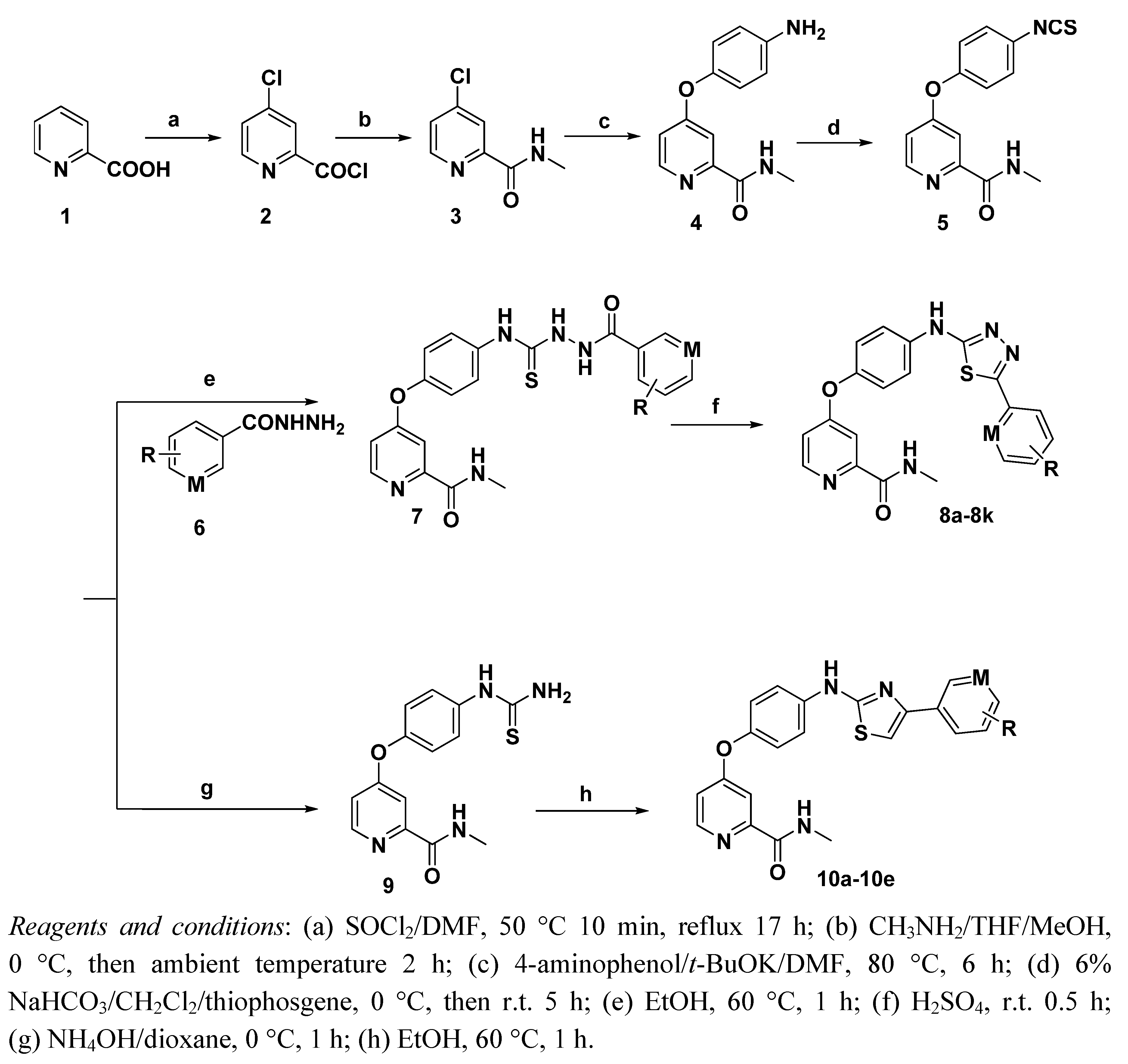
3.2.6. General procedure for the preparation of compound 8a–8k
4-(4-(2-arylhydrazinecarbothioamido)phenoxy)-N-methylpicolinamides 7a–7k (0.26 mmol) were added portionwise to 3 mL concentrated sulfuric acid and stirred at room temperature for 30 min. Then crushed ice (10 g) was add to the mixture, followed by ammonia was used to neutralize the solution. The crude product precipitated was purified by chromatography on silica gel using 30:1 MeOH/CH2Cl2 as eluent.
