Methoxymethyl (MOM) Group Nitrogen Protection of Pyrimidines Bearing C-6 Acyclic Side-Chains


Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry


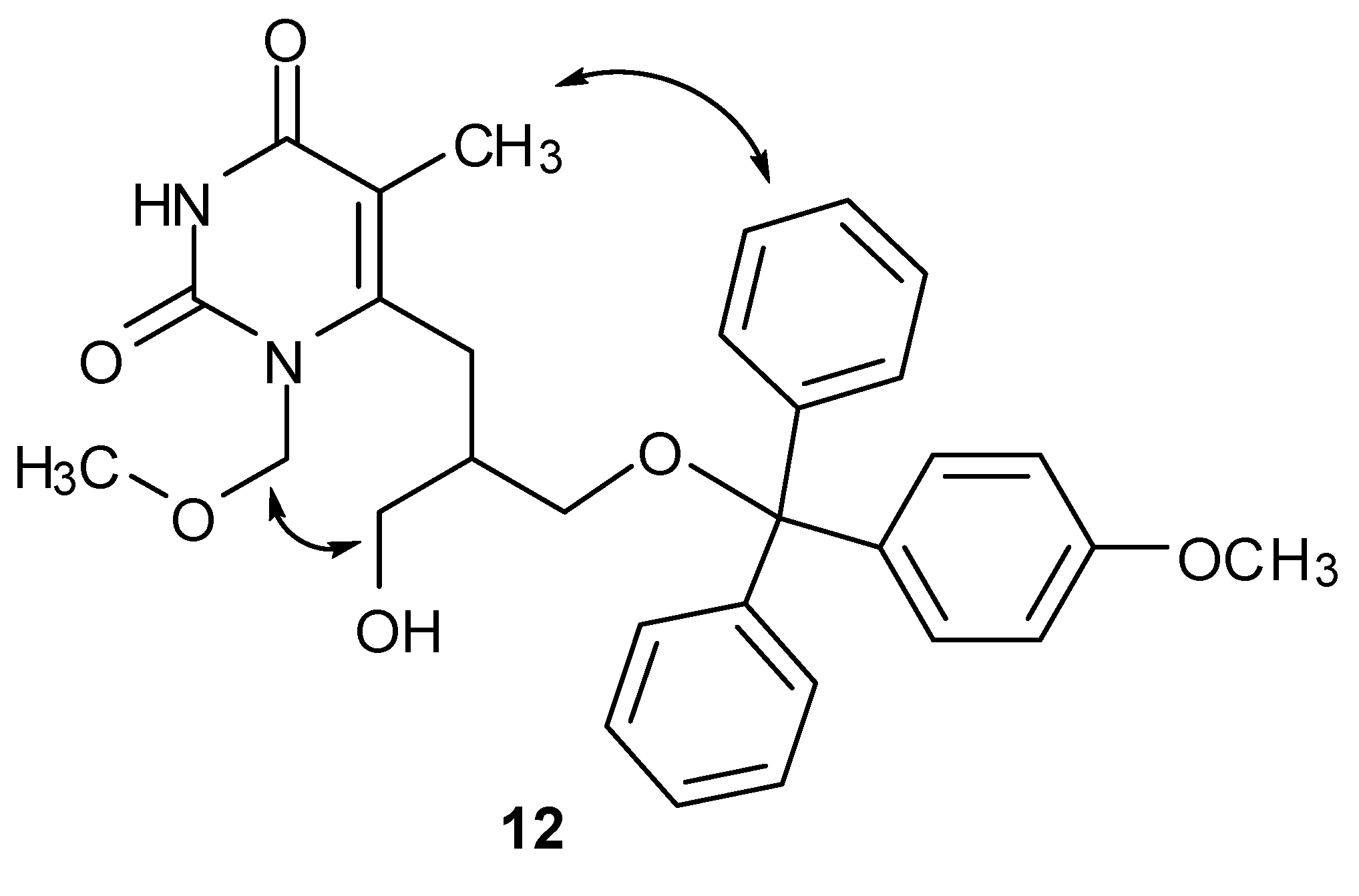{kind=link}
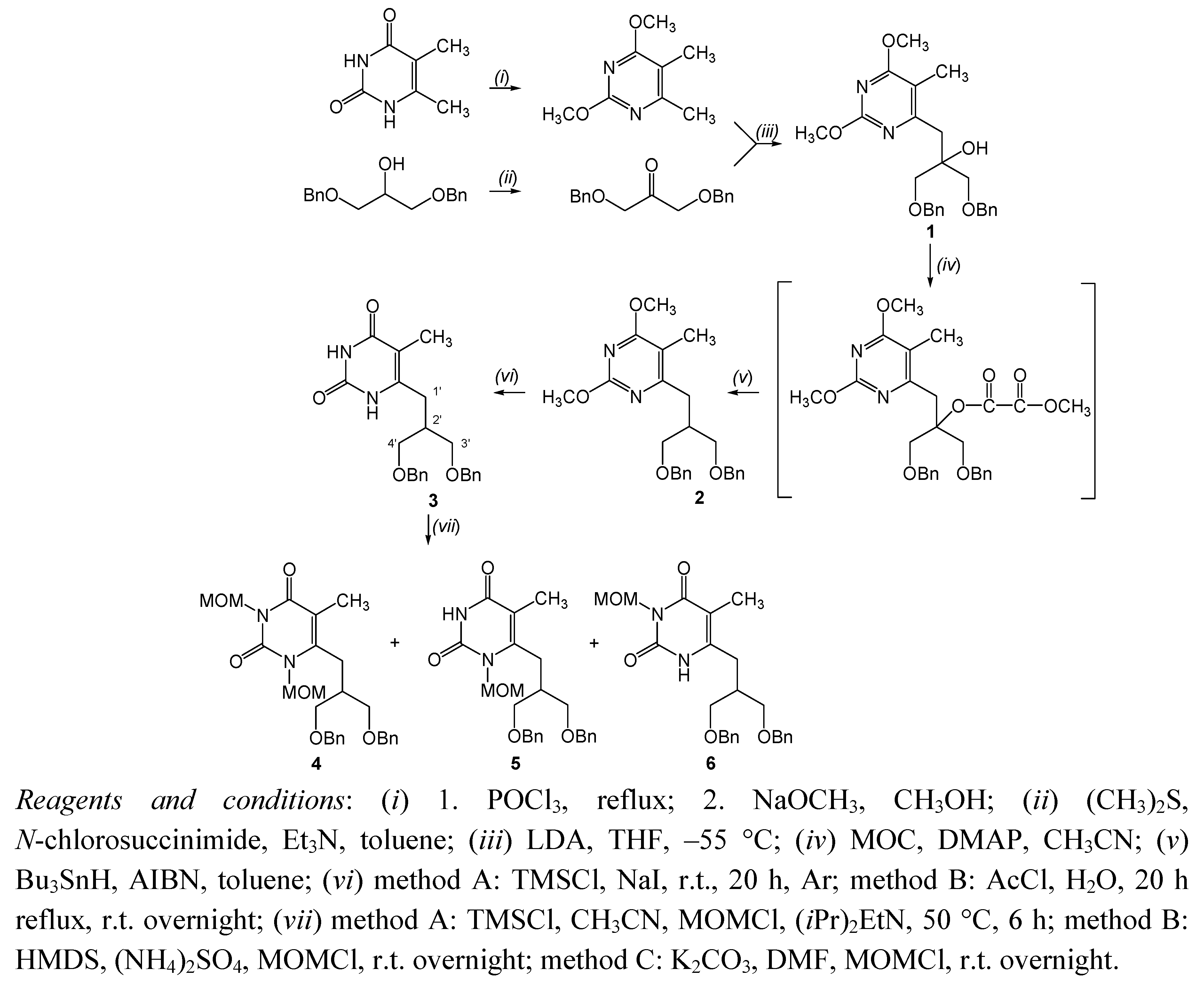{kind=link}
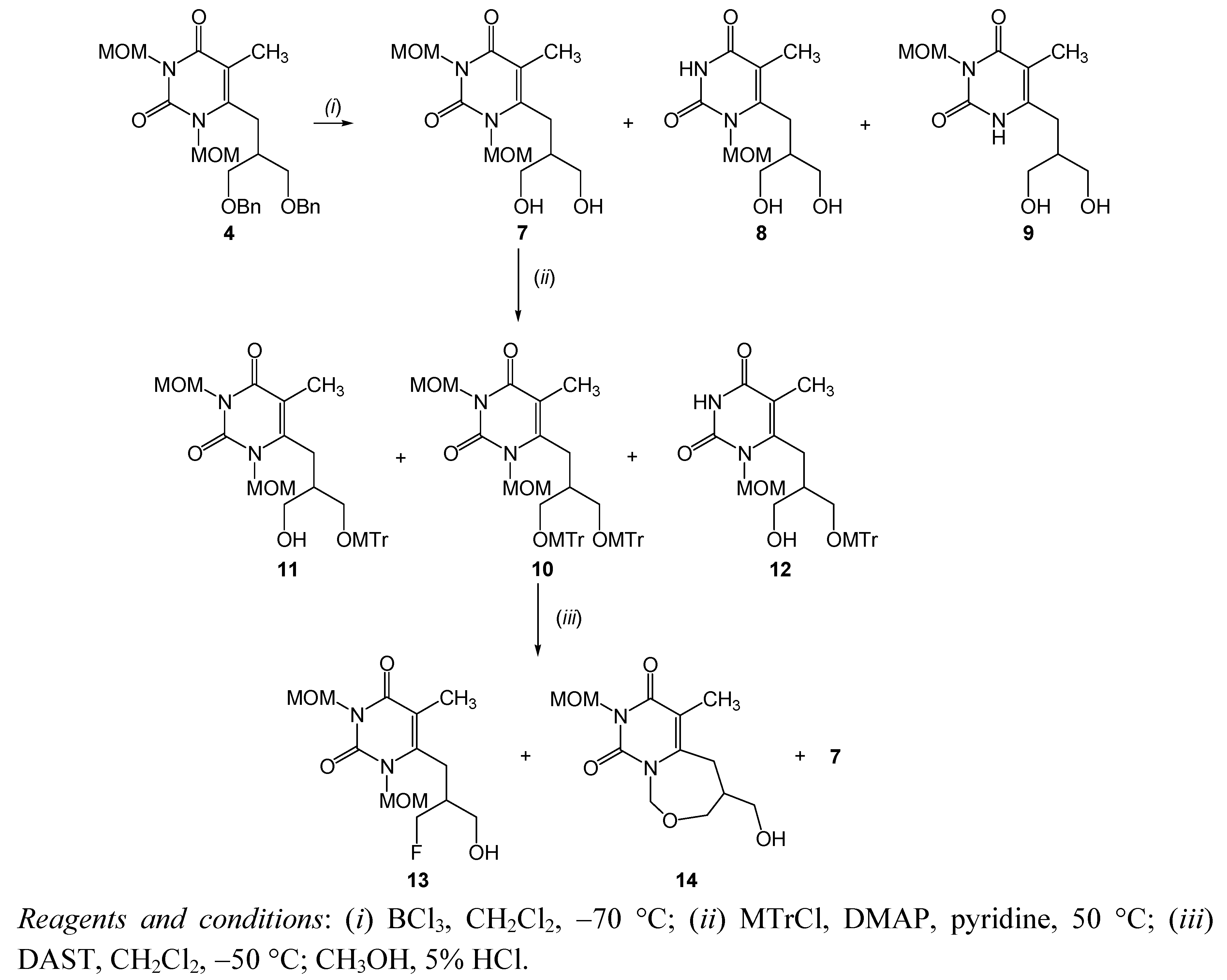{kind=link}
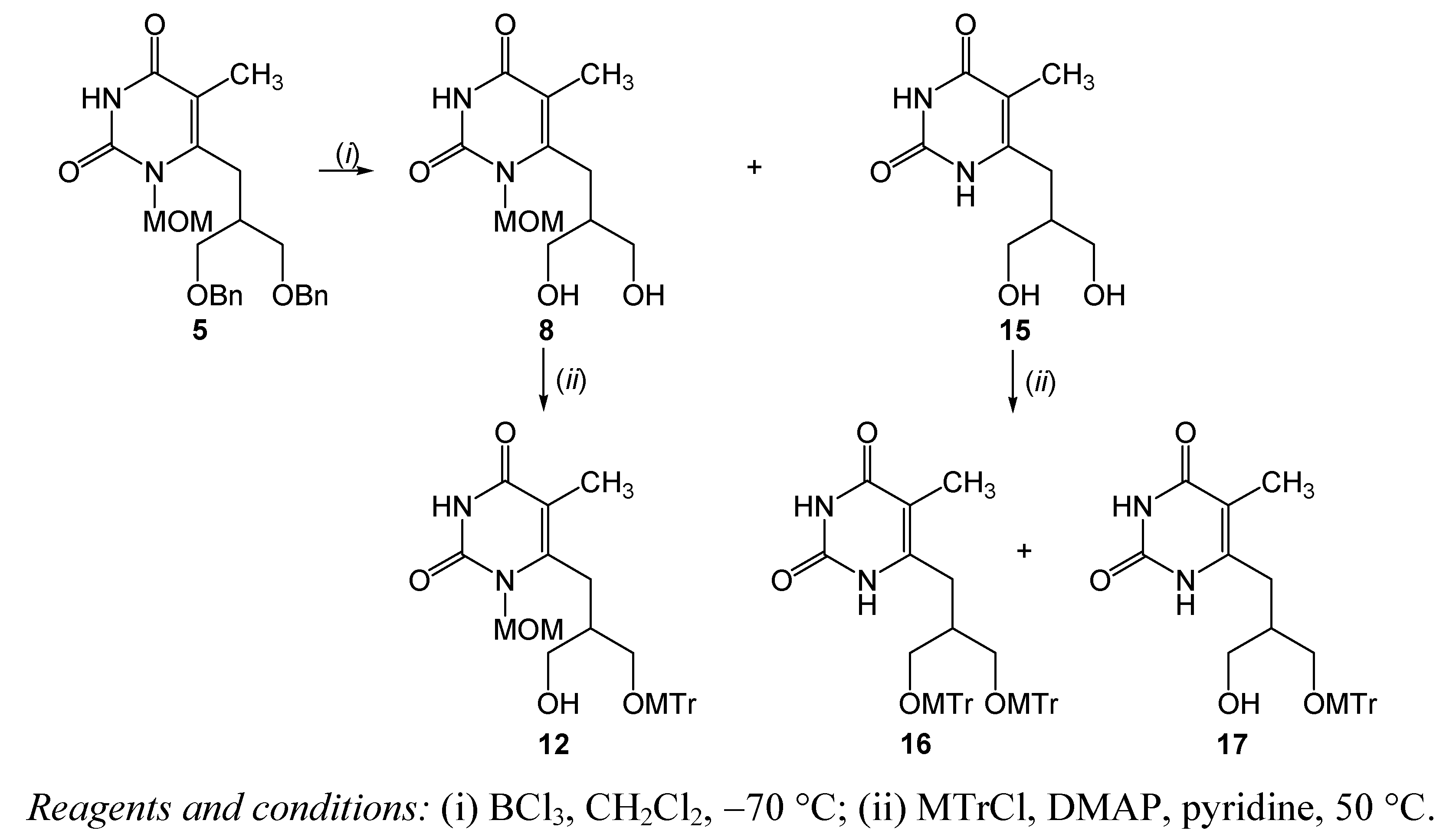{kind=link}
| Entry | Reaction | Starting compd | Reagents and conditions | Product | Yield (%) |
|---|---|---|---|---|---|
| 1 | demethoxylation | 2 | TMSCl, NaI, CH3CN, r.t., 20 h | 3 | 18.3 |
| AcCl, reflux, 20 h, H2O, r.t., overnight | 67.8 | ||||
| 2 | methoxymethylation | 3 | TMSCl, CH3CN, reflux, 2 h, ( iPr)2EtN, MOMCl, 50 °C, 6 h | 4 | 22.4 |
| 5 | 13.4 | ||||
| 6 | 11 | ||||
| HMDS, (NH4)2SO4, reflux, 1 h, MOMCl, r.t., overnight | 4 | 2.9 | |||
| 5 | 21.4 | ||||
| K2CO3, DMF, MOMCl, r.t., overnight | 4 | 28.5 | |||
| 5 | 12.1 | ||||
| 6 | 15.4 | ||||
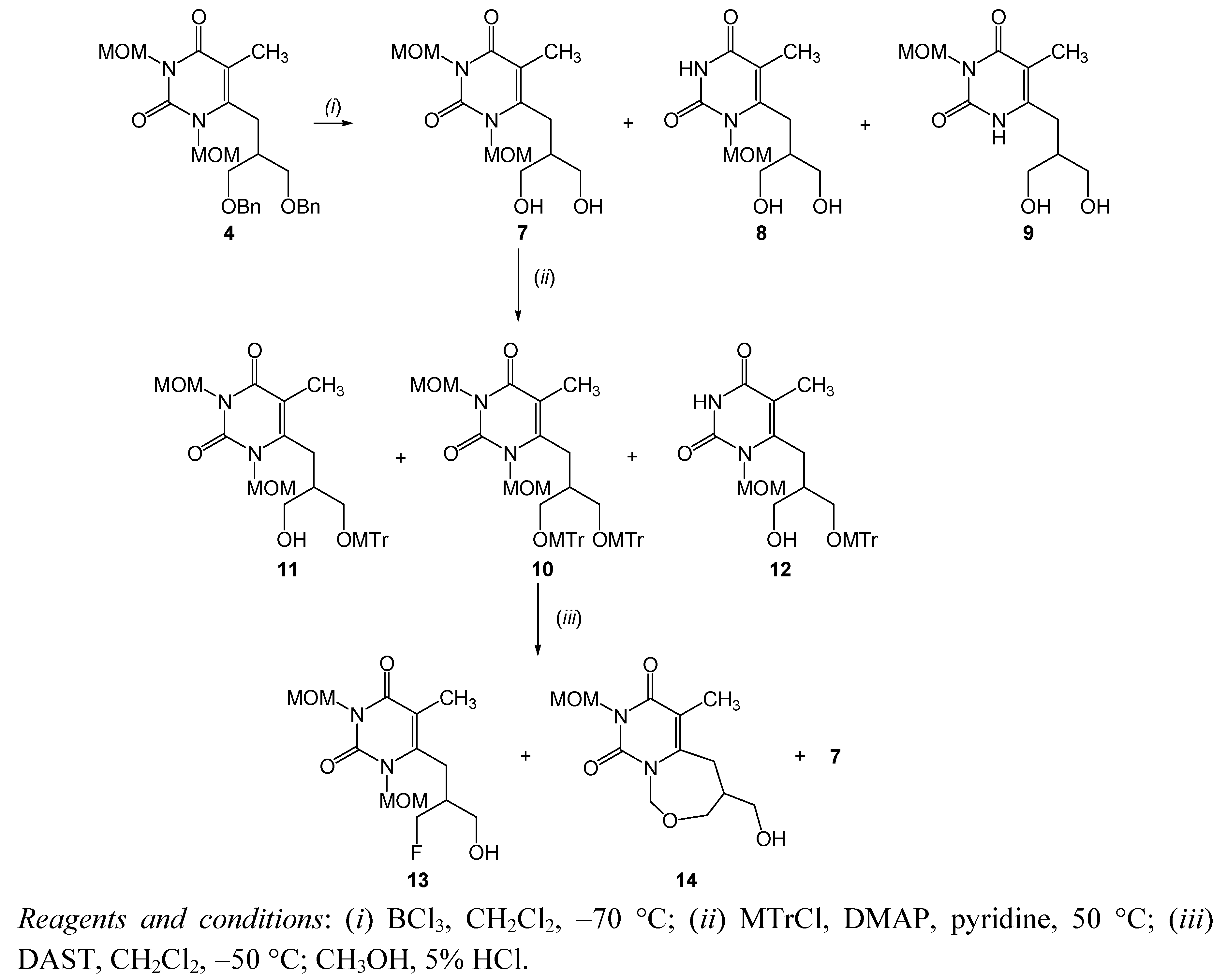
| 3 | debenzylation | 4 | BCl3, CH2Cl2, −70 °C, 1h | 7 | 33.4 |
| 8 and 9 | 24.8 | ||||
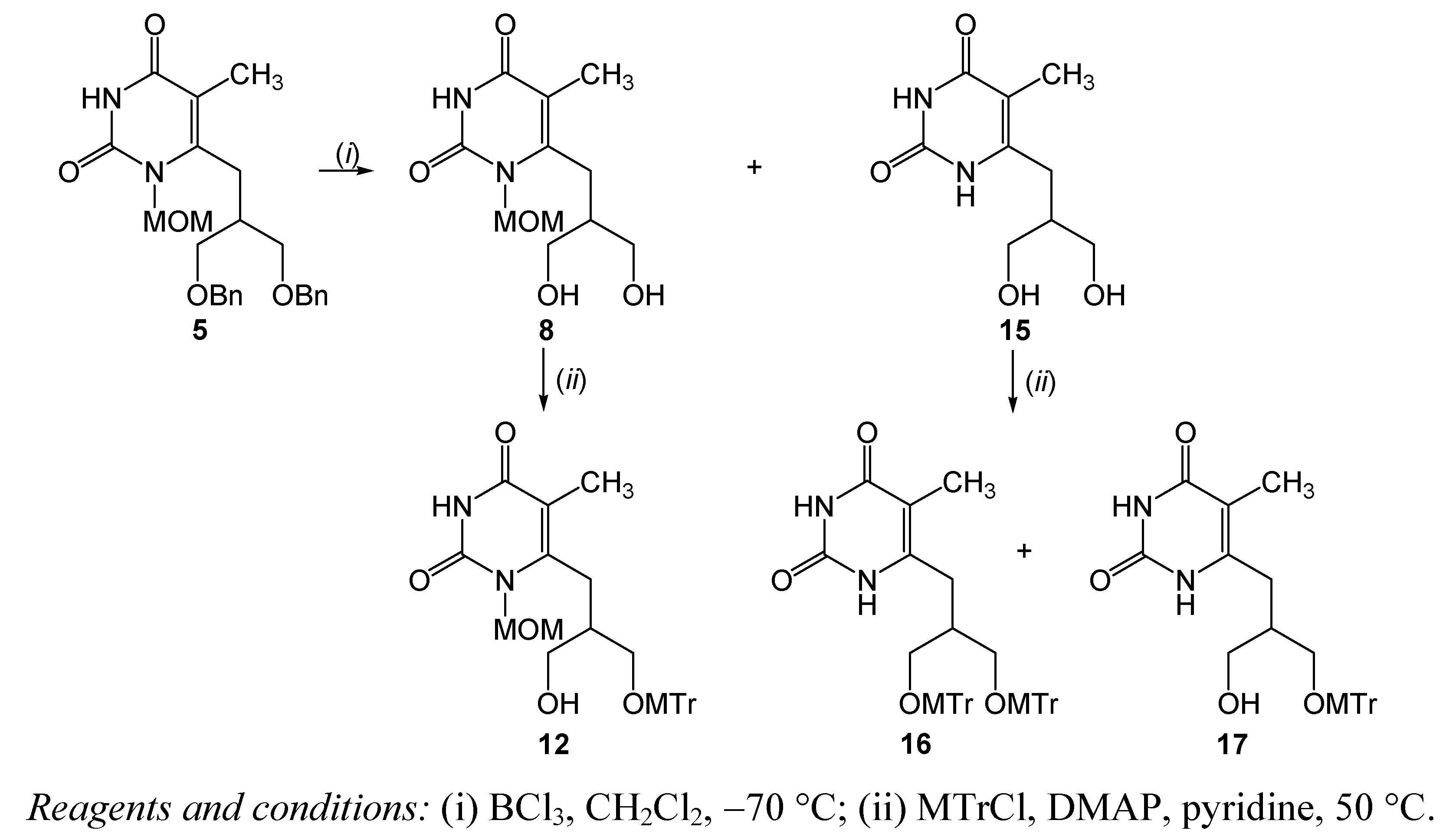
| 4 | 5 | BCl3, CH2Cl2, −70 °C, 4 h | 8 | 18.6 | |
| 15 | 60 | ||||
| BCl3, CH2Cl2, −70 °C, 2 h | 8 | 31.7 | |||
| 5 | tritylation | 7 | MTrCl, DMAP, DMF, r.t., overnight | 10 | 46.4 |
| 11 | 37.5 | ||||
| 12 | 5.5 | ||||
| 6 | 8 | 12 | 30.8 | ||
| 7 | 15 | 16 | 26.12 | ||
| 17 | 9.4 |
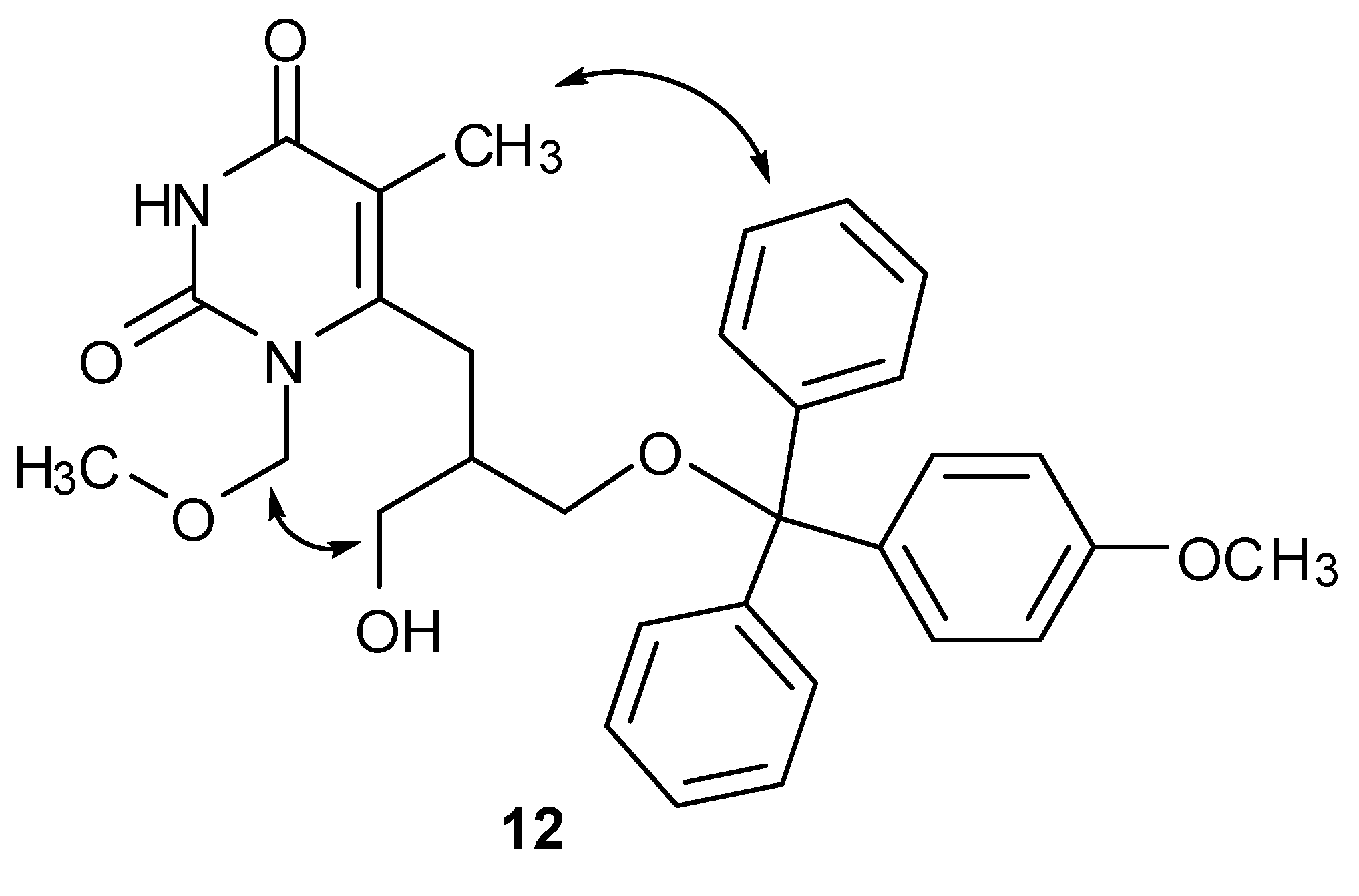
2.2. NMR Studies
3. Experimental
3.1. General
3.2. Procedures for the Preparation of Compounds
3.2.1. 6-[(3-Benzyloxy-2-benzyloxymethyl)propyl]-5-methylpyrimidin-2,4-dione (3)
3.2.2. 6-[(3-Benzyloxy-2-benzyloxymethyl)propyl]-1,3-dimethoxymethyl-5-methylpyrimidin-2,4-dione (4), 6-[(3-benzyloxy-2-benzyloxymethyl)propyl]-1-methoxymethyl-5-methylpyrimidin-2,4-dione (5) and 6-[(3-benzyloxy-2-benzyloxymethyl)propyl]-3-methoxymethyl-5-methylpyrimidin-2,4-dione (6)
