Microwave Assisted Synthesis of Some New Fused 1,2,4-Triazines Bearing Thiophene Moieties With Expected Pharmacological Activity


Abstract
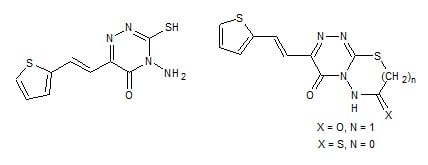
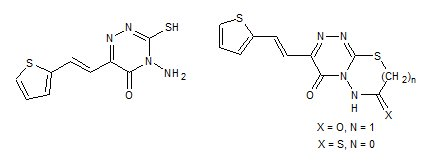
:
1. Introduction
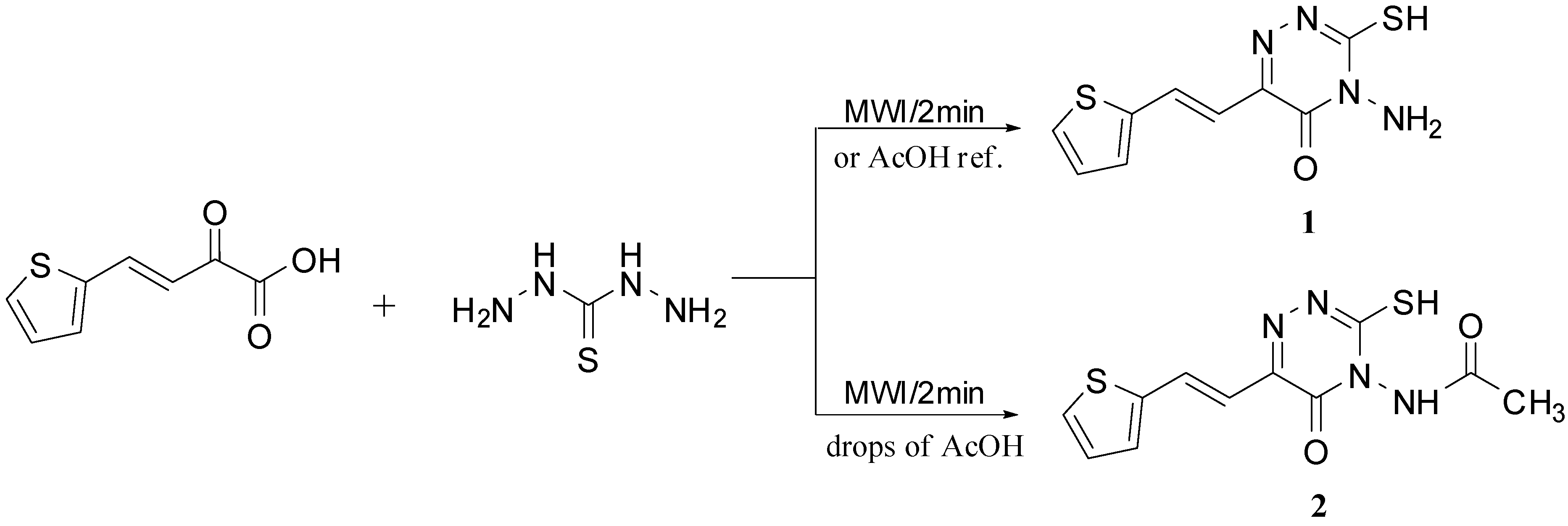
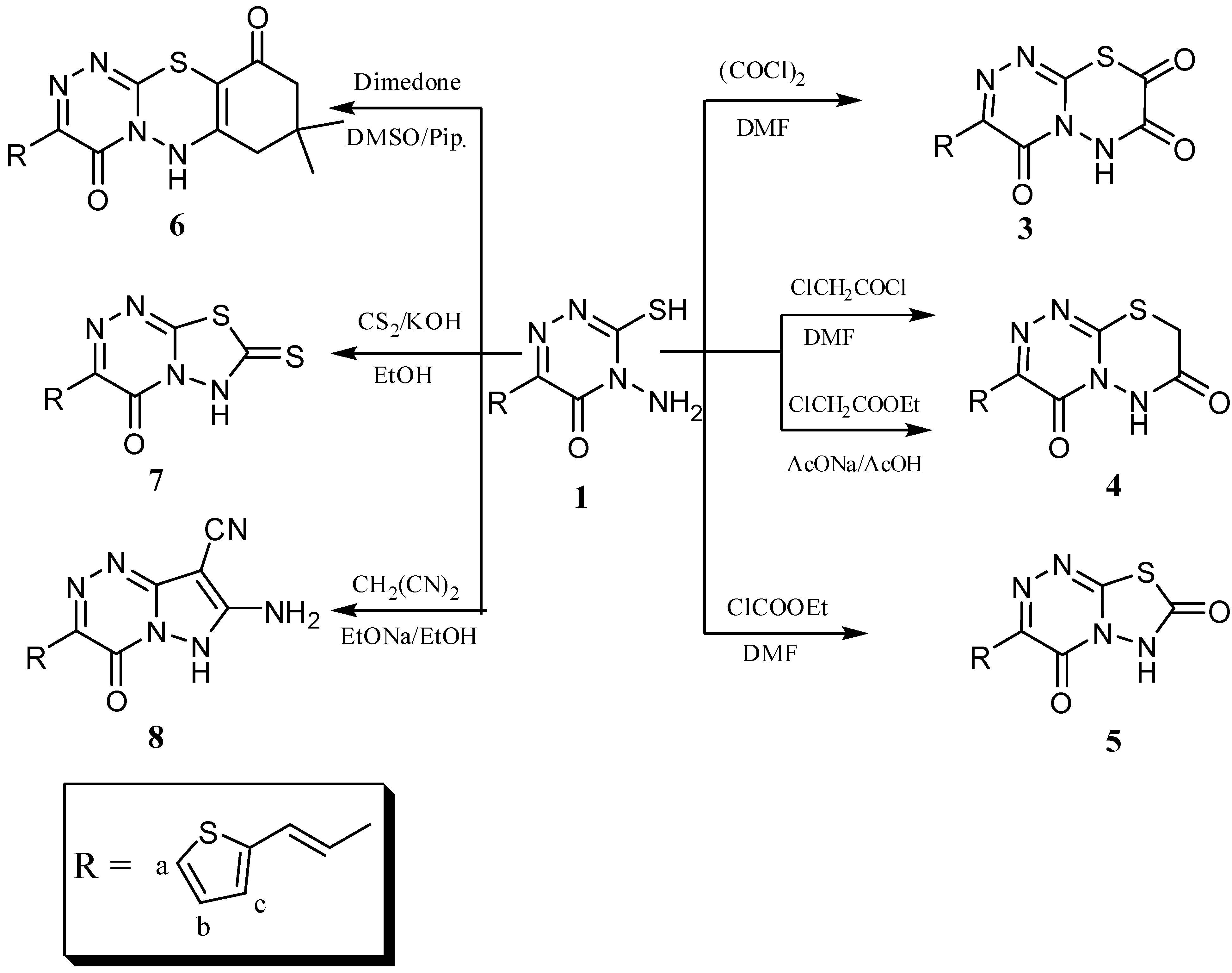
2. Results and Discussion



3. Pharmacological Studies
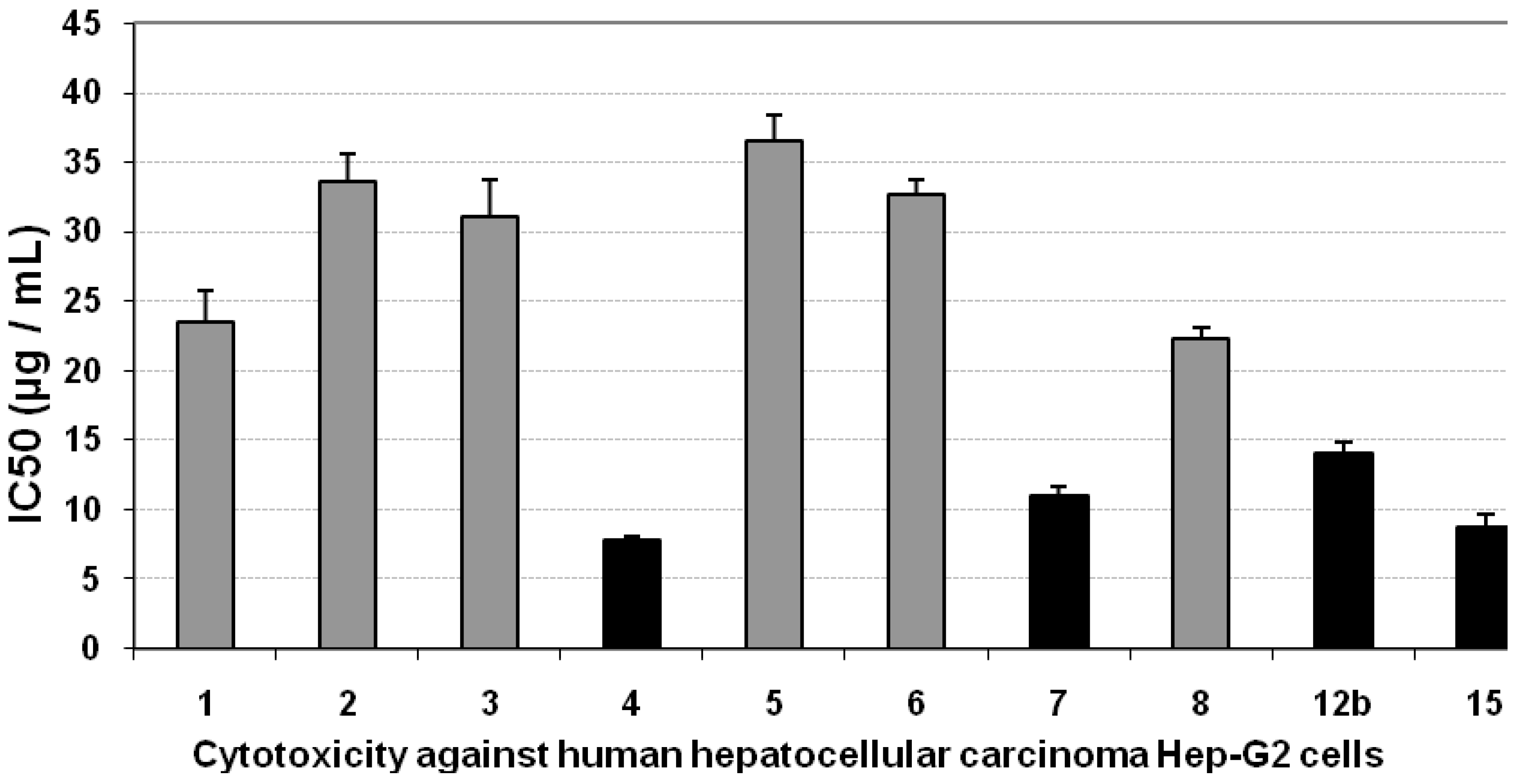
3.1. Cytotoxicity of the Compounds against Hep-G2 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
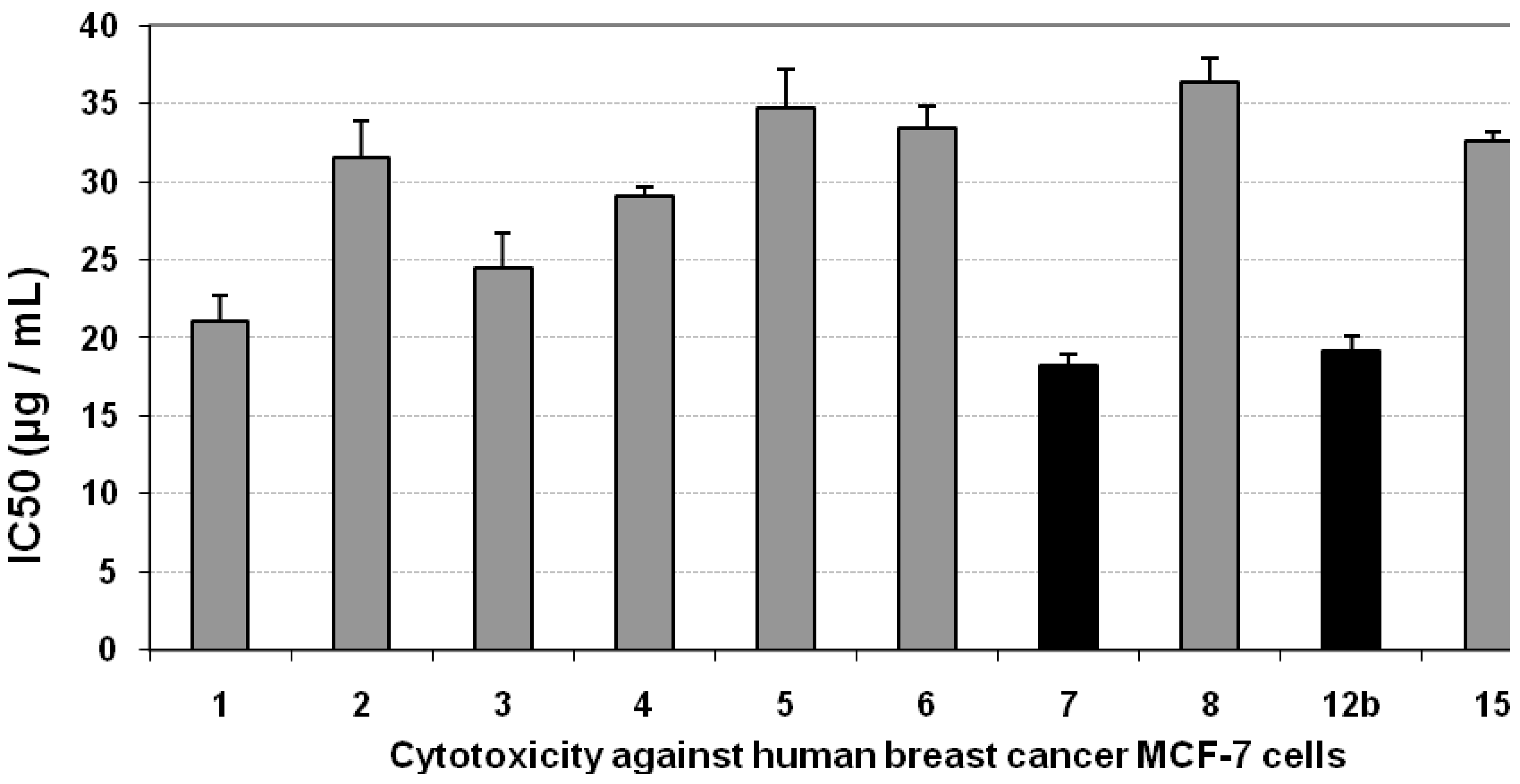
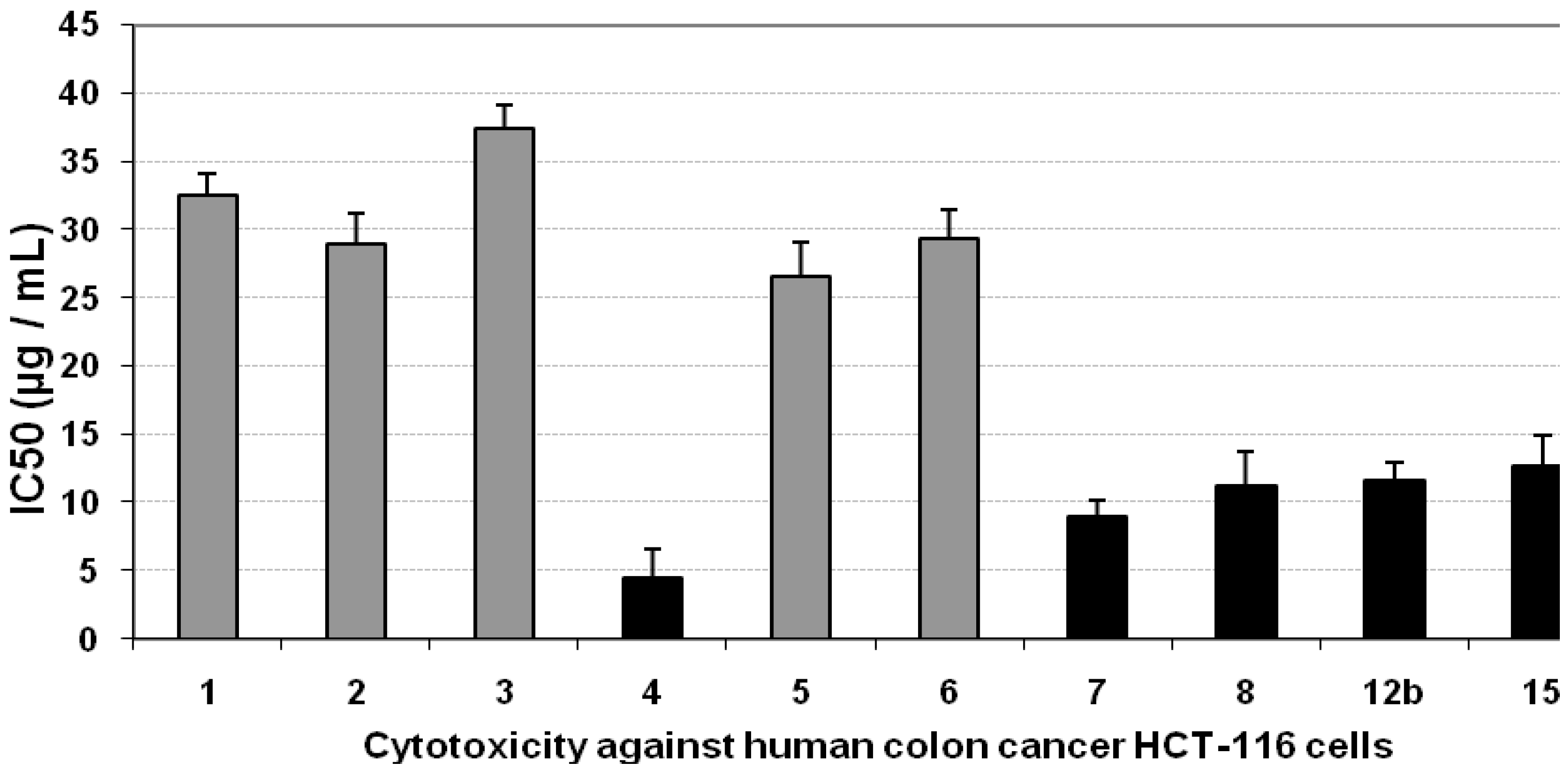
| Compd. no. | Mean IC50 (µg/mL) | SE | ||||
|---|---|---|---|---|---|---|
| Hep-G2 cells | MCF-7 cells | HCT-116 cells | Hep-G2 cells | MCF-7 cells | HCT-116 cells | |
| 1 | 23.51 | 21.04 | 32.6 | 2.25 | 1.62 | 1.45 |
| 2 | 33.6 | 31.56 | 29.01 | 2.00 | 2.32 | 2.18 |
| 3 | 31.11 | 24.5 | 37.4 | 2.58 | 2.15 | 1.69 |
| 4 | 7.76 | 29.11 | 4.52 | 0.31 | 0.54 | 2.01 |
| 5 | 36.5 | 34.73 | 26.65 | 1.84 | 2.52 | 2.40 |
| 6 | 32.6 | 33.4 | 29.4 | 1.13 | 1.45 | 2.11 |
| 7 | 11.03 | 18.2 | 8.93 | 0.62 | 0.76 | 1.26 |
| 11 | 22.34 | 36.41 | 11.2 | 0.77 | 1.54 | 2.51 |
| 12b | 14.05 | 19.16 | 11.61 | 0.80 | 0.97 | 1.32 |
| 15 | 8.79 | 32.54 | 12.67 | 0.87 | 0.61 | 2.25 |

3.2. Cytotoxicity of the Compounds against MCF-7 Cells