N-Methyl-4-(4-(5-phenyl-1,3,4-thiadiazol-2-ylamino)phenoxy)picolinamide (8a). Yield: 76%. m.p.: 233–234 °C. MS [MH−] (m/z): 404.2 (M−1); 1H-NMR δ: 10.71 (s, 1H, –NH), 8.77–8.75 (m, 1H, amide–NH), 8.52 (d, J = 6.0 Hz, 1H, pyridine–6H), 7.88–7.86 (m, 2H, phenyl–3H,5H), 7.81 (d, J = 9.0 Hz, 2H, phenyl–3H,5H), 7.52–7.51 (m, 3H, phenyl–2H,4H,6H), 7.40 (d, J = 3.0 Hz, 1H, pyridine–3H), 7.28 (d, J = 9.0 Hz, 2H, phenyl–2H,6H), 7.17 (q, J = 3.0, 6.0 Hz, 1H, pyridine–5H), 2.79 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 171.5 (C), 164.3 (C), 159.7 (C), 151.0 (C), 146.6 (C), 141.2 (CH), 138.9 (C), 134.4 (C), 130.2 (C), 127.8 (CH), 127.8 (CH), 126.3 (CH), 126.3 (CH), 125.8 (CH), 123.5 (CH), 123.5 (CH), 119.8 (CH), 119.8 (CH), 110.1 (CH), 107.3 (CH), 26.1 (CH3); Anal. Calcd for C21H17N5O2S (%): C, 64.08; H, 4.38; N, 15.64; found C 63.01, H 4.05, N 15.21.
N-Methyl-4-(4-(5-(4-fluorophenyl)-1,3,4-thiadiazol-2-ylamino)phenoxy)picolinamide (8b). Yield: 52%. m.p.: 219–220 °C. MS [MH−] (m/z): 422.2 (M−1); 1H-NMR δ: 10.77 (s, 1H, –NH), 8.77–8.75 (br, 1H, amide–NH), 8.51 (d, J = 6.0 Hz, 1H, pyridine–6H), 7.95–7.90 (m, 2H, phenyl–3H,5H), 7.82 (d, J = 9.0 Hz, 2H, phenyl–3H,5H), 7.40–7.33 (m, 3H, phenyl–2H,6H, pyridine-3H), 7.26 (d, J = 9.0 Hz, 2H, phenyl-2H,6H), 7.16–7.14 (m, 1H, pyridine–5H), 2.79 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 173.3 (C), 163.8 (C), 162.5 (C), 151.2 (C), 148.8 (C), 143.5 (CH), 140.6 (C), 137.1 (C), 130.2 (C), 129.7 (CH), 127.3 (CH), 127.3 (CH), 125.6 (CH), 125.6 (CH), 123.5 (CH), 123.5 (CH), 119.8 (CH), 119.8 (CH), 111.9 (CH), 110.7 (CH), 26.1 (CH3); Anal. Calcd for C21H16 FN5O2S (%): C, 59.85; H, 3.83; N, 16.62; found C 60.03, H 3.99, N 16.83.
N-Methyl-4-(4-(5-(4-chlorophenyl)-1,3,4-thiadiazol-2-ylamino)phenoxy)picolinamide (8c). Methylene Yield: 69%. m.p.: 245–246 °C. MS [MH−] (m/z): 439.2 (M−1); 1H-NMR δ: 10.77 (s, 1H, –NH), 8.78–8.74 (br, 1H, amide–NH), 8.50 (d, J = 6.0 Hz, 1H, pyridine–6H), 7.89 (d, J = 9.0 Hz, 2H, phenyl–3H,5H), 7.80 (d, J = 9.0 Hz, 2H, phenyl–3H,5H), 7.58 (d, 2H, J = 9.0 Hz, phenyl–2H,6H), 7.39 (s, 1H, pyridine–3H), 7.25 (d, J = 9.0 Hz, 2H, phenyl–2H,6H), 7.15–7.14 (br, 1H, pyridine–5H), 2.78 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 173.5 (C), 164.3 (C), 160.6 (C), 152.1(C), 150.8(C), 144.3 (CH), 141.9 (C), 136.4 (C), 130.9 (C), 126.3 (CH), 126.3 (CH), 125.7 (CH), 125.7 (CH), 123.8 (CH), 121.3 (CH), 121.3 (CH), 119.6 (CH), 119.6 (CH), 109.9 (CH), 107.3 (CH), 26.1 (CH3); Anal. Calcd for C21H16 ClN5O2S (%): C, 57.60; H, 3.68; N, 15.99; found C 57.82, H 3.75, N 16.18.
