -211 ± 6 | −161 ± 2 | 4 ± 4 |
| JNK2 b | 2 ± 3 | 9 ± 5 | -1 ± 9 | 6 ± 3 | 4 ± 2 | 1 ± 6 | 4 ± 3 | 6 ± 2 | 6 ± 6 | 5 ± 6 | 8 ± 1 | 3 ± 5 | 6 ± 4 |
| KDR a | 1 ± 3 | 16 ± 2 | 6 ± 4 | 33 ± 5 | 3 ± 3 | -1 ± 1 | 4 ± 1 | 4 ± 1 | 11 ± 1 | 2 ± 1 | 14 ± 6 | 1 ± 4 | 4 ± 1 |
| MAPK1 a | -15 ± 2 | -3 ± 1 | -3 ± 2 | 21 ± 1 | -4 ± 1 | -7 ± 1 | -11 ± 3 | -4 ± 1 | 2 ± 3 | 2 ± 5 | 3 ± 4 | -4 ± 1 | -35 ± 2 |
| MAPK11 b | -4 ± 5 | 8 ± 7 | 3 ± 6 | -8 ± 8 | 0 ± 1 | -8 ± 11 | -8 ± 2 | -6 ± 7 | 0 ± 1 | -7 ± 1 | 1 ± 4 | 0 ± 10 | 2 ± 5 |
| MAPK14 b | 1 ± 3 | 17 ± 5 | 11 ± 1 | 14 ± 3 | -2 ± 7 | 3 ± 4 | 7 ± 4 | 9 ± 2 | 15 ± 4 | 13 ± 6 | 4 ± 7 | 11 ± 4 | 18 ± 2 |
| MAPK3 a | -16 ± 5 | 51 ± 1 | 18 ± 8 | 13 ± 20 | 14 ± 13 | -6 ± 4 | -8 ± 10 | -6 ± 11 | -6 ± 2 | -8 ± 6 | 10 ± 3 | 2 ± 4 | -8 ± 11 |
| PDGFRb a | -14 ± 9 | 18 ± 6 | -3 ± 2 | -8 ± 6 | 4 ± 1 | 5 ± 2 | 21 ± 8 | -32 ± 15 | -5 ± 1 | -8 ± 2 | -11 ± 10 | -34 ± 3 | 31 ± 3 |
| PKA b | 0 ± 1 | 2 ± 1 | 3 ± 1 | 0 ± 4 | 0 ± 1 | 1 ± 1 | 3 ± 2 | 7 ± 6 | 5 ± 1 | 4 ± 3 | 5 ± 2 | 2 ± 2 | 5 ± 1 |
| PKCA a | 18 ± 8 | -3 ± 6 | 2 ± 4 | -16 ± 3 | -8 ± 2 | 25 ± 4 | -1 ± 1 | -6 ± 4 | 11 ± 3 | 9 ± 9 | -2 ± 19 | -18 ± 13 | -10 ± 4 |
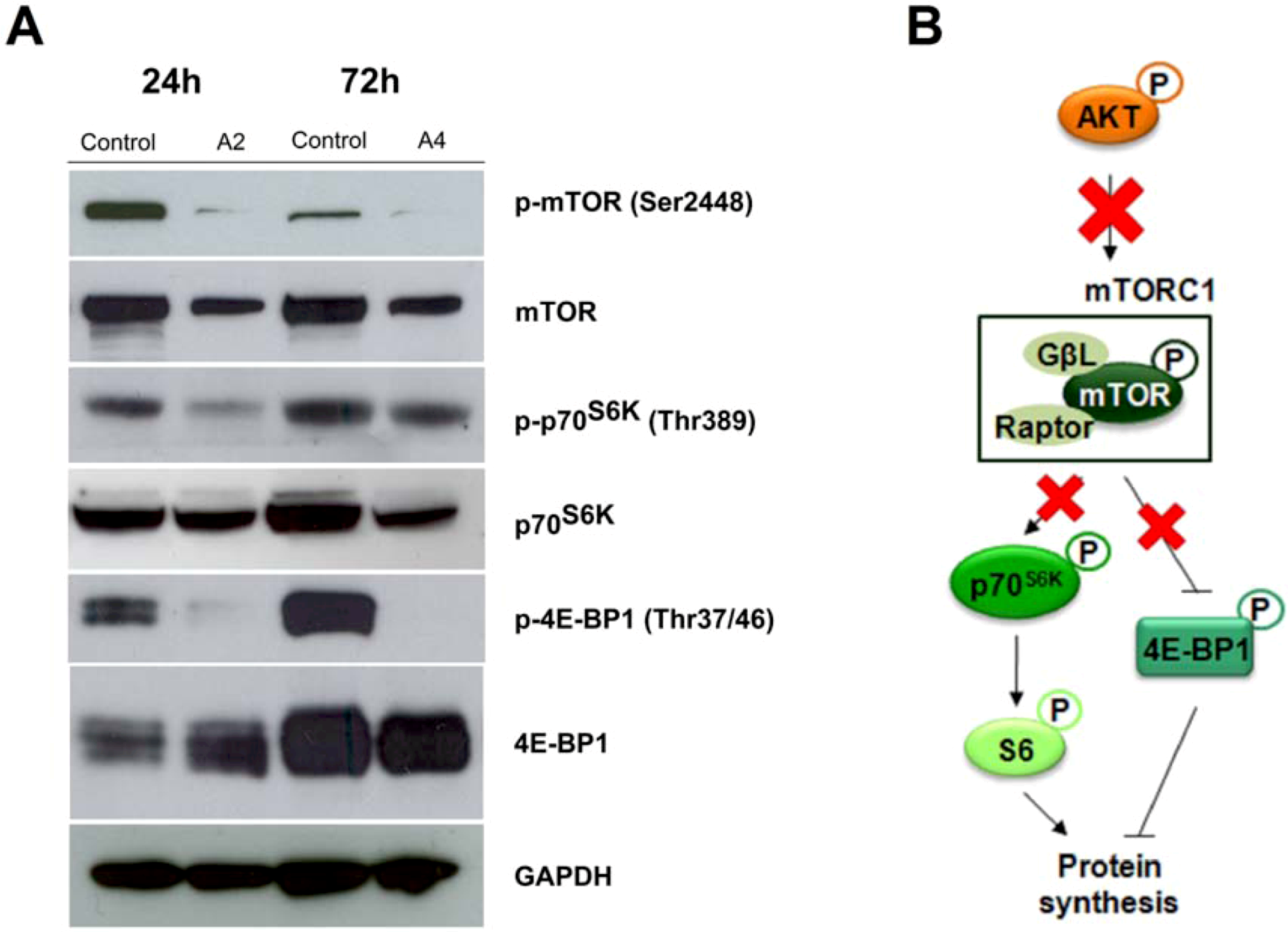
2.2.2. Effect of compounds A2 and A4 on PI3K/AKT and MAPK pathways
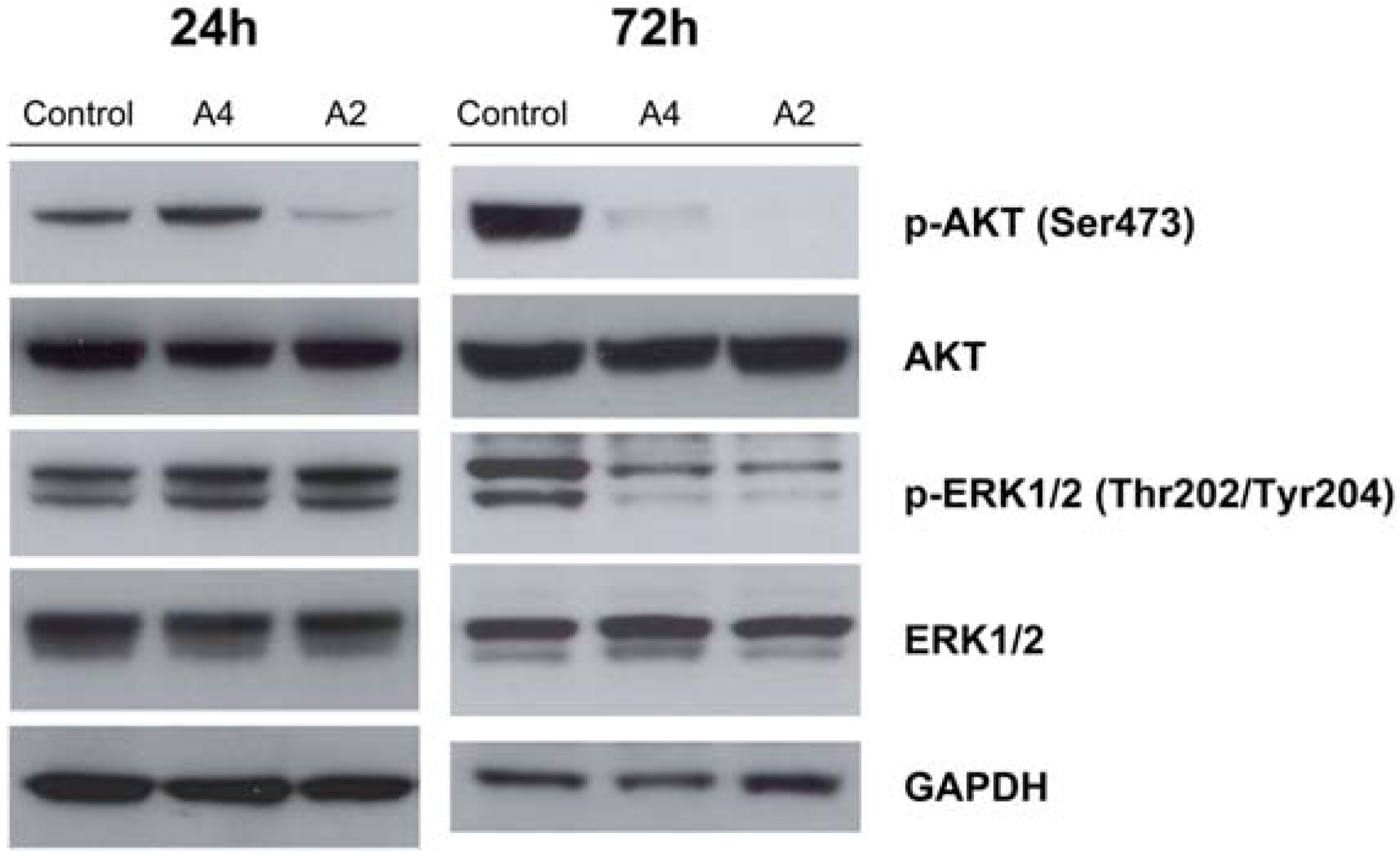
2.2.3. mTOR signaling alteration by compounds A2 and A4
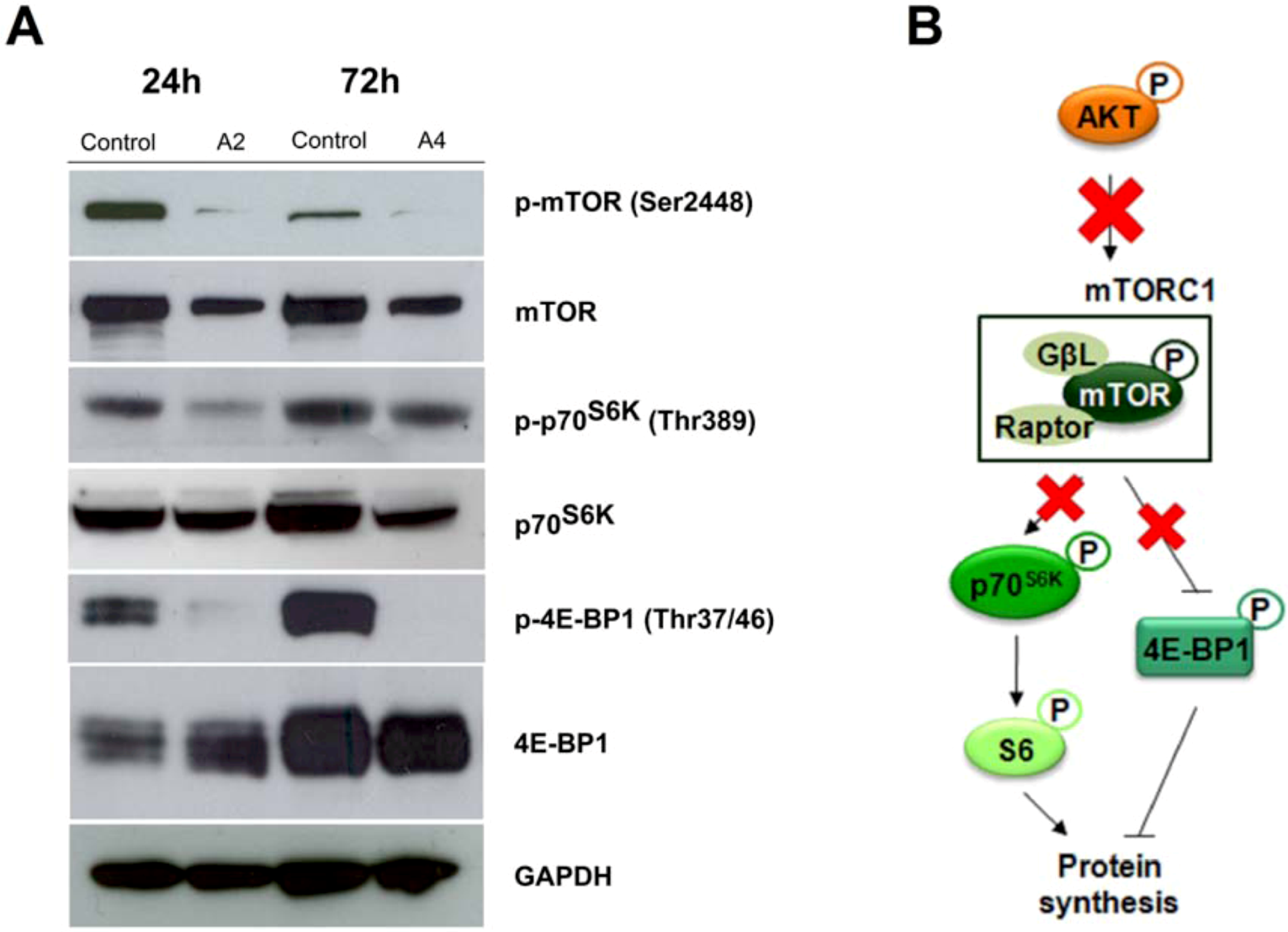
3. Experimental
3.1. Synthetic Chemistry and Compound Characterization
3.2. Kinase Inhibition Screening Assay
3.3. Cell Lines and Cell Culture
3.4. Western Blot Analysis
4. Conclusions
- Behavior 1: Compounds that do not inhibit kinases significantly. The compounds are A5, B1, B2, C1 and C2.
- Behavior 2: Compounds that inhibit, with similar intensity, two kinases and activate one kinase. These compounds, which are representative of D series, are D1 and D2, that caused Aur A and ErbB4 inhibition and IGF-1R activation.
- Behavior 3: Compounds from different series which inhibited the same kinase activity. For instance, compounds A3, C3 and E inhibited GSK3a; compounds A1, D1 and D2 AurA and compounds A3, D1 and D2 ErbB4.