3.3. Cytotoxicity of the Compounds against HCT-116 Cells

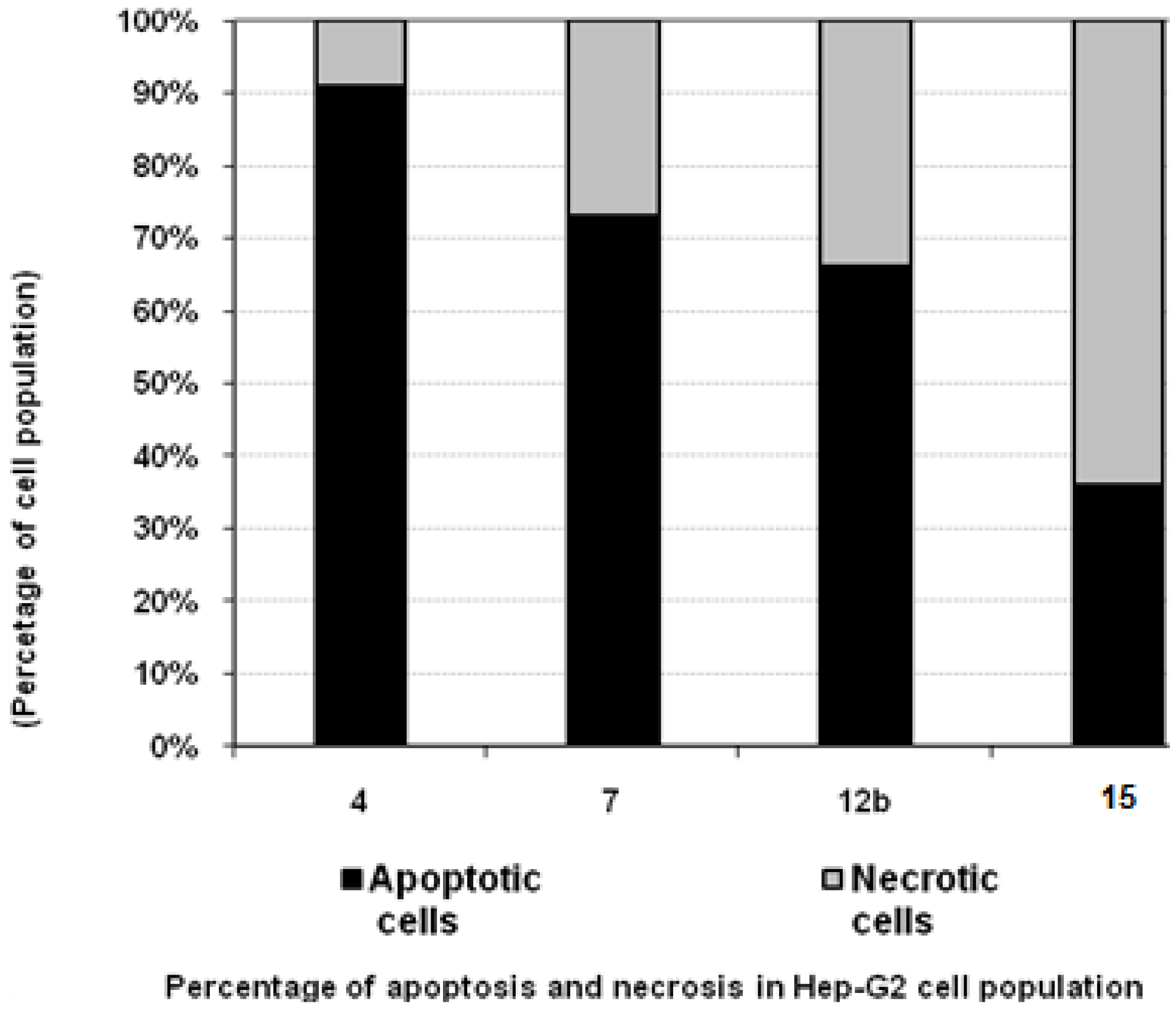
3.4. Percentage of Induced Apoptotic and Necrotic Cells in Hep-G2 Cells

| Compd. No. | Hep-G2 | |
|---|---|---|
| Apoptotic cells | Necrotic cells | |
| 4 | 91 | 9 |
| 7 | 73 | 27 |
| 12b | 66 | 34 |
| 15 | 36 | 64 |
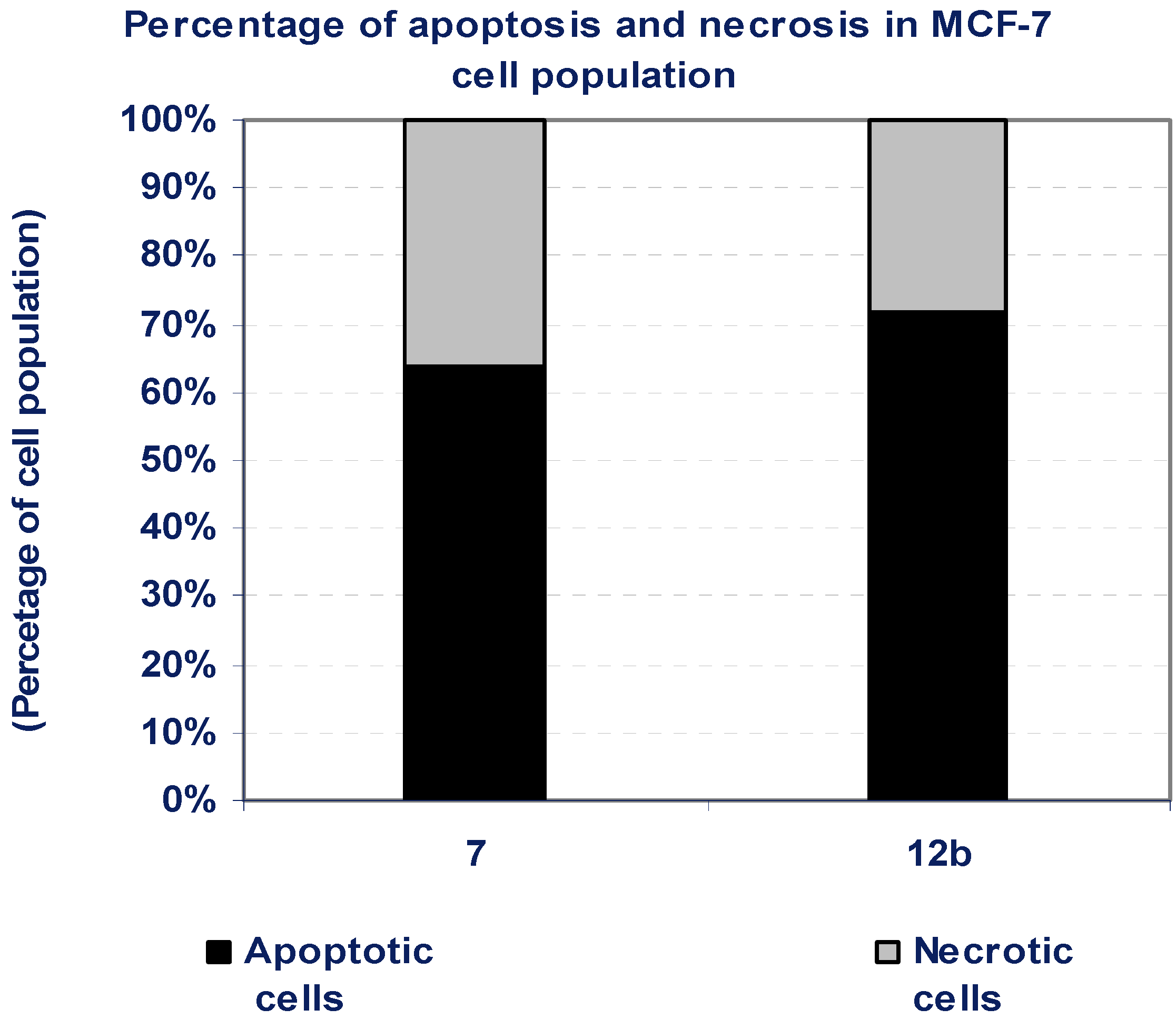
3.5. Percentage of Induced Apoptotic and Necrotic Cells in MCF-7 Cells

| Compd. No. | MCF-7 | |
|---|---|---|
| Apoptotic cells | Necrotic cells | |
| 7 | 64 | 36 |
| 12b | 72 | 28 |
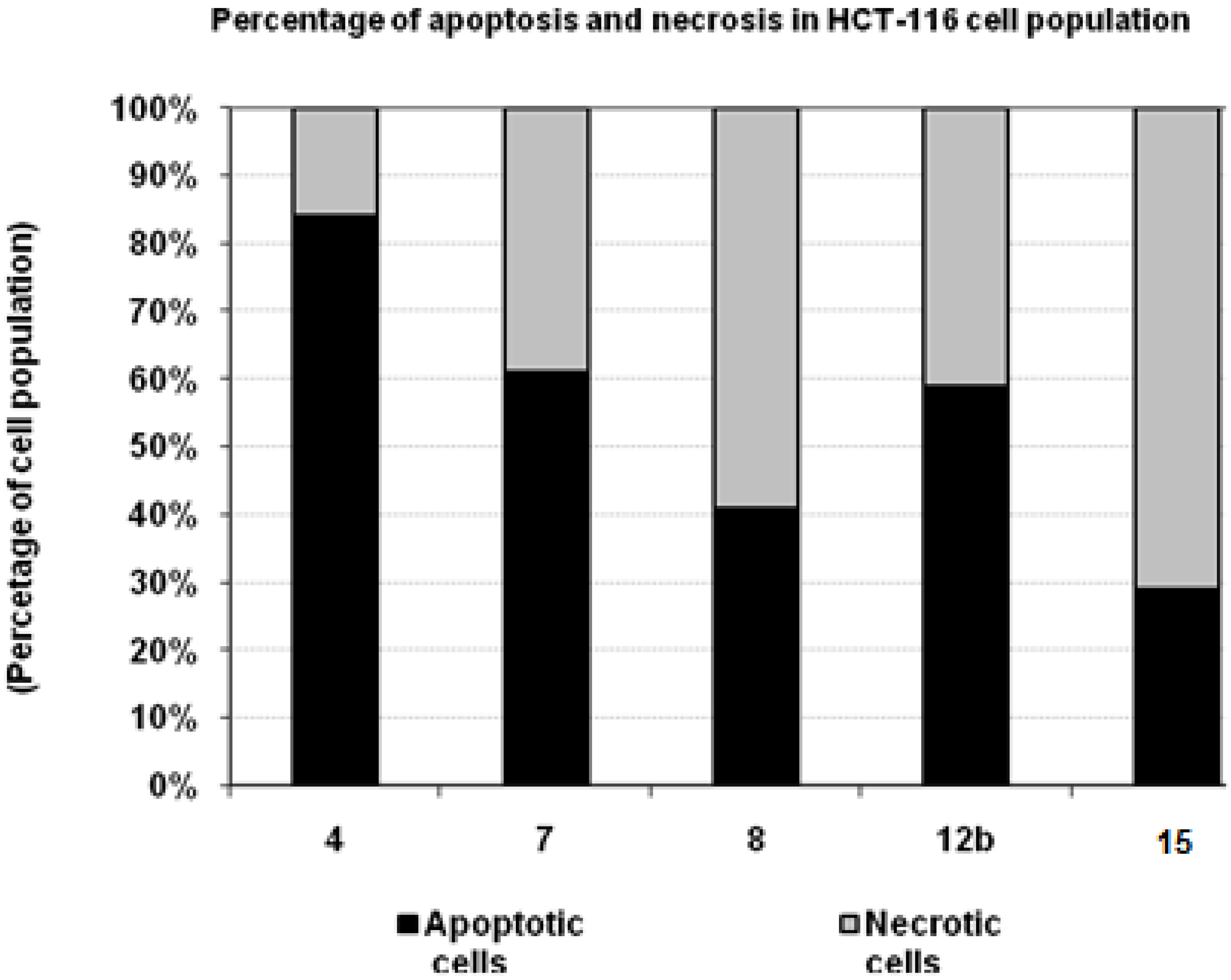
3.6. Percentage of Induced Apoptotic and Necrotic Cells in HCT-116 Cells

| Compd. No. | HCT-116 | |
|---|---|---|
| Apoptotic cells | Necrotic cells | |
| 4 | 84 | 16 |
| 7 | 61 | 39 |
| 11 | 41 | 59 |
| 12b | 59 | 41 |
| 15 | 29 | 71 |
3.6. Material and Methods
3.6.1. Cell Culture
3.6.2. Cytotoxicity Assay
3.6.2.1. Reagent Preparation
3.6.2.2. Procedure
3.6.2.3. Calculations
3.6.3. Apoptosis and Necrosis Staining
4. Conclusions
5. Experimental
5.1. General
5.2. General Procedure for the Synthesis of Compounds 3, 4 and 5
5.3. General Procedure for Preparation of 9 and 10
5.4. General Procedure for Preparation of 12a and 12b
Acknowledgments
References