N-Methyl-4-(4-(5-(3,4-difluorophenyl)-1,3,4-thiadiazol-2-ylamino)phenoxy)picolinamide (8d). Yield: 40%. m.p.: 225–226 °C. MS [MH−] (m/z): 440.7 (M−1); 1H-NMR δ: 10.83 (s, 1H, –NH), 8.78–8.75 (br, 1H, amide–NH), 8.51 (d, J = 6.0 Hz, 1H, pyridine–6H), 7.99–7.93 (m, 1H, phenyl–5H), 7.81–7.71 (m, 3H, phenyl–3H,5H, phenyl–2H), 7.63–7.54 (q, J = 6.0, 9.0 Hz, 1H, phenyl–6H), 7.39 (s, 1H, pyridine–3H), 7.26 (d, J = 9.0 Hz, 2H, phenyl–2H,6H), 7.14 (q, J = 3.0, 6.0 Hz, 1H, pyridine–5H), 2.79 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 171.9 (C), 164.8 (C), 161.1 (C), 151.4(C), 150.6(C), 148.4 (C), 147.3 (C), 145.9(CH), 141.6 (C), 135.8 (C), 130.3 (C), 128.9 (CH), 124.3 (CH), 124.3 (CH), 121.5 (CH), 121.5 (CH), 119.6 (CH), 119.6 (CH), 111.4 (CH), 109.1 (CH), 26.1 (CH3); Anal. Calcd for C21H15F2N5O2S (%): C, 57.40; H, 3.44; N, 15.94; found C 58.21, H 3.63, N 16.27.
N-Methyl-4-(4-(5-(3-(trifluoromethyl)phenyl)-1,3,4-thiadiazol-2-ylamino)phenoxy)picolinamide (8e). Yield: 55%. m.p.: 242–243 °C. MS [MH+] (m/z): 472.6 (M+1); 1H-NMR δ: 10.85 (s, 1H, –NH), 8.76–8.75 (br, 1H, amide–NH), 8.51 (d, J = 6.0 Hz, 1H, pyridine–6H), 8.16 (br, 2H, pyridine–2H,4H), 7.87–7.76 (m, 4H, phenyl–3H,5H, phenyl–5H,6H), 7.40 (s, 1H, pyridine–3H), 7.27 (d, J = 9.0 Hz, 2H, phenyl–2H,6H), 7.16–7.13 (br, 1H, pyridine–5H), 2.78 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 173.3 (C), 165.2 (C), 160.5 (C), 154.6(C), 149.9(C), 144.2 (CH), 141.3 (C), 133.5(C), 133.1 (CH), 132.9 (C), 130.5 (C) 129.5 (CH), 126.5 (CH), 125.1 (CH), 124.4 (CH), 121.6 (CH), 121.6 (CH), 119.6 (CH), 119.6 (CH), 112.7 (CH), 108.9 (CH), 26.2 (CH3); Anal. Calcd for C22H16F3N5O2S (%): C, 56.05; H, 3.42; N, 14.85; found C 56.21, H 3.69, N 15.03.
