3. Experimental
3.1. General
Unless otherwise stated, all chemicals and reagents were purchased from Sigma-Aldrich Chemical Co. Melting points were determined on an Electrothermal MEL-TEMP apparatus and are uncorrected. 1H-NMR and 13C-NMR spectra were recorded in DMSO-d6 on a Bruker 500 MHz instrument and the chemical shift (δ) values are reported in parts per million (ppm)relative to TMS. A Thermo Scientific LTQ Linear Trap LC/MS/MS system was used for mass spectrometry. UV spectra were recorded on a Beckman-Coulter DU-800 spectrophotometer. Analytical TLC was carried out on Sigma-Aldrich (cat # Z122785-25EA), 0.2 mm percolated silica gel polyester sheets with UV indicator. Elemental analysis was carried out by M-H-W Laboratories, Phoenix, AZ. Analysis of C, H, N were within ± 0.4% of theoretical values. The carbon numbering was shown for representative monomer 20a and for one representative dimer 20b. All others were referred similarly on the 13C-NMR assignments.
3.2. General Procedure-A for N9-[(Z)-4'-chloro-2'-butenyl-1'-yl]-6-methoxypurine (6)
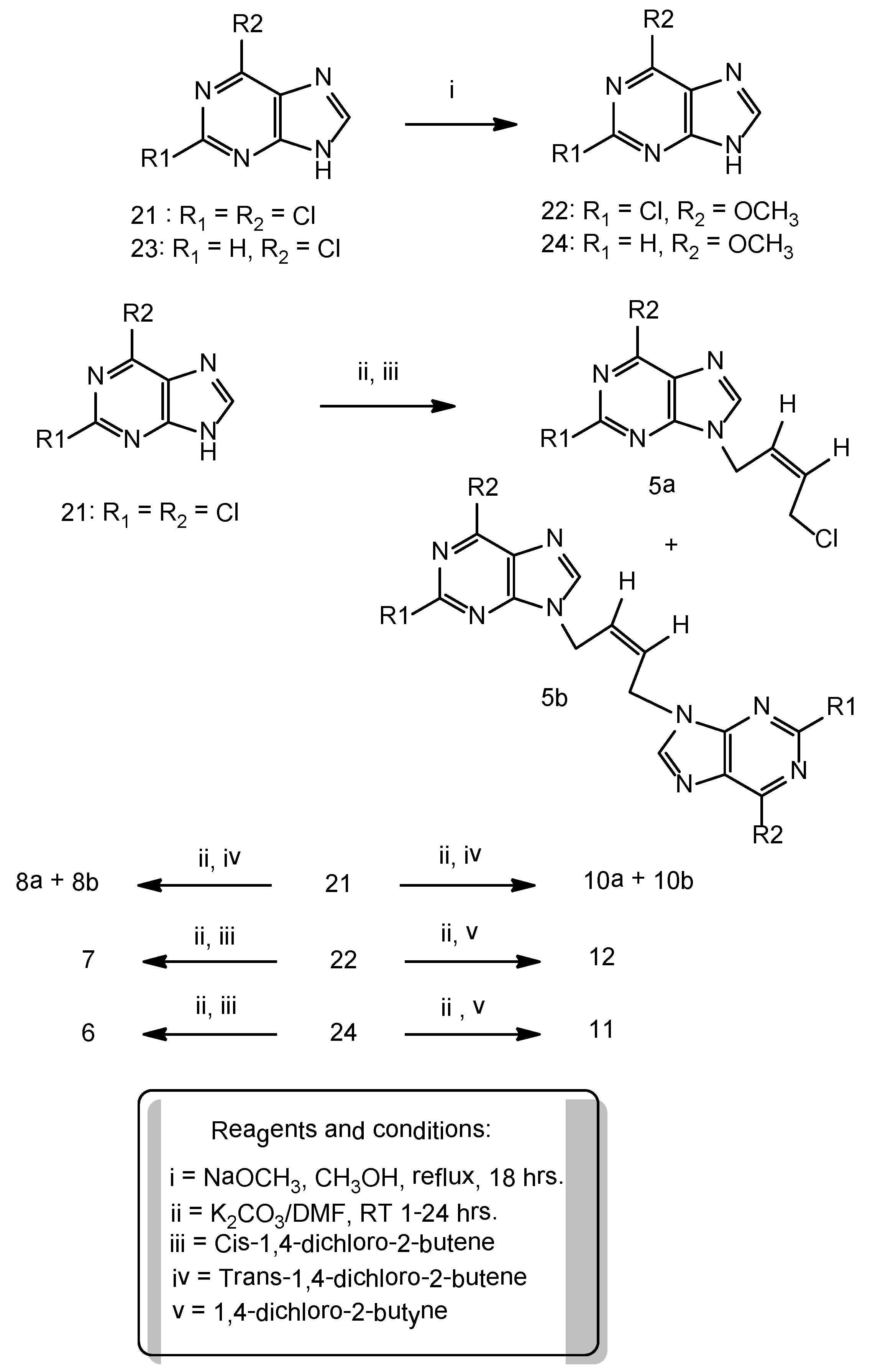
Synthesis of 6-methoxypurine: To a stirred suspension of 6-chloropurine (5.0 g, 32 mmol) in anhydrous methanol (240 mL) was slowly added 30 W% sodium methoxide in methanol (17.3 g, 320 mmol) at room temperature and then the reaction mixture was refluxed for 18 h. It was cooled to room temperature, neutralized with glacial acetic acid to pH 7.5–8.0 and then evaporated in a rotary evaporator to remove the solvent. The residue was treated with cold water (5 °C) (100 mL), the resulting solid was filtered, thoroughly washed with DI water. The product was crystallized from methanol as brownish white solid (3.5 g), 72% yield. 1H-NMR: δ 13.39 (1H, br s, NH), 8.50 (1H, s, H-8), 4.10 (3H, s, OCH3).
To a suspension of 6-methoxypurine (1.51 g, 10 mmol), anhydrous potassium carbonate (2.1 g, 15 mmol) in DMF (50 mL) at room temperature, was added cis-1,4-dichloro-2-butene (1.25 g, 10 mmol) and the contents stirred at room temperature for 5 h. The reaction mixture was filtered to remove the potassium carbonate that was also washed with DMF (25 mL). The filtrate and washings combined and evaporated under vacuum. The residue was chromatographed on a column of silica gel and the product was eluted with ethyl acetate: hexane 1:1 v/v. Evaporation of the homogeneous fractions resulted in a residue that was further crystallized from ethyl acetate-hexane as cream white rosettes, (1.3 g), 54% yield, m.p. 104–106 °C. 1H-NMR: δ 8.54 (1H, s, H-2), 8.36 (1H, s, H-8), 5.89–5.84 (2H, m, HC=CH), 5.4 (2H, s, N-CH2), 4.10 (3H, s, OCH3), 2.71 (2H, m, CH2Cl), 1.10 (3H, t, J = 7.5 Hz, OCH3). 13C-NMR: δ 160.22 (C-6), 151.76 (C-4), 151.49 (C-2), 129.66 (C-3'), 128.11 (C-2'), 120.51 (C-5), 53.84 (OCH3), 40.01 (C-1'), 39.85 (C-4'). Anal. Calcd. for C10H11N4OCl: C 50.32, H 4.65, N 23.47; Found: C 50.20, H 4.74, N 23.55.