- Chow, W.S.; Chan, T.H. Microwave-assisted solvent-free N-arylation of imidazole and pyrazole. Tetrahedron Lett. 2009, 50, 1286–1289. [Google Scholar] [CrossRef]
- Abdul Rauf, S.S.; Gangal, S. Microwave assisted efficient one-pot synthesis of 3,5,6-trisubstituted-1,2,4-triazines from fatty acid hydrazides under solvent-free conditions and their antimicrobial activity. ARKIVOC 2007, xvi, 137–147. [Google Scholar]
- Kappe, C.O.; Dallinger, D. The impact of microwave synthesis on drug discovery. Nat. Rev. Drug Discov. 2006, 5, 51–64. [Google Scholar] [CrossRef]
- Shipe, W.D.; Wolkenberg, S.E.; Lindsley, C.W. Accelerating lead development by microwave-enhanced medicinal chemistry. Drug Discov. Today Tech. 2005, 2, 155–161. [Google Scholar]
- Leadbeater, N.E. Fast, easy, clean chemistry by using water as a solvent and microwave heating: The Suzuki coupling as an illustration. Chem. Commun. 2005, 2881–2902. [Google Scholar] [CrossRef]
- Caddick, S. Microwave assisted organic reactions. Tetrahedron 1995, 51, 10403–10432. [Google Scholar] [CrossRef]
- Kidwai, M.; Sapra, P.; Bhushan, K.R.; Saxena, R.K.; Gupta, R.; Singh, M. Microwave assisted stereoselective synthesis and antibacterial activity of new beta-lactam derivatives. Montash Chemie. 2000, 131, 85–90. [Google Scholar]
- Hermkens, P.H.H.; Ottenheijm, H.C.J.; Rees, D.C. Solid-phase organic reactions II. Tetrahedron 1997, 53, 5643–5678. [Google Scholar]
