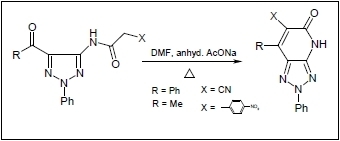
2. Results and Discussion
Readily obtainable (5-amino-2-phenyl-
2H-1,2,3-triazol-4-yl)phenylmethanone (
1a) [
19], 1-(5-amino-2-phenyl-
2H-1,2,3-triazol-4-yl)ethanone (
1b) [
20] and 4-amino-3-benzoyl-1-phenyl-1
H-pyrazole-5-carbonitrile (
2) [
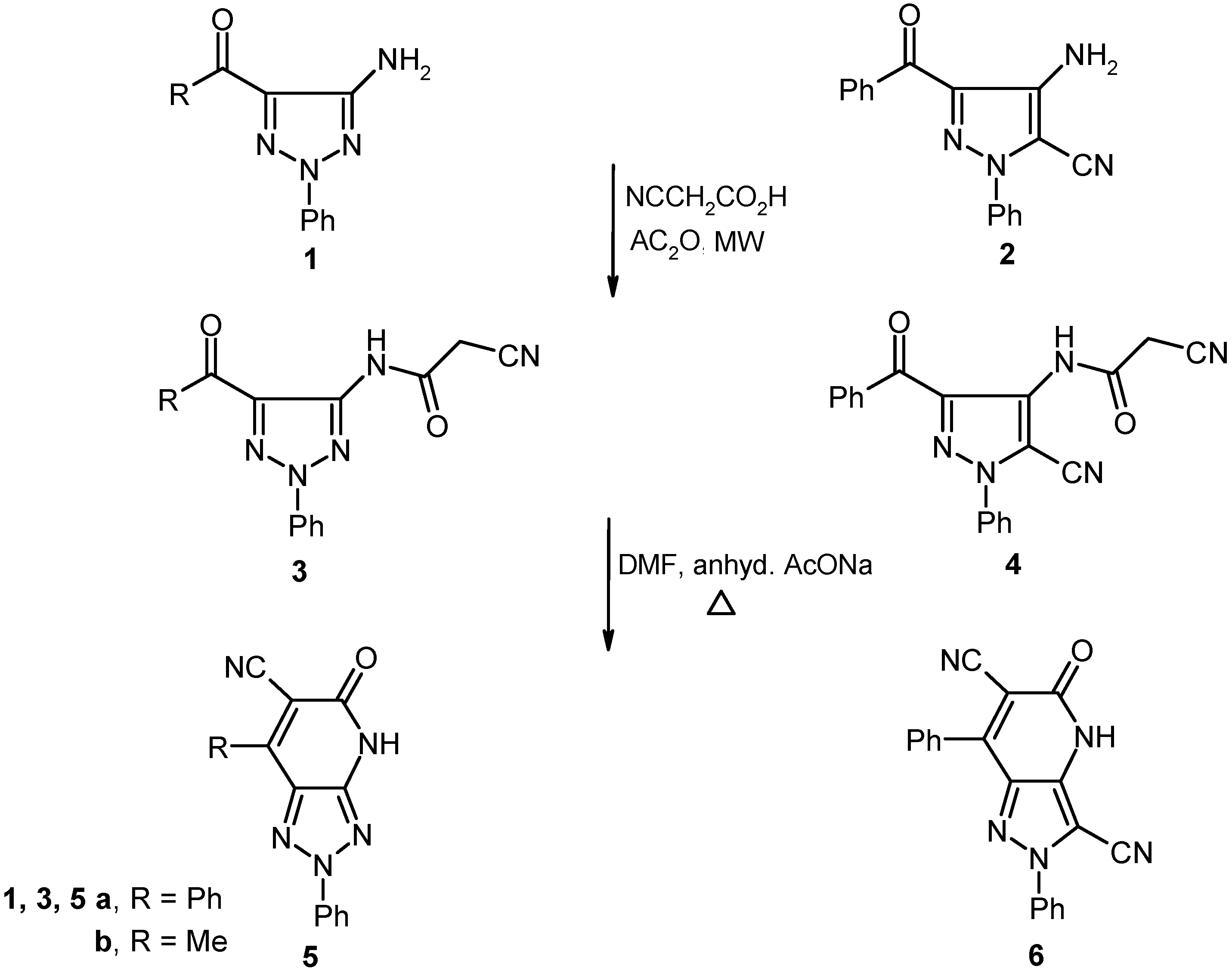
21] were found to react with a preheated mixture of acetic anhydride and cyanoacetic acid under microwave irradiation conditions to yield the corresponding cyanoacetamides
3 and
4 in excellent yields. These substances undergo cyclization to generate the respective fused pyridones
5 and
6 upon stirring at reflux for 30 min in DMF containing anhydrous sodium acetate (cf.
Scheme 1). The structure of
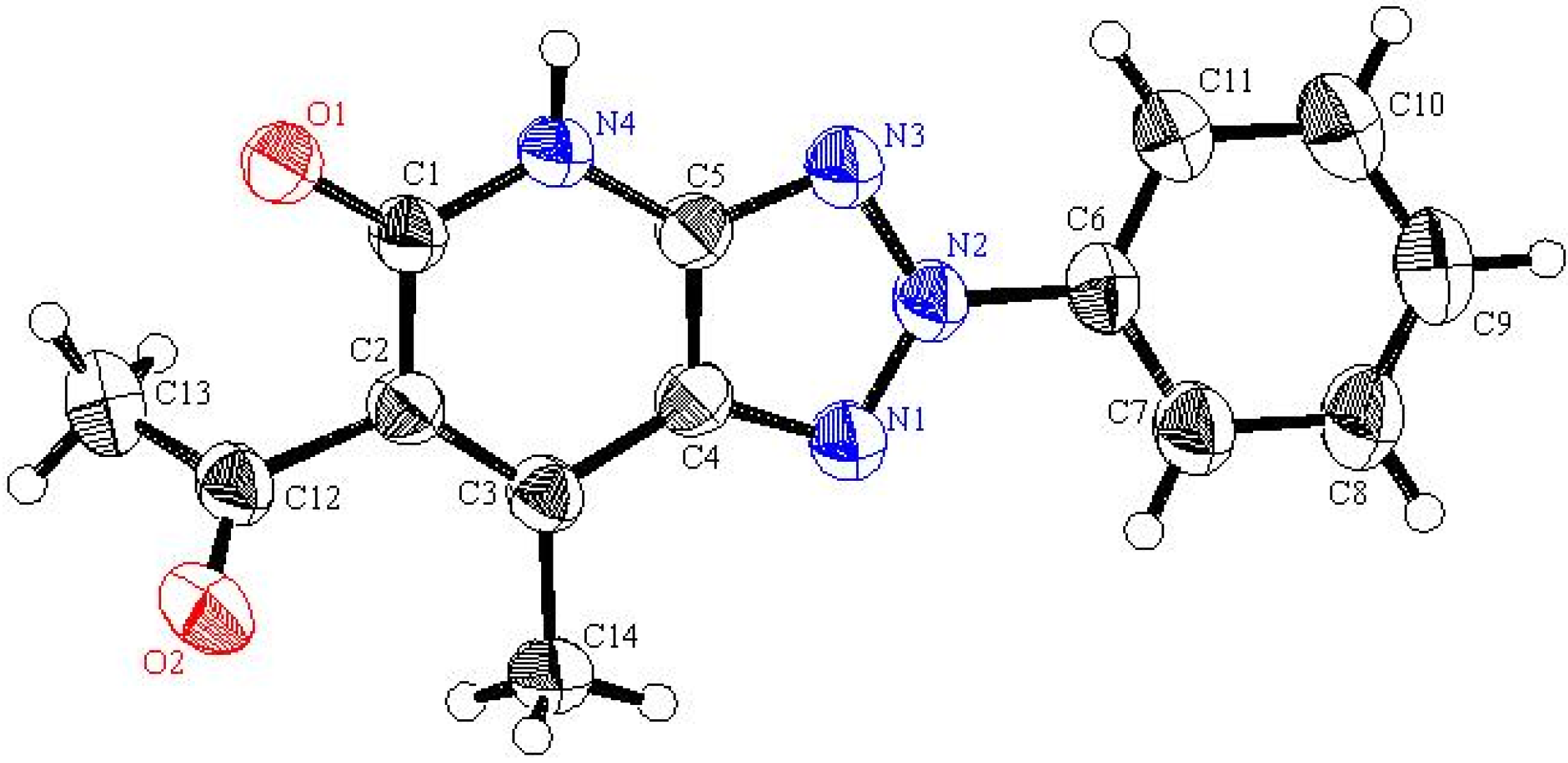
5a was assigned by using X-ray crystallographic analysis (cf.
Table 1 and
Figure 1).
Scheme 1.
Reaction of 5-amino-1,2,3-triazoles and 4-aminopyrazole with cyanoacetic acid.
Scheme 1.
Reaction of 5-amino-1,2,3-triazoles and 4-aminopyrazole with cyanoacetic acid.
Figure 1.
ORTEP plot of the x-ray crystallographic data determined for
5a. Crystallo- graphic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 804721 [
22].
Figure 1.
ORTEP plot of the x-ray crystallographic data determined for
5a. Crystallo- graphic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 804721 [
22].
Table 1.
Selected bond lengths and bond angles for 5a.
Table 1.