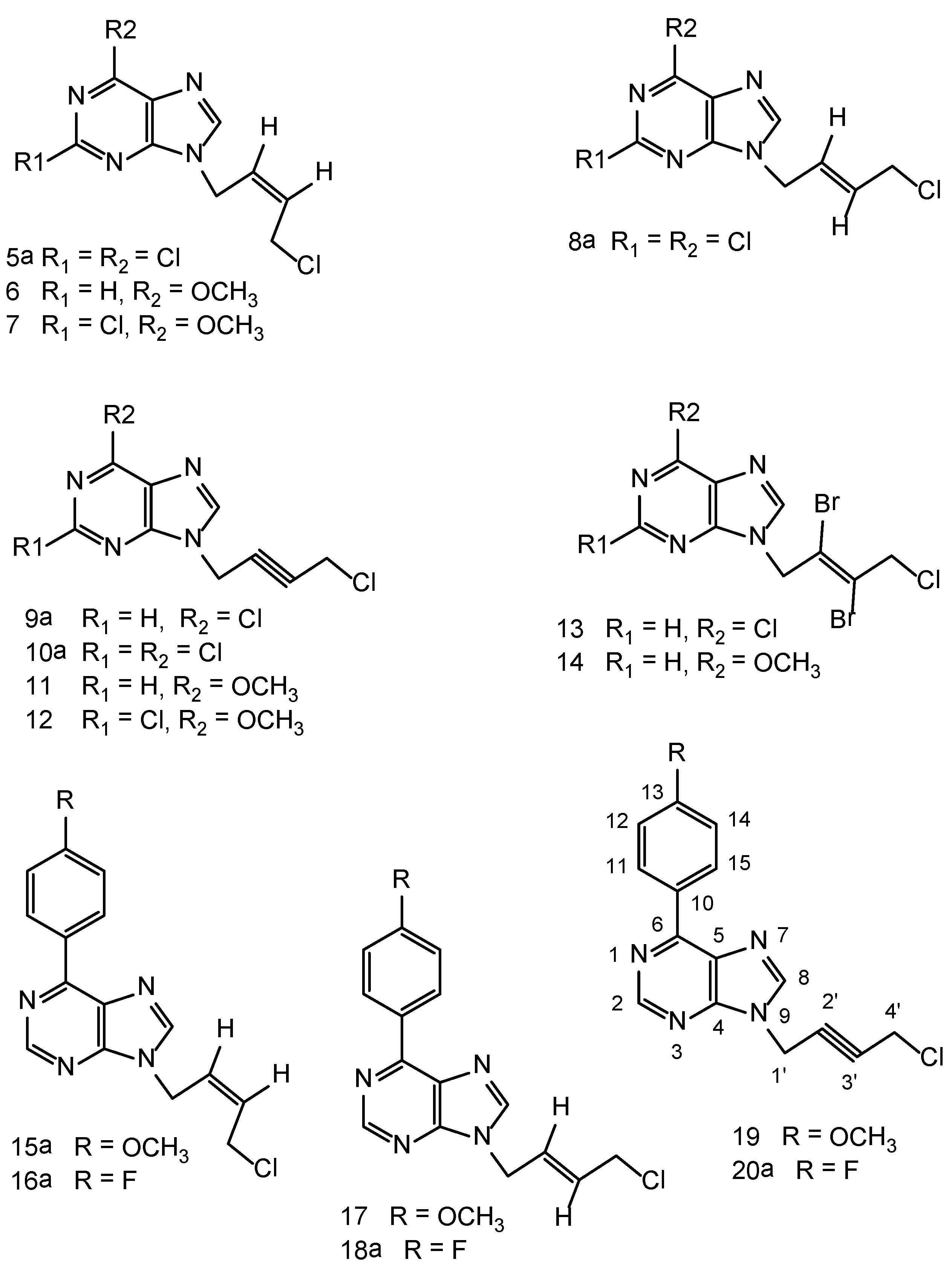
N9-[(Z)-4'-Chloro-2'-butenyl-1'-yl]-2,6-dichloropurine (5a). A suspension of 2,6-dichloropurine (2.0 g, 11 mmol), anhydrous potassium carbonate (4.6 g, 34 mmol), 1,4-dichloro-2-butyne (2.1 g, 17 mmol) in anhydrous DMF was stirred at r.t. for 6 h. The reaction has been worked-up and purified on a column of silica gel as described in the general procedure-A , to give a major product 5a and a minor dimeric product 5b. Compound 5a was isolated as a cream white solid (1.1g), 46% yield, m.p. 65–67 °C. 1H-NMR: δ 8.67 (1H, s, H-8), 5.93–5.91 (2H, m, HC=CH), 5.10–5.09 (2H, m, N-CH2), 4.54–4.52 (2H, m, CH2Cl). 13C-NMR: δ 153.23 (C-2), 150.95 (C-6), 149.59 (C-4), 148.0 (C-8), 130.44 (C-5), 130.24 (C-3'), 127.16 (C-2'), 40.53 (C-1'), 39.13 (C-4'). LC-MS (m/z): 277 [M+1]+, 100%. Anal. Calcd. forC9H7N4Cl3: C 38.95, H 2.54, N 20.19; Found: C 39.84, H 2.45, N 20.25.
N9,N9'' -bis[(Z)-2'-Butenyl-1',4'-diyl]-2,6-dichloropurine (5b). Compound 5b was isolated in the above reaction as a brownish white solid (0.45g), 15% yield, m.p. 226–228 °C. 1H-NMR: δ 8.65 (2H, H-8, H-8''), 5.97 (m, 2H, HC=CH), 4.97 (4H, m, 2 × N-CH2). 13C-NMR: δ 153.33 (C-2, C-2''), 150.93 (C-6, C-6''), 149.60 (C-4, C-4''), 148.17 (C-8, C-8''), 130.52 (C-5, C-5''), 127.69 (C-2', C-3'), 40.95 (C-1', C-4'). LC-MS (m/z): 431 [M+1]+, 100%. Anal. Calcd. forC14H8N8Cl4: C 39.10, H 1.88, N 26.05; Found: C 39.0, H 2.0, N 25.95.
3.3. N9-[(Z)-4'-Chloro-2'-butenyl-1'-yl]-2-chloro-6-methoxypurine (7)
Synthesis of 2-chloro-6-methoxypurine: 2,6-dichloropurine (5.0 g, 27 mmol) was reacted with sodium methoxide (30 wt.% 14.6 g, 270 mmol) in anhydrous methanol (250 mL) under reflux for 16 h. The reaction has been worked-up as described above under general procedure-A The crude product was crystallized from methanol as snow white solid (4.0g), 82% yield. 1H-NMR: δ 13.52 (1H, br s, NH), 8.42 (1H, s, H-8), 4.1 (3H, s, OCH3).
2-Chloro-6-methoxypurine (1.9 g, 10 mmol), anhydrous potassium carbonate (2.1 g, 15 mmol), cis- 1,4-dichloro-2-butene (1.5 g, 12mmol) were stirred in DMF (50 mL) at room temperature for 4 h. The crude product was isolated as described under the general procedure-A. The product was chromatographed on a column of silica gel, eluent ethyl acetate/hexane 1:1 v/v. Cream white solid (1.8g) 64% yield, m.p. 136–138 °C. 1H-NMR: δ8.38 (1H, s, H-8), 5.9–5.83 (2H, m, HC=CH), 5.02 (2H, d, J = 5.5 Hz, N-CH2), 4.5 (2H, d, J = 5.5 Hz, CH2Cl), 4.1 (3H, s, OCH3). 13C-NMR: δ 160.73 (C-6), 152.94 (C-2), 151.32 (C-4), 144.04 (C-8), 129.96 (C-3'), 127.64 (C-2'), 119.77 (C-5), 54.87 (OCH3), 40.05 (C-1'), 39.15(C-4'). Anal. Calcd. for C10H10N4OCl2: C 43.93, H 3.69, N 20.51; Found: C 43.85, H 3.75, N 20.45.
N9-[(E)-4'-Chloro-2'-butenyl-1'-yl]-2,6-dichloropurine (8a). A suspension of 2,6-dichloropurine (2.0 g, 11 mmol), anhydrous potassium carbonate (4.6 g, 34 mmol), trans-1,4-dichloro-2-butene (2.15 g, 17 mmol) in anhydrous DMF was stirred at r.t. for 10 h. The reaction was worked-up as described in the general procedure-A. Chromatography of the resulting crude product on a column of silica gel yielded one major product 8a and a minor dimeric product 8b. Compound 8a was isolated as a cream white solid (1.2 g), 40% yield, m.p. 70–72 °C. 1H-NMR: δ 8.78 (1H, H-8), 6.42 (1H, m, HC=CH), 5.80 (1H, m, HC=CH), 5.0 (2H, d, J = 5.5 Hz, N-CH2), 4.21 (2H, m, CH2Cl). LC-MS (m/z): 277 [M+1]+, 100%. Anal. Calcd. for C9H7N4Cl3: C 38.95, H 2.54, N 20.19; Found C 38.82, H 2.65, N 20.25.