- Behavior 4: Multikinase inhibitor compounds, which include A2 and A4.
Supplementary Materials
Supplementary File 1Acknowledgments
Conflict of Interest
References and Notes
- Ventura, J.J.; Nebreda, A.R. Protein kinases and phosphatases as therapeutic targets in cancer. Clin. Transl. Oncol. 2006, 8, 153–160. [Google Scholar] [CrossRef]
- Lahiry, P.; Torkamani, A.; Schork, N.J.; Hegele, R.A. Kinase mutations in human disease: Interpreting genotype-phenotype relationships. Nat. Rev. Genet. 2010, 11, 60–74. [Google Scholar] [CrossRef]
- Garuti, L.; Roberti, M.; Bottegoni, G. Irreversible protein kinase inhibitors. Curr. Med. Chem. 2011, 18, 2981–2994. [Google Scholar] [CrossRef]
- Hanks, S.K.; Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: Knase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar]
- Zhang, Q.; Feng, W.; Zhou, H.; Yan, B. Advances in preclinical small molecules for the treatment of NSCLC. Expert Opin. Ther. Pat. 2009, 19, 731–751. [Google Scholar] [CrossRef]
- Sathornsumetee, S.; Reardon, D.A. Targeting multiple kinases in glioblastoma multiforme. Expert Opin. Investig. Drugs 2009, 18, 277–292. [Google Scholar] [CrossRef]
- Wu, G. Recent progress in phosphoinositide 3-kinases: Oncogenic properties and prognostic and therapeutic implications. Curr. Protein Pept. Sci. 2010, 11, 425–435. [Google Scholar] [CrossRef]
- Morphy, R. Selectively nonselective kinase inhibition: Striking the right balance. J. Med. Chem. 2010, 53, 1413–1437. [Google Scholar] [CrossRef]
- Ciraolo, E.; Morello, F.; Hirsch, E. Present and future of PI3K pathway inhibition in cancer: Perspectives and limitations. Curr. Med. Chem. 2011, 18, 2674–2685. [Google Scholar]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- Hoshino, R.; Chatani, Y.; Yamori, T.; Tsuruo, T.; Oka, H.; Yoshida, O.; Shimada, Y.; Ari-i, S.; Wada, H.; Fujimoto, J.; et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene 1999, 18, 813–822. [Google Scholar] [CrossRef]