Selected bond lengths and bond angles for 5a.
| Bond | Bond length | Bond | Bond angle |
|---|
| N1-N2 | 1.327 | N1-N2-C2 | 102.5 |
| N1-N3 | 1.356 | N1-N3-C1 | 100.4 |
| N3-C1 | 1.333 | N2-N1-N3 | 117.5 |
| N2-C2 | 1.350 | N3-C1-C2 | 111.6 |
| C1-C2 | 1.384 | N2-C2-C1 | 108.0 |
| C1-N4 | 1.375 | C1-C2-C3 | 121.4 |
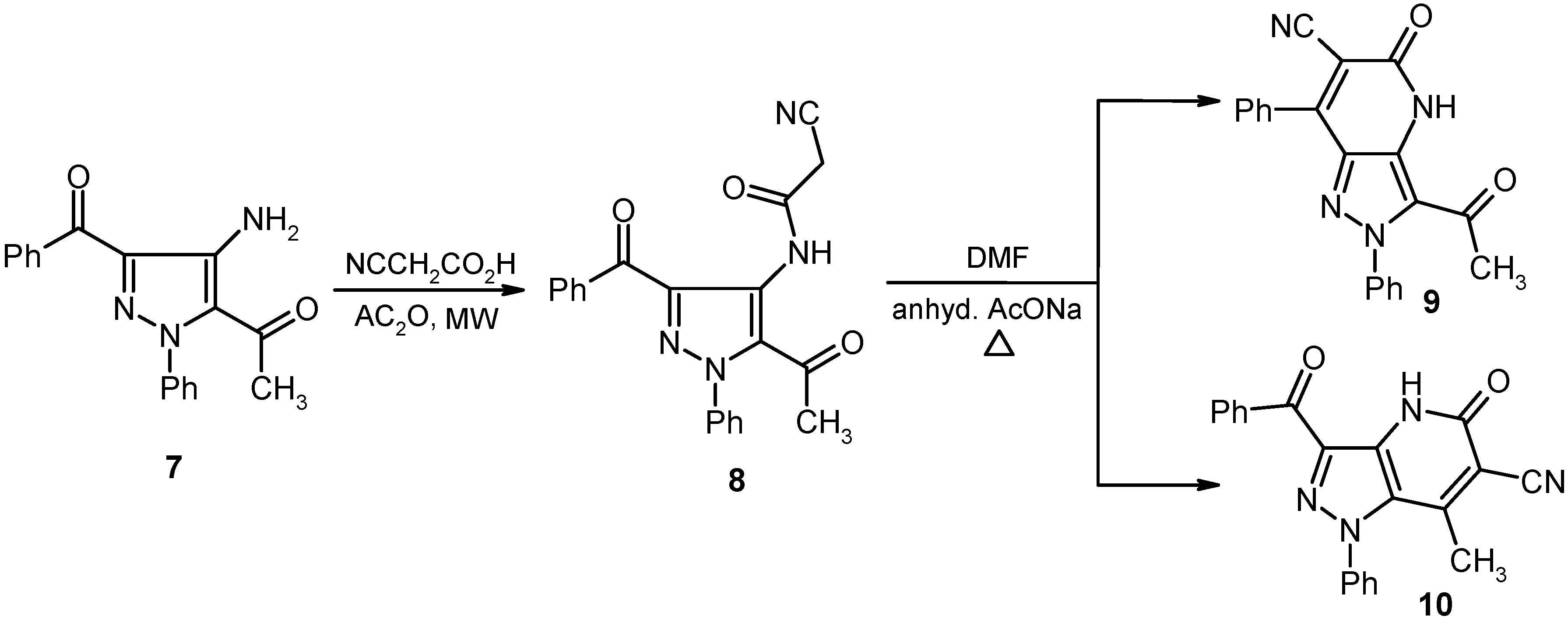
The acylpyrazole 7 underwent ready cyanoacylation to afford the cyanoacetamide 8 in 93% yield. Heating a solution of 8 in DMF containing anhydrous sodium acetate leads to production of a substance whose structure should be either 9 or its isomer 10.
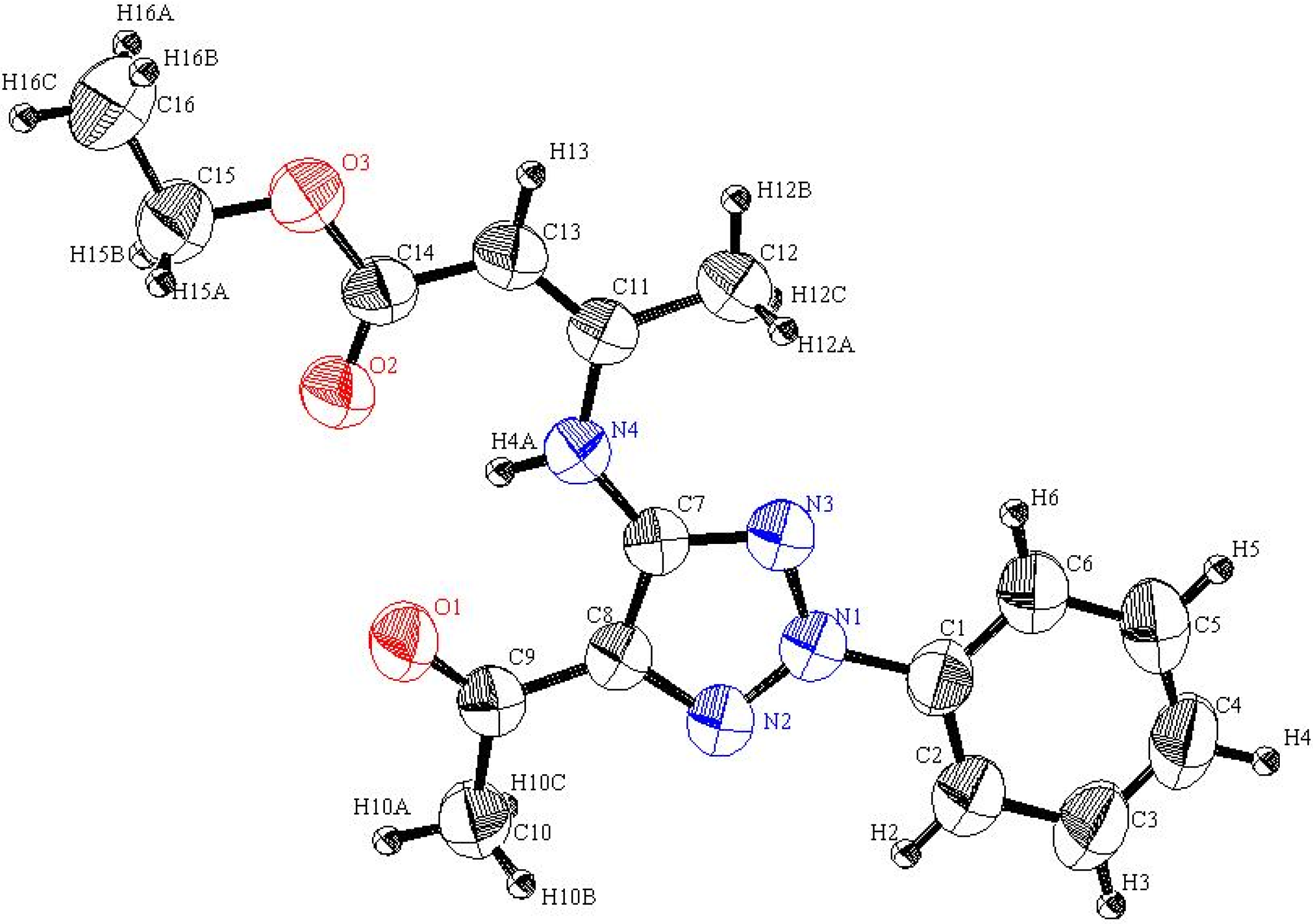
The actual structure of the product was assigned as 10 based on its
13C-NMR spectroscopic data which showed the absence of an acetyl carbonyl carbon resonance and its replacement by a peak at 186.19 ppm. Moreover, the methyl protons’ resonance at
δ = 2.12 ppm displays a HMBC cross peak with the carbon peak at
δ = 105.72 ppm that is assigned as C-6. In addition the X-ray crystallographic analysis of this product demonstrated that it has the structure represented by 10 (cf.
Scheme 2 and
Figure 2).