N9,N9''-bis[(E)-2'-Butenyl-1',4'-diyl]-2,6-dichloropurine (8b). Pale brown solid (0.6g), 13% yield, m.p. 240 °C, decomposes. 1H-NMR: δ 8.77 (2H, s, H-8), 5.97 (2H, m, HC=CH), 4.96 (4H, 2 × N-CH2). LC-MS (m/z): 431 [M+1]+,100%. Anal. Calcd. for C14H8N8Cl4: C 39.10, H 1.88, N 26.05; Found: C 39.20, H 2.0, N 26.15.
N9-[4'-Chloro-2'-butynyl-1'-yl]-6-chloropurine (9a). A suspension of 6-chloropurine (2.5 g, 16 mmol), anhydrous potassium carbonate (3.5 g, 25 mmol) and 1,4-dichlorobutyne (3.0 g, 24 mmol) in DMF (150 mL) was stirred at room temperature for 3.0 h. The crude product was isolated as described in the general procedure-A. It was chromatographed on a column of silica gel using ethyl acetate/hexane 1:1 (v/v)as eluents to furnish 9a as a brownish white solid, 2.3 g, 59% yield, m.p. 92–94 °C. 1H-NMR: δ 8.83 (1H, s, H-2), 8.77 (1H, s, H-8), 5.32 (2H, t, J = 2.0 Hz, N-CH2), 4.49 (2H, t, J = 2.0 Hz, CH2Cl). 13C-NMR: δ 151.79 (C-2), 151.35 (C-6), 149.23 (C-4), 146.74 (C-8), 130.72 (C-5), 80.76 (C-3'), 79.71 (C-2'), 33.43 (C-1'), 30.47 (C-4'). Anal. Calcd. for C9H6N4Cl2: C 44.84, H 2.51, N 23.24; Found: C 44.75, H 2.64, N 23.15. From the above column, the ethyl acetate/hexane 2:1 v/v and 100% ethyl acetate eluents furnished the minor dimeric product N9,N9'' -bis[2'-butynyl-1',4'-diyl]-6-chloropurine (9b) as a pale brown solid. 0.7 g, 12% yield, m.p. 220 °C decomposes. 1H-NMR: δ 8.95 (2H, s, H-2, H-2, H-2''), 8.75 (2H, s, H-8, H-8''), 5.24 (4H, 2 × N-CH2). 13C-NMR: δ 151.9 (C-2, C-2''), 151.5 (C-6,C-6''), 149.4 (C-4, C-4''), 146.9 (C-8, C-8''), 130.8 (C-5, C-5''), 80.5 (C-2', C-3'), 40.9 (C-1',C-4'). Anal. Calcd. for C14H8N8Cl2: C 46.82, H 2.25, N 31.20; Found: C 46.92, H 2.14, N 31.25.
N9-[4'-Chloro-2'-butynyl-1'-yl]-2,6-dichloropurine (10a). A suspension of 2,6-dichloropurine (1.9 g, 10 mmol), anhydrous K2CO3 (4.2 g, 30 mmol) and 1,4-dichloro-2-butyne (1.85 g, 15 mmol) was stirred in DMF at r.t. for 8 h under argon. The reaction has been worked-up as described in the general procedure-A. The resulting product was chromatographed on a column of silica gel tofurnish 10a as the major product and 10b as a minor dimeric product.Compound 10a cream white solid (1.4 g) 50% yield, m.p. 80–82 °C. 1H-NMR: δ 8.79 (1H, s, H-8), 5.29 (2H, t, J = 2.0 Hz, N-CH2), 4.50 (2H, t, J = 2.0 Hz, CH2Cl). 13C-NMR: δ 152.87 (C-2), 151.23 (C-6), 149.91 (C-4), 147.66 (C-8), 130.41 (C-5), 81.11 (C-3'), 79.32 (C-2'), 33.66 (C-1'), 30.46 (C-4')). LC-MS (m/z): 275 [M+1]+, 100%.Anal. Calcd. for C9H5N4Cl3: C 39.23, H 1.83, N 20.33; Found: C 39.10, H 1.70, N 20.15.
N9, N9'' -bis[2'-Butynyl-1',4'-diyl]-2,6-dichloropurine (10b). It was isolated as a brownish white solid (0.6g), 14% yield, m.p. 210 °C decomposes. 1H-NMR: δ 8.78 (2H, s, H-8, H-8''), 5.25 (4H, s, 2 × N-CH2). LC-MS (m/z): 429 [M+1]+, 100%. Anal. Calcd. for C14H6N8Cl4: C 39.28, H 1.41, N 26.18; Found: C 39.12, H 1.55, N 26.25.
N9-[4'-Chloro-2'-butynyl-1'-yl]-6-methoxypurine (11). 6-Methoxypurine (1.0 g, 7.0 mmol), anhydrous potassium carbonate (1.6 g, 12.0 mmol), 1,4-dichlorobutyne (1.1 g, 9.0 mmol) was stirred at r.t. for 4 h. The crude product was isolated as described in the general procedure-A. Further purification on a column of silica gel using ethyl acetate/hexane 1:1 v/v as the eluent furnished a brownish white solid (1.1g), 70% yield, m.p. 141–143 °C. 1H-NMR: δ 8.57 (1H, s, H-2), 8.45 (1H, s, H-8), 5.25 (2H, t, J = 2.0 Hz, N-CH2), 4.48 (2H, t, J = 2.0 Hz, CH2Cl), 4.10 (3H, s, OCH3). 13C-NMR: δ 160.31 (C-6), 151.79 (C-2), 151.50 (C-4), 143.07 (C-8), 120.45 (C-5), 80.30 (C-2', C-3'), 53.95 (OCH3), 32.92 (C-1'), 30.52 (C-4'). Anal. Calcd. for C10H9N4OCl: C 50.75, H 3.83, N 23.67; Found: C 50.60, H 3.90, N 23.55.
N9-[4'-chloro-2'-butynyl-1'-yl]-2-chloro-6-methoxypurine (12). Brownish white solid, 61% yield, m.p. 143–145 °C. 1H-NMR: δ 8.48 (1H, s, H-8), 5.23 (2H, t, J = 2.0 Hz, N-CH2), 4.45 (2H, t, J = 2.0 Hz, CH2Cl), 4.11 (3H, s, OCH3). 13C-NMR: δ 160.85 (C-6), 152.64 (C-2), 151.61 (C-4), 143.82 (C-8), 119.75 (C-5), 80.66 (C-3'), 79.88 (C-2'), 54.98 (OCH3), 33.18 (C-1'), 30.50 (C-4'). Anal. Calcd. for C10H8N4OCl2: C 44.30, H 2.97, N 20.67; Found: C 44.15, H 3.15, N 20.55.