- Wu, Y.; Zu, K.; Warren, M.A.; Wallace, P.K.; Ip, C. Delineating the mechanism by which selenium deactivates Akt in prostate cancer cells. Mol. Cancer Ther. 2006, 5, 246–252. [Google Scholar]
- Li, S.; Zhou, Y.; Wang, R.; Zhang, H.; Dong, Y.; Ip, C. Selenium sensitizes MCF-7 breast cancer cells to doxorubicin-induced apoptosis through modulation of phosphor-Akt and its downstream substrates. Mol. Cancer Ther. 2007, 6, 1031–1038. [Google Scholar]
- Lee, J.H.; Shin, S.H.; Kang, S.; Lee, Y.S.; Bae, S. A novel activation-induced suicidial degradation mechanism for Akt by selenium. Int. J. Mol. Med. 2008, 21, 91–97. [Google Scholar]
- Abdulah, R.; Faried, A.; Kobayasiti, K.; Yamazaki, C.; Suradji, E.W.; Ito, K.; Suzuki, K.; Murakami, M.; Kuwano, H.; Koyama, H. Selenium enrichment of broccoli sprout extract increases chemosensitivity and apoptosis of LNCaP prostate cancer cells. BMC Cancer 2009, 9, 414. [Google Scholar] [CrossRef]
- Ren, Y.; Huang, F.; Liu, Y.; Yang, Y.; Jiang, Q.; Xu, C. Autophagy inhibition through PI3K/Akt increases apoptosis by sodium selenite in NB4 cells. BMB Rep. 2009, 42, 599–604. [Google Scholar]
- Zhao, R.; Xiang, N.; Domann, F.E.; Zhong, W. Effects of selenite and genistein on G2/M cell cycle arrest and apoptosis in human prostate cancer cells. Nutr. Cancer 2009, 61, 397–407. [Google Scholar]
- Berggren, M.; Sittadjody, S.; Song, Z.; Samira, J.L.; Burd, R.; Meuillet, E.J. Sodium selenite increases the activity of the tumor suppressor protein, PTEN, in DU-145 prostate cancer cells. Nutr. Cancer 2009, 61, 322–331. [Google Scholar] [CrossRef]
- Lee, Y.K.; Park, S.Y.; Kim, Y.M.; Kim, D.C.; Lee, W.S.; Surh, Y.J.; Park, O.J. Suppression of mTOR via Akt-dependent and -independent mechanisms in selenium-treated colon cancer cells: Involvement of AMPKalpha1. Carcinogenesis 2010, 31, 1092–1099. [Google Scholar]
- Chung, C.Y.; Madhunapantula, S.V.; Desai, D.; Amin, S.; Robertson, G.P. Melanoma prevention using topical PBISe. Cancer Prev. Res. 2011, 4, 935–948. [Google Scholar] [CrossRef]