3.2.3. 6-[(3-Hydroxy-2-hydroxymethyl)propyl]-1,3-dimethoxymethyl-5-methylpyrimidin-2,4-dione (7), 6-[(3-hydroxy-2-hydroxymethyl)propyl]-1-methoxymethyl-5-methylpyrimidin-2,4-dione (8) and 6-[(3-hydroxy-2-hydroxymethyl)propyl]-3-methoxymethyl-5-methylpyrimidin-2,4-dione (9)
3.2.4. 6-[2,3-Bis(4-methoxytriphenylmethoxymethyl)propyl]-1,3-dimethoxymethyl-5-methylpyrimidin-2,4-dione (10), 6-[3-hydroxy-2-(4-methoxytriphenylmethoxymethyl)propyl]-1,3-dimethoxymethyl-5-methylpyrimidin-2,4-dione (11) and 6-[3-hydroxy-2-(4-methoxytriphenylmethoxymethyl)propyl]-1-methoxymethyl-5-methylpyrimidin-2,4-dione (12)
3.2.5. 6-[(3-Fluoro-2-hydroxymethyl)propyl]-1,3-dimethoxymethyl-5-methylpyrimidin-2,4-dione (13), 6-[(3-hydroxy-2-hydroxymethyl)propyl]-1,3-dimethoxymethyl-5-methylpyrimidin-2,4-dione (7) and 4-hydroxymethyl-8-methoxymethyl-6-methyl-4,5-dihydro-pyrimido[1,6-c][1,3]oxazepine-7,9-dione (14)
3.2.6. 6-[(3-Hydroxy-2-hydroxymethyl)propyl]-1-methoxymethyl-5-methylpyrimidin-2,4-dione (8) and 6-[(3-hydroxy-2-hydroxymethyl)propyl]-5-methylpyrimidin-2,4-dione (15)
3.2.7. 6-[3-Hydroxy-2-(4-methoxytriphenylmethoxymethyl)propyl]-1-methoxymethyl-5-methyl-pyrimidin-2,4-dione (12)
3.2.8. 6-[2,3-Bis(4-methoxytriphenylmethoxymethyl)propyl]-5-methylpyrimidin-2,4-dione (16) and 6-[3-hydroxy-2-(4-methoxytriphenylmethoxymethyl)propyl]-5-methylpyrimidin-2,4-dione (17)
4. Conclusions
Acknowledgements
References and Notes
- Hurst, D.T. Introduction to the Chemistry and Biochemistry of Pyrimidines, Purines and Pteridines; John Wiley and Sons: New York, NY, USA, 1980. [Google Scholar]
- Choudhury, A.; Chen, H.; Nilsen, C.N.; Sorgi, K.L. Microwave-assisted synthesis of pyrazolo[3,4-d]pyrimidines from 2-amino-4,6-dichloropyrimidine-5-carbaldehyde under solvent-free conditions. Tetrahedron Lett. 2008, 49, 102–105. [Google Scholar] [CrossRef]
- Braendvang, M.; Gundersen, L.L. Efficient and regioselective N-1 alkylation of 4-chloropyrazolo[3,4-d]pyrimidine. Tetrahedron Lett. 2007, 48, 3057–3059. [Google Scholar] [CrossRef]
- Peng, Z.-H.; Journet, M.; Humphrey, G. A highly regioselective amination of 6-aryl-2,4-dichloropyrimidine. Org. Lett. 2006, 18, 395–398. [Google Scholar]