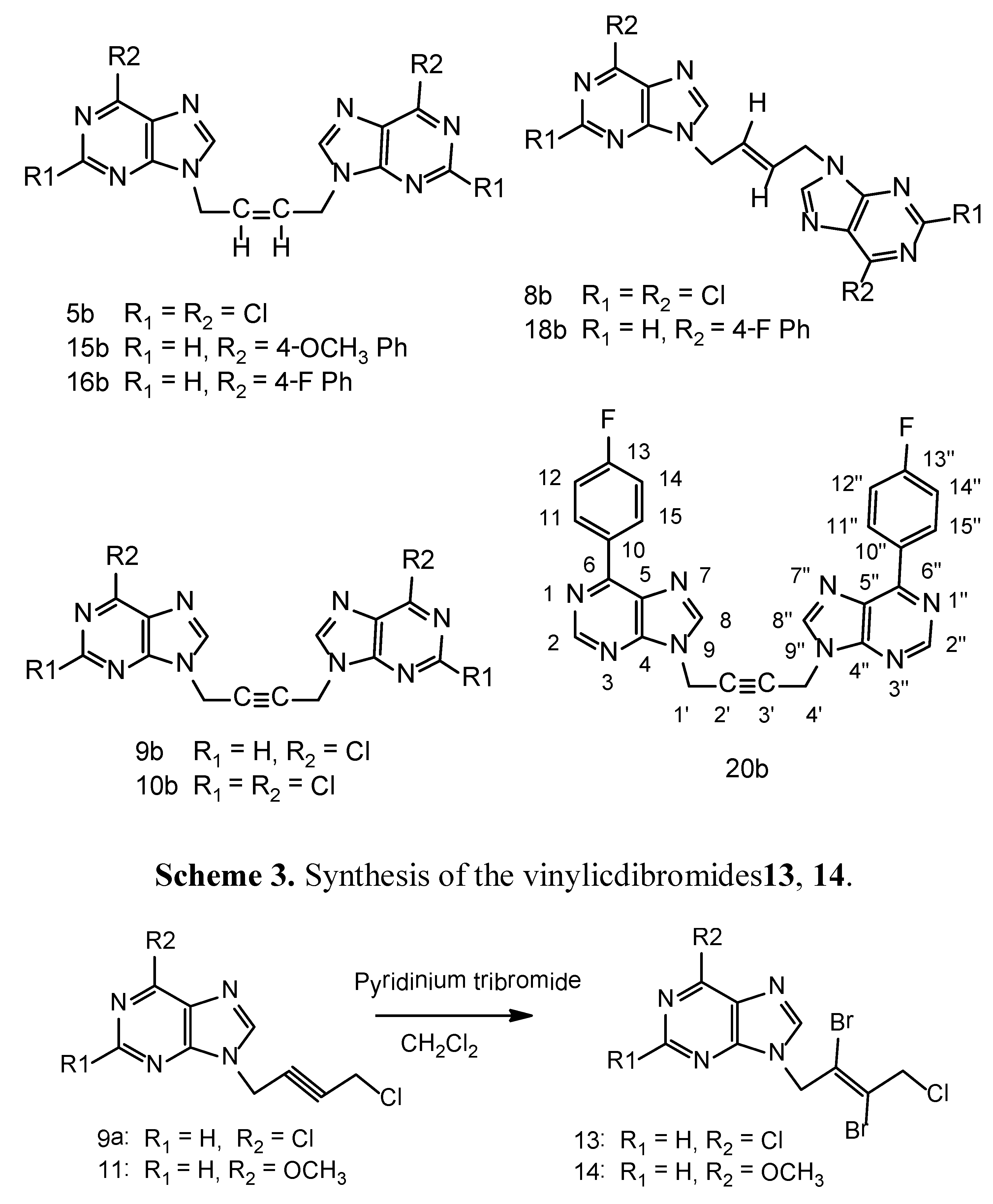
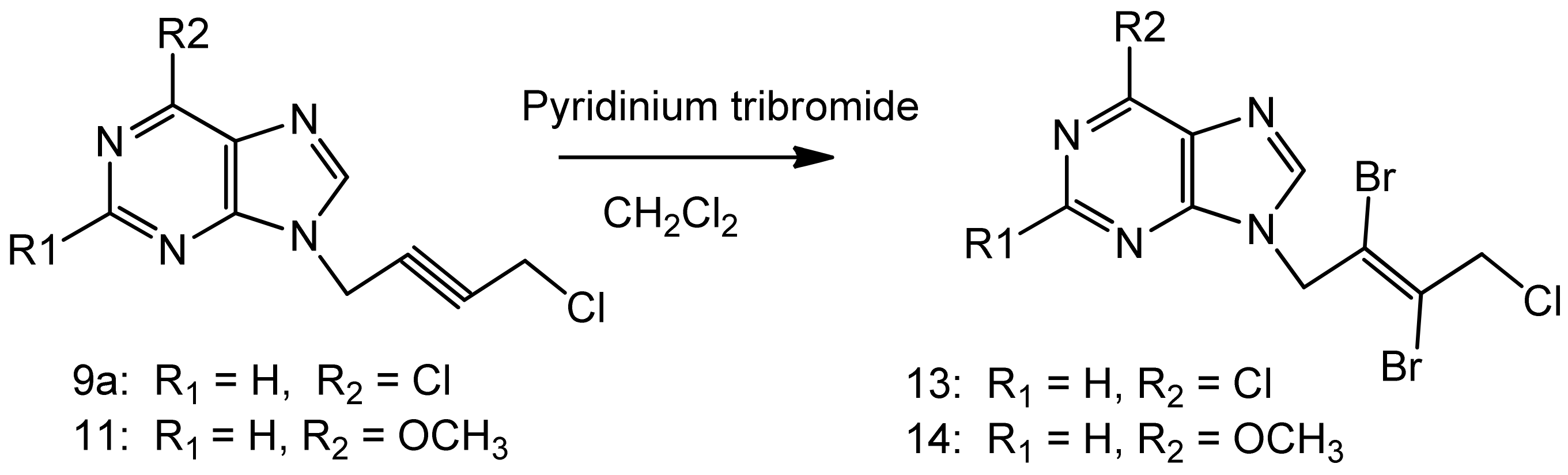
3.4.General procedure B for N9-[(E)-2',3'-dibromo)-4'-chloro-2'-butenyl-1'-yl]-6-chloropurine (13)
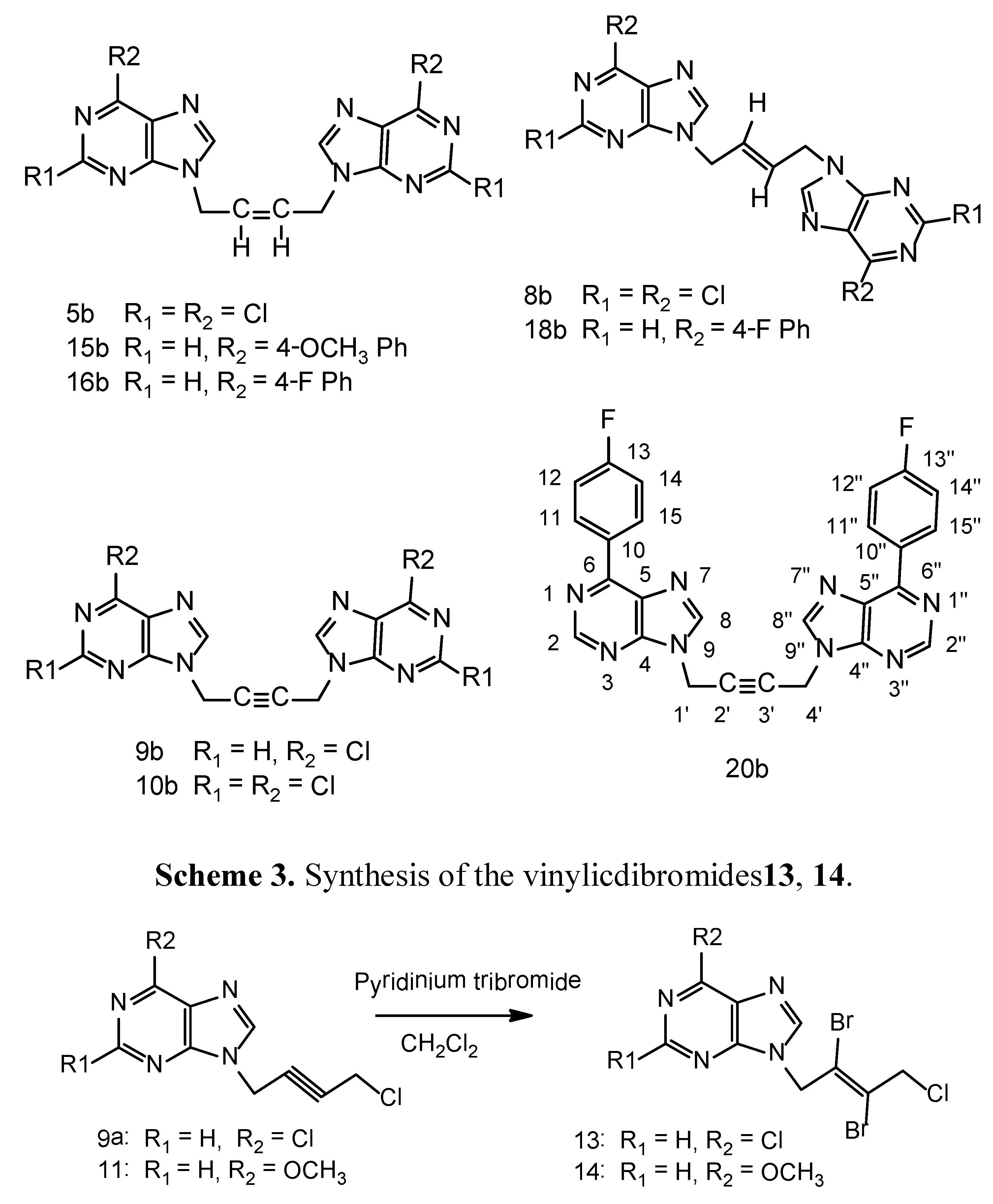
A suspension of 9a (0.242 g, 1.0 mmol), pyridiniumtribromide (0.4 g, 1.3 mmol) in anhydrous dichloromethane (100 mL) was cooled to −10 °C while stirring. Anhydrous methanol (50 mL) was added drop wise during 15–20 minutes. The reaction mixture was allowed to warm-up and stirred for 20 h in a fume hood. The clarified reaction mixture was evaporated on a rotary evaporator at 30–35 °C without any quenching with sodium thiosulfate. The product was purified on a column of silica gel using ethyl acetate - light petroleum ether 1:1, 2:1 v/v as the eluents. It was further crystallized from ethyl acetate-light petroleum ether as a white crystalline solid 0.22 g, 54% yield, m.p. 88–90 °C. 1H-NMR: δ 8.80 (1H, s, H-2), 8.70 (1H, s, H-8), 5.40 (2H, s, N-CH2), 4.71 (2H, s, CH2Cl). 13C-NMR: δ 151.70 (C-2), 151.30 (C-6), 149.0 (C-4), 146.7 (C-8), 130.70 (C-5), 121.30 (C-3'), 119.40 (C-2'), 50.40 (C-1'), 49.70 (C-4'). Anal. Calcd. for C9H6N4Br2Cl2: C 26.96, H 1.51, N 13.98; Found: C 26.80, H 1.65, N 14.15. The pyridinium salts were retained on the column.
N9-[(E)-2',3'-Dibromo-4'-chloro-2'-butenyl-1'-yl]-6-methoxypurine (14). Compound 11 was used as the starting material and the above bromination procedure was followed. A crystalline white solid resulted, 65% yield, m.p. 127–129 °C. 1H-NMR: δ 8.56 (1H, s, H-2), 8.41 (1H, s, H-8), 5.45 (2H, s, N-CH2), 4.72 (2H, s, CH2Cl), 4.11 (3H, s, OCH3). 13C-NMR: δ 160.29 (C-6), 152.01 (C-4), 151.94 (C-2), 143.93 (C-8), 121.59 (C-5), 121.33 (C-3'), 119.46 (C-2'), 53.97 (OCH3), 50.42 (C-1'), 49.75 (C-4'). LC-MS (m/z): 397[M+1]+, 100%. Anal. Calcd. for C10H9N4OBr2Cl: C 30.29, H 2.29, N 14.13; Found: C 30.10, H 2.34, N 14.05.