- Nguyen, N.; Sharma, A.; Nguyen, N.; Sharma, A.K.; Desai, D.; Huh, S.J.; Amin, S.; Meyers, C.; Robertson, G.P. Melanoma chemoprevention in skin reconstructs and mouse xenografts using isoselenocyanate-4. Cancer Prev. Res. 2011, 4, 248–258. [Google Scholar] [CrossRef]
- Kong, L.; Yuan, Q.; Zhu, H.; Li, Y.; Guo, Q.; Wang, Q.; Bi, X.; Gao, X. The suppression of prostate LNCaP cancer cells growth by selenium nanoparticles through Akt/Mdm2/AR controlled apoptosis. Biomaterials 2011, 32, 6515–6522. [Google Scholar] [CrossRef]
- Sharma, A.K.; Kline, C.L.; Berg, A.; Amin, S.; Irby, R.B. The Akt inhibitor ISC-4 activates prostate apoptosis response protein-4 and reduces colon tumor growth in a nude mouse model. Clin. Cancer Res. 2011, 17, 4474–4483. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, A.K.; Madhunapantula, S.V.; Desai, D.; Huh, S.J.; Mosca, P.; Amin, S.; Robertson, G.P. Targeting Akt3 signaling in malignant melanoma using isoselenocyanates. Clin. Cancer Res. 2009, 15, 1674–1685. [Google Scholar] [CrossRef]
- Nguyen, N.; Sharma, A.; Nguyen, N.; Sharma, A.K.; Desai, D.; Huh, S.J.; Amin, S.; Meyers, C.; Robertson, G.P. Melanoma chemoprevention in skin reconstructs and mouse xenografts using isoselenocyanate-4. Cancer Prev. Res. 2011, 4, 248–258. [Google Scholar] [CrossRef]
- Kato, M.A.; Finley, D.J.; Lubitz, C.C.; Zhu, B.; Moo, T.A.; Loeven, M.R.; Ricci, J.A.; Zarnegar, R.; Katdare, M.; Fahey, T.J. Selenium decreases thyroid cancer cell growth by increasing expression of GADD153 and GADD34. Nutr. Cancer 2009, 62, 66–73. [Google Scholar] [CrossRef]
- Wang, Z.; Lee, H.J.; Chai, Y.; Hu, H.; Wang, L.; Zhang, Y.; Jiang, C.; Lü, J. Persistent p21Cip1 induction mediates G(1) cell cycle arrest by methylseleninic acid in DU145 prostate cancer cells. Curr. Cancer Drug Targets 2010, 10, 307–318. [Google Scholar] [CrossRef]
- Zeng, H.; Botnen, J.H.; Briske-Anderson, M. Deoxycholic acid and selenium metabolite methylselenol exert common and distinct effects on cell cycle, apoptosis, and MAP kinase pathway in HCT116 human colon cancer cells. Nutr. Cancer 2009, 62, 85–92. [Google Scholar] [CrossRef]