N-Methyl-4-(4-(5-(3-methoxyphenyl)-1,3,4-thiadiazol-2-ylamino)phenoxy)picolinamide (8f). Yield: 82%. m.p.: 237–238 °C. MS [MH−] (m/z): 434.2 (M−1); 1H-NMR δ: 10.61 (s, 1H, –NH), 8.77–8.75 (m, 1H, amide–NH), 8.51 (d, J = 6.0 Hz, 1H, pyridine–6H), 7.82 (d, J = 9.0 Hz, 2H, phenyl–3H,5H), 7.74(d, J = 6.0 Hz, J = 9.0 Hz, 2H, phenyl–4H,6H), 7.40 (d, J = 3.0 Hz, 1H, pyridine–3H),7.25 (d, J = 9.0 Hz, 2H, phenyl–2H,6H), 7.16 (q, J = 3.0, 6.0 Hz, 1H, pyridine–5H), 7.00 (s, 2H, phenyl–2H,5H), 3.81 (s, 3H, CH3), 2.79 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 173.5 (C), 162.8 (C), 160.3 (C), 160.0 (C), 153.3 (C), 152.6 (CH), 145.6 (CH), 141.7 (C), 135.8 (C), 133.7 (C), 131.9 (CH) 122.2 (CH), 120.9 (CH), 120.9 (CH), 118.9 (CH), 118.9 (CH), 113.6 (CH), 113.1 (CH), 111.3 (CH), 108.6 (CH), 55.6 (CH3), 26.1 (CH3); Anal. Calcd for C22H19N5O3S (%): C, 60.96; H, 4.42; N, 16.16; found C 60.57, H 4.49, N 16.13.
N-Methyl-4-(4-(5-(2,4-dichlorophenyl)-1,3,4-thiadiazol-2-ylamino)phenoxy)picolinamide (8g). Yield: 68%. m.p.: 251–252 °C. MS [MH+] (m/z): 473.0 (M+1); 1H-NMR δ: 10.95 (s, 1H, –NH), 8.77–8.75 (m, 1H, amide–NH), 8.51 (d, J = 6.0 Hz, 1H, pyridine–6H), 8.11 (d, J = 9.0 Hz, 1H, phenyl–3H), 7.86–7.81 (m, 3H, phenyl–3H,5H, phenyl–5H), 7.62 (q, J = 3.0, 9.0 Hz, 2H, phenyl–6H), 7.39 (d, 1H, J = 3.0 Hz, pyridine–3H), 7.26 (d, J = 9.0 Hz, 2H, phenyl–2H,6H), 7.16 (q, J = 3.0, 6.0 Hz, 1H, pyridine–5H), 2.79 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 173.7 (C), 163.6 (C), 160.9 (C), 151.7 (C), 151.0 (C), 146.2 (CH), 142.4 (C), 136.4 (C), 135.7 (C), 135.0 (C), 133.6 (C), 130.9 (CH), 130.3 (CH) 127.4 (CH), 120.5 (CH), 120.5 (CH), 119.1 (CH), 119.1 (CH), 113.9 (CH), 108.6 (CH), 26.1 (CH3); Anal. Calcd for C21H15Cl2N5O2S (%): C, 53.40; H, 3.20; N, 14.83; found C 54.07, H 3.31, N 15.01.
N-methyl-4-(4-(5-(2,6-dichlorophenyl)-1,3,4-thiadiazol-2-ylamino)phenoxy)picolinamide (8h). Yield: 72%. m.p.: 261–262 °C. MS [MH−] (m/z): 473.8 (M−1); 1H-NMR δ: 10.90 (s, 1H, –NH), 8.77–8.75 (m, 1H, amide–NH), 8.51 (d, J = 6.0 Hz, 1H, pyridine–6H), 7.84 (d, J = 9.0 Hz, 2H, phenyl–3H,5H), 7.69 (d, J = 9.0 Hz, 2H, phenyl–3H,5H), 7.62–7.57 (m, 1H, phenyl–4H), 7.41 (d, J = 3.0 Hz, 1H, pyridine–3H), 7.27 (d, J = 9.0 Hz, 2H, phenyl–2H, 6H), 7.16 (q, J = 3.0, 6.0 Hz, 1H, pyridine–5H), 2.80 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 173.5 (C), 162.9 (C), 160.3 (C), 151.8 (C), 150.6 (C), 145.5 (CH), 141.7 (C), 138.0 (C), 136.3 (C), 134.7 (C), 134.7 (C), 131.5 (CH), 127.4 (CH), 127.4 (CH) 121.3 (CH), 121.3 (CH), 120.1 (CH), 120.1 (CH), 113.9 (CH), 108.6 (CH), 26.1 (CH3); Anal. Calcd for C21H15Cl2N5O2S (%): C, 53.40; H, 3.20; N, 14.83; found C 53.67, H 3.34, N 14.89.
