Proline-Catalysed Amination Reactions in Cyclic Carbonate Solvents

School of Chemistry, Research Centre in Catalysis and Intensified Processing, Bedson Building, University of Newcastle, Newcastle upon Tyne, NE1 7RU, UK
*
Author to whom correspondence should be addressed.
Molecules 2011, 16(4), 3420-3432; https://doi.org/10.3390/molecules16043420
Submission received: 6 April 2011
/
Revised: 19 April 2011
/
Accepted: 20 April 2011
/
Published: 21 April 2011
(This article belongs to the Special Issue Catalytic Asymmetric Synthesis)
Abstract
:Propylene carbonate is shown to be an environmentally friendly and sustainable replacement for dichloromethane and acetonitrile in proline-catalysed α-hydrazinations of aldehydes and ketones. Enantioselectivities comparable to those obtained in conventional solvents or ionic liquids can be obtained, even when using a lower catalyst loading.
1. Introduction
Proline (1) is unique amongst the proteinogenic amino acids in that it is a secondary amine. This leads to it having a special role in peptides and proteins as it cannot act as a hydrogen-bond donor, but is often found in β-turns [1,2,3]. Proline and its derivatives have also found many applications in synthetic chemistry as chiral-auxiliaries [4], chiral reagents [5,6] and most recently as chiral catalysts [7,8,9,10,11,12] where it was developments in proline-catalysed enamine chemistry that sparked the recent resurgence of interest in asymmetric organocatalysis [13,14,15,16,17]. Proline is undoubtedly the most sustainable of the organocatalysts as it is directly available from biological sources without any need for chemical transformations. Proline-catalysed reactions are however, commonly carried out in traditional solvents such as DMSO, DMF and chlorinated solvents [7,8,9,10,11,12] which severely undermine the green credentials of proline-catalysed reactions due to their toxicity. Although both water [18,19] and ionic liquids [20] have also been used as solvents for proline-catalysed reactions, the green credentials of both of these solvents have been questioned [21,22] and water has been shown to inhibit proline-catalysed aldol reactions [23]. In some cases, proline-catalysed reactions can also be carried out without a solvent [24,25]. This limited solvent compatibility of proline-catalysed reactions is one of the reasons that so much effort has been put into developing other asymmetric organocatalysts which are soluble in a broader range of solvents. However, we have adopted a different approach, namely the use of alternative, sustainable and non-toxic polar-aprotic solvents in proline-catalysed reactions.
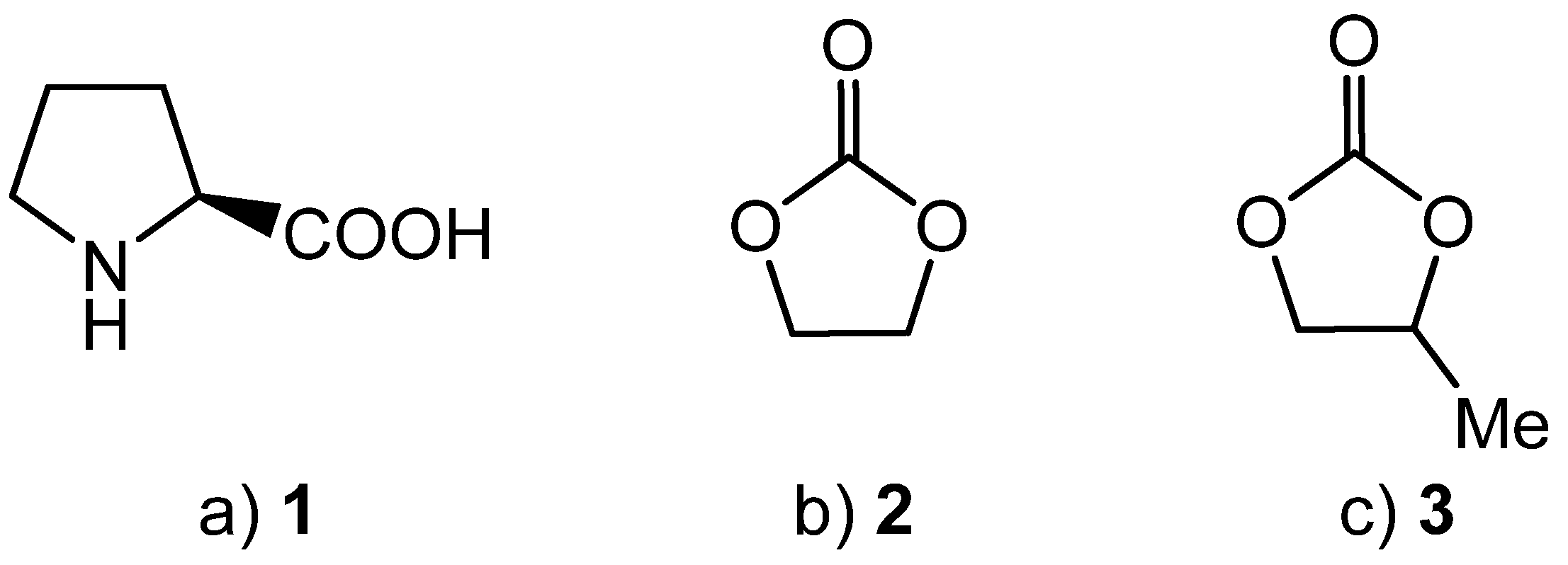
Cyclic carbonates (Figure 1), especially ethylene carbonate (2) and propylene carbonate (3), are attracting increasing interest as green solvents for metal-catalysed reactions [26,27,28]. Ethylene carbonate (2) is a solid at room temperature (m.p. 36 °C, b.p. 248 °C), though its melting point is depressed on addition of reactants. Propylene carbonate (3) has a very wide liquid range (m.p. −49 °C, b.p. 242 °C), making it a suitable solvent for reactions carried out below, at, or above room temperature. As a result of their high boiling points, cyclic carbonates 2 and 3 have very low vapour pressures. Compounds 2 and 3 are biodegradable, have high flash points and low odour levels [29]. They are used commercially as degreasers, paint strippers and in cleaning applications, their toxicities have been assessed and compound 3 is licensed for use in cosmetics [26]. Ethylene and propylene carbonate have high dielectric constants, so they can be considered as sustainable replacements for traditional polar aprotic solvents such as DMF, DMSO, HMPA and NMP. There is however, a significant difference in polarity between the two cyclic carbonates: ethylene carbonate 2 has a dielectric constant of 90, whilst propylene carbonate 3 has a dielectric constant of 65 [30]. Thus, the solvent polarity can be tuned by choice of the appropriate cyclic carbonate or by use of a mixture of the two solvents.
Cyclic carbonates are prepared by the 100% atom economical reaction between carbon dioxide and ethylene or propylene oxide (Scheme 1) [31,32,33,34,35]. We have shown that, in the presence of an appropriate catalyst, the synthesis of cyclic carbonates 2 and 3 can be achieved at atmospheric pressure and room temperature for batch processes [36,37,38,39] and at temperatures of 100 °C or below in a gas phase flow reactor [40,41].
This opens the possibility of utilizing waste carbon dioxide from major fixed site producers such as power stations [42], oil refineries and chemical plants in the production of cyclic carbonates, especially as the catalysts have been shown to tolerate the impurities present in power station flue gas [41].
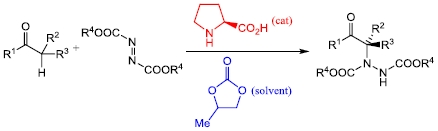
In recent papers we have shown that cyclic carbonates 2 and 3 make excellent solvents for proline-catalysed aldol reactions [43,44,45]. In this manuscript, we extend that study to the proline-catalysed α-hydrazination of aldehydes and ketones by diazodicarboxylates (Scheme 2). Previous work on this extensively utilized, proline-catalysed reaction has employed dichloromethane [46,47,48,49,50,51,52,53], acetonitrile [54,55,56,57,58,59,60,61,62] or an ionic liquid [63] as the solvent.