- Girreser, U.; Heber, D.; Schütt, M. Synthesis of 6-substituted 7-aryl-5,6-dihydropyrido[2,3-d]pyrimidine(1H,3H)-2,4-diones using the Vilsmeier reaction. Tetrahedron 2004, 60, 11511–11517. [Google Scholar] [CrossRef]
- Mishra, R.; Tomar, I. Pyrimidine: the molecule of diverse biological and medicinal importance. Int. J. Pharm. Sci. Res. 2011, 2, 758–771. [Google Scholar]
- Rani, U.; Oturak, H.; Sudha, S.; Sundaraganesan, N. Molecular structure, harmonic and anharmonic frequency calculations of 2,4-dichloropyrimidine and 4,6-dichloropyrimidine by HF and density functional methods. Spectrochim. Acta A 2011, 78, 1467–1475. [Google Scholar] [CrossRef]
- Boncel, S.; Gondela, A.; Walczak, K. Uracil as a target for nucleophilic and electrophilic reagents. Curr. Org. Synth. 2008, 5, 365–396. [Google Scholar] [CrossRef]
- Larsen, J.S.; Pedersen, E.B.; Nielsen, C. Synthesis of N-1-alkylated 6-benzyluracil-5-carboxylic esters as potential non-nucleoside reverse transcriptase inhibitors. Synthesis 2004, 11, 1874–1878. [Google Scholar]
- Romero, D.L.; Olmsted, R.A.; Poel, T.J.; Morge, R.A.; Biles, C.; Keiser, B.J.; Kopta, L.A.; Friis, J.M.; Hosley, J.D. Targeting delavirdine/atevirdine resistant HIV-1: Identification of (alkylamino)piperidine-containing bis(heteroaryl)piperazines as broad spectrum HIV-1 reverse transcriptase inhibitors. J. Med. Chem. 1996, 39, 3769–3789. [Google Scholar] [CrossRef]
- Sudbeck, E.A.; Mao, C.; Vig, R.; Venkatachalam, T.K.; Tuel-Ahlgren, L.; Uckun, F.M. Structure-based design of novel dihydroalkoxybenzyloxopyrimidine derivatives as potent nonnucleoside inhibitors of the human immunodeficiency virus reverse transcriptase. Antimicrob. Agents Chemother. 1998, 4, 3225–3233. [Google Scholar]
- Loksha, Y.M.; Globisch, D.; Loddo, R.; Collu, G.; La Colla, P.; Pedersen, E.B. A novel synthetic route for the anti-HIV drug MC-1220 and its analogues. ChemMedChem 2010, 5, 1847–1849. [Google Scholar] [CrossRef] [Green Version]