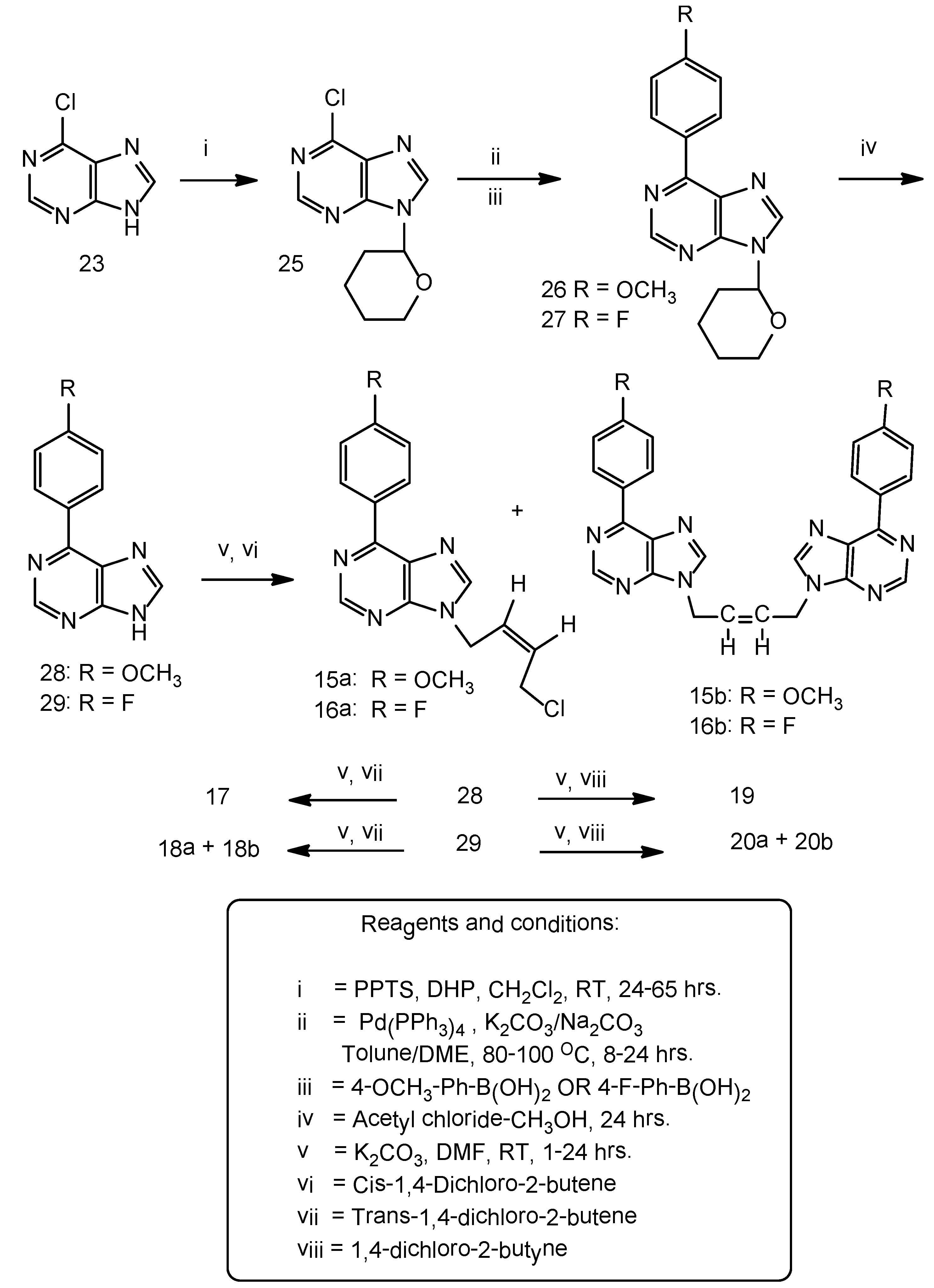
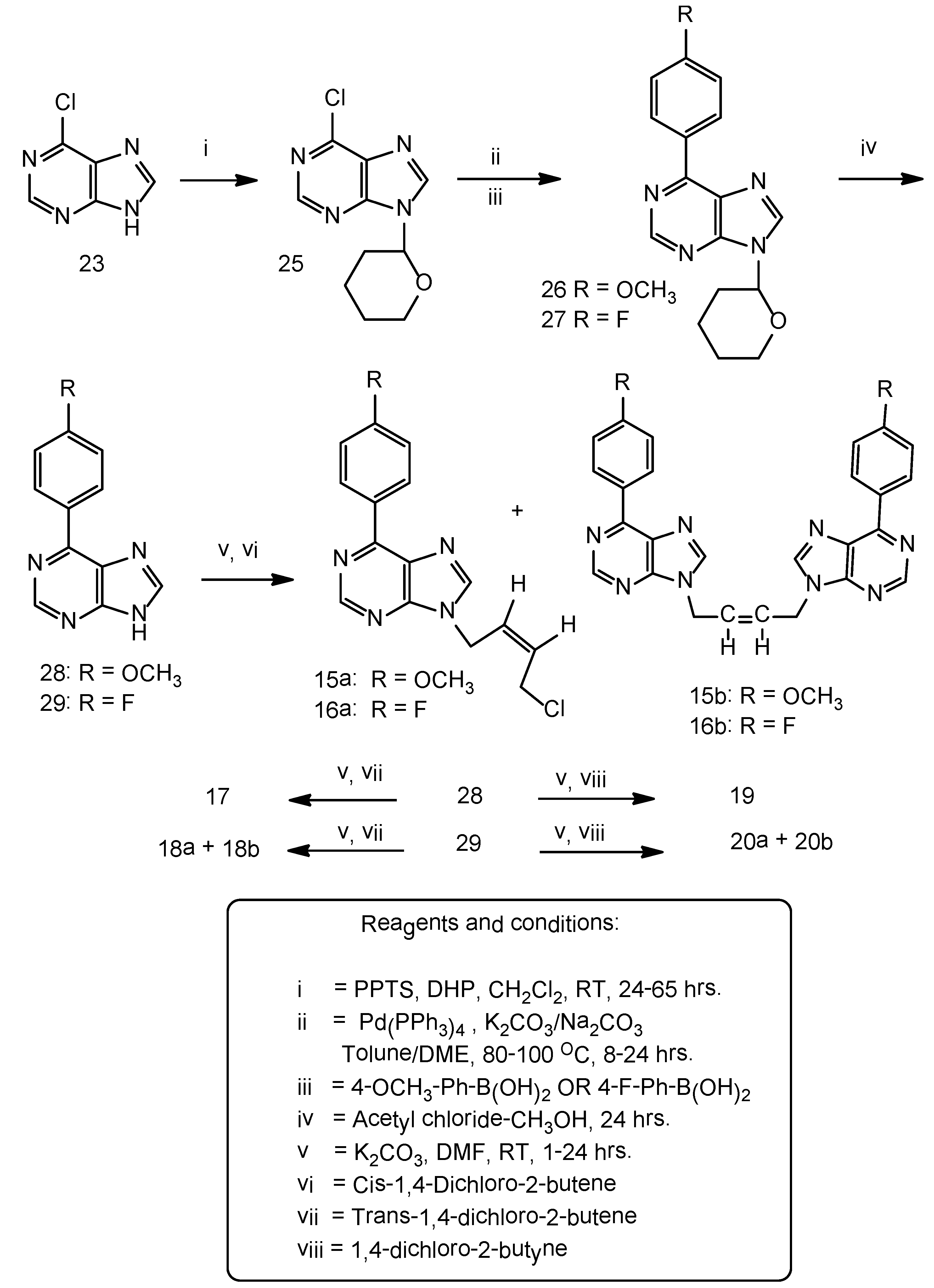
Synthesis of N9-(tetrahydropyran-2-yl)-6-chloropurine (25). A suspension of 6-chloropurine (5.0 g, 32 mmol), 3,4-dihydro-2H-pyran (5.4 g, 64 mmol) and pyridinium-p-toluenesulfonate (PPTS) (3.3 g, 13 mmol) in anhydrous dichloromethane (250 mL) was stirred at room temperature in an atmosphere of argon for 64 h. The original suspension transformed in to a clear liquid and the starting material disappeared. The reaction mixture was evaporated and the residue was chromatographed on a column of silica gel. The THP ether was eluted with 100% ethyl acetate and that was isolated as a semi-solid. It solidified as a pale yellow soft solid upon cooling in a freezer (7.0 g), 91% yield. Homogeneous on TLC on silica gel plate, Rf 0.58, mobile phase 100% ethyl acetate. The starting material 6-chloropurine under the same conditions was slow moving, Rf 0.12. 1H-NMR: δ 8.92 (1H, s, H-2), 8.81 (1H, H-8), THP protons: 5.81–5.77 (1H, m), 4.01–3.99 (1H, m), 3.77–3.68 (1H, m), 2.37–2.28 (1H, m), 2.04–1.96 (2H, m), 2.04 (1H, m), 2.03 (2H, m).
3.5. General Procedure C for Synthesis of N9-(tetrahydropyran-2-yl)-6-(4-methoxyphenyl)purine (26)
Suzuki-Miyaura cross coupling reaction: A stirred suspension of 6-chloro-9-(tetrahydropyran-2-yl) purine 25, (3.5 g, 15 mmol), 4-methoxyphenyl boronic acid (3.35 g, 22 mmol), anhydrous potassium carbonate (3.1 g, 22 mmol),tetrakis(triphenylphosphine)palladium (0), Pd (PPh3)4, (0.85 