2. Results and Discussion
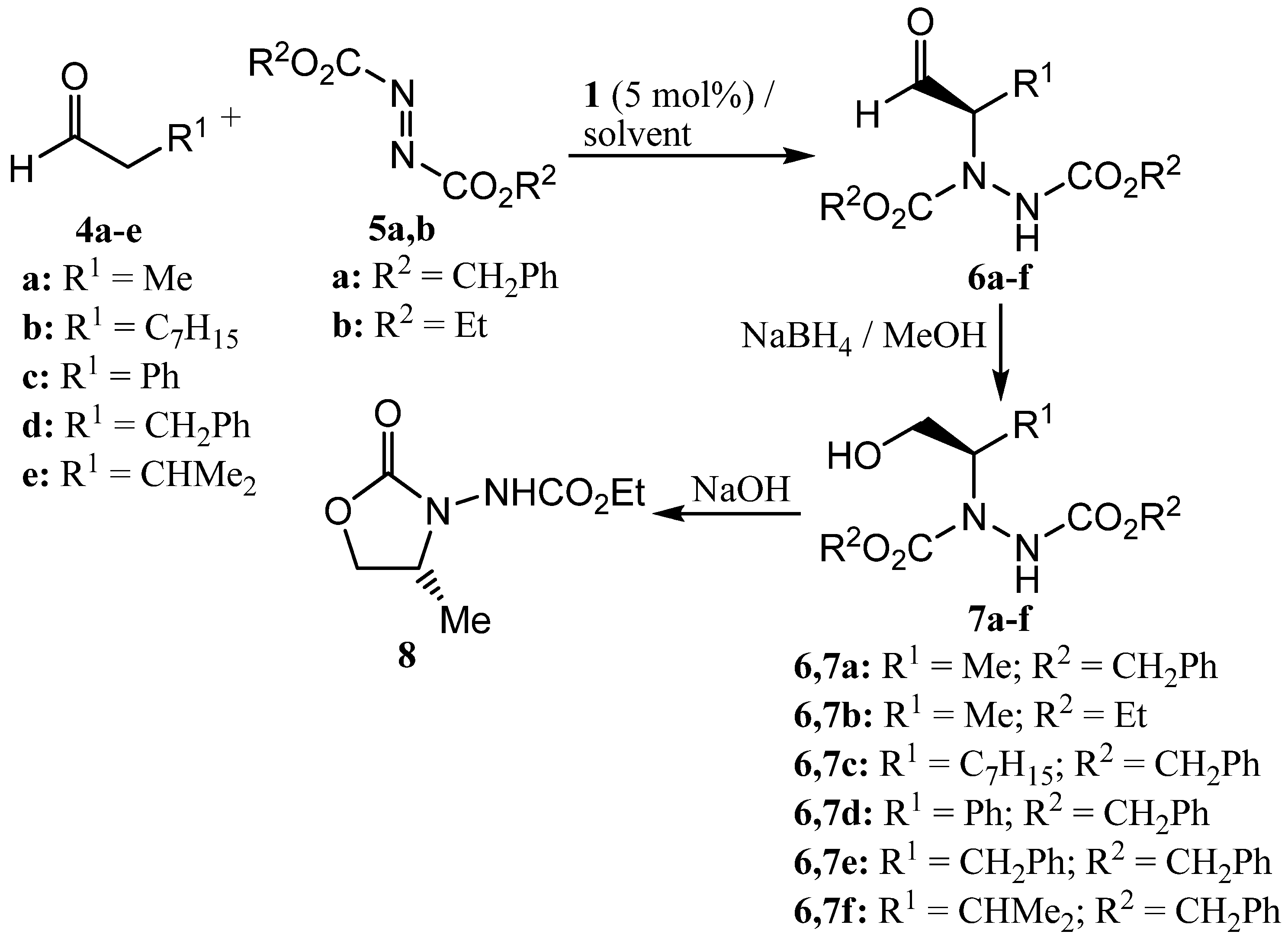
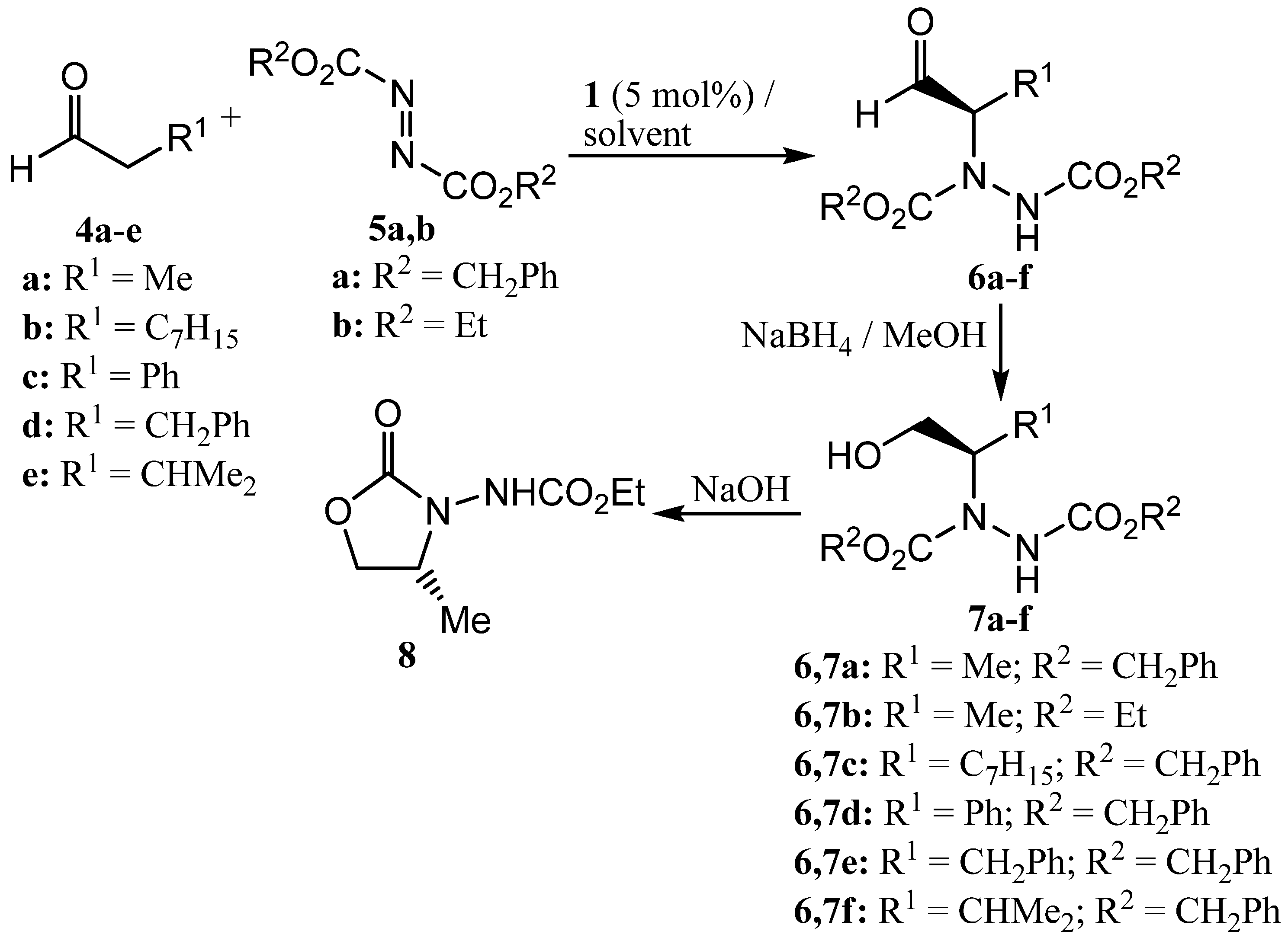
For initial studies, the reaction between propanal (4a) and dibenzyl azodicarboxylate (5a) catalysed by (S)-proline (1, 5 mol%) was selected since there is literature precedent for this reaction [54] and the benzyl groups provided a convenient chromophore for our chiral HPLC system. The initially produced aldehyde 6a was immediately reduced to the more stable alcohol 7a by treatment with sodium borohydride (Scheme 3) and all yields and enantioselectivities refer to the formation of compound 7a. The results are presented in Table 1. Entry 1 of Table 1 shows the result of a control experiment carried out in dichloromethane which confirmed that alcohol 7a was obtained in excellent yield and enantioselectivity under these conditions. The absolute configuration of alcohol 7a was determined to be R- on the basis of the chiral HPLC retention times [64].