- Medina-Franco, J.L.; Martínez-Mayorga, K.; Juárez-Gordiano, C.; Castillo, R. Pyridin-2(1H)-ones: A promising class of HIV-1 non-nucleoside reverse transcriptase inhibitors. ChemMedChem 2007, 2, 1141–1147. [Google Scholar] [CrossRef]
- Dolle, V.; Aubertin, A.M.; Ludwig, O.; Nguyen, C.H.; Bisagni, E.; Legraverend, M. Synthesis of new non-nucleoside inhibitors of HIV-1. Bioorg. Med. Chem. Lett. 1996, 6, 173–178. [Google Scholar] [CrossRef]
- Kulikowski, T. Structure-activity relationships and conformational features of antiherpetic pyrimidine and purine nucleoside analogues. Pharm. World Sci. 1994, 16, 127–138. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Grill, S.P.; Dutschman, G.E.; Nakayama, K.; Bastow, K.F. Metabolism of 9-(1,3-dihydroxy-2-propoxymethyl)guanine, a new anti-herpes virus compound, in herpes simplex virus-infected cells. J. Biol. Chem. 1983, 258, 12460–12464. [Google Scholar]
- Germershausen, J.; Bostedor, R.; Field, A.K.; Perry, H.; Liou, R.; Bull, H.; Tolman, R.L.; Karkas, J.D. A comparison of the antiviral agents 2'-nor-2'-deoxyguanosine and acyclovir: Uptake and phosphorylation in tissue culture and kinetics of in vitro inhibition of viral and cellular DNA polymerases by their respective triphosphates. Biochem. Biophys. Res. Commun. 1983, 116, 360–367. [Google Scholar] [CrossRef]
- Martin, J.C.; McGee, D.P.C.; Jeffrey, G.A.; Hobbs, D.W.; Smee, D.F.; Matthews, T.R.; Verheyden, J.P. Synthesis and anti-herpes-virus activity of acyclic 2'-deoxyguanosine analogues related to 9-[(1,3-dihydroxy-2-propoxy)methyl]guanine. J. Med. Chem. 1986, 29, 1384–1389. [Google Scholar] [CrossRef]
- Degrève, B.; Andrei, G.; Izquierdo, M.; Piette, J.; Morin, K.; Knaus, E.E.; Wiebe, L.I.; Basrah, I.; Walker, R.T.; De Celrcq, E.; Balzarini, J. Varicella-zoster virus thymidine kinase gene and antiherpetic pyrimidine nucleoside analogues in a combined gene/chemotherapy treatment for cancer. Gene Ther. 1997, 4, 1107–1114. [Google Scholar]