- Villemin, D.; Alloumn, A.B. Dry reaction under microwave: Condensation of sulfones with aldehydes on KF-alumina. Synth. Commun. 1991, 21, 63–68. [Google Scholar] [CrossRef]
- Loupy, A.; Pegion, P.; Ramdani, M.; Jacquault, P. Solid-liquid phase transfer catalysis without solvent coupled with microwave irradiation: A quick and efficient method for saponification of esters. Synth. Commun. 1994, 24, 159–165. [Google Scholar]
- Kidwai, M.; Sapra, P. Ring closure reactions of chalcones using microwave technolog. Synth. Commun. 1999, 29, 3237–3250. [Google Scholar] [CrossRef]
- Abdel-Rahman, R.M. Chemistry of uncondensed 1,2,4-triazines: Part II sulfur containing 5-oxo-1,2,4–triazin-3-yl moiety. An overview. Phosphorus Sulfur Silicon Relat. Elem. 2000, 166, 315–357. [Google Scholar] [CrossRef]
- Abdel-Rahman, R.M. Chemistry of uncondensed 1,2,4-triazines. Part III. Synthesis and chemistry of fluorine containing bioactive 1,2,4-triazines: An overview. Pharmazie 1999, 54, 791–804. [Google Scholar]
- Saad, H.A.; Allimony, H.A.; El-Mariah, F.A.A. Synthesis and antimicrobial activity of some nitrogen heterobicyclic systems. Part 2. Indian J. Chem. 1998, 37B, 1142–1148. [Google Scholar]