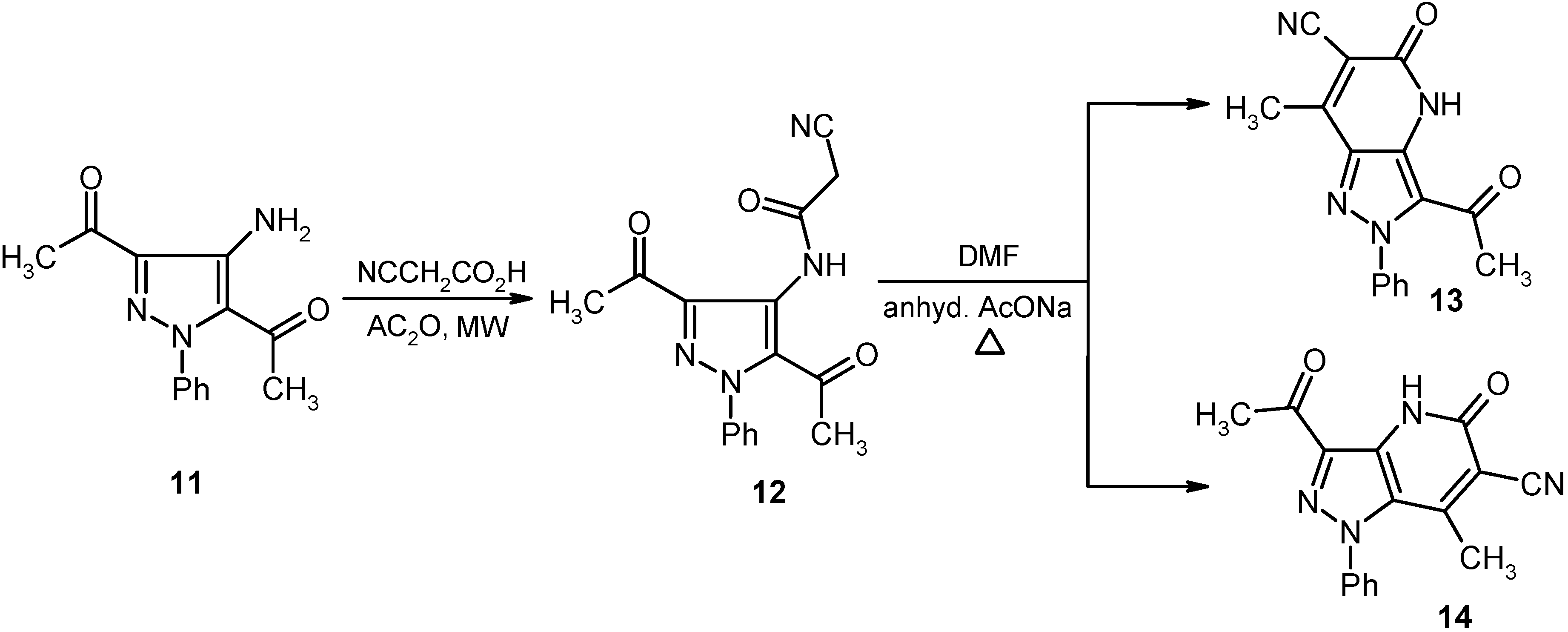
In a similar manner, bis-acetylpyrazole
11 is readily cyanoacylated to afford the corresponding cyanoacetamide
12 in excellent yield. Heating a DMF solution of
12 containing anhydrous sodium acetate afforded a product that may also have the isomeric structures represented by
13 and
14. As before, the actual structure of the product was shown to be
14 based on its
13C-NMR spectrum, which contained a carbonyl resonance at 202.41 ppm. This chemical shift is expected for an acyl carbonyl at the C-3 position of the pyrazole ring and not at C-5 since in the latter case shielding provided by the N-lone pair should make the resonance appear at a higher field (cf.
Scheme 3).
Scheme 2.
Synthesis of pyrazolo[4,3-b]pyridine-6-carbonitrile 10.
Scheme 2.
Synthesis of pyrazolo[4,3-b]pyridine-6-carbonitrile 10.
Figure 2.
ORTEP plot of the x-ray crystallographic data determined for
10 containing one DMSO molecule. Crystallographic data have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 816562 [
23].
Figure 2.
ORTEP plot of the x-ray crystallographic data determined for
10 containing one DMSO molecule. Crystallographic data have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 816562 [
23].
Scheme 3.
Synthesis of pyrazolo[4,3-b]pyridine-6-carbonitrile 14.
Scheme 3.
Synthesis of pyrazolo[4,3-b]pyridine-6-carbonitrile 14.
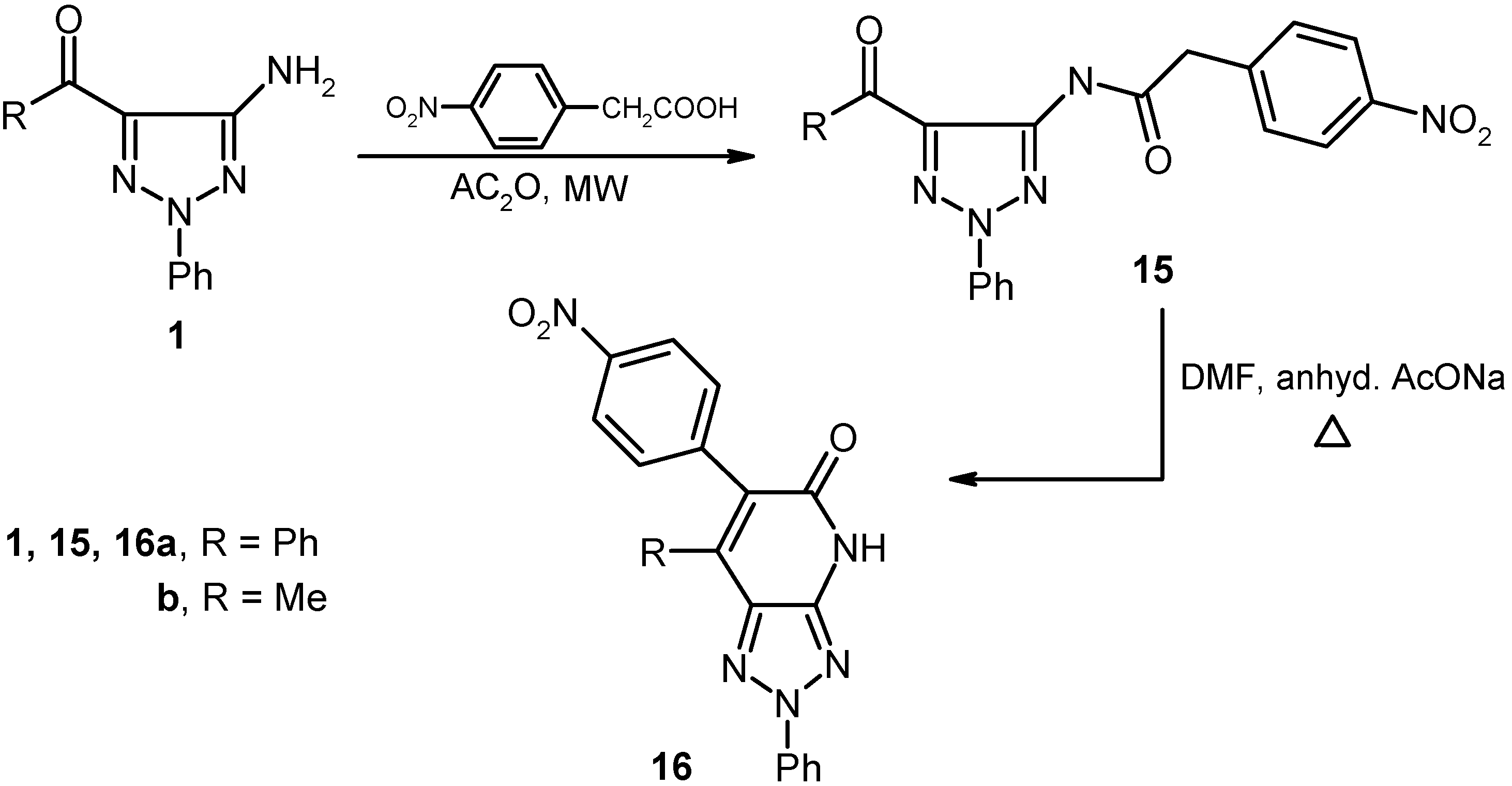
We have previously [
10] suggested that in the mechanistic pathway for the process described by Slatt [
9], the mixed anhydride is formed initially and then it reacts by nucleophilic addition of an amine or electron rich aromatic system at the more electron deficient cyanoacetyl carbonyl. As a consequence of this proposal, we believed that other mixed anhydrides could be used as arylacetamide precursors provided that the reactions occur at the more electron deficient aroyl carbonyl. In fact, heating
p-nitrophenylacetic acid with acetic anhydride, followed by addition of either
1a or
1b and heating the mixture in a microwave oven for 60 s, afforded the corresponding amides
15a and
15b that are readily cyclized to form the respective triazolo[4,5-
b]pyridines derivatives
16a and
16b upon heating in DMF containing anhydrous sodium acetate (cf.
Scheme 4).
Scheme 4.
Reaction of 5-amino-1,2,3-triazoles with p-nitrophenylacetic acid.
Scheme 4.
Reaction of 5-amino-1,2,3-triazoles with p-nitrophenylacetic acid.