- Grignet-Debrus, C.; Calberg-Bacq, C.-M. Potential of Varicella zoster virus thymidine kinase as a suicide gene in breast cancer cells. Gene Ther. 1997, 4, 560–569. [Google Scholar]
- Culver, K.W.; Van Gilder, J.; Link, C.J.; Carlstrom, T.; Buroker, T.; Yuh, W.; Koch, K.; Schabold, K.; Doombas, S.; Wetjen, B. Gene therapy for the treatment of malignant brain tumors with in vivo tumor transduction with the herpes simplex thymidine kinase gene/ganciclovir system. Hum. Gene Ther. 1994, 5, 343–379. [Google Scholar] [CrossRef]
- Alauddin, M.M.; Conti, P.S. Synthesis and preliminary evaluation of 9-(4-[18F]fluoro-3-hydroxymethylbutyl)guanine ([18F]FHBG): A new potential imaging agent for viral infection and gene therapy using PET. Nucl. Med. Biol. 1998, 25, 175–180. [Google Scholar] [CrossRef]
- de Vries, E.F.J.; van Waarde, A.; Harmsen, M.C.; Mulder, N.H.; Vaalburg, W.; Hospers, G.A.P. [(11)C]FMAU and [(18)F]FHPG as PET tracers for herpes simplex virus thymidine kinase enzyme activity and human cytomegalovirus infections. Nucl. Med. Biol. 2000, 27, 113–119. [Google Scholar] [CrossRef]
- Hospers, G.A.K.; Calogero, A.; van Waarde, A.; Doze, P.; Vaalburg, W.; Mulder, N.H.; de Vries, E.F. Monitoring of herpes simplex virus thymidine kinase enzyme activity using positron emission tomography. Cancer Res. 2000, 60, 1488–1491. [Google Scholar]
- MacLaren, D.C.; Toyokuni, T.; Cherry, S.R.; Barrio, J.R.; Phelps, M.E.; Herschman, H.R.; Gambhir, S.S. PET imaging of transgene expression. Biol. Psychiat. 2000, 48, 337–348. [Google Scholar] [CrossRef]
- Baba, M.; De Clercq, E.; Tanaka, H.; Ubasawa, M.; Takashima, H.; Sekiya, K.; Nitta, I.; Umezu, K.; Walker, R.T.; Mori, S. Highly potent and selective inhibition of human immunodeficiency virus type 1 by a novel series of 6-substituted acyclouridine derivatives. Mol. Pharmacol. 1991, 39, 805–810. [Google Scholar]