- El-Mariah, F.A.A.; Saad, H.A.; Allimony, H.A.; Abdel-Rahman, R.M. Synthesis and antimicrobial activity of some nitrogen heterobicyclic systems. Part 3. Indian J. Chem. 2000, 39B, 36–41. [Google Scholar]
- Salimon, J.; Salih, N. Synthesis, characterization and biological activity of some new 1,2,4-triazine derivatives. Int. J. Pharm. Tech. Res. 2010, 2, 1041–1045. [Google Scholar]
- Sangshetti, J.N.; Shinde, D.B. One pot synthesis and SAR of some novel 3-substituted 5,6-diphenyl-1,2,4-triazines as antifungal agents. Bioorg. Med. Chem. Lett. 2010, 20, 742–745. [Google Scholar]
- Abdel-Rahman, R.M. Role of uncondensed 1,2,4-triazine compounds and related heterocyclic systems as therapeutic agents: A review. Pharmazie 2001, 56, 18–22. [Google Scholar]
- El-Gendy, Z.; Morsy, J.M.; Allimony, H.A.; Abdel-Monem, W.R.; Abdel-Rahman, R.M. Synthesis of heterobicyclic nitrogen systems bearing the 1,2,4-triazine moiety as anti-HIV and anticancer drugs. Part III. Pharmazie 2001, 56, 376–383. [Google Scholar]
- Gill, C.; Jadhav, G.; Shaikh, M.; Kale, R.; Ghawalkar, A.; Nagargoje, D.; Shiradkar, M. Clubbed [1,2,3]triazoles by fluorine benzimidazole: A novel approach to H 37Rv inhibitors as a potential treatment for tuberculosis. Bioorg. Med. Chem. Lett. 2008, 18, 6244–6247. [Google Scholar] [CrossRef]