- Fang, W.; Han, A.; Bi, X.; Xiong, B.; Yang, W. Tumor inhibition by sodium selenite is associated with activation of c-Jun NH2-terminal kinase 1 and suppression of beta-catenin signalling. Int. J. Cancer 2010, 127, 32–42. [Google Scholar] [CrossRef]
- Sanmartín, C.; Plano, D.; Font, M.; Palop, J.A. Kinase regulation by sulfur and selenium containing compounds. Curr. Cancer Drug Targets 2011, 11, 496–523. [Google Scholar] [CrossRef]
- Juang, S.H.; Lung, C.C.; Hsu, P.C.; Hsu, K.S.; Li, Y.C.; Hong, P.C.; Shiah, H.S.; Kuo, C.C.; Huang, C.W.; Wang, Y.C.; et al. D-501036, a novel selenophene-based triheterocycle derivative, exhibits, potent in vitro and in vivo antitumoral activity which involves DNA damage and ataxia telangiectasia-mutated nuclear protein kinase activation. Mol. Cancer Ther. 2007, 6, 193–202. [Google Scholar] [CrossRef]
- Sharma, A.K.; Sharma, A.; Desai, D.; Madhunapantula, S.V.; Huh, S.J.; Robertson, G.P.; Amin, S. Synthesis and anticancer activity comparison of phenylalkyl isoselenocyanates with corresponding naturally occurring and synthetic isothiocyanates. J. Med. Chem. 2008, 51, 7820–7826. [Google Scholar] [CrossRef]
- Plano, D.; Sanmartín, C.; Moreno, E.; Prior, C.; Calvo, A.; Palop, J.A. Novel potent organo-selenium compounds as cytotoxic agents in prostate cancer cells. Bioorg. Med. Chem. Lett. 2007, 17, 6853–6859. [Google Scholar] [CrossRef]
- Sanmartín, C.; Plano, D.; Domínguez, E.; Font, M.; Calvo, A.; Prior, C.; Encío, I.; Palop, J.A. Synthesis and pharmacological screening of several aroyl and heteroaroyl selenylacetic acid derivatives as cytotoxic and antiproliferative agents. Molecules 2009, 14, 3313–3338. [Google Scholar] [CrossRef]
- Plano, D.; Moreno, E.; Font, M.; Encío, I.; Palop, J.A.; Sanmartín, C. Synthesis and in vitro anticancer activities of some selenadiazole derivatives. Arch. Pharm. 2010, 343, 680–691. [Google Scholar] [CrossRef]
- Plano, D.; Ibáñez, E.; Palop, J.A.; Sanmartín, C. Synthesis, characterization, crystal structure and cytotoxicity of 2,4-bis(selenomethyl)quinazoline. Struct. Chem. 2011. [Google Scholar] [CrossRef]