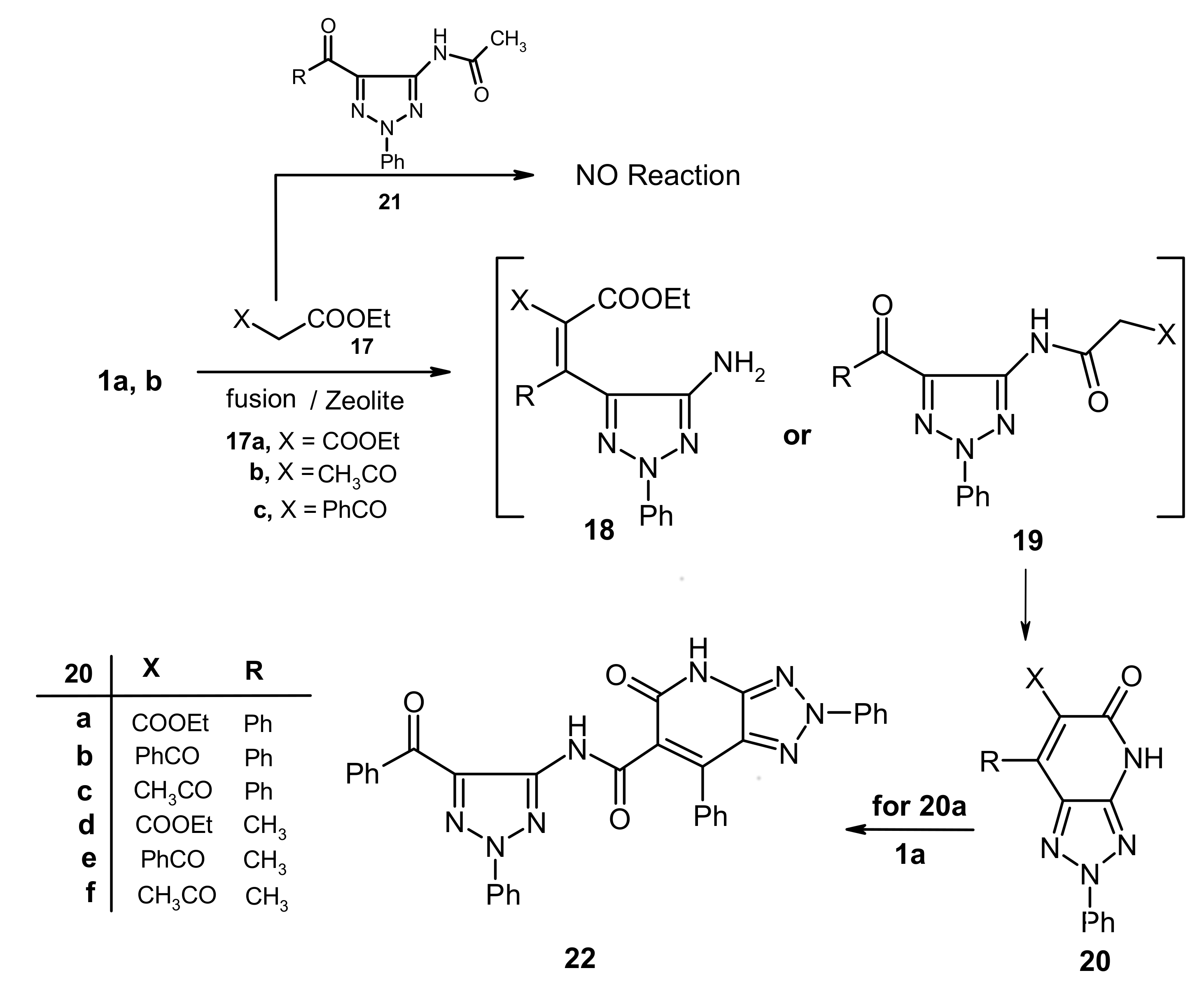
We have explored a possible extension of this methodology, which relies on conversion of the acetamide derivatives of 1,2,3-triazoles to their corresponding 1,2,3-triazolo[4,5-
b]pyridines, by probing the reactivity
1a,b with other active methylene compounds like
17a-c. The results of this study showed that
1a,b underwent condensation with
17a-c in presence of zeolite catalysts, followed by heating to produce the corresponding 1,2,3-triazolo[4,5-
b]pyridine derivatives
20a-f. The structure of
20f was assigned by X-ray crystallographic analysis. Although these products could potentially formed
via the intermediacy of either triazole
18 or
19, it is almost certain that
19 is the intermediate as attempts to condense
N-acetyl-1,2,3-triazole derivatives
21 with active methylene compounds failed. Moreover the reaction of
20a with another molecule of
1a afforded
22, which is generated
via elimination of ethanol (cf.
Figure 3 and
Scheme 5).
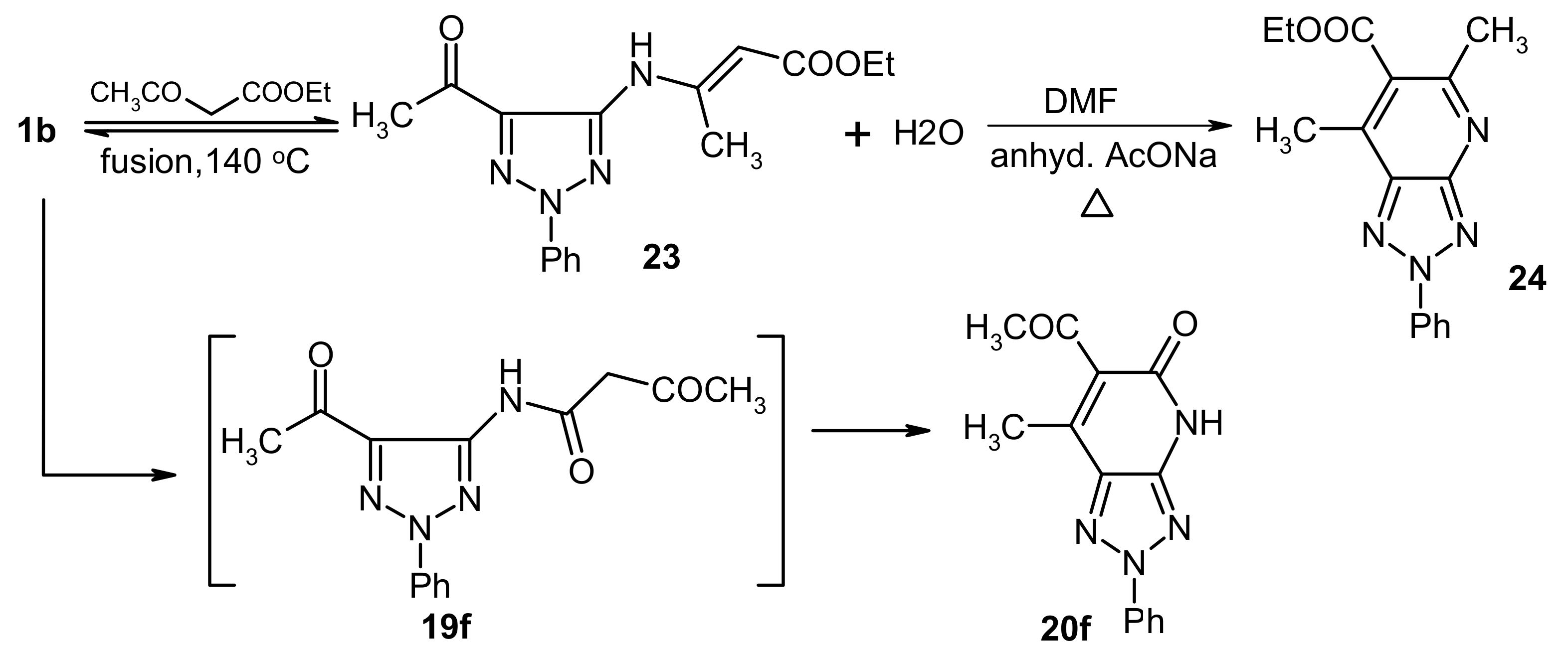
In contrast,
1b was found to react with ethyl acetoacetate (
17b) in absence of zeolite to yield a condensation product that arises by elimination of one molecule of water. X-ray crystallographic analysis of this substance demonstrated that it has the structure represented by
23, a product that is formed
via initial addition of the amine to the carbonyl carbon of
17b (
Figure 4,
Table 2).The fact that
1b reacts with ethyl acetoacetate (
17b) to yield either the intermediate
23 or
19f demonstrates the effect of the zeolite, a microporous catalyst that favors formation of slim molecules like
19 rather than bulky ones like
23, so the latter is formed in absence of such
a catalyst, Also,
23 separated from the reaction mixture underwent cyclization in refluxing DMF containing anhydrous sodium acetate to form
24 via loss of another molecule of water. (cf.
Scheme 6).
Figure 3.
ORTEP plot of the x-ray crystallographic data determined for
20f. Crystallo- graphic data have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 815380 [
24].
Figure 3.
ORTEP plot of the x-ray crystallographic data determined for
20f. Crystallo- graphic data have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 815380 [
24].
Scheme 5.
Reaction of 5-amino-1,2,3-triazoles with active methylene compounds.
Scheme 5.
Reaction of 5-amino-1,2,3-triazoles with active methylene compounds.
Scheme 6.
Reaction of 1b with ethyl acetoacetate to afford 24.
Scheme 6.
Reaction of 1b with ethyl acetoacetate to afford 24.
Figure 4.
ORTEP plot of the x-ray crystallographic data determined for
23. Crystallo- graphic data have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 805282 [
25].
Figure 4.
ORTEP plot of the x-ray crystallographic data determined for
23. Crystallo- graphic data have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 805282 [
25].
Table 2.
Selected bond lengths and bond angles for 23.
Table 2.