g, 0.74 mmol) in anhydrous toluene (150 mL) was gradually heated to 100 °C during 1 hr and then maintained at that temperature for 18 h. The TLC, silica gel plate, indicated the disappearance of the starting material Rf 0.45 and the formation of the new product Rf 0.54, mobile phase ethyl acetate-hexane 7:3 v/v. The reaction mixture was filtered while warm to remove the potassium and boron salts. The filtrate was concentrated and the resulting residue was crystallized from ethyl acetate-methanol 1:1 v/v as a brownish white solid (4.0g), 88% yield, m.p. 146–148 °C. 1H-NMR: δ 8.93 (1H, s, H-2), 8.87–8.82 (2H, m, Ar-H), 8.70 (1H, s, H-8), 7.18–7.13 (2H, m, Ar-H), 4.06–4.03 (1H, m, THP), 3.87 (3H, s,OCH3), 3.76–3.71 (1H, m, THP), 2.37–2.31 (1H, m, THP), 2.04–1.99 (2H, m, THP), 1.81–1.73 (1H, m, THP), 1.67–1.79 (2H, m, THP).
Synthesis of 6-(4-Methoxyphenyl)purine (28). To a suspension of compound 26 (3.2 g, 10 mmol) in methanol (100 mL) was added acetyl chloride (0.2 mL, 2.8 mmol) and the contents stirred overnight at room temperature for 24 h. The reaction mixture was evaporated, the residue was treated with water (100 mL) and the pH was adjusted to 7.5–8.0 with a saturated solution of sodium bicarbonate in water. The resulting solid was filtered, washed with DI water (100 mL) followed by cold ethanol (25 mL), hexane (25 mL) and dried under vacuum, brownish white solid (1.9 g), 81% yield. No further purification was necessary as the product was found to be homogeneous on silica gel TLC, Rf 0.3, mobile phase ethyl acetate-hexane 7:3 v/v. 1H-NMR: δ 8.97 (1H, s, H-2), 8.82–8.79 (2H, m, Ar-H), 8.75 (1H, s, H-8), 8.0 (1H, broad, NH), 7.2–7.17 (3H, m, Ar-H), 3.88 (3H, s, OCH3).