- Balzarini, J.; Karlsson, A.; De Clercq, E. Human immunodeficiency virus type 1 drug-resistance patterns with different 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine derivatives. Mol. Pharmacol. 1993, 44, 694–701. [Google Scholar]
- Mai, A.; Artico, M.; Sbardella, G.; Quartarone, S.; Massa, S.; Loi, A.G.; De Montis, A.; Scintu, F.; Putzolu, M.; La Colla, P. Dihydro(alkylthio) (naphthylmethyl)oxopyrimidines: Novel non-nucleoside reverse transcriptase inhibitors of the S-DABO series. J. Med. Chem. 1997, 40, 1447–1454. [Google Scholar] [CrossRef]
- Kundu, N.G.; Das, P.; Balzarini, J.; De Clercq, E. Palladium-catalyzed synthesis of [E]-6-(2-acylvinyl)uracils and [E]-6-(2-acylvinyl)-1-[(2-hydroxyethoxy)methyl]uracils-their antiviral and cytotoxic activities. Bioorg. Med. Chem. 1997, 5, 2011–2018. [Google Scholar] [CrossRef]
- Gazivoda Kraljević, T.; Krištafor, S.; Šuman, L.; Kralj, M.; Ametamey, S.M.; Cetina, M.; Raić-Malić, S. Synthesis, X-ray crystal structure study and antitumoral evaluations of 5,6-disubstituted pyrimidine derivatives. Bioorg. Med. Chem. 2010, 18, 2704–2712. [Google Scholar]
- Prekupec, S.; Makuc, D.; Plavec, J.; Šuman, L.; Kralj, M.; Pavelić, K.; Balzarini, J.; De Clercq, E.; Mintas, M.; Raić-Malić, S. Novel C-6 fluorinated acyclic side chain pyrimidine derivatives: Synthesis, H-1 and C-13 NMR conformational studies, and antiviral and cytostatic evaluations. J. Med. Chem. 2007, 50, 3037–3045. [Google Scholar] [CrossRef]
- Krištafor, S.; Gazivoda Kraljević, T.; Makuc, D.; Plavec, J.; Šuman, L.; Kralj, M.; Raić-Malić, S. Synthesis, structural studies and antitumoral evaluation of C-6 alkyl and alkenyl side chain pyrimidine derivatives. Molecules 2009, 14, 4866–4879. [Google Scholar] [CrossRef]
- Prekupec, S.; Makuc, D.; Plavec, J.; Kraljević, S.; Kralj, M.; Pavelić, K.; Andrei, G.; Snoeck, R.; Balzarini, J.; De Clercq, E.; Raić-Malić, S.; Mintas, M. Antiviral and cytostatic evaluation of the novel 6-acyclic chain substituted thymine derivatives. Antivir. Chem. Chemother. 2005, 16, 327–338. [Google Scholar]