Entries 2 and 3 of Table 1 show the results of experiments carried out in cyclic carbonates 2 and 3 under conditions which are otherwise identical to entry 1. In both cases, alcohol 7a was obtained in good yield, but with lower enantiomeric excess than that obtained in dichloromethane. To improve the enantioselectivity of the reaction, the effect of lowering the reaction temperature was investigated. This was only possible with propylene carbonate (3) as solvent, and at 0 °C the reaction in propylene carbonate gave alcohol 7a with excellent enantioselectivity, but in low yield after a standard reaction time of two hours (Table 1, entry 4). To increase the chemical yield, the reaction time was extended to 24 hours (Table 1, entry 5) and under these conditions alcohol 7a was obtained in good yield and with excellent enantioselectivity. An attempt to use diethyl azodicarboxylate (5b) in place of the dibenzyl derivative was not particularly successful as alcohol 7b could not be detected on our HPLC system, so the alcohol was allowed to cyclise to oxazolidinone 8 [46,63] which was obtained in overall 39% yield from propanal. The absolute configuration and enantiomeric excess of compound 8 could be determined by chiral GC analysis, but the latter was significantly lower than that of alcohol 7a (Table 1, entry 6).
Taking the conditions of Table 1, entry 5 as optimal, the applicability of the chemistry to four other aldehydes was investigated (Table 1, entries 7-10). Nonanal (4b) gave alcohol 7c with good enantioselectivity but only moderate yield (Table 1, entry 7). In contrast, phenylacetaldehyde (4c) gave alcohol 7d in good yield, but with a low enantiomeric excess, almost certainly due to the extremely facile racemization of the initially formed aldehyde 6d (Table 1, entry 8). Aldehydes 4d and 4e gave the corresponding alcohols 7e and 7f in both good yield and with excellent enantiomeric excesses. The enantiomeric excesses of alcohols 7a,d,e,f compare favourably with those reported in the literature for the corresponding product prepared using 10 mol% of (S)-proline in dichloromethane [46] or acetonitrile [54] and in each case the (R)-enantiomer of the alcohol was formed predominantly [64].
Having demonstrated that propylene carbonate was a suitable solvent for the asymmetric α-hydrazination of aldehydes, the use of ketones as substrates was investigated. There are only two previous reports of proline-catalysed ketone hydrazination [55,63], indicating that this is a more difficult undertaking than the use of aldehydes as substrates. Cyclohexanone (9a) was chosen as the first test substrate and initial experiments showed that whilst reaction with dibenzyl azodicarboxylate (5a) did indeed occur under the standard conditions developed for aldehyde substrates, the reaction was much slower and required a reaction time of 72 hours to produce a reasonable yield of α-hydrazinoketone 10a (Scheme 4). Compound 10a was found to be more stable than the corresponding aldehyde adducts 6a-f and could be isolated and characterized without the need to reduce the ketone to the corresponding alcohol. Two other ketones 9b,c were converted into α-hydrazinoketones 10b,c under the same conditions and the results are shown in Table 2.
Cyclohexanones 9a,b were found to be reasonable substrates for proline catalysed α-hydrazination in propylene carbonate, giving products 10a,b in good yield and with respectable enantiomeric excesses (Table 2, entries 1 and 2). Butanone (9c) was not as good a substrate, giving only a moderate yield of product 10c and with lower enantioselectivity than that observed for the cyclohexanone derivatives (Table 2, entry 3). Notably however, this substrate did react regioselectively, as no evidence for formation of the product derived from reaction at the methyl group of ketone 9c was observed.
In all the results discussed so far, racemic propylene carbonate has been used as solvent. We have previously shown that for proline-catalysed aldol reactions, use of enantiomerically pure propylene carbonate as solvent can affect both the yield and stereoselectivity of the transformation with matched and mismatched pair effects being seen for the stereochemistry of the catalyst and solvent [45]. Therefore, reactions involving cyclohexanone (9a) as substrate were carried out in enantiomerically pure (R)-propylene carbonate in the presence of both (S)- and (R)-proline as catalyst (Table 2, entries 4 and 5). However, in this case, no significant difference between the reactions carried out in racemic and enantiomerically pure solvent was observed, nor was there a difference between reactions carried out using the two enantiomers of proline in (R)-propylene carbonate.
3. Experimental
3.1. Chemicals and instrumentation
Propylene carbonate (3) was distilled from CaH2 under reduced pressure and stored over molecular sieves. Other commercially available products (Aldrich, Acros) were used as received. Chromatographic separations were performed using silica gel 60 (230–400 mesh, Davisil). Infrared spectra were recorded at room temperature on a Varian 800 FT-IR Scimitar series spectrometer. Optical rotation measurements were conducted on a Polaar 2001 Optical Activity automatic polarimeter at the sodium D-line using a 0.25 dm thermostated cell and a suitable solvent that is reported along with the concentration (g/100 mL). High- and low-resolution electrospray ionization (ES) mass spectra were recorded on a Waters LCT Premier LCMS spectrometer using direct injection of the sample in MeOH. 1H- and 13C-NMR were recorded in CDCl3 on a Bruker Avance 300 spectrometer at 300 and 75 MHz respectively. 13C-NMR spectra were also recorded at 125 MHz on a JEOL Lambda 500 spectrometer. Chiral gas chromatrogaphy was performed on a Varian450-GC instrument with a TCD detector using a Supelco Gamma DEX 120 fused silica capillary column (30 m × 0.25 mm × 0.25 µm film thickness) with hydrogen as the carrier gas (flow rate 3.5 mL/min). Analyses were performed with an initial column temperature of 165 °C, followed by a ramp rate 0.8 °C/min to 210 °C, then hold at 210 °C for 80 minutes. Chiral HPLC was performed using a Varian ProStar system comprising binary pumping modules, a diode array detector and an autosampler and equipped with a Daicel Chiralcel OD-H, AD-H or AS-H column (25 cm by 4.6 mm), using a mixture of isopropanol and hexane as eluent. The HPLC retention times of the enantiomeric products were determined using racemic samples of compounds 7a-f and 10a-c which were prepared from reactions catalysed by (R,S)-proline in dichloromethane or propylene carbonate at room temperature.
3.2. Synthesis of compound 8 [46,63]
To a stirred solution of diethyl azodicarboxylate (5b, 0.18 mL, 1.00 mmol) and propanal (4a, 0.11 mL, 1.50 mmol) in propylene carbonate (3, 3 mL) was added (S)-proline (1, 5.9 mg, 0.05 mmol). The reaction was stirred at 0 °C for 24 hours, then quenched by the addition of H2O (5 mL), extracted with Et2O (20 mL), washed with H2O (4 × 10 mL) and dried (Na2SO4). The solvent and excess propanal were evaporated in vacuo and the residue was dissolved in ethanol (10 mL) and NaBH4 (40.0 mg, 1.05 mmol) was added. The reaction was stirred for 10 minutes at 0 °C, then aqueous ammonium chloride solution (10 mL) and EtOAc (20 mL) were added. The organic layer was separated, dried (Na2SO4) and the solvent evaporated in vacuo. The residue was purified by silica gel chromatography eluting with hexane:EtOAc (85:15) to give alcohol 7b. Compound 7b was dissolved in MeOH (2 mL) and 0.5 M aqueous NaOH (2.5 mL) and the reaction stirred for 2 hours. MeOH was then evaporated in vacuo and the aqueous phase extracted with EtOAc (3 × 15 mL). The combined organic layers were dried (MgSO4) and the solvent evaporated in vacuo to give compound 8 (0.91 g, 39%) as a yellow oil. νmax(neat) 3362 w, 3167 w, 2968 m, 2927 m, 2879 m, 1721 s and 1538 cm−1 m; [α]D20 -11.0 (c=0.4, CHCl3) (lit [65]. [α]D28 -19.9 (c=1.4, CHCl3)); δH 1.3–1.4 (6H, m, 2xCH3), 3.87 (1H, t J 8.4 Hz, CH2CH), 4.1–4.2 (1H, m, CHN), 4.25 (2H, q J 6.9 Hz, CH2CH3), 4.51 (1H, t J 8.4 Hz, CH2CH), 6.5–6.6 (1H, m, NH); δC(75 MHz) 14.6 (CH3), 16.8 (CH3), 53.2 (CHN), 63.2 (CH2O), 69.1 (CH2O), 155.6 (C=O), 157.9 (C=O); Chiral GC retention times 11.1 (minor) and 12.7 (major) minutes.