N-Methyl-4-(4-(5-(benzo[d][1,3]dioxol-5-yl)-1,3,4-thiadiazol-2-ylamino)phenoxy)picolinamide (8i). Yield: 47%. m.p.: 243–244 °C. MS [MH+] (m/z): 448.2 (M+1); 1H-NMR δ: 10.65 (s, 1H, -NH), 8.77–8.76 (br, 1H, amide–NH), 8.51 (d, J = 6.0 Hz, 1H, pyridine–6H), 7.79 (d, J = 9.0 Hz, 2H, phenyl–3H,5H), 7.43–7.33 (m, 3H, phenyl–2H, 6H, pyridine–3H), 7.25 (d, J = 9.0 Hz, 2H, phenyl–2H,6H), 7.05 (d, 1H, J = 9.0 Hz, phenyl–5H), 6.77–6.71 (m, 1H, pyridine–5H), 2.79 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 174.1 (C), 164.8 (C), 161.0 (C), 152.7 (C), 151.0 (C), 146.6 (C), 146.2 (CH), 145.2 (C), 142.4 (C), 136.4 (C), 123.1 (C), 122.5 (CH), 121.7 (CH), 121.7 (CH), 120.8 (CH), 120.0 (CH), 120.0 (CH), 115.3 (CH), 113.9 (CH), 109.6 (CH), 101.5 (CH2), 26.1 (CH3); Anal. Calcd for C22H17N5O4S (%): C, 59.05; H, 3.83; N, 15.65; found C 58.73, H 3.81, N 15.59.
N-Methyl-4-(4-(5-(2,5-dimethoxyphenyl)-1,3,4-thiadiazol-2-ylamino)phenoxy)picolinamide (8j). Yield: 63%. m.p.: 267–268 °C. MS [MH−] (m/z): 464.7 (M−1); 1H-NMR δ: 10.57 (s, 1H, -NH), 8.76–8.75 (br, 1H, amide–NH), 8.51 (d, J = 6.0 Hz, 1H, pyridine–6H), 7.82 (d, J = 9.0 Hz, 2H, phenyl–3H,5H), 7.74 (d, J = 3.0 Hz, 1H, phenyl–6H), 7.40 (s, 1H, pyridine–3H), 7.25–7.18 (m, 3H, phenyl–2H,6H, phenyl–2H), 7.16–7.14 (br, 1H, pyridine–5H), 7.10 (q, J = 3.0 Hz, 6.0 Hz, 1H, pyridine–5H), 2.93 (s, 3H, CH3), 3.79 (s, 3H, CH3), 2.79 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 173.8 (C), 164.2 (C), 160.5 (C), 151.9 (C), 151.8 (C), 150.3 (C), 148.7 (C), 144.1 (CH), 141.3 (C), 135.8 (C), 121.1 (C), 121.0 (CH), 121.0 (CH), 118.9 (CH), 118.9 (CH), 115.3 (CH), 113.7 (CH), 112.3 (CH), 111.9 (CH), 109.6 (CH), 56.1 (CH3), 55.8 (CH3), 26.1 (CH3); Anal. Calcd for C23H21N5O4S (%): C, 59.60; H, 4.57; N, 15.11; found C 59.28, H 4.45, N 14.89.
N-Methyl-4-(4-(5-(3,5-dimethoxyphenyl)-1,3,4-thiadiazol-2-ylamino)phenoxy) picolinamide (8k). Yield: 72%. m.p.: 250–251 °C. MS [MH+] (m/z): 464.3 (M+1); 1H-NMR δ: 10.79 (s, 1H, -NH), 8.75–8.74 (br, 1H, amide–NH), 8.50 (d, J = 6.0 Hz, 1H, pyridine–6H), 7.80 (d, J = 9.0 Hz, 2H, phenyl–3H,5H), 7.39 (s, 1H, pyridine–3H), 7.24 (d, 2H, J = 9.0 Hz, phenyl–2H, 6H), 7.15–7.13 (br, 1H, pyridine–5H), 6.97 (s, 2H, phenyl–2H,6H), 6.62 (s, 1H, phenyl–4H), 3.81 (s, 3H, CH3), 2.78 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 173.5 (C), 164.2 (C), 160.6 (C), 158.9 (C), 158.9 (C), 151.9 (C), 151.0 (C), 145.3 (CH), 142.8 (C), 135.9 (CH), 134.9 (C), 121.0 (CH), 121.0 (CH), 118.9 (CH), 118.9 (CH), 113.8 (CH), 108.6 (CH), 102.9 (CH), 102.9 (CH), 100.1 (CH), 55.7 (CH3) , 55.3 (CH3) , 26.1 (CH3). Anal. Calcd for C23H21N5O4S (%): C, 59.60; H, 4.57; N, 15.11; found C 60.18, H 4.73, N 15.26.