- Raić-Malić, S.; Johayem, A.; Ametamey, S.M.; Batinac, S.; De Clercq, E.; Folkers, G.; Scapozza, L. Synthesis, 18F-radiolabelling and biological evaluations of C-6 alkylated pyrimidine nucleoside analogues. Nucleos. Nucleot. Nucleic Acids 2004, 23, 1707–1721. [Google Scholar] [CrossRef]
- Johayem, A.; Raić-Malić, S.; Lazzati, K.; Schubiger, P.A.; Scapozza, L.; Ametamey, S.M. Synthesis and characterization of a C(6) nucleoside analogue for the in vivo imaging of the gene expression of Herpes Simplex Virus type 1 Thymidine Kinase (HSV1 TK). Chem. Biodiv. 2006, 3, 274–283. [Google Scholar] [CrossRef]
- Raić-Malić, S.; Gazivoda, T.; Krištafor, S.; Ametamey, S.M. New 6-substituted pyrimidine derivatives. WO 2011/036505, 31 May 2011. [Google Scholar]
- Martic, M.; Pernot, L.; Westermaier, Y.; Perozzo, R.; Gazivoda Kraljevic, T.; Kristafor, S.; Raic-Malic, S.; Scapozza, L.; Ametamey, S. Synthesis, crystal structure and in vitro biological evaluation of C-6 pyrimidine derivatives: New lead structures for monitoring gene expression in vivo. Nucleos. Nucleot. Nucleic Acids 2011. In Press.. [Google Scholar]
- Krištafor, S.; Gazivoda Kraljević, T.; Ametamey, S.M.; Cetina, M.; Ratkaj, I.; Tandara Haček, R.; Kraljević Pavelić, S.; Raić-Malić, S. Syntheses and antitumor evaluation of C(6) isobutyl and isobutenyl substituted pyrimidines and dihydropyrrolo[1,2-c]pyrimidin-1,3-diones. Chem. Biodiv. 2011. In Press. [Google Scholar] [CrossRef]
- Hsu, L.Y.; Wise, D.S.; Kucera, L.S.; Drach, J.C.; Townsend, L.B. Synthesis of anti-restricted pyrimidine acyclic nucleosides. J. Org. Chem. 1992, 57, 3354–3358. [Google Scholar] [CrossRef]
- Muschalek, B.; Weidner, I.; Butenschön, H. Synthesis of tricarbonyl(N-methylisatin)chromium(0) and an unanticipated transformation of a N-MEM to a N-MOM group. J. Organomet. Chem. 2007, 692, 2415–2424. [Google Scholar] [CrossRef]
- Zhang, F.; Kulesza, A.; Rani, S.; Bernet, B.; Vasella, A. 6-(Diazomethyl)-1,3-bis(methoxymethyl)uracil, synthesis and transformation into annulated pyrimidinediones. Helv. Chim. Acta 2008, 91, 1201–1218. [Google Scholar] [CrossRef]