3.3. General procedure for the synthesis of alcohols 7
To a stirred solution of dibenzyl azodicarboxylate (5a, 0.3 g, 1.0 mmol) and an aldehyde 4a–e (1.5 mmol) in propylene carbonate (3, 1 mL) was added (S)-proline (1, 5.9 mg, 0.05 mmol). The reaction was stirred at 0 °C for 24 hours, then quenched by the addition of H2O (5 mL), extracted with Et2O (20 mL), washed with H2O (4 × 10 mL) and dried (Na2SO4). Volatiles were removed by evaporation in vacuo and the residue was dissolved in ethanol (10 mL) and NaBH4 (40.0 mg, 1.05 mmol) was added. The reaction was stirred for 10 minutes at 0 °C, then aqueous ammonium chloride solution (10 mL) and EtOAc (20 mL) were added. The organic layer was separated, dried (Na2SO4) and the solvent evaporated in vacuo. The residue was purified by silica gel chromatography eluting with hexane:EtOAc (85:15) to give alcohols 7a,c–f.
Dibenzyl (R)-1-(1-methyl-2-hydroxyethyl)hydrazine-1,2-dicarboxylate (7a) [54]. Obtained as a yellow oil (0.27 g, 76%). νmax(neat) 3271 br, 2955 s and 1715 cm-1 s; [α]D20 -32.0 (c = 0.4, CHCl3); δH 0.85 (3H, d J = 6.9 Hz, CH3), 3.4–3.5 (2H, m, CH2OH), 4.0–4.5 (2H, m, CH and OH), 4.9–5.2 (4H, m, 2 × CH2Ph), 7.0–7.4 (10H, m, ArH); δC(75 MHz) 15.5 (CH3), 56.4 (CH) 63.6 (CH2OH), 66.1(OCH2Ph) 68.6 (OCH2Ph), 128.5 (ArCH), 128.6 (ArCH), 128.9 (ArCH), 129.0 (ArCH), 135.8 (ArC), 136.3 (ArC), 157.0 (NCO2), 159.0 (NCO2); HPLC (Chiralpak AS-H using hexane: iPrOH (95:5) as solvent at a flow rate of 0.5 mL/min) retention times 22.2 (major) and 24.7 (minor) minutes.
Dibenzyl (R)-1-(1-heptyl-2-hydroxyethyl)hydrazine-1,2-dicarboxylate (7c). Obtained as a colourless oil (0.18 g, 41%). νmax(neat) 3267 br, 2963 s and 1716 cm-1 s; [α]D20 -26.0 (c = 0.4, CHCl3); δH 0.7–0.9 (3H, m, CH3), 1.0–1.4 (12H, m, Me(CH2)6), 3.2–3.5 (2H, m, CH2OH), 4.0–4.4 (2H, m, CH and OH), 5.0–5.2 (4H, m, 2×CH2Ph), 6.90 (1H, s, NH), 7.0–7.4 (10H, m, ArH); δC(75 MHz) 13.9 (CH3), 22.5 (CH2), 26.0 (CH2), 27.9 (CH2), 29.0 (CH2), 29.3 (CH2), 31.6 (CH2), 60.8 (NCH), 62.1 (CH2OH), 68.2 (2 × OCH2Ph), 127.7 (ArCH), 128.0 (ArCH), 128.1 (ArCH), 128.2 (ArCH), 128.4 (ArCH), 128.5 (ArCH), 135.3 (ArC), 135.9 (ArC), 157.4 (NCO2), 159.1 (NCO2); m/z(ES+) 465 (M+Na+, 10); Found (ES+) 465.2348, C25H34N2O5Na, (M+Na+) requires 465.2365; HPLC (Chiralpak AS-H using hexane: iPrOH (90:10) as solvent at a flow rate of 0.5 mL/min) retention times 20.4 (minor) and 43.0 (major) minutes.
Dibenzyl (R)-1-(1-phenyl-2-hydroxyethyl)hydrazine-1,2-dicarboxylate (7d). Obtained as a white solid (0.29 g, 70%). m.p. 140–141 °C; νmax(neat) 3273 br, 2946 s and 1711 cm-1 s; [α]D20 -38.0 (c = 0.4, CHCl3); δH 3.2–3.5 (2H, m, CH2OH), 4.0–4.5 (2H, m, CH and OH), 4.8–5.1 (4H, m, 2 × CH2Ph) 7.0–7.4 (15H, m, ArH); δC(75 MHz) 56.7 (CH), 63.5 (CH2OH), 65.5 (CH2Ph), 70.2 (CH2Ph), 127.8 (ArCH), 127.9 (ArCH), 128.2 (ArCH), 128.6 (ArCH), 129.0 (ArCH), 135.7 (ArC), 136.2 (ArC), 156.3 (NCO2), 159.3 (NCO2); HPLC (Chiralpak AD-H using hexane: iPrOH (90:10) as solvent at a flow rate of 0.5 mL/min) retention times 46.2 (minor) and 48.7 (major) minutes.
Dibenzyl (R)-1-(1-benzyl-2-hydroxyethyl)hydrazine-1,2-dicarboxylate (7e) [54]. Obtained as a white solid (0.33 g, 76%). m.p. 105–106 °C (lit [68]. 110–116 °C); νmax(neat) 3272 br, 2963 s and 1716 cm−1 s; [α]D20 +11.1 (c = 0.9, CHCl3) (lit [68] [α]D25 +11.4 (c = 1.8, CHCl3)); δH 2.4–2.7 (2H, m, CH2Ph), 3.3–3.6 (2H, m, CH2OH) 4.4–4.6 (2H, m, CH and OH), 4.9–5.2 (4H, m, 2 × OCH2Ph), 6.8–7.4 (15H, m, ArH); δC(75 MHz) 34.5 (CH2), 60.2 (CH), 61.8 (CH2OH), 68.0 (OCH2Ph), 68.3 (OCH2Ph), 128.2 (ArCH), 128.5 (ArCH), 128.7 (ArCH), 128.8 (ArCH), 128.9 (ArCH), 129.0 (ArCH), 129.1 (ArCH), 129.2 (ArCH), 129.4 (ArCH), 135.1 (ArC), 135.7 (ArC), 137.3 (ArC), 156.7 (NCO2), 158.8 (NCO2); m/z(ES+) 443 (M+Na+, 50), 863 (2M+Na+), 1283 (3M+Na+); Found (ES+) 443.1550, C24H24N2O5Na, (M+Na+) requires 443.1583; HPLC (Chiralpak AS-H using hexane: iPrOH (85:15) as solvent at a flow rate of 1.0 mL/min) retention times 13.4 (major) and 31.5 (minor) minutes.