Selected bond lengths and bond angles for 23.
| Bond | Bond length | Bond | Bond angle |
|---|
| N1-N2 | 1.317 | N1-N2-C8 | 103.8 |
| N1-N3 | 1.359 | N1-N3-C7 | 102.7 |
| N3-C7 | 1.330 | N2-N1-N3 | 116.1 |
| N2-C8 | 1.336 | N3-C7-C8 | 109.0 |
| C8-C9 | 1.462 | N2-C8-C7 | 108.5 |
| N4-C7 | 1.379 | O1-C9-C8 | 119.5 |
Inspection of the crystallographically determined bond angles and lengths of the 1,2,3-triazole rings in both
5a,
20f and
23 indicate that the N3-N1-N2 bond angles deviate significantly from typical sp
3 nitrogen values and are close to those that are associated with sp
2 nitrogens. However, the N2-C8-C7 or N3-C7-C8 bond angles are close to those expected for sp
3 carbons. Similar observations, made earlier in studies of very similar systems by Elnagdi
et al., [
19] have been taken as evidence for the significant contribution of charge separated resonance forms delocalizing N-1 lone pairs to the ring carbons. Importantly, both
5a,
20f and
23 are planar substances, a fact that adds further support to the conclusion that resonance delocalization of N-1 lone pair occurs in these systems.
3. Experimental
3.1. General
Melting points were recorded on a Griffin melting point apparatus and are reported uncorrected. IR spectra were recorded using KBr disks using a Perkin-Elmer System 2000 FT-IR spectrophoto- meter.
1H-NMR (400 MHz) and
13C-NMR (100 MHz) spectra were recorded at 25 °C in CDCl
3 or DMSO-
d6 as solvent with TMS as internal standard on a Bruker DPX 400 super-conducting NMR spectrometer. Chemical shifts are reported in ppm. Mass spectra were measured using a high resolution GC-MS (DFS) thermo spectrometers with EI (70 EV). Microanalyses were performed on a LECO CHNS-932 Elemental Analyzer. Reactions were conducted under microwave irradiation in heavy-walled Pyrex tubes (capacity 10 mL) fitted with PCS caps. Microwave heating was carried out with a single mode cavity Explorer Microwave synthesizer (CEM Corporation, 3100 Smith Farm Road, Matthews, NC, USA). The zeolite (≤ 45 µm) was purchased from Fluka Company with product No. 96096. The crystal structures were determined by a Rigaku R-AXIS RAPID diffractometer using filtered Mo-Kα radiation at Kuwait University. Compounds
1a,b, 2 and
21 were prepared using literature procedures [
19,
20,
21].
3.2. General Procedure for the Preparation of Cyanoacetamides 3, 4, 8 and 12
A solution of cyanoacetic acid (0.45 g, 5 mmol) in Ac2O (5 mL) was heated in the microwave oven at 85 °C for 10 s then compounds 1, 2, 7 or 11 (5 mmol) were added and the reaction mixture was heated for further 30 s at 100 °C. The reaction mixture was allowed to cool to room temperature and the formed crystalline solid was separated by filtration and washed with cold ethanol and then hot ethanol to afford 3, 4, 8 and 12, respectively, as pure substances.
N-(5-Benzoyl-2-phenyl-2H-1,2,3-triazol-4-yl)-2-cyanoacetamide (3a). Creamy white crystals, yield: 98%, m.p. 210 °C; IR (KBr): ν/cm−1 3291 (NH), 2257 (CN), 1692, 1634 (2CO); 1H-NMR (DMSO-d6): δ = 4.04 (s, 2H, CH2), 7.50 (t, J = 7.6 Hz, 1H, Ar-H), 7.56–7.64 (m, 4H, Ar-H), 7.73 (t, J = 7.6 Hz, 1H, Ar-H), 8.01 (d, J = 7.6 Hz, 2H, Ar-H), 8.10 (d, J =7.6 Hz, 2H, Ar-H) and 11.23 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 26.03 (CH2), 115.35 (CN), 118.76, 128.64, 128.68, 129.91, 129.97, 133.77, 136.04, 137.22, 138.48, 144.19, 161.40 and 185.82 ppm (Ar-C and CO); MS (EI): m/z (%) 331 (M+, 74.35), 332 (M++1, 16.90). Anal. calcd. for C18H13N5O2 (331.34): C, 65.25; H, 3.95; N, 21.14. Found: C, 65.28; H, 4.02; N, 21.20.
N-(5-Acetyl-2-phenyl-2H-1,2,3-triazol-4-yl)-2-cyanoacetamide (3b). Buff crystals, yield: 95%, m.p. 205 °C; IR (KBr): ν/cm−1 3302 (NH), 2262 (CN), 1685, 1638 (2CO); 1H-NMR (DMSO-d6): δ 2.62 (s, 3H, CH3), 4.11 (s, 2H, CH2), 7.51 (t, J = 8.0 Hz, 1H, Ar-H), 7.62 (t, J = 8.0 Hz, 2H, Ar-H), 8.03 (d, J = 8.0 Hz, 2H, Ar-H) and 10.67 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 26.28 (CH2), 27.71 (CH3), 115.51 (CN), 118.79, 128.85, 129.90, 137.79, 138.46, 143.32, 161.66 and 191.69 ppm (Ar-C and CO); MS (EI): m/z (%) 269 (M+, 100), 270 (M++1, 25.6). Anal. calcd. for C13H11N5O2 (269.26): C, 57.99; H, 4.12; N, 26.01. Found: C, 58.04; H, 4.06; N, 25.93.
N-(3-Benzoyl-5-cyano-1-phenyl-1H-pyrazol-4-yl)-2-cyanoacetamide (4). Creamy white crystals, yield: 93%, m.p. 234 °C; IR (KBr): ν/cm−1 3259 (NH), 2227, 2263 (2CN), 1712, 1631 (2CO); 1H-NMR (DMSO-d6, 25 °C): δ = 4.13 (s, 2H, CH2), 7.57–7.72 (m, 6H, Ar-H), 7.83 (d, J = 7.6 Hz, 2H, Ar-H), 8.14 (d, J = 7.6 Hz, 2H, Ar-H) and 10.83 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 25.84 (CH2), 109.64, 110.24, 115.41, 124.20, 128.60, 128.75, 129.76, 130.08, 130.20, 133.74, 135.94, 137.70, 141.38, 161.56 and 186.71 ppm (2CN, Ar-C and CO); MS (EI): m/z (%) 355 (M+, 73.20), 356 (M++1, 20.35). Anal. calcd. for C20H13N5O2 (355.36): C, 67.60; H, 3.69; N, 19.71. Found: C, 67.57; H, 3.75; N, 19.74.
N-(5-Acetyl-3-benzoyl-1-phenyl-1H-pyrazol-4-yl)-2-cyanoacetamide (8). Orange crystals, yield: 93%, m.p. above 300 °C; IR (KBr): ν/cm−1 3297 (NH), 22114 (CN), 1687 (br), 1636 (3CO); 1H-NMR (DMSO-d6): δ = 2.40 (s, 3H, CH3), 4.02 (s, 2H, CH2), 7.53–7.60 (m, 7H, Ar-H), 7.69 (t, J = 7.2 Hz, 1H, Ar-H), 8.10 (d, J = 7.6 Hz, 2H, Ar-H) and 10.45 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 25.72 (CH2), 29.87 (CH3), 115.54 (CN), 122.13, 125.61, 128.48, 129.13, 129.17, 130.14, 133.42, 135.78, 136.26, 139.65, 142.76, 162.14, 187.20 and 189.62 ppm (Ar-C and CO); MS (EI): m/z (%) 372 (M+, 30.20), 373 (M++1, 8.57). Anal. calcd. for C21H16N4O3 (372.39): C, 67.73; H, 4.33; N, 15.05. Found: C, 67.69; H, 4.31; N, 15.10.
2-Cyano-N-(3,5-diacetyl-1-phenyl-1H-pyrazol-4-yl)acetamide (12). Creamy white crystals, yield: 89%, m.p. 230 °C; IR (KBr): ν/cm−1 3284 (NH), 2260 (CN), 1685 (br), 1637 (3CO); 1H-NMR (DMSO-d6): δ = 2.33 (s, 3H, CH3), 2.57 (s, 3H, CH3), 4.03 (s, 2H, CH2), 7.49–7.57 (m, 5H, Ar-H) and 10.34 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 26.16 (CH2), 27.72 (CH3), 30.22 (CH3), 116.01 (CN), 125.95, 127.94, 129.63, 129.68, 137.18, 140.05, 143.17, 162.64, 190.18 and 193.80 ppm (Ar-C and CO); MS (EI): m/z (%) 310 (M+, 55.1), 311 (M++1, 10.75). Anal. calcd. for C16H14N4O3 (310.31): C, 61.93; H, 4.55; N, 18.05. Found: C, 61.88; H, 4.57; N, 17.98.
3.3. General Procedure for the Cyclization of Cyanoacetamides to Azolo Pyridines 5, 6, 10 and 14
Independent solutions of cyanoacetamides 3, 4, 8 and 12 (5 mmol), in DMF (10 mL) containing anhydrous sodium acetate (1 g) were stirred at reflux for 1 h. Then, the reaction mixture was cooled to room temperature and poured into ice cold water. The formed crude products were collected by filtration, washed with water and recrystallized from the appropriate solvent to afford the corresponding azolo pyridine derivatives 5, 6, 10 and 14, respectively.