- Edstrom, E.D.; Feng, X.; Tumkevicius, S. Development of methylthiomethyl (MTM) protection for N1 of pyrrolo[2,3-d]pyrimidin-2,4-diones. Tetrahedron Lett. 1996, 37, 759–762. [Google Scholar] [CrossRef]
- Su, T.-L.; Huang, J.-T.; Burchenal, J.H.; Watanabe, K.A.; Fox, J.J. Synthesis and biological activities of 5-deaza analogues of aminopterin and folic acid. J. Med. Chem. 1986, 29, 709–715. [Google Scholar] [CrossRef]
- Heintzelman, G.R.; Fang, W.-K.; Keen, S.P.; Wallace, G.A.; Weinreb, S.M. Stereoselective total synthesis of the cyanobacterial hepatotoxin 7-Epicylindrospermopsin: revision of the stereochemistry of cylindrospermopsin. J. Am. Chem. Soc. 2001, 123, 8851–8853. [Google Scholar] [CrossRef]
- Batinac, S.; Mrvoš Sermek, D.; Cetina, M.; Pavelić, K.; Mintas, M.; Raić-Malić, S. Synthesis of the novel bicyclic oxepinopyrimidine and fluorinated pyrrolidinopyrimidines. Heterocycles 2004, 63, 2523–2536. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1−17 are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kraljević, T.G.; Petrović, M.; Krištafor, S.; Makuc, D.; Plavec, J.; Ross, T.L.; Ametamey, S.M.; Raić-Malić, S. Methoxymethyl (MOM) Group Nitrogen Protection of Pyrimidines Bearing C-6 Acyclic Side-Chains. Molecules 2011, 16, 5113-5129. https://doi.org/10.3390/molecules16065113
Kraljević TG, Petrović M, Krištafor S, Makuc D, Plavec J, Ross TL, Ametamey SM, Raić-Malić S. Methoxymethyl (MOM) Group Nitrogen Protection of Pyrimidines Bearing C-6 Acyclic Side-Chains. Molecules. 2011; 16(6):5113-5129. https://doi.org/10.3390/molecules16065113
Chicago/Turabian StyleKraljević, Tatjana Gazivoda, Martina Petrović, Svjetlana Krištafor, Damjan Makuc, Janez Plavec, Tobias L. Ross, Simon M. Ametamey, and Silvana Raić-Malić. 2011. "Methoxymethyl (MOM) Group Nitrogen Protection of Pyrimidines Bearing C-6 Acyclic Side-Chains" Molecules 16, no. 6: 5113-5129. https://doi.org/10.3390/molecules16065113