3.2.8. General procedure for preparation of compound (10a–10e)
A mixture of N-methyl-4-(4-thioureidophenoxy)picolinamide (9, 0.16 g, 0.5 mmol) and 2-bromo-1-arylethanone (0.5 mmol) was refluxed in anhydrous ethanol (5 mL) for 1h. Then mixture was allowed to precipitate enough crude product at room temperature for 1 h and compound 10a–10e was obtained after filtration and recrystallation in ethanol.
N-Methyl-4-(4-(4-phenylthiazol-2-ylamino)phenoxy)picolinamide (10a). 2-bromo-1-phenylethanone. Yield: 79%. m.p.: 271–272 °C. MS [MH+] (m/z): 403.2 (M+1); 1H-NMR δ: 10.52 (s, 1H, –NH), 9.03–9.01 (m, 2H, amide–NH, pyridine–6H), 8.57–8.55 (m, 1H, pyridine–5H), 7.92–7.89 (m, 3H, thiazole–H, phenyl–3H, 5H), 7.81 (d, J = 3.0 Hz, 1H, pyridine–3H), 7.47–7.36 (m, 4H, phenyl–2H, 6H, phenyl–2H, 6H), 7.29–7.22 (m, 3H, phenyl–3H, 4H, 5H), 2.79 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 163.9 (C), 160.2 (C), 160.0 (C), 150.3 (C), 149.1 (C), 145.6 (CH), 141.5 (C), 136.0 (C), 133.9 (C), 129.0 (CH), 129.0 (CH), 128.1 (CH), 127.1 (CH), 127.1 (CH), 121.2 (CH), 121.2 (CH), 119.3 (CH), 119.3 (CH), 113.5 (CH), 109.2 (CH), 105.0 (CH), 26.1 (CH3); Anal. Calcd for C22H18N4O2S (%): C, 65.65; H, 4.51; N, 13.92; found C 65.88, H 4.39, N 14.02.
N-Methyl-4-(4-(4-(4-chlorophenyl)thiazol-2-ylamino)phenoxy)picolinamide (10b). 2-bromo-1-(4-chlorophenyl)ethanone. Yield: 88%. m.p.: 278–279 °C. MS [MH+] (m/z): 437.3 (M+1); 1H-NMR δ: 10.67 (s, 1H, –NH), 8.99–8.92 (br, 1H, amide–NH), 8.55–8.53 (m, 1H, pyridine–6H), 7.94 (d, J = 9.0 Hz, 2H, phenyl–3H,5H), 7.88 (d, 1H, J = 3.0 Hz, pyridine–3H), 7.78 (d, 2H, J = 9.0 Hz, phenyl–3H,5H), 7.61–7.58 (br, 1H, pyridine–5H), 7.53 (d, 2H, J = 9.0 Hz, phenyl–2H, 6H), 7.24–7.21 (m, 3H, phenyl–2H,6H, thiazole–H), 2.79 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 163.8 (C), 161.0 (C), 160.3 (C), 150.2 (C), 148.9 (C), 146.2 (CH), 142.1 (C), 135.4 (C), 133.8 (C), 131.0 (C), 129.3 (CH), 129.3 (CH), 128.5 (CH), 128.5 (CH), 121.2 (CH), 121.2 (CH), 119.6 (CH), 119.6 (CH), 113.2 (CH), 109.8 (CH), 105.1 (CH), 26.3 (CH3); Anal. Calcd for C22H17 ClN4O2S (%): C, 60.48; H, 3.92; N, 12.82; found C 60.73, H 3.97, N 13.03.