- Schmitz, W.D.; Brenner, A.B.; Bronson, J.J.; Ditta, J.L.; Li, Y.; Lodge, N.J.; Molski, T.F.; Olson, R.E.; Griffin, C.R.; Zhuo, X.; et al. 5-Arylamino-1,2,4-triazin-6(1H)-one CRF1 receptor antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 3579–3583. [Google Scholar]
- Rankovic, Z.; Cai, J.; Fradera, X.; Dempster, M.; Mistry, A.; Long, C.; Hamilton, E.; King, A.; Boucharens, S.; Jamieson, C.; et al. Dioxo-triazines as a novel series of cathepsin K inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1488–1490. [Google Scholar]
- Mullick, P.; Khan, S.A.; Begum, T.; Verma, S.; Kaushik, D.; Alam, O. Synthesis of 1,2,4-triazine derivatives as potential anti-anxiety and anti-inflammatory agents. Acta Pol. Pharm. Drug Res. 2009, 66, 379–385. [Google Scholar]
- Hynes, J.; Kanner, S.B.; Yang, X.; Tokarski, J.S.; Schieven, G.L.; Dyckman, A.J.; Lonial, H.; Zhang, R.; Sack, J.S.; Lin, S.; et al. Design, synthesis, and anti-inflammatory properties of orally active 4-(phenylamino)-pyrrolo[2,1-f][1,2,4]triazine p38α mitogen-activated protein kinase Inhibitors. J. Med. Chem. 2008, 51, 4–16. [Google Scholar]
- Abdel-Rahman, R.M. Role of uncondensed 1,2,4-triazine derivatives as biocidal plant protection agents: A review. Pharmazie 2001, 56, 195–204. [Google Scholar]