5-Oxo-2,7-diphenyl-4,5-dihydro-2H-[1,2,3]triazolo[4,5-b]pyridine-6-carbonitrile (5a). Recrystallized from a EtOH/dioxane (1:1) mixture as yellow crystals, yield: 87%, m.p. above 300 °C; IR (KBr): ν/cm−1 3437 (NH), 2230 (CN), 1651 (CO); 1H-NMR (DMSO-d6): δ 7.47 (t, J = 7.6 Hz, 1H, Ar-H), 7.56 (t, J = 8.0 Hz, 2H, Ar-H), 7.65–7.67 (m, 3H, Ar-H), 7.89 (d, J = 7.6 Hz, 2H, Ar-H), 7.97 (d, J = 8.0 Hz, 2H, Ar-H) and 13.30 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 105.22, 115.73 (CN), 119.10, 128.81, 129.21, 129.53, 129.95, 130.53, 131.14, 131.51, 138.62, 148.07, 151.23 and 160.14 ppm (Ar-C and CO); MS (EI): m/z (%) 313 (M+, 100), 314 (M++1, 21.40). Anal. calcd. for C18H11N5O (313.32): C, 69.00; H, 3.54; N, 22.35. Found: C, C, 69.03; H, 3.48; N, 22.39.
3.3.1. Crystallographic Analysis for 5a
The crystals were mounted on a glass fiber. All measurements were performed on a Rigaku R-AXIS RAPID diffractometer using filtered Mo-Kα radiation. The data were collected at a temperature of 20 ± 1 °C to a maximum 2θ value of 55.0° using the ω scanning technique. The structure was solved by charge flipping method and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model.
3.3.2. Crystal Data
C
18H
11N
5O, M = 313.32, triclinic, a = 6.861(4)Å, b = 11.229(6)Å, c = 12.987(7)Å, V = 869.3(8)Å
3, α = 110.560(9)°, β = 103.584(9)°, γ = 100.858(9)°, space group: P-1, Z = 2, D
calc = 1.365 g cm
−3, No. of reflection measured 3956, 2θ
max = 55.0°, R1 = 0.12.
Figure 1 illustrates the structure as determined. Full data can be obtained on request from the CCDC [
22].
7-Methyl-5-oxo-2-phenyl-4,5-dihydro-2H-[1,2,3]triazolo[4,5-b]pyridine-6-carbonitrile (5b). Recrystallized from an EtOH/dioxane (2:1) mixture as yellow crystals, yield: 83%, m.p. above 300 °C; IR (KBr): ν/cm−1 3410 (NH), 2228 (CN), 1658 (CO); 1H-NMR (DMSO-d6,): δ = 2.64 (s, 3H, CH3), 7.48 (t, J = 8.0 Hz, 1H, Ar-H), 7.58 (t, J = 8.0 Hz, 2H, Ar-H), 8.00 (d, J = 8.0 Hz, 2H, Ar-H), and 13.04 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ = 16.94 (CH3), 107.37, 115.30 (CN), 119.39, 129.57, 130.41, 131.96, 139.07, 147.66, 152.55 and 160.07 ppm (Ar-C and CO); MS (EI): m/z (%) 251 (M+, 100), 252 (M++1, 30.58). Anal. calcd. for C13H9N5O (251.25): C, 62.15; H, 3.61; N, 27.87. Found: C, 62.19; H, 3.55; N, 27.91.
5-Oxo-2,7-diphenyl-4,5-dihydro-2H-pyrazolo[4,3-b]pyridine-3,6-dicarbonitrile (6). Recrystallized from a EtOH/dioxane (1:1) mixture as beige crystals, yield: 80%, m.p. above 300 °C; IR (KBr): ν/cm−1 3375 (NH), 2227 (br, 2CN), 1656 (CO); 1H-NMR (DMSO-d6): δ 7.64–7.66 (m, 6H, Ar-H), 7.79 (d, J = 7.6 Hz, 2H, Ar-H), 7.85 (d, J = 7.2 Hz, 2H, Ar-H) and 13.30 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ = 100.37, 105.99, 109.67, 115.83, 124.37, 129.02, 129.91, 130.18, 130.77, 131.22, 131.71, 133.12, 135.46, 138.11, 152.78 and 159.81 ppm (2CN, Ar-C and CO); MS (EI): m/z (%) 337 (M+, 100), 338 (M++1, 25.0). Anal. calcd. for C20H11N5O (337.34): C, 71.21; H, 3.29; N, 20.76. Found: 71.19; H, 3.36; N, 20.83.
3-Benzoyl-7-methyl-5-oxo-1-phenyl-4,5-dihydro-1H-pyrazolo[4,3-b]pyridine-6-carbonitrile (10).Rec- rystallized from DMSO as brown crystals, yield: 77%, m.p. 238–240 °C; IR (KBr): ν/cm−1 3381 (NH), 2223 (CN), 1681, 1658 (2CO); 1H-NMR (DMSO-d6): δ 2.12 (s, 3H, CH3), 7.56–7.81 (m, 8H, Ar-H), 8.22 (d, J = 7.2 Hz, 2H, Ar-H) and 12.02 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 18.23 (CH3), 105.72, 115.34 (CN), 126.28, 127.04, 127.57, 128.59, 129.15, 129.45, 129.60, 130.07, 130.63, 133.52, 136.00, 138.95, 159.17 and 186.19 ppm (Ar-C and CO); MS (EI): m/z (%) 354 (M+, 100), 355 (M++1, 27.1). Anal. calcd. for C21H14N4O2 (354.37): C, 71.18; H, 3.95; N, 15.81. Found: 71.24; H, 4.02; N, 15.77.
3.3.3. Crystallographic Analysis for 10
The crystals were mounted on a glass fiber. All measurements were performed on a Rigaku R-AXIS RAPID diffractometer using filtered Mo-Kα radiation. The data were collected at a temperature of 20 ± 1 °C to a maximum 2θ value of 55.0° using the ω scanning technique. The structure was solved by charge flipping method and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model.
3.3.4. Crystal Data
C
21H
14N
4O
2+ one DMSO molecule, M = 354.37, monoclinic, a = 11.518(2) Å, b = 9.386(2)Å, c = 19.655(3)Å, V = 2123.5(5) Å
3, α = γ = 90.00°, β = 92.069(7)°, space group: P2
1/n, Z = 4, D
calc = 1.353g cm
−3, No. of reflection measured 4731, 2θ
max = 55.0 °, R1 = 0.1088.
Figure 2 illustrates the structure as determined. Full data can be obtained on request from the CCDC [
23].
3-Acetyl-7-methyl-5-oxo-1-phenyl-4,5-dihydro-1H-pyrazolo[4,3-b]pyridine-6-carbonitrile (14). Recrystallized from DMF as buff crystals, yield: 80%, m.p. above 300 °C; IR (KBr): ν/cm−1 3183 (NH), 2221 (CN), 1673, 1639 (2CO); 1H-NMR (DMSO-d6): δ 2.15 (s, 3H, CH3), 2.60 (s, 3H, CH3), 7.49–7.71 (m, 5H, Ar-H) and 11.91 ppm (s, 1H, NH); 13C-NMR (TFA-d): δ 19.82 (CH3), 27.25 (CH3), 107.63, 116.18 (CN), 125.08, 129.39, 131.18, 131.62, 132.45, 134.48, 138.03, 140.46, 154.60 and 202.41 ppm (Ar-C and CO); MS (EI): m/z (%) 292 (M+, 100), 293 (M++1, 20.45). Anal. calcd. for C16H12N4O2 (292.30): C, 65.75; H, 4.14; N, 19.17. Found: C, 65.81; H, 4.17; N, 19.19.
3.4. General Procedure for the Preparation of 15
Independent solutions of p-nitrophenylacetic acid (0.9 g, 5 mmol) in Ac2O (5 mL) were heated in a microwave oven at 100 °C for 20 s. To these mixtures, 1a,b (5 mmol) were added and the mixtures were heated for further 60 s at the same temperature. The reaction mixtures were cooled to room temperature the crystalline solid formed were separated by filtration and washed by cold ethanol and then hot ethanol to afford 15a,b, respectively, as pure substances.
N-(5-Benzoyl-2-phenyl-2H-1,2,3-triazol-4-yl)-2-(4-nitrophenyl)acetamide (15a). White crystals, yield: 90%, m.p. 196 °C; IR (KBr): ν/cm−1 3294 (NH), 1690, 1632 (2CO); 1H-NMR (CDCl3): δ 4.02 (s, 2H, CH2), 7.43 (t, J = 7.6 Hz, 1H, Ar-H), 7.50–7.62 (m, 6H, Ar-H), 7.68 (t, J = 7.6 Hz, 1H, Ar-H), 8.15 (d, J = 8.0 Hz, 2H, Ar-H), 8.27 (d, J = 7.6 Hz, 2H, Ar-H), 8.43 (d, J = 8.0 Hz, 2H, Ar-H) and 10.02 ppm (s, 1H, NH); 13C-NMR (CDCl3): δ 43.90 (CH2), 119.47, 124.05, 128.59, 128.76, 129.41, 130.41, 130.54, 132.99, 133.95, 135.98, 138.99, 141.04, 147.40, 148.35, 166.42 and 187.60 ppm (Ar-C and CO); MS (EI): m/z (%) 427 (M+, 56.75), 428 (M++1, 15.55). Anal. calcd. for C23H17N5O4 (427.42): C, 64.63; H, 4.01; N, 16.39. Found: 64.59; H, 3.94; N, 16.43.