- Plano, D.; Baquedano, Y.; Ibáñez, E.; Jiménez, I.; Palop, J.A.; Spallholz, J.E.; Sanmartín, C. Antioxidant-prooxidant properties of a new organoselenium compound library. Molecules 2010, 15, 7292–7312. [Google Scholar] [CrossRef]
- Plano, D.; Baquedano, Y.; Moreno, D.; Font, M.; Jiménez-Ruiz, A.; Palop, J.A.; Sanmartín, C. Selenocyanates and diselenides: A new class of potent antileishmanial agents. Eur. J. Med. Chem. 2011, 46, 3315–3323. [Google Scholar]
- Pittoni, P.; Piconese, S.; Tripodo, C.; Colombo, M.P. Tumor-intrinsic and -extrinsic roles of c-Kit: Mast cells as the primary off-target of tyrosine kinase inhibitors. Oncogene 2011, 30, 757–769. [Google Scholar] [CrossRef]
- Sánchez-Martín, R.; Campos, J.M.; Conejo-García, A.; Cruz-López, O.; Báñez-Coronel, M.; Rodríguez-González, A.; Gallo, M.A.; Lacal, J.C.; Espinosa, A. Symmetrical bis-quinolinium compounds: new human choline kinase inhibitors with antiproliferative activity against the HT-29 cell line. J. Med. Chem. 2005, 48, 3354–3363. [Google Scholar]
- Sanmartín, C.; Font, M.; Palop, J.A. Molecular symmetry: A structural property frequently present in new cytotoxic and proapoptotic drugs. Mini Rev. Med. Chem. 2006, 6, 639–650. [Google Scholar]
- Cho, S.D.; Chintharlapalli, S.; Abdelrahim, M.; Papineni, S.; Liu, S.; Guo, J.; Lei, P.; Abudayyeh, A.; Safe, S. 5,5’-Dibromo-bis(3’-indolyl)methane induces Kruppel-like factor 4 and p21 in colon cancer cells. Mol. Cancer Ther. 2008, 7, 2109–2120. [Google Scholar] [CrossRef]
- Xiao, Q.; Liu, Y.; Qiu, Y.; Yao, Z.; Zhou, G.; Yao, Z.J.; Jiang, S. Design, synthesis of symmetrical bivalent mimetics of annonaceous acetogenins and their cytotoxicities. Bioorg. Med. Chem. Lett. 2011, 21, 3613–3615. [Google Scholar] [CrossRef]
- Lorenzo, C.; Liao, Q.; Hardwicke, M.A.; Ducommun, B. Pharmacological inhibition of aurora-A but not aurora-B impairs interphase microtubule dynamics. Cell Cycle 2009, 8, 1733–1737. [Google Scholar] [CrossRef]
- Morshed, M.N.; Cho, Y.S.; Seo, S.H.; Han, K.C.; Yang, E.G.; Pae, A.N. Computational approach to the identification of novel aurora-A inhibitors. Bioorg. Med. Chem. 2011, 19, 907–916. [Google Scholar] [CrossRef]