Dibenzyl (R)-1-(1-(1-methyl)ethyl-2-hydroxyethyl)hydrazine-1,2-dicarboxylate (7f) [54] Obtained as a yellow oil (0.34 g, 87%). νmax(neat) 3272 br, 2963 s and 1716 cm−1 s; [α]D20 -21.0 (c = 0.4, CHCl3); δH 0.6–1.0 (6H, m, 2 × CH3), 1.4–1.7 (1H, m, CHMe2) 3.3–4.4 (4H, m, NCHCH2OH) 4.9–5.2 (4H, m, 2 × CH2Ph) 6.7–6.9 (1H, br s, NH) 7.1–7.4 (10H, m, ArH); δC (75 MHz) 19.3 (CH3), 20.0 (CH3), 27.5 (CH), 60.4 (CH), 67.2 (CH2OH), 68.4 (2 × OCH2Ph), 127.7 (ArCH), 128.0 (ArCH), 128.1 (ArCH), 128.2 (ArCH), 128.4 (ArCH), 128.6 (ArCH), 135.2 (ArC), 135.9 (ArC), 156.4 (NCO2), 157.4 (NCO2); HPLC (Chiralpak OD-H using hexane: iPrOH (90:10) as solvent at a flow rate of 1.0 mL/min) retention times 14.4 (major) and 17.7 (minor) minutes.
3.4. General procedure for the synthesis of ketones 10a-d
To a stirred solution of dibenzyl azodicarboxylate (5a, 0.3 g, 1.0 mmol) and a ketone (1.5 mmol) in propylene carbonate (3, 1 mL), was added (S)-proline (1, 5.9 mg, 0.05 mmol). The reaction was stirred at 0 °C for 24 hours, then quenched by the addition of H2O (5 mL), extracted with Et2O (10 mL), washed with H2O (4 × 10 mL) and dried (Na2SO4). Volatiles were removed by evaporation in vacuo and the residue was purified by silica gel chromatography eluting with hexane:EtOAc (70:30) and recrystallized with cold isopropanol to give ketones 10-d.
Dibenzyl (R)-1-(2-oxocyclohexyl)hydrazine-1,2-dicarboxylate (10a) [66]. Obtained as a colourless oil (0.28 g, 71%). νmax(neat) 3271 br, 2965 s, 1797 s, 1743 s and 1679 cm−1 m; [α]D20 -21.0 (c = 0.1, CH2Cl2) (lit [66]. [α]D20 -24.1 (c=0.1, CH2Cl2)); δH 1.6–2.5 (8H, m, (CH2)4), 5.1-5.2 (5H, m, 2 × CH2Ph and NCH), 6.88 (1H, br, NH), 7.3–7.5 (10H, m, ArH); δC (75 MHz) 24.4 (CH2), 26.7 (CH2), 30.8 (CH2), 41.2 (CH2), 66.5 (2 × OCH2Ph), 67.9 (NCH), 127.7 (ArCH), 128.0 (ArCH), 128.1 (ArCH), 128.2 (ArCH), 128.5 (2 × ArCH), 135.9 (2 × ArC), 156.3 (2 × NCO2), 206.9 (CO); HPLC (Chiralpak OD-H using hexane: iPrOH (90:10) as solvent at a flow rate of 0.9 mL/min) retention times 16.9 (minor) and 22.6 (major) minutes.
Dibenzyl (R)-1-(2-oxo-4-oxacyclohexyl)hydrazine-1,2-dicarboxylate (10b). Obtained as a colourless oil (0.20 g, 51%). νmax(neat) 3272 br, 2965 s, 1798 s and 1716 cm−1 s; [α]D20 -25.0 (c = 0.4, CHCl3); δH 2.4–2.6 (2H, m, CH2CO), 3.4–3.6 (2H, m, CH2O), 4.1–4.5 (2H, m, CH2O), 4.6–5.3 (5H, m, 2 × CH2Ph and NCH), 6.5–6.7 (1H, br, NH), 7.2–7.4 (10H, m, ArH); δC (125 MHz) 25.3 (CH2), 42.3 (CH2), 64.2 (CH2), 67.4 (CH), 67.8 (CH2), 67.9 (CH2), 128.0 (ArCH), 128.1 (ArCH), 128.2 (ArCH), 128.3 (ArCH), 128.4 (ArCH), 128.5 (ArCH), 135.4 (ArC), 135.6 (ArC), 155.4 (NCO2), 155.7 (NCO2), 203.4 (CO); m/z(ES+) 421 (M+Na+, 100), 819 (2M+Na+, 85), 1117 (3M+Na+, 32); Found (ES+) 819.2853, C42H44N4O12Na, (2M+Na+) requires 819.2812; HPLC (Chiralpak OD-H using hexane: iPrOH (90:10) as solvent at a flow rate of 0.9 mL/min) retention times 37.1 (minor) and 57.3 (major) minutes.
Dibenzyl (R)-1-(1-methyl-2-oxopropyl)hydrazine-1,2-dicarboxylate (10c) [67,69]. Obtained as colourless needles (0.09 g, 31%). m.p. 59–60 °C; νmax(neat) 3255 m, 2958 w, 2884 w, 1749 s, 1703 s and 1512 cm−1 m; [α]D20 -16.0 (c = 0.4, CHCl3); δH 1.36 (3H, d J = 16.6 Hz, CH3CH), 2.01 (3H, s, CH3CO), 4.9–5.1 (5H, m, 2xCH2 and NCH), 6.6–6.8 (1H, br, NH), 7.1–7.4 (10H, m, ArH); δC (75MHz): 13.1 (CH3), 26.3 (CH3), 67.7 (OCH2Ph), 67.9 (OCH2Ph), 68.5 (NCH), 127.8 (ArCH), 128.0 (ArCH), 128.1 (ArCH), 128.2 (ArCH), 135.7 (ArC), 135.8 (ArC), 156.1 (NCO2), 156.4 (NCO2), 206.4 (CO); HPLC (Chiralpak OD-H using hexane: iPrOH (93:7) as solvent at a flow rate of 0.9 mL/min) retention times 12.5 (minor) and 15.0 (major) minutes.
4. Conclusions
Propylene carbonate is a sustainable and environmentally friendly replacement for dichloromethane and acetonitrile in proline catalysed α-hydrazinations of aldehydes and ketones. Comparable enantioselectivities to those seen with conventional solvents can be obtained, even with a lower catalyst loading. Use of enantiomerically pure propylene carbonate is not advantageous for this reaction.
Acknowledgements
The authors thank Shasun Pharma Solutions Ltd for a generous donation of (R)-propylene carbonate.