N-(5-Acetyl-2-phenyl-2H-1,2,3-triazol-4-yl)-2-(4-nitrophenyl)acetamide (15b). Beige crystals, yield: 87%, m.p. 207 °C; IR (KBr): ν/cm−1 3358 (NH), 1686, 1641(2CO); 1H-NMR (CDCl3): δ 2.69 (s, 3H, CH3), 3.99 (s, 2H, CH2), 7.43 (t, J = 7.6 Hz, 1H, Ar-H), 7.51 (t, J = 7.6 Hz, 2H, Ar-H), 7.60 (d, J = 8.0 Hz, 2H, Ar-H), 8.12 (d, J = 7.6 Hz, 2H, Ar-H), 8.26 (d, J = 8.0 Hz, 2H, Ar-H) and 9.51 ppm (s, 1H, NH); 13C-NMR (CDCl3): δ 27.02 (CH3), 43.81 (CH2), 119.38, 124.06, 128.74, 129.42, 130.56, 133.67, 138.99, 140.97, 146.14, 147.43, 166.43 and 195.23 ppm (Ar-C and CO); MS (EI): m/z (%) 365 (M+, 27.35), 366 (M++1, 6.91). Anal. calcd. for C18H15N5O4 (365.35): C, 59.18; H, 4.14; N, 19.17. Found: C, 59.24; H, 4.07; N, 19.14.
3.5. General Procedure for the Preparation of 16
Independent solutions of amides 15a,b (5 mmol) in DMF (10 mL) containing anhydrous sodium acetate (1 g) were stirred at reflux for 1 h. Then, the reaction mixtures were cooled to rt and poured onto ice cold water. The crude products were collected by filtration, washed with water and recrystallized from the appropriate solvent to afford the corresponding azolo pyridine derivatives 16a,b respectively.
6-(4-Nitrophenyl)-2,7-diphenyl-2,4-dihydro[1,2,3]triazolo[4,5-b]pyridin-5-one (16a). Recrystallized from an EtOH/dioxane (1:1) mixture as yellow crystals, yield: 82%, m.p. 302 °C; IR (KBr): ν/cm−1 3435 (NH), 1645 (CO); 1H-NMR (DMSO-d6): δ 7.32–7.35 (m, 5H, Ar-H), 7.44 (d, J = 8.0 Hz, 3H, Ar-H), 7.56 (t, J = 7.6 Hz, 2H, Ar-H), 8.00 (d, J = 8.0 Hz, 2H, Ar-H), 8.10 (d, J = 8.0 Hz, 2H, Ar-H) and 12.90 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 118.62, 122.51, 128.18, 128.25, 128.89, 129.82, 129.86, 130.66, 132.29, 132.64, 133.14, 139.02, 140.68, 142.80, 146.36, 146.65 and 161.83 ppm (Ar-C and CO); MS (EI): m/z (%) 409 (M+, 100), 410 (M++1, 30.72). Anal. calcd. for C23H15N5O3 (409.41): C, 67.48; H, 3.69; N, 17.11. Found: C, 67.51; H, 3.70; N, 17.08.
7-Methyl-6-(4-Nitrophenyl)-2-phenyl-2,4-dihydro[1,2,3]triazolo[4,5-b]-pyridin-5-one (16b). Recrysta- llized from an EtOH/dioxane (2:1) mixture as buff crystals, yield: 85%, m.p. 298 °C; IR (KBr): ν/cm−1 3285 (NH), 1643 (CO); 1H-NMR (DMSO-d6): δ = 2.30 (s, 3H, CH3), 7.46 (t, J = 7.6 Hz, 1H, Ar-H), 7.58–7.63 (m, 4H, Ar-H), 8.05 (d, J = 8.0 Hz, 2H, Ar-H), 8.30 (d, J = 8.0 Hz, 2H, Ar-H), and 12.67 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 15.16 (CH3), 118.48, 122.99, 128.13, 129.84, 131.14, 131.86, 133.08, 138.08, 139.10, 142.38, 145.94, 146.81 and 161.63 ppm (Ar-C and CO); MS (EI): m/z (%) 347 (M+, 100), 348 (M++1, 21.87). Anal. calcd. for C18H13N5O3 (347.34): C, 62.25; H, 3.77; N, 20.16. Found: C, 62.19; H, 3.81; N, 20.12.
3.6. General Procedure for the Preparation of 20a-f
Independent mixtures of 5-amino-1,2,3-triazoles 1a,b (10 mmol), active methylene compounds 17a-c (15 mmol) and zeolite (10% by weight) were heated at 150 °C for 1 h. Dioxane was added to the reaction mixtures followed by filtration to remove the zeolite. The crystals formed upon cooling the filtrates were collected by filtration and washed with methanol.
Ethyl-5-oxo-2,7-diphenyl-4,5-dihydro-2H-[1,2,3]triazolo[4,5-b]pyridine-6-carboxylate (20a). Pale orange crystals, yield: 76%, m.p. 205 °C; IR (KBr): ν/cm−1 3391(NH), 1733, 1650 (2CO); 1H-NMR (DMSO-d6): δ 1.30 (t, J = 7.2 Hz, 3H, CH3CH2), 4.13 (q, J = 7.2 Hz, 2H, CH3CH2), 7.46 (t, J = 7.6 Hz, 1H, Ar-H), 7.56–7.60 (m, 5H, Ar-H), 7.65–7.67 (m, 2H, Ar-H), 8.00 (d, J = 7.6 Hz, 2H, Ar-H) and 13.00 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 13.65 (CH3CH2), 61.17 (CH3CH2), 118.81, 128.54, 128.57, 128.67, 129.86, 129.87, 130.10, 130.85, 132.11, 138.97, 140.27, 146.93, 160.14 and 165.01 ppm (Ar-C and CO); MS (EI): m/z (%) 360 (M+, 100), 361 (M++1, 24.55). Anal. Calcd. for C20H16N4O3 (360.38): C, 66.66; H, 4.48; N, 15.55. Found: C, 66.72; H, 4.40; N, 15.49.
6-Benzoyl-2,7-diphenyl-2,4-dihydro[1,2,3]triazolo[4,5-b]pyridin-5-one (20b). Canary yellow crystals, yield: 80%, m.p. above 300 °C; IR (KBr): ν/cm−1 3423 (NH), 1672, 1644 (2CO); 1H-NMR (DMSO-d6): δ 7.39–7.41 (m, 3H, Ar-H), 7.46 (t, J = 8.0 Hz, 3H, Ar-H), 7.52–7.62 (m, 5H, Ar-H), 7.89 (d, J = 8.0 Hz, 2H, Ar-H), 8.02 (d, J = 8.0 Hz, 2H, Ar-H) and 12.99 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ = 118.79, 128.14, 128.45, 128.88, 129.01, 129.73, 129.89, 131.40, 131.67, 132.08, 133.17, 133.90, 136.41, 139.06, 140.49, 147.13, 161.25 and 193.91 ppm (Ar-C and CO); MS (EI): m/z (%) 392 (M+, 100), 393 (M++1, 27.84). Anal. calcd. for C24H16N4O2 (392.42): C, 73.46; H, 4.11; N, 14.28. Found: C, 73.52; H, 4.08; N, 14.36.
6-Acetyl-2,7-diphenyl-2,4-dihydro[1,2,3]triazolo[4,5-b]pyridin-5-one (20c). Pale yellow crystals, yield: 79%, m.p. 249 °C; IR (KBr): ν/cm−1 3299 (NH), 1709, 1646 (2CO); 1H-NMR (DMSO-d6): δ 2.33 (s, 3H, CH3), 7.47 (t, J = 7.6 Hz, 1H, Ar-H), 7.53-7.60 (m, 7H, Ar-H), 7.99 (d, J = 7.6 Hz, 2H, Ar-H) and 12.96 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 31.57 (CH3), 118.72, 128.43, 128.60, 128.96, 129.76, 129.80, 131.38, 132.13, 133.69, 138.95, 139.20, 146.64, 160.80 and 201.47 ppm (Ar-C and CO); MS (EI): m/z (%) 330 (M+, 100), 331(M++1, 19.84). Anal. calcd. for C19H14N4O2 (330.35): C, 69.08; H, 4.27; N, 16.96. Found: C, 68.98; H, 4.35; N, 16.88.
Ethyl-7-methyl-5-oxo-2-phenyl-4,5-dihydro-2H-[1,2,3]triazolo[4,5-b]pyridine-6-carboxylate (20d). Yellow crystals, yield: 82%, m.p. 219 °C; IR (KBr): ν/cm−1 3263 (NH), 1735, 1659 (2CO); 1H-NMR (DMSO-d6): δ 1.31 (t, J = 7.2 Hz, 3H, CH3CH2), 2.44 (s, 3H, CH3), 4.33 (q, J = 7.2 Hz, 2H, CH3CH2), 7.47 (t, J = 8.0 Hz, 1H, Ar-H), 7.60 (t, J = 8.0 Hz, 2H, Ar-H), 8.04 (d, J = 8.0 Hz, 2H, Ar-H) and 12.74 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 14.04 (CH3CH2), 14.31(CH3), 61.28 (CH3CH2), 118.52, 127.22, 128.33, 129.61, 129.76, 131.86, 138.90, 146.12, 159.82 and 165.08 ppm (Ar-C and CO); MS (EI): m/z (%) 298 (M+, 61.90), 299 (M++1, 12.75). Anal. calcd. for C15H14N4O3 (298.30): C, 60.40; H, 4.73; N, 18.78. Found: C, 60.33; H, 4.84; N, 18.76.