- Grunt, T.W.; Tomek, K.; Wagner, R.; Puckmair, K.; Zielinski, C.C. The DNA-binding epidermal growth factor-receptor inhibitor PD153035 and other DNA-intercalating cytotoxic drugs reactivate the expression of the retinoic acid receptor-beta tumor-suppressor gene in breast cancer cells. Differentiation 2007, 75, 883–890. [Google Scholar] [CrossRef]
- Klutchko, S.R.; Zhou, H.; Winters, R.T.; Tran, T.P.; Bridges, A.J.; Althaus, I.W.; Amato, D.M.; Elliott, W.L.; Ellis, P.A.; Meade, M.A.; et al. Tyrosine kinase inhibitors. 19. 6-Alkynamides of 4-anilinoquinazolines and 4-anilinopyrido[3,4-d]pyrimidines as irreversible inhibitors of the erbB family of tyrosine kinase receptors. J. Med. Chem. 2006, 49, 1475–1485. [Google Scholar] [CrossRef]
- Yanochko, G.M.; Eckhart, W. Type I insulin-like growth factor receptor over-expression induces proliferation and anti-apoptotic signaling in a three-dimensional culture model of breast epithelial cells. Breast Cancer Res. 2006, 8, R18. [Google Scholar] [CrossRef]
- Zhao, D.; Bakirtzi, K.; Zhan, Y.; Zeng, H.; Koon, H.W.; Pothoulakis, C. Insulin-like growth factor-1 receptor transactivation modulates the inflammatory and proliferative responses of neurotensin in human colonic epithelial cells. J. Biol. Chem. 2011, 286, 6092–6099. [Google Scholar]
- Smith, T.J. Insulin-like growth factor-I regulation of immune function: A potential therapeutic target in autoimmune diseases? Pharmacol. Rev. 2010, 62, 199–236. [Google Scholar] [CrossRef]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest. 2008, 118, 3065–3074. [Google Scholar]
- Brachmann, S.M.; Hofmann, I.; Schnell, C.; Fritsch, C.; Wee, S.; Lane, H.; Wang, S.; García-Echeverría, C.; Maira, S.M. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 22299–22304. [Google Scholar]