- Abdel-Rahman, R.M.; El-Gendy, Z.; Mahmoud, M.B. Synthesis of some new 3-substituted 1,2,4-triazino-indolederivatives and related compounds of potential antifungalactivity. Indian J. Chem. 1990, 29B, 352–358. [Google Scholar]
- Abdel-Rahman, R.M.; Abdel-Malik, M.S. Synthesis of some new 3,6-dihydroaryl-1,2,4-triazin-5-ones and their effect on amylolytic activity of some fungi. Pak. J. Sci. Ind. Res. 1990, 33, 142–147. [Google Scholar]
- Abdel-Rahman, R.M.; Seada, M.; El-Gendy, Z.; Islam, I.E.; Mahmoud, M.B. Synthesis of some new 4,6-disubstituted 1,2,4-triazin-3,5(2H)-diones and related compounds of potential antifungal activity. Farmaco 1993, 48, 407–416. [Google Scholar]
- Rusinov, V.L.; Ulomsky, E.N.; Chupakhin, O.N.; Zubairov, M.M.; Kapustin, A.B.; Mitin, N.I.; Ziravetskii, M.I.; Vinograd, I.A. Synthesis and antiviral activity of 6-Nitro-7-oxo-4,7-dihydroazolo-[5,1-c][1,2,4]triazines. Pharm. Chem. J. 1990, 24, 640–646. [Google Scholar]
- Chupakhin, O.N.; Rusinov, V.L.; Ulomsky, E.N.; Charushin, V.N.; Petrov, A.Y.; Kiselev, O.N. 2-Methylthio-6-nitro-1,2,4-triazolo[5,1-C]-1,2,4-triazine-7(4H)-one. Sodium salt dihydrate possessing antiviral activity. Russian Patent RU 2294936, 2007. [Google Scholar]
- Deeva, E.G.; Rusinov, V.L. International Conference on “Preparedness for the Influenza Pandemic – an International Outlook”, Saint-Petersburg, Russia, March 15-17th, 2007; pp. 35–36.