N-Methyl-4-(4-(4-(pyridin-3-yl)thiazol-2-ylamino)phenoxy)picolinamide (10c). 2-bromo-1-(pyridin-3-yl)ethanone. Yield: 82%. m.p.: 266–267 °C. MS [MH+] (m/z): 404.2 (M+1); 1H-NMR δ: 10.72 (s, 1H, –NH), 9.37(s, 1H, pyridine–3H), 9.02–9.01 (br, 1H, amide–NH), 8.85–8.82 (m, 2H, pyridine–6H, pyridine–6H), 8.53 (d, J = 3.0 Hz, 1H, pyridine–4H), 8.11 (q, J = 3.0, 6.0 Hz, 1H, pyridine–5H), 7.93–7.89 (m, 3H, phenyl–3H,5H, pyridine–3H), 7.47 (s, 1H, thiazole–H), 7.23–7.19 (m, 3H, phenyl–2H,6H, pyridine–5H), 2.78 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 163.4 (C), 160.0 (C), 158.2 (C), 149.9 (C), 146.7 (CH), 147.3 (CH), 146.1 (CH), 142.0 (C), 136.1 (C), 135.3 (C), 133.8 (CH), 133.8 (C), 124.0 (CH), 121.1 (CH), 121.1 (CH), 120.1 (CH), 120.1 (CH), 113.5 (CH), 109.6 (CH), 109.6 (CH), 26.3 (CH3); Anal. Calcd for C21H17 N5O2S (%): C, 62.52; H, 4.25; N, 17.36; found C 62.84, H 4.47, N 17.39.
N-Methyl-4-(4-(4-(4-cyanophenyl)thiazol-2-ylamino)phenoxy)picolinamide (10d). 4-(2-bromoacetyl)benzonitrile. Yield: 75%. m.p.: 281–282 °C. MS [MH+] (m/z): 428.6 (M+1); 1H-NMR δ: 10.56 (s, 1H, –NH), 8.88–8.87 (br, 1H, amide–NH), 8.53–8.51 (m, 1H, pyridine–6H), 8.12–8.09 (m, 2H, phenyl–3H,5H), 7.94–7.76 (m, 4H, phenyl–2H,3H,5H,6H), 7.67 (s, 1H, thiazole–H), 7.52–7.49 (m, 1H, pyridine–3H), 7.52–7.49 (m, 1H, phenyl–2H,6H, pyridine–5H), 2.79 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 164.0 (C), 161.1 (C), 160.5 (C), 151.2 (C), 149.7 (C), 146.5 (CH), 142.3 (C), 136.9 (C), 136.2 (C), 132.1 (CH), 132.1 (CH), 126.0 (CH), 126.0 (CH), 121.3 (CH), 121.3 (CH), 120.0 (CH), 120.0 (CH), 118.5 (CH), 113.7 (CH), 112.8 (CH), 109.3 (CH), 105.2 (CH), 26.3 (CH3); Anal. Calcd for C23H17 N5O2S (%): C, 64.62; H, 4.01; N, 16.38; found C 64.48, H 4.06, N 16.41.
N-Methyl-4-(4-(4-(4-hydroxyphenyl)thiazol-2-ylamino)phenoxy)picolinamide (10e). 2-bromo-1-(4-hydroxyphenyl)ethanone. Yield: 79%. m.p.: 275–276 °C. MS [MH+] (m/z): 417.3 (M+1); 1H-NMR δ: 10.39 (s, 1H, –NH), 8.91–8.85 (br, 1H, amide–NH), 8.53 (d, J = 3.0 Hz, 1H, pyridine–6H), 7.88 (m, 2H, phenyl–3H,5H), 7.88 (m, 2H, phenyl–2H,6H), 7.53–7.49 (m, 1H, pyridine–3H), 7.23–7.21 (m, 3H, phenyl–2H,6H, pyridine–5H), 7.07 (s, 1H, thiazole–H), 6.79 (m, 1H, phenyl–3H,5H), 2.79 (d, J = 6.0 Hz, 3H, CH3); 13C-NMR δ: 163.9 (C), 160.1 (C), 159.3 (C), 157.5 (C), 150.0 (C), 148.9 (C), 147.3 (CH), 141.6 (C), 134.9 (C), 128.2 (CH), 128.2 (CH), 125.1 (C), 121.4 (CH), 121.4 (CH), 120.8 (CH), 120.8 (CH), 116.1 (CH), 116.1 (CH), 113.3 (CH), 109.2 (CH), 103.9 (CH), 26.3 (CH3); Anal. Calcd for C22H18 N4O3S (%): C, 63.14; H, 4.34; N, 13.39; found C 63.39, H 4.42, N 13.52.




{kind=link}

