6-Benzoyl-7-methyl-2-phenyl-2,4-dihydro[1,2,3]triazolo[4,5-b]pyridin-5-one (20e). Creamy white crystals, yield: 79%, m.p. 298 °C; IR (KBr): ν/cm−1 3429 (NH), 1668, 1641 (2CO); 1H-NMR (DMSO-d6): δ 2.30 (s, 3H, CH3), 7.47 (t, J = 7.6 Hz, 1H, Ar-H), 7.55 (t, J = 8.0 Hz, 2H, Ar-H), 7.61 (t, J = 7.6 Hz, 2H, Ar-H), 7.69 (t, J = 7.6 Hz, 1H, Ar-H), 7.92 (d, J = 7.6 Hz, 2H, Ar-H), 8.06 (d, J = 8.0 Hz, 2H, Ar-H) and 12.75 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 14.18(CH3), 118.62, 128.34, 129.01, 129.04, 129.89, 131.90, 132.57, 134.12, 136.24, 138.83, 139.08, 146.45, 161.04 and 194.41 ppm (Ar-C and CO); MS (EI): m/z (%) 330 (M+, 100), 331 (M++1, 22.88). Anal. calcd. for C19H14N4O2 (330.35): C, 69.08; H, 4.27; N, 16.96. Found: C, 69.15; H, 4.21; N, 17.02.
6-Acetyl-7-methyl-2-phenyl-2,4-dihydro[1,2,3]triazolo[4,5-b]pyridin-5-one (20f). Yellow crystals, yield: 86%, m.p. 242 °C; IR (KBr): ν/cm−1 3435 (NH), 1694, 1648 (2CO); 1H-NMR (DMSO-d6): δ 2.36 (s, 3H, CH3), 2.47 (s, 3H, CH3), 7.44 (t, J = 8.0 Hz, 1H, Ar-H), 7.57 (t, J = 8.0 Hz, 2H, Ar-H), 8.00 (d, J = 8.0 Hz, 2H, Ar-H) and 12.71 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 13.98 (CH3), 30.98 (CH3), 118.57, 128.34, 129.79, 132.46, 133.31, 138.53, 138.95, 146.09, 160.99 and 201.93 ppm (Ar-C and CO); MS (EI): m/z (%) 268 (M+, 76.45), 269 (M++1, 13.89). Anal. calcd. for C14H12N4O2 (268.28): C, 62.68; H, 4.51; N, 20.88. Found: C, 62.74; H, 4.47; N, 20.94.
3.6.1. Crystallographic Analysis for 20f
The crystals were mounted on a glass fiber. All measurements were performed on a Rigaku R-AXIS RAPID diffractometer using filtered Mo-Kα radiation. The data were collected at a temperature of 20 ± 1 °C to a maximum 2θ value of 55.0° using the ω scanning technique. The structure was solved by charge flipping method and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model.
3.6.2. Crystal Data
C
14H
12N
4O
2, M = 268.28, triclinic, a = 4.003(4)Å, b = 13.07(2)Å, c = 13.49(2)Å, V = 643(1)Å
3, α = 113.65(1)°, β = 91.13(2)°, γ = 94.89(2)°, space group: P-1, Z= 2,
Dcalc = 1.385 g cm
−3, No. of reflection measured 2935, 2θ
max= 55.0°, R1= 0.053.
Figure 3 illustrates the structure as determined. Full data can be obtained on request from the CCDC [
24].
3.7. N-(5-Benzoyl-2-phenyl--2H-1,2,3-triazol-4-yl)-5-oxo-2,7-diphenyl-4,5-dihydro-2H-[1,2,3]- triazolo[4,5-b]pyridine-6-carboxamide (22)
A mixture of 5-amino-1,2,3-triazole 1a (0.66 g, 2.5 mmol), pyrazolo[4,3-b]pyridine 20a (0.9 g, 2.5 mmol) and zeolite (10% by weight) in dioxane (10 mL) was stirred at reflux for 2 h, filtered to remove the zeolite, and cooled to room temperature. The solid which formed was collected by filtration, washed with ethanol, and recrystallized from dioxane giving beige crystals, yield: 68%, m.p. 271 °C; IR (KBr): ν/cm−1 3404, 3314 (2NH), 1711, 1663, 1635 (3CO); 1H-NMR (DMSO-d6): δ = 7.49–7.73 (m, 14H, Ar-H), 8.01-–8.12 (m, 6H, Ar-H), 11.48 (s, 1H, NH) and 12.93 ppm (s, 1H, NH); 13C-NMR (DMSO-d6): δ 118.83, 128.52, 128.60, 128.70, 129.04, 129.75, 129.90, 130.87, 131.39, 132.26, 133.67, 136.35, 136.46, 138.61, 139.01, 140.66, 144.50, 147.02, 161.14, 162.51, and 185.99 ppm (Ar-C and CO); MS (EI): m/z (%) 578 (M+, 46.25), 579 (M++1, 17.30). Anal. calcd. for C33H22N8O3 (578.60): C, 68.51; H, 3.83; N, 19.37. Found: C, 68.44; H, 3.87; N, 19.42.
3.8. (E)-Ethyl-3-(5-acetyl-2-phenyl-2H-1,2,3-triazol-4-ylamino)but-2-enoate (23)
A mixture of 5-amino-1,2,3-triazole 1b (2.02 g, 10 mmol) and ethyl acetoacetate (1.95 g, 15 mmol) was fused at 140 °C for 20 min. The mixture was poured into water and cooled to room temperature. The crude solid which formed was collected by filtration, washed with cold ethanol, and recrystallized from ethanol to give creamy white crystals, yield: 71%, m.p. 158 °C; IR (KBr): ν/cm−1 3435 (NH), 1672, 1620 (2CO); 1H-NMR (CDCl3): δ 1.30 (t, J = 7.2 Hz, 3H, CH3CH2), 2.50 (s, 3H, CH3), 2.68 (s, 3H, CH3CO), 4.27 (q, J = 7.2 Hz, 2H, CH3CH2), 4.93 (s, 1H, olefinic CH), 7.38(t, J = 8.0 Hz, 1H, Ar-H), 7.49 (t, J = 8.0 Hz, 2H, Ar-H), 8.05 (d, J = 8.0 Hz, 2H, Ar-H) and 11.95 ppm (s, 1H, NH); 13C-NMR (CDCl3): δ 14.55 (CH3CH2), 22.75 (CH3), 26.84 (CH3), 59.40 (CH3CH2), 92.61, 118.76, 128.05, 129.35, 133.81, 139.16, 149.00, 154.87, 168.94 and 193.51 ppm (Ar-C, olefinic C and CO); MS (EI): m/z (%) 314 (M+, 100), 315 (M++1, 22.85). Anal. Calcd. for C16H18N4O3 (314.35): C, 61.14; H, 5.77; N, 17.82. Found: C, 61.17; H, 5.75; N, 17.86.
3.8.1. Crystallographic Analysis for 23
The crystals were mounted on a glass fiber. All measurements were performed on a Rigaku R-AXIS RAPID diffractometer using filtered Mo-Kα radiation. The data were collected at a temperature of 20 ± 1 °C to a maximum 2θ value of 55.0° using the ω scanning technique. The structure was solved by charge flipping method and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model.
3.8.2. Crystal Data
C
16H
18N
4O
3, M = 314.35, monoclinic, a = 4.949(2)Å, b = 14.130(4)Å, c = 23.315(7)Å, V = 1629.14(9)Å
3, α = γ = 90.00°, β = 92.190(2)°, space group: P2
1/n, Z = 4, D
calc = 1.282 g cm
−3, No. of reflection measured 3733, 2θ
max= 55.0°, R1= 0.0807.
Figure 4 illustrates the structure as determined. Full data can be obtained on request from the CCDC [
25].
3.9. Ethyl-5,7-dimethyl-2-phenyl-2H-[1,2,3]triazolo[4,5-b]pyridine-6-carboxylate (24)
A solution of 23 (1.57 g, 5 mmol) in DMF (10 mL) containing anhydrous sodium acetate (1 g) was stirred at reflux for 1 h. The mixture was cooled to room temperature and poured into ice cold water. The formed solid was collected by filtration, washed with water and recrystallized from EtOH/H2O (2:1) to give pale brown crystals, yield: 71%, m.p. 78 °C; IR (KBr): ν/cm−1 1719 (CO); 1H-NMR (DMSO-d6): δ 1.46 (t, J = 7.2 Hz, 3H, CH3CH2), 2.74 (s, 3H, CH3), 2.75 (s, 3H, CH3), 4.50 (q, J = 7.2 Hz, 2H, CH3CH2), 7.49 (t, J = 8.0 Hz, 1H, Ar-H), 7.57 (t, J = 8.0 Hz, 2H, Ar-H) and 8.40 ppm (d, J = 8.0 Hz, 2H, Ar-H); 13C-NMR (DMSO-d6): δ 14.27 (CH3), 14.71(CH3), 24.42 (CH3), 61.89 (CH2), 120.59, 128.72, 129.50, 129.55, 136.67, 137.78, 140.14, 154.96, 158.88 and 168.10 pm (Ar-C and CO); MS (EI): m/z (%) 296 (M+, 100), 297 (M++1, 29.8). Anal. calcd. for C16H16N4O2 (296.33): C, 64.85; H, 5.44; N, 18.91. Found: C, 64.78; H, 5.51; N, 18.94.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