- Huang, H.; Cheville, J.C.; Pan, Y.; Roche, P.C.; Schmidt, L.J.; Tindall, D.J. PTEN induces chemosensitivity in PTEN-mutated prostate cancer cells by suppression of Bcl-2 expression. J. Biol. Chem. 2001, 276, 38830–38836. [Google Scholar]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Wander, S.A.; Hennessy, B.T.; Slingerland, J.M. Next-generation mTOR inhibitors in clinical oncology: How pathway complexity informs therapeutic strategy. J. Clin. Invest. 2011, 121, 1231–1241. [Google Scholar] [CrossRef]
- Schmelzle, T.; Hall, M.N. TOR, a central controller of cell growth. Cell 2000, 103, 253–262. [Google Scholar] [CrossRef]
- Card, A.; Caldwell, C.; Min, H.; Lokchander, B.; Hualin, X.; Sciabola, S.; Kamath, A.V.; Clugston, S.L.; Tschantz, W.R.; Leyu, W.; et al. High-throughput biochemical kinase selectivity assays: panel development and screening applications. J. Biomol. Screen. 2009, 14, 31–42. [Google Scholar]
- Cao, S.; Leng, Y.; Huang, W.; Liu, X.; Kufe, D. Glutathione peroxidase 1 is regulated by the c-Abl and Arg tyrosine kinases. J. Biol. Chem. 2003, 278, 39609–39614. [Google Scholar]
- Wu, M.; Kang, M.M.; Schoene, N.W.; Cheng, W.H. Selenium compounds activate early barriers of tumorigenesis. J. Biol. Chem. 2010, 285, 12055–12062. [Google Scholar] [CrossRef]
- Qi, Y.; Schoene, N.W.; Lartey, F.M.; Cheng, W.H. Selenium compounds activate ATM-dependent DNA damage response via the mismatch repair protein hMLH1 in colorectal cancer cells. J. Biol. Chem. 2010, 285, 33010–33017. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds A1–A5, B1–B2, C1–C3, D1–D2 and E are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Plano, D.; Ibáñez, E.; Calvo, A.; Palop, J.A.; Sanmartín, C. Novel Library of Selenocompounds as Kinase Modulators. Molecules 2011, 16, 6349-6364. https://doi.org/10.3390/molecules16086349
Plano D, Ibáñez E, Calvo A, Palop JA, Sanmartín C. Novel Library of Selenocompounds as Kinase Modulators. Molecules. 2011; 16(8):6349-6364. https://doi.org/10.3390/molecules16086349
Chicago/Turabian StylePlano, Daniel, Elena Ibáñez, Alfonso Calvo, Juan Antonio Palop, and Carmen Sanmartín. 2011. "Novel Library of Selenocompounds as Kinase Modulators" Molecules 16, no. 8: 6349-6364. https://doi.org/10.3390/molecules16086349