- Sharma, N.; Kumar, Y.; Sahi, S. Priyanka, 3D QSAR studies of pyrrolo[2,1-f][1,2,4]triazines as tyrosine kinase inhibitors. Int. J. Pharmacy Pharm. Sci. 2010, 2, 118–121. [Google Scholar]
- Giordanetto, F.; Karlsson, O.; Lindberg, J.; Larsson, L.O.; Linusson, A.; Evertsson, E.; Morgan, D.G.A.; Inghardt, T. Discovery of cyclopentane- and cyclohexane-trans-1,3-diamines as potent melanin-concentrating hormone receptor 1 antagonists. Bioorg. Med. Chem. Lett. 2007, 17, 5222–5231. [Google Scholar] [CrossRef]
- Bonini, C.; Chiummiento, L.; de Bonis, M.; Funicello, M.; Lupattelli, P.; Suanno, G.; Berti, F.; Campaner, P. Synthesis, biological activity and modelling studies of two novel anti HIV PR inhibitors with a thiophene containing hydroxyethylamino core. Tetrahedron 2005, 61, 6580–6589. [Google Scholar] [CrossRef]
- Brault, L.; Migianu, E.; Néguesque, A.; Battaglia, E.; Bagrel, D.; Kirsch, G. New thiophene analogues of kenpaullone: Synthesis and biological evaluation in breast cancer cells. Eur. J. Med. Chem. 2005, 40, 757–736. [Google Scholar]
- Kumar, P.R.; Raju, S.; Goud, P.S.; Sailaja, M.; Sarma, M.R.; Reddy, G.O.; Kumar, M.P.; Reddy, V.V.; Suresha, T.; Hegdeb, P. Synthesis and biological evaluation of thiophene [3,2-b] pyrrole derivatives as potential anti-inflammatory agents. Bioorg. Med. Chem. 2004, 12, 1221–1230. [Google Scholar] [CrossRef]
- Graff, J.; Harder, S.; Wahl, O.; Scheuermann, E.H.; Gossmann, J. Anti-inflammatory effects of clopidogrel intake in renal transplant patients: Effects on platelet-leukocyte interactions, platelet CD40 ligand expression, and proinflammatory biomarkers. Clin. Pharmacol. Ther. 2005, 78, 468–476. [Google Scholar] [CrossRef]
- Hymete, A.; Rohloff, J.; Kjosen, H.; Iversen, T.H. Acetylenic thiophenes from the roots of Echinops ellenbeckii from Ethiopia. Nat. Prod. Res. 2005, 19, 755–761. [Google Scholar]
- Tapia, R.A.; Alegria, L.; Pessoa, C.D.; Salas, C.; Cortes, M.J.; Valderrama, J.A.; Sarciron, M.E.; Pautet, F.; Walchshofer, N.; Fillion, H. Synthesis and antiprotozoal activity of naphthofuranquinones and naphthothiophenequinones containing a fused thiazole ring. Bioorg. Med. Chem. 2003, 11, 2175–2182. [Google Scholar]
- Dallemagne, P.; Khanh, L.P.; Alsaidi, A.; Varlet, I.; Collot, V.; Paillet, M.; Bureau, R.; Rault, S. Synthesis and biological evaluation of five-membered heterocycles fused to cyclopenta[c]thiophene as new antitumor agents. Bioorg. Chem. 2003, 11, 1161–1167. [Google Scholar]
- Caridha, D.; Kathcart, A.K.; Jirage, D.; Waters, N.C. Activity of substituted thiophene sulfonamides against malarial and mammalian cyclin dependent protein kinases. Bioorg. Med. Chem. Lett. 2010, 20, 3863–3867. [Google Scholar]
- Kumar, M.P.; Panda, G.; Manju, Y.K.; Chaturvedi, V.; Sinha, S. Thiophene containing triarylmethanes as antitubercular agents. Bioorg. Med. Chem. Lett. 2008, 18, 289–292. [Google Scholar] [CrossRef]
- Gazit, A.; Yaish, P.; Gilon, C.; Levitzki, A. Tyrphostins I: Synthesis and biological activity of protein tyrosine kinase inhibitors. J. Med. Chem. 1989, 32, 2344–2352. [Google Scholar] [CrossRef]
- Al-Juwaiser, I.A.; Ibrahim, M.R.; Al-Awadi, N.A.; Ibrahim, Y.A. An improved direct synthetic approach to anhydronucleosides (II). Tetrahedron 2008, 64, 8206–8212. [Google Scholar] [CrossRef]
- Saha, G.C.; Khayer, K.; Islam, R.; Chowdhury, S.K. Syntheses of thiocarbohydrazide, some thiocarbohydrazones and their cyclized products as probes for pharmacological studies. Indian J. Chem. 1992, 31, 547–550. [Google Scholar]
- Varshney, V.; Mishra, N.N.; Shukla, P.K.; Sahu, D.P. Novel 4-N-substituted aryl but-3-ene-1,2-dione derivatives of piperazinyloxazolidinones as antibacterial agents. Bioorg. Med. Chem. Lett. 2009, 19, 6810–6812. [Google Scholar]
- Dornow, A.; Menzel, H.; Marx, P. Synthesen stickstoffhaltiger Heterocyclen, XXVII. Über 1,2,4-Triazine, I Darstellung einiger neuer s-Triazolo[3.2-c]-as-triazine. Chem. Ber. 1964, 97, 2173–2178. [Google Scholar] [CrossRef]
- El-Sayed, H.A.; Moustafa, A.H.; Haikal, A.Z.; El-Ashry, E.S.H. Synthesis and antibacterial activity of some glucosyl- and ribosyl-pyridazin-3-ones. Nucleosides Nucleotides Nucleic Acids 2009, 28, 184–192. [Google Scholar]
- Saad, H.A.; Moustafa, A.H. Synthesis of fused heteropolycyclic systems containing an indole moiety. J. Chem. Res. 2006, 318–323. [Google Scholar]
- Hansen, M.B.; Nielsen, S.E.; Berg, K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods 1989, 119, 203–210. [Google Scholar]
- Ribble, D.; Goldstein, N.B.; Norris, D.A.; Shellman, Y.G. A simple technique for quantifying apoptosis in 96-well plates. PMC Biotechnol. 2005, 10, 5–12. [Google Scholar]
- Martin, D.; Leonardo, M. Related isolation procedures and functional assays. In Current Protocols in Immunology; Coligan, J.E., Kruisbeek, A.M., Margulies, D.H., Shevach, E.M., Strober, W., Eds.; John Wiley & Sons: New York, NY, USA, 1998; Volume 1.3, pp. 3–17. [Google Scholar]
- Sample Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Saad, H.A.; Youssef, M.M.; Mosselhi, M.A. Microwave Assisted Synthesis of Some New Fused 1,2,4-Triazines Bearing Thiophene Moieties With Expected Pharmacological Activity. Molecules 2011, 16, 4937-4957. https://doi.org/10.3390/molecules16064937
Saad HA, Youssef MM, Mosselhi MA. Microwave Assisted Synthesis of Some New Fused 1,2,4-Triazines Bearing Thiophene Moieties With Expected Pharmacological Activity. Molecules. 2011; 16(6):4937-4957. https://doi.org/10.3390/molecules16064937
Chicago/Turabian StyleSaad, Hosam A., Mohamed M. Youssef, and Mosselhi A. Mosselhi. 2011. "Microwave Assisted Synthesis of Some New Fused 1,2,4-Triazines Bearing Thiophene Moieties With Expected Pharmacological Activity" Molecules 16, no. 6: 4937-4957. https://doi.org/10.3390/molecules16064937