References and Notes
- Hayashi, T.; Asai, T.; Ogoshi, H. Conformational Analysis of β-Turn Structure in Tetrapeptides Containing Proline or Proline Analogs. Tetrahedron Lett. 1997, 38, 3039–3042. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. β-Turns in Proteins. J. Mol. Biol. 1997, 115, 135–175. [Google Scholar] [CrossRef]
- Jones, I.G.; Jones, W.; North, M. Conformational Analysis of Peptides and Pseudopeptides Incorporating an endo-(2S,3R)-Norborn-5-ene Residue as a Turn Inducer. J. Org. Chem. 1998, 63, 1505–1513. [Google Scholar] [CrossRef]
- Belokon, Y.N.; Bulychev, A.G.; Vitt, S.V.; Struchkov, Y.T.; Batsanov, A.S.; Timofeeva, T.V.; Tsyryapkin, V.A.; Ryzhov, M.G.; Lysova, L.A.; Bakhmutov, V.I.; Belikov, V.M. General method of diastereo- and enantio-selective synthesis of β-hydroxy-α-amino acids by condensation of aldehydes and ketones with glycine. J. Am. Chem. Soc. 1985, 107, 4252–4259. [Google Scholar] [CrossRef]
- North, M.; Zagotto, G. Asymmetric Desymmetrisation of Meso-Norbornene Anhydrides Utilising Methyl Prolinate as a Chiral Reagent. Synlett 1995, 639–640. [Google Scholar] [CrossRef]
- Albers, T.; Biagini, S.C.G.; Hibbs, D.E.; Hursthouse, M.B.; Malik, K.M.A.; North, M.; Uriarte, E.; Zagotto, G. Desymmetrisation of Meso-Anhydrides utilising (S)-Proline Derivatives. Synthesis 1996, 393–398. [Google Scholar] [CrossRef]
- List, B. Asymmetric Aminocatalysis. Synlett 2001, 1675–1686. [Google Scholar] [CrossRef]
- Gröger, H.; Wilken, J. The Application of L-Proline as an Enzyme Mimic and Further New Asymmetric Syntheses Using Small Organic Molecules as Chiral Catalysts. Angew. Chem. Int. Ed. 2001, 40, 529–532. [Google Scholar] [CrossRef]
- List, B. Proline-catalyzed asymmetric reactions. Tetrahedron 2002, 58, 5573–5590. [Google Scholar] [CrossRef]
- Jarvo, E.R.; Miller, S.J. Amino acids and peptides as asymmetric organocatalysts. Tetrahedron 2002, 58, 2481–2495. [Google Scholar] [CrossRef]
- List, B. Enamine Catalysis Is a Powerful Strategy for the Catalytic Generation and Use of Carbanion Equivalents. Acc. Chem. Res. 2004, 37, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Kazmaier, U. Amino Acids–Valuable Organocatalysts in carbohydrate Synthesis. Angew. Chem. Int. Ed. 2005, 44, 2186–2188. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H. Asymmetric organocatalysis. Tetrahedron 2007, 63, 9267–9331. [Google Scholar] [CrossRef]
- Gaunt, M.J.; Johansson, C.C.C.; McNally, A.; Vo, N.T. Enantioselective organocatalysis. Drug Discov. Today 2007, 12, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A.; Massi, A. Asymmetric Organocatalysis: From Infancy to Adolescence. Angew. Chem. Int. Ed. 2008, 47, 4638–4660. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Asymmetric Aminocatalysis—Gold Rush in Organic Chemistry. Angew. Chem. Int. Ed. 2008, 47, 6138–6171. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H. Recent Developments in Asymmetric Organocatalysis; RSC Publishing: Cambridge, UK, 2010. [Google Scholar]
- Brogan, A.P.; Dickerson, T.J.; Janda, K.D. Enamine-Based Aldol Organocatalysis in Water: Are They Really “All Wet”? Angew. Chem. Int. Ed. 2006, 45, 8100–8102. [Google Scholar] [CrossRef] [PubMed]
- Gruttadauria, M.; Giacalone, F.; Noto, R. Water in Stereoselective Organocatalytic Reactions. Adv. Synth. Catal. 2009, 351, 33–57. [Google Scholar] [CrossRef]
- Toma, Š.; Mečiarova, M.; Šebesta, R. Are Ionic Liquids Suitable Media for Organocatalytic Reactions? Eur. J. Org. Chem. 2009, 321–327. [Google Scholar] [CrossRef]
- Blackmond, D.G.; Armstrong, A.; Coombe, V.; Wells, A. Water in Organocatalytic Processes: Debunking the Myths. Angew. Chem. Int. Ed. 2007, 46, 3798–3800. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Liu, W.; Zhang, Y.; Wang, H. Do We Understand the Recyclability of Ionic Liquids? Chem. Eur. J. 2009, 15, 1804–1810. [Google Scholar] [CrossRef] [PubMed]
- Zotova, N.; Franzke, A.; Armstrong, A.; Blackmond, D.G. Clarification of the Role of Water in Proline-Mediated Aldol Reactions. J. Am. Chem. Soc. 2007, 129, 15100–15101. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, B.; Rantanen, T.; Bolm, C. Solvent-Free Asymmetric Organocatalysis in a Ball Mill. Angew. Chem. Int. Ed. 2006, 46, 6924–6926. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, B.; Bruckmann, A.; Bolm, C. A Highly Efficient Asymmetric Organocatalytic Aldol Reaction in a Ball Mill. Chem. Eur. J. 2007, 13, 4710–4722. [Google Scholar] [CrossRef] [PubMed]
- Schäffner, B.; Schäffner, F.; Verevkin, S.P.; Börner, A. Organic Carbonates as Solvents in Synthesis and Catalysis. Chem. Rev. 2010, 110, 4554–4581. [Google Scholar] [CrossRef] [PubMed]
- North, M.; Omedes-Pujol, M. Catalytic, asymmetric cyanohydrin synthesis in propylene carbonate. Tetrahedron Lett. 2009, 50, 4452–4454. [Google Scholar] [CrossRef]
- North, M.; Omedes-Pujol, M. Kinetics and mechanism of vanadium catalysed asymmetric cyanohydrin synthesis in propylene carbonate. Belstein J. Org. Chem. 2010, 6, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.H. Reactive Applications of Cyclic Alkylene Carbonates. Ind. Eng. Chem. Res. 2003, 42, 663–674. [Google Scholar] [CrossRef]
- Silva, L.B.; Freitas, L.C.G. Structural and thermodynamic properties of liquid ethylene carbonate and propylene carbonate by Monte Carlo Simulations. J. Mol. Structure Theochem. 2007, 806, 23–34. [Google Scholar] [CrossRef]
- Yoshida, M.; Ihara, M. Novel Methodologies for the Synthesis of Cyclic Carbonates. Chem. Eur. J. 2004, 10, 2886–2893. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.-L.; Luo, S.-L.; Yin, S.-F.; Au, C.-T. The direct transformation of carbon dioxide to organic carbonates over heterogeneous catalysts. Appl. Cat. A 2009, 366, 2–12. [Google Scholar] [CrossRef]
- Sakakura, T.; Kohno, K. The synthesis of organic carbonates from carbon dioxide. Chem. Commun. 2009, 1312–1330. [Google Scholar] [CrossRef] [PubMed]
- North, M.; Pasquale, R.; Young, C. Synthesis of Cyclic Carbonates from Epoxides and CO2. Green Chem. 2010, 12, 1514–1539. [Google Scholar] [CrossRef]
- Ballivet-Tkatchenko, D.; Dibenedetto, A. Synthesis of Linear and Cyclic Carbonates. In Carbon Dioxide as Chemical Feedstock; Aresta, M., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp. 169–212. [Google Scholar]
- Meléndez, J.; North, M.; Pasquale, R. Synthesis of cyclic carbonates from atmospheric pressure carbon dioxide using exceptionally active aluminium(salen) complexes as catalysts. Eur. J. Inorg. Chem. 2007, 3323–3326. [Google Scholar] [CrossRef]
- Meléndez, J.; North, M.; Pasquale, R. Mechanism of cyclic carbonate synthesis from epoxides and CO2. Angew. Chem., Int. Ed. 2009, 48, 2946–2948. [Google Scholar]
- Meléndez, J.; North, M.; Villuendas, P. One-component catalysts for cyclic carbonate synthesis. Chem. Commun. 2009, 2577–2579. [Google Scholar] [CrossRef] [PubMed]
- Clegg, W.; Harrington, R.W.; North, M.; Pasquale, R. Cyclic carbonate synthesis catalysed by bimetallic aluminium(salen) complexes. Chem. Eur. J. 2010, 16, 6828–6843. [Google Scholar] [CrossRef] [PubMed]
- North, M.; Villuendas, P.; Young, C. A gas phase flow reactor for ethylene carbonate synthesis from waste CO2. Chem. Eur. J. 2009, 11454–11457. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, J.; North, M.; Villuendas, P.; Young, C. One-component bimetallic aluminium(salen)-based catalysts for cyclic carbonate synthesis and their immobilization. Dalton Trans. 2011, 40, 3885–3902. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, I.S.; North, M.; Pasquale, R.; Thursfield, A. An integrated approach to energy and chemicals production. Energy Environ. Sci. 2010, 3, 212–215. [Google Scholar] [CrossRef]
- North, M.; Pizzato, F.; Villuendas, P. Organocatalytic, asymmetric aldol reactions with a sustainable catalyst in a green solvent. ChemSusChem 2009, 2, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Clegg, W.; Harrington, R.W.; North, M.; Pizzato, F.; Villuendas, P. Cyclic carbonates as sustainable solvents for proline-catalysed aldol reactions. Tetrahedron: Asymmetry 2010, 21, 1262–1271. [Google Scholar] [CrossRef]
- North, M.; Villuendas, P. A Chiral Solvent Effect in Asymmetric Organocatalysis. Org. Lett. 2010, 12, 2378–2381. [Google Scholar] [CrossRef] [PubMed]
- Bøgevig, A.; Juhl, K.; Kumaragurubaran, N.; Zhuang, W.; Jørgensen, K.A. Direct Organo-Catalytic Asymmetric α-Amination of Aldehydes–A Simple Approach to Optically Active α-Amino Aldehydes, α-Amino Alcohols, and α-Amino Acids. Angew. Chem. Int. Ed. 2002, 41, 1790–1793. [Google Scholar] [CrossRef]
- Iwamura, H.; Mathew, S.P.; Blackmond, D.G. In Situ Catalyst Improvement in the Proline-Mediated α-Amination of Aldehydes. J. Am. Chem. Soc. 2004, 126, 11770–11771. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, H.; Wells, D.H., Jr.; Mathew, S.P.; Klussmann, M.; Armstrong, A.; Blackmond, D.G. Probing the Active Catalyst in Product-Accelerated Proline-Mediated Reactions. J. Am. Chem. Soc. 2004, 126, 16312–16313. [Google Scholar] [CrossRef] [PubMed]
- Umbreen, S.; Brockhaus, M.; Ehrenberg, H.; Schmidt, B. Norstatines from Aldehydes by Sequential Organocatalytic α-Amination and Passerini Reaction. Eur. J. Org. Chem. 2006, 4585–4595. [Google Scholar] [CrossRef]
- Baumann, T.; Vogt, H.; Bräse, S. The Proline-Catalyzed Asymmetric Amination of Branched Aldehydes. Eur. J. Org. Chem. 2007, 266–282. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Kitajima, M.; Takayama, H. First Asymmetric Total Syntheses of Cernuane-Type Lycopodium Alkaloids, Cernuine, and Cermizine D. Org. Lett. 2008, 10, 1987–1990. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Peng, F.; Zhang, H.; Shao, Z. Organocatalytic Synthesis of Terminal Propargylamine Derivatives by Tandem Amination–Alkynylation. Synlett 2009, 3287–3290. [Google Scholar]
- Blackmond, D.G.; Moran, A.; Hughes, M.; Armstrong, A. Unusual Reversal of Enantioselectivity in the Proline-Mediated α-Amination of Aldehydes Induced by Tertiary Amine Additives. J. Am. Chem. Soc. 2010, 132, 7598–7599. [Google Scholar] [CrossRef] [PubMed]
- List, B. Direct Catalytic Asymmetric α-Amination of Aldehydes. J. Am. Chem. Soc. 2002, 124, 5656–5657. [Google Scholar] [CrossRef] [PubMed]
- Kumaragurubaran, N.; Juhl, K.; Zhuang, W.; Bøgevig, A.; Jørgensen, K.A. Direct L-Proline-Catalyzed Asymmetric α-Amination of Ketones. J. Am. Chem. Soc. 2002, 124, 6254–6255. [Google Scholar] [CrossRef] [PubMed]
- Suri, J.T.; Steiner, D.D.; Barbas, C.F., III. Organocatalytic Enantioselective Synthesis of Metabotropic Glutamate Receptor Ligands. Org. Lett. 2005, 7, 3885–3888. [Google Scholar] [CrossRef] [PubMed]
- Kotkar, S.P.; Chavan, V.B.; Sudalai, A. Organocatalytic Sequential α-Amination-Horner-Wadsworth-Emmons Olefination of Aldehydes: Enantioselective Synthesis of γ-Amino-α,β-Unsaturated Esters. Org. Lett. 2007, 9, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Baumann, T.; Bächle, M.; Hartmann, C.; Bräse, S. Thermal Effects in the Organocatalytic Asymmetric α-Amination of Disubstituted Aldehydes with Azodicarboxylates: A High-Temperature Organocatalysis. Eur. J. Org. Chem. 2008, 2207–2212. [Google Scholar] [CrossRef]
- Hartmann, C.E.; Gross, P.J.; Nieger, M.; Bräse, S. Towards an asymmetric synthesis of the bacterial peptide deformylase (PDF) inhibitor fumimycin. Org. Biomol. Chem. 2009, 7, 5059–5062. [Google Scholar] [CrossRef] [PubMed]
- Ait-Youcef, R.; Sbargoud, K.; Moreau, X.; Greck, C. Asymmetric α-Amination of Chiral Protected β-Hydroxyaldehydes Catalyzed by Proline. Synlett 2009, 3007–3010. [Google Scholar]
- Kalch, D.; De Rycke, N.; Moreau, X.; Greck, C. Efficient syntheses of enantioenriched (R)-pipecolic acid and (R)-proline via electrophilic organocatalytic amination. Tetrahedron Lett. 2009, 50, 492–494. [Google Scholar] [CrossRef]
- Hartmann, C.E.; Baumann, T.; Bächle, M.; Bräse, S. Asymmetric synthesis of deuterated and fluorinated aromatic α,α-disubstituted amino acid derivatives. Tetrahedron: Asymmetry 2010, 21, 1341–1349. [Google Scholar] [CrossRef]
- Kotrusz, P.; Alemayehu, S.; Toma, Š.; Schmalz, H.-G.; Adler, A. Enantioselective Organocatalysis in Ionic Liquids: Addition of Aliphatic Aldehydes and Ketones to Diethyl Azodicarboxylate. Eur. J. Org. Chem. 2005, 4904–4911. [Google Scholar] [CrossRef]
- Liu, P.-M.; Chang, C.; Reddy, R.J.; Ting, Y.-F.; Kuan, H.-H.; Chen, K. Remarkable Reaction Rate and Excellent Enantioselective Direct α-Amination of Aldehydes with Azodicarboxylates Catalyzed by Pyrrolidinylcamphor-Derived Organocatalysts. Eur. J. Org. Chem. 2010, 42–46. [Google Scholar] [CrossRef]
- Gosiewska, S.; Soni, R.; Clarkson, G.J.; Wills, M. Synthesis and use of a stable aminal derived from TsDPEN in asymmetric organocatalysis. Tetrahedron Lett. 2010, 51, 4214–4217. [Google Scholar] [CrossRef]
- Lacoste, E.; Vaique, E.; Berlande, M.; Pianet, I.; Vincent, J.-M.; Landais, Y. Benzimidazole-pyrrolidine/H+ (BIP/H+), a Highly Reactive Organocatalyst for Asymmetric Processes. Eur. J. Org. Chem. 2007, 167–177. [Google Scholar] [CrossRef]
- Thomassigny, C.; Primm, D.; Geck, C. Amino acid-catalyzed asymmetric α-amination of carbonyls. Tetrahedron Lett. 2006, 47, 1117–1119. [Google Scholar] [CrossRef]
- George, S.; Suryavanshi, G.S.; Sudalai, A. A short enantioselective synthesis of (–)-bestatin via L-proline-catalyzed α-amination of an aldehyde. Tetrahedron Lett. 2008, 49, 6791–6793. [Google Scholar] [CrossRef]
- Ait-Youcef, R.; Kalch, D.; Moreau, X.; Thomassigny, C.; Greck, C. Asymmetric α-Amination of Aldehydes and Ketones Catalyzed by tert-Butoxy-L-Proline. Lett. Org. Chem. 2009, 6, 377–380. [Google Scholar] [CrossRef]
Sample Availability: Not Available. |
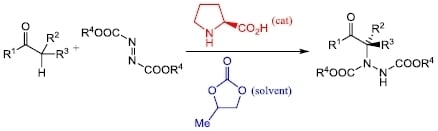
Figure 1.
(a) (S)-Proline 1. (b) Ethylene carbonate 2. (c) Propylene carbonate 3.
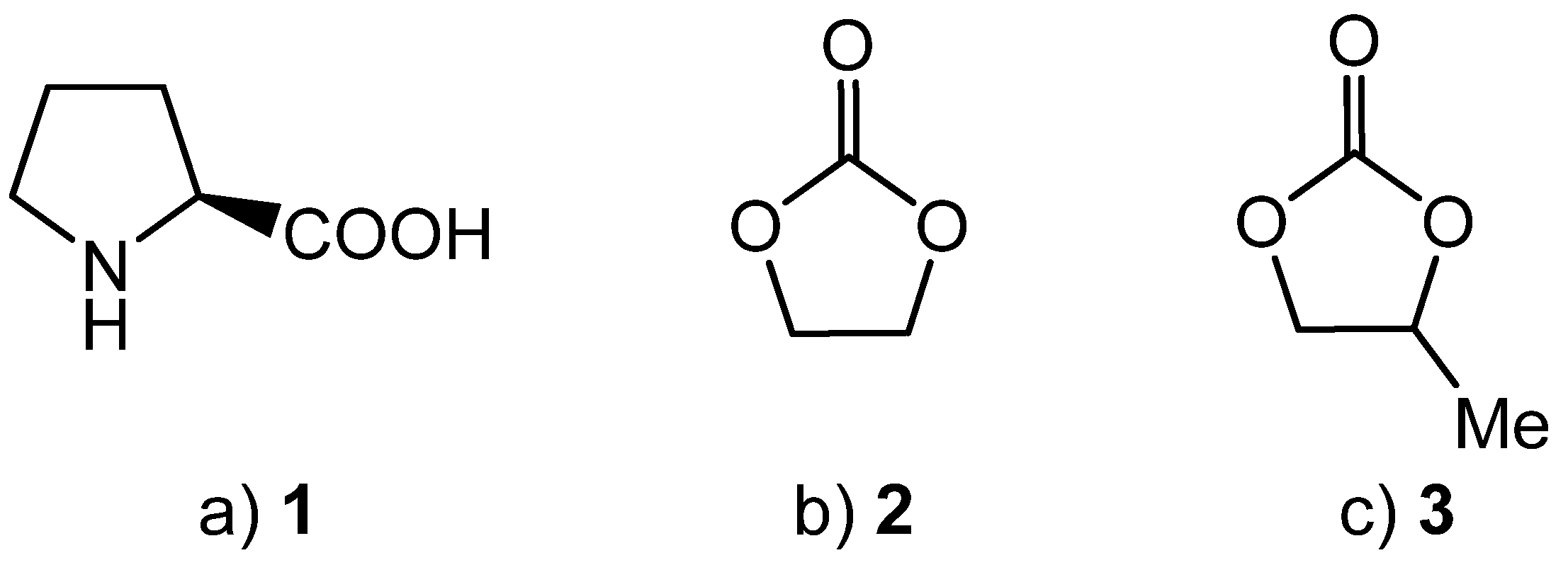
Scheme 1.
Synthesis of cyclic carbonates from epoxides and carbon dioxide.
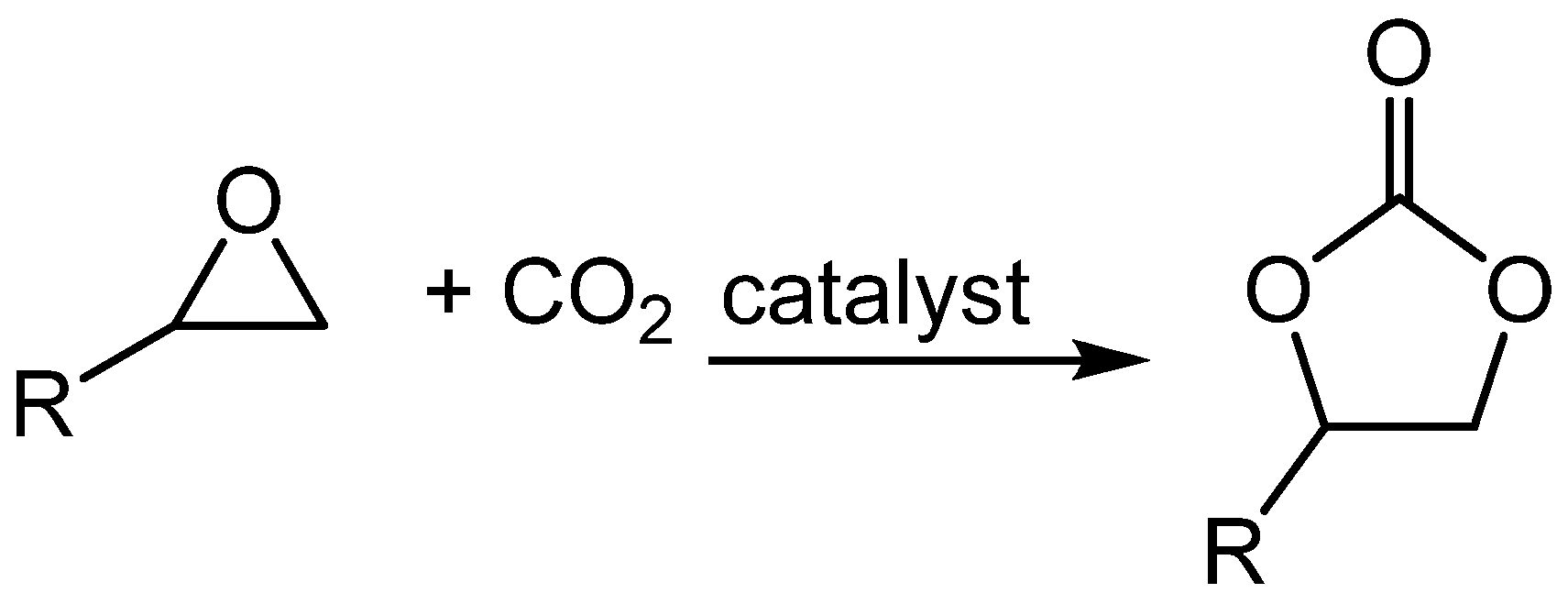
Scheme 2.
Proline-catalysed α-hydrazination of carbonyl compounds.
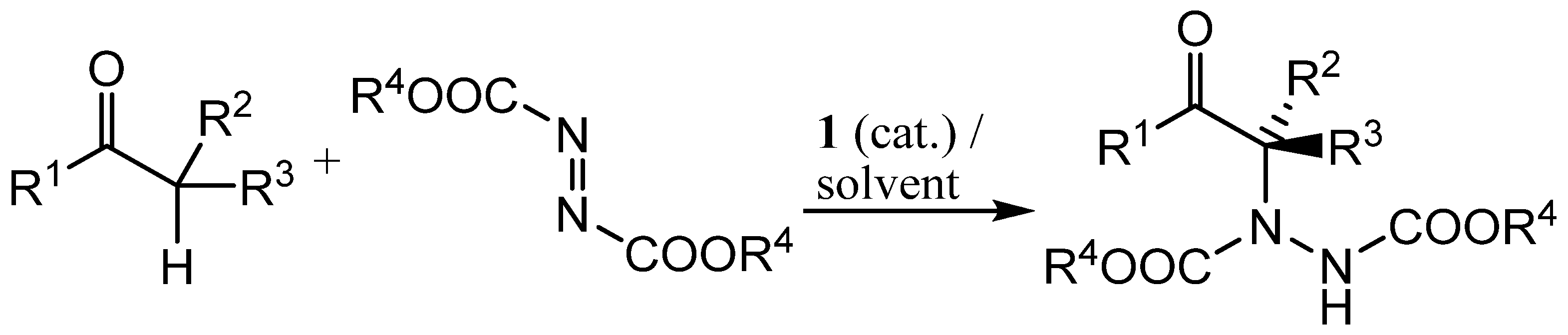
Scheme 3.
Proline-catalysed α-hydrazination of aldehydes in cyclic carbonate solvents.
Scheme 4.
Proline-catalysed α-hydrazination of ketones in propylene carbonate.