N9-[(Z)-4'-Chloro-2'-butenyl-1'-yl]-6-(4-methoxyphenyl)purine (15a). A suspension of 6-(4-methoxy-phenyl)purine (0.68 g, 3.0 mmol), anhydrous potassium carbonate (1.0 g, 7.2 mmol), cis-1,4-dichloro-2-butene (0.42 g, 3.4 mmol) was stirred at room temperature under argon atmosphere for 4h. The reaction mixture was worked-up as described in the general procedure-A. The resulting residue was chromatographed on a column of silica gel. Fractions 3–4 (100 mL) each from ethyl acetate-hexane (1:1) v/v furnished a brownish white solid 15a (0.41 g), 43% yield, m.p. 129-131 °C. 1H-NMR: δ 8.92 (1H, s, H-2), 8.87–8.82 (2H, m, Ar-H), 8.61 (1H, s, H-8), 7.16–7.13 (2H, s, Ar-H), 5.92–5.89 (2H, m, HC=CH), 5.09–5.07 (2H, d, J = 9.0 Hz, N-CH2), 4.54–4.52 (2H, m, CH2Cl), 3.86 (3H, s, OCH3). 13C-NMR: δ 161.61 (C-13), 152.42 (C-4), 151.86 (C-6), 145.44 (C-8), 131.07 (C-11, C-15), 129.74 (C-3'), 129.62 (C-10), 128.08 (C-2'), 127.82 (C-5), 114.05 (C-12, C-14), 55.32 (OCH3), 39.68 (C-1'), 39.22 (C-4'). LC-MS (m/z): 315 [M+1]+, 100%. Anal. Calcd. for C16H15N4OCl: C 61.05, H 4.80, N 17.80; Found: C 61.10, H 4.92, N 17.90.
N9,N9''-bis[(Z)-2'-Butenyl-1'4'-diyl]-6-(4-methoxyphenyl)purine (15b). The fractions 5–8 from the above column chromatography yielded a brownish white solid (0.1 g), 11% yield, m.p. 163–165 °C. 1H-NMR: δ 8.93 (2H, s, H-2, H-2''), 8.89–8.84 (4H, m, Ar-H), 8.72 (2H, s, H-8, H-8''), 7.17–7.14 (4H, m, Ar-H), 5.99–5.96 (2H, m, HC=CH), 5.29–5.27 (4H, d, J = 10.0 Hz, 2 × N-CH2), 3.87 (6H, s, C-13-OCH3, C-13''-OCH3). 13C-NMR: δ 161.63 (C-13, C-13''), 152.45 (C-4, C-4''), 151.98 (C-6, C-6''), 151.73 (C-2, C-2''), 145.66 (C-8, C-8''), 131.10 (C-11, C-11'', C-15, C-15''), 129.70 (C-10, C-10''), 128.03 (C-2', C-3'), 127.85 (C-5, C-5''), 114.09 (C-12, C-12'', C-14, C-14''), 55.34 (C-13-OCH3, C-13''-OCH3), 40.20 (C-1', C-4'). LC-MS (m/z): 505 [M+1]+, 100%. Anal. Calcd. for C28H24N8O2: C 66.65, H 4.80, N 22.21; Found: C 66.52, H 4.92, N 22.33.
3.6. Synthesis of N9-(tetrahydropyran-2-yl)-6-(4-fluorophenyl)purine (27)
Suzuki-Miyaura cross coupling reaction: 9-(Tetrahydropyran-2-yl)-6-chloropurine (4.8 g, 20 mmol), 4-fluorophenyl boronic acid (4.2 g, 30 mmol), Pd(PPh3)4, (1.0 g, 0.9 mmol) in anhydrous dimethoxy-ethane (200 mL) was added a 2.7 molar saturated Na2CO3 solution in water (11.2 mL, 30 mmol). The reaction mixture was gradually heated to reflux during 2 h in an oil bath and then maintained at reflux for 7 h. Reaction was worked-up and purified as above to yield a brownish white solid (5.0 g), 83% yield. m.p. 146–148 °C. 1H-NMR: δ 9.0 (1H, s, H-2), 8.93–8.89 (2H, m, Ar-H), 8.88 (1H, s, H-8), 7.46–7.42 (2H, m, Ar-H), THP protons: 5.85–5.82 (1H, m), 4.06–4.03 (1H, m), 3.77–3.72 (1H, m), 2.38–2.35 (1H, m), 2.06–1.99 (2H, m), 1.84–1.72 (1H, m), 1.63–1.61 (2H, m).
6-(4-Fluorophenyl)purine (29). To a stirred suspension of 9-(tetrahydropyran-2-yl)-6-(4-fluorophenyl) purine (27, 3.75 g, 13 mmol) in methanol (200 mL) at room temperature was added acetyl chloride (0.4 mL, 5.6 mmol) and the reaction was worked-up as described in 26. A brownish white solid resulted (2.6 g), 96% yield. The product was found homogeneous on TLC on a silica gel plate, Rf 0.3, mobile phase ethyl acetate-hexane (7:3) v/v. 1H-NMR: δ 8.94 (3H, s, H-2 and 2H Ar-H), 8.92 (1H, br, NH), 8.65 (1H, s, H-8), 7.45–7.40 (2H, m, Ar-H).
N9-[(Z)-4'-Chloro-2'-butenyl-1'-yl]-6-(4-fluorophenyl)purine (16a). A suspension of 6-(4-fluorophenyl) purine 29 (0.65 g, 3.0 mmol), anhydrous potassium carbonate (1.0 g, 7.2 mmol), cis-1,4-dichloro-2-butene (0.413 g, 3.3 mmol) in DMF (50 mL) was stirred under argon at room temperature for 3.5 h. The reaction was worked-up as described in general procedureA.The crude reaction mixture contained a major product 16a and a minor dimeric product 16b. The above crude product upon crystallization from ethyl acetate-methanol furnished the dimeric product 16b as feathery brownish white needeles. The mother liquor was concentrated and chromatographed on a column of silica gel using ethyl acetate-light petroleum ether (1:1, 2:1v/v)as the eluents that furnished a brownish white solid of 16a (0.4 g), 43% yield, m.p. 94–96 °C. 1H-NMR: δ 8.99 (1H, s, H-2), 8.93–8.90 (2H, m, Ar-H), 8.67 (1H, 2, H-8), 7.45–7.41 (2H, m, Ar-H), 5.93–5.91 (2H, m, HC=CH), 5.10–5.09 (2H, d, J = 5.5 Hz, N-CH2), 4.54–4.52 (2H, m, CH2Cl). 13C-NMR: δ 164.80 (d, 1JCF = 249 Hz, C-13), 152.13 (C-4), 151.75 (C-2), 151.39 (C-6), 146.09 (C-8), 131.85 (d, 4JCF = 3.8 Hz, C-10), 131.75 (d, 3JCF = 8.8 Hz, C-11, C-15), 129.99 (C-5), 129.82 (C-3'), 127.97 (C-2'), 115.73 (d, 2JCF = 21.4 Hz, C-12, C-14), 39.74 (C-1'), 39.21 (C-4'). LC-MS (m/z): 303 [M+1]+, 10%, 267 [M+1]+-HCl, 100%. Anal. Calcd. for C15H12N4FCl: C 59.51, H 4.0, N 18.51; Found: C 59.40, H 4.10, N 18.45.
N9,N9''-bis[(Z)-2'-Butenyl-1',4'-diyl]-6-(4-fluorophenyl)purine (16b). 0.27 g, 18% yield, m.p. 198–200 °C. 1H-NMR: δ 8.99 (2H, s, H-2, H-2''), 8.94–8.92 (4H, m, Ar-H), 8.77 (2H, s, H-8,H-8''), 7.46–7.42 (4H, m, Ar-H), 6.01–5.99 (2H, m, HC=CH), 5.31–3.30 (4H, d, J = 5.5 Hz, 2 × N-CH2). 13C-NMR: δ 164.83 (d, 1JCF = 249 Hz, C-13, C-13''), 152.27 (C-4, C-4''), 151.76 (C-2, C-2''), 151.44 (C-6, C-6''), 146.34 (C-8, C-8''), 131.90 (d, 4JCF = 3.8 Hz, C-10, C-10''), 131.79 (d, 3JCF = 18.9 Hz, C-11, C-11'', C-15, C-15''), 130.09 (C-5, C-5''), 128.04 (C-2', C-3'), 115.80 (d, 2JCF = 21.4 Hz, C-12, C-12'', C-14,C-14''), 40.29 (C-1', C-4'). LC-MS (m/z): 481 [M+1]+, 100%. Anal. Calcd. for C26H18N8F2: C 64.99, H 3.78, N 23.32; Found: C 65.10, H 3.82, N 23.45.
N9-[(E)-4'-Chloro-2'-butenyl-1'-yl]-6-(4-methoxyphenyl)purine (17). Cream white needles, 63% yield, m.p. 112–114 °C. 1H-NMR: δ 8.92 (1H, s, H-2), 8.87–8.82 (2H, m, Ar-H), 8.61 (1H, s, H-8), 7.16–7.13 (2H, s, Ar-H), 6.19–6.09 (1H, m, HC=CH), 5.83–5.73 (1H, m, HC=CH), 4.99–4.98 (2H, d, J = 9.0 Hz, N-CH2), 4.23–4.20 (2H, m, CH2Cl), 3.86 (3H, s, OCH3). 13C-NMR: δ 161.62 (C-13), 152.47 (C-4), 151.91 (C-6), 151.82 (C-2), 145.66 (C-8), 131.09 (C-11, C-15), 129.59 (C-10), 129.50 (C-3'), 129.02 (C-2'), 127.83 (C-5), 114.06 (C-12,14), 55.33 (C-13-OCH3), 44.08 (C-1'), 43.78 (C-4'). LC-MS (m/z): 277 [M+1]+, 100%. Anal. Calcd. for C16H15N4OCl: C 61.05, H 4.80, N 17.80; Found: C 61.15, H 4.95, N 17.95.
N9-[(E)-4'-Chloro-2'-butenyl-1'-yl]-6-(4-fluorophenyl)purine (18a). A suspension of 6-(4-fluoro-phenyl) purine (0.5 g, 2.3 mmol), anhydrous potassium carbonate (0.65 g, 4.7 mmol), trans-1,4-dichloro-2-butene (0.33 g, 2.6 mmol) in DMF (40 mL) was stirred at room temperature under argon for 22 h. The reaction has been worked-up as described in the general procedureA.The resulting product was chromatographed on a column of silica gel using ethyl acetate - light petroleumether (1:1) v/v as an eluent. Fractions of 100 mL were collected. The fractions 3-4 yielded 18a as a cream white solid, homogeneous on silicagel TLC, mobile phase ethyl acetate-hexane 7:3 v/v, Rf 0.69 (0.4g), 57% yield, m.p. 73–75 °C. 1H-NMR: δ 8.98 (1H, s, H-2), 8.94–8.91 (2H, m, Ar-H), 8.68 (1H, s, C-8), 7.45–7.42 (2H, m, Ar-H), 6.16–6.12 (1H, m, HC=CH), 5.82–5.79 (1H, m, HC=CH), 5.01–5.0 (2H, d, J = 5.0 Hz, N-CH2), 4.22–4.20 (2H, m, CH2Cl). 13C-NMR: δ 164.80 (d, 1JCF = 250 Hz, C-13), 152.16 (C-4), 151.80 (C-2), 151.43 (C-6),146.27 (C-8), 131.85 (d, 4JCF = 2.52 Hz, C-10), 131.75 (d, 3JCF = 8.8 Hz, C-11, C-15),129.96 (C-5), 129.59 (C-3'), 128.87 (C-2'),115.71 (d, 2JCF = 21.4 Hz, C-12, C-14), 44.04 (C-1'), 43.85(C-4'). LC-MS (m/z): 303 [M+1]+, 100%. Anal. Calcd. for C15H12N4FCl: C 59.51, H 4.0, N 18.51; Found: C 59.25, H 4.16, N 18.65.
N9,N9''-bis[(E)-2'-Butenyl-1',4'-diyl]-6-(4-fluorophenyl)purine (18b). From the above chromatography, fractions 6–10 yielded this dimer 18b, Rf 0.1, brownish white solid, (0.15 g), 13% yield, m.p. 217–219 °C. 1H-NMR: δ 8.95 (2H, s, H-2, H-2''), 8.93–8.90 (4H, m, Ar-H), 8.65 (2H, s, H-8, H-8''), 7.46–7.42 (4H, m, Ar-H), 5.98 (2H, m, HC=CH), 4.97–4.96 (4H, m, 2 × N-CH2). 13C-NMR: δ 164.86 (d, 1JCF = 252 Hz, C-13, C-13''), 152.61 (C-4, C-4''), 151.75 (C-6, C-6''), 151.61 (C-2, C-2''), 144.04 (C-8, C-8''), 131.44 (d, 3JCF = 8.8 Hz, C-11, C-11'', C-15, C-15''), 131.32 (d, 4JCF = 2.52 Hz, C-10, C-10''), 130.06 (C-5, C5''), 127.90 (C-2', C-3'), 115.06 (d, 2JCF = 21.4 Hz, C-12, C-12'' C-14, C-14''), 43.88 (C-1', C-4'). LC-MS (m/z): 481 [M+1]+, 100%. Anal. Calcd. for C26H18N8F2: C 64.99, H 3.78, N 23.32; Found: C 64.82, H 3.85, N 23.45.
N9-[4'-Chloro-2'-butynyl-1'-yl]-6-(4-methoxyphenyl)purine (19). White solid, 80% yield, m.p. 119–121 °C. 1H-NMR: δ 8.93 (1H, s, H-2), 8.87–8.82 (2H, m, Ar-H), 8.70 (1H, s, H-8), 7.18–7.13 (2H, m, Ar-H), 5.31 (2H, t, J = 3.5 Hz, N-CH2), 4.50 (2H, m, CH2Cl), 3.87 (3H, s, OCH3). 13C-NMR: δ 161.70 (C-13), 152.67 (C-4), 152.0 (C-2), 151.58 (C-6), 145.19 (C-8), 131.13 (C-11, C-15), 129.49 (C-10), 127.67 (C-5), 114.11 (C-12, C-14), 80.34 (C-3'), 80.28 (C-2'), 55.34 (OCH3), 32.77 (C-1'), 30.55 (C-4'). LC-MS (m/z): 313 [M+1]+, 100%. Anal. Calcd. for C16H13N4OCl: C 61.44, H 4.19, N 17.91; Found: C 61.35, H 4.30, N 17.85.
N9-[4'-Chloro-2'-butynyl-1'-yl]-6-(4-fluorophenyl)purine (20a). Abrownish white solid, 64% yield, m.p. 128–130 °C. 1H-NMR: δ 9.02 (1H, s, H-2), 8.92–8.89 (2H, m, Ar-H), 8.75 (1H, s, H-8), 7.46–7.42 (2H, m, Ar-H), 5.33 (2H, t, J = 2.0 Hz, N-CH2), 4.50 (2H, t, J = 2.0 Hz, CH2Cl). 13C-NMR: δ 164.86 (d, 1JCF = 249 Hz, C-13), 151.99 (C-2), 151.83 (C-4), 151.65 (C-6), 145.80 (C-8), 131.79 (d, 3JCF = 8.8 Hz, C-11, C15), 131.68 (d, 4JCF = 2.52 Hz, C-10), 129.87 (C-5), 115.75 (d, 2JCF = 21.4 Hz, C-12, C-14), 80.43 (C-3'), 80.14 (C-2'), 32.86 (C-1'), 30.52 (C-4'). LC-MS (m/z): 301 [M+1]+, 100%. Anal. Calcd. for C15H10N4FCl: C 59.91, H 3.35, N 18.63; Found: C 60.0, H 3.40, N 18.55.
N9,N9''-[2'-butynyl-1',4'-diyl]-6-(4-fluorophenyl)purine (20b). A light brown solid, 11% yield, m.p. 225–227 °C. 1H-NMR: δ 8.99 (2H, s, H-2, H-2''), 8.93–8.90 (4H, m, Ar-H), 8.76 (2H, s, H-8, H-8''), 7.47–7.43 (4H, m, Ar-H), 6.01–5.30 (4H, s, 2 × N-CH2). LC-MS (m/z): 479 [M+1]+, 100%.Anal. Calcd. for C26H16N8F2: C 65.27, H 3.37, N 23.42; Found: C 65.15, H 3.45, N 23.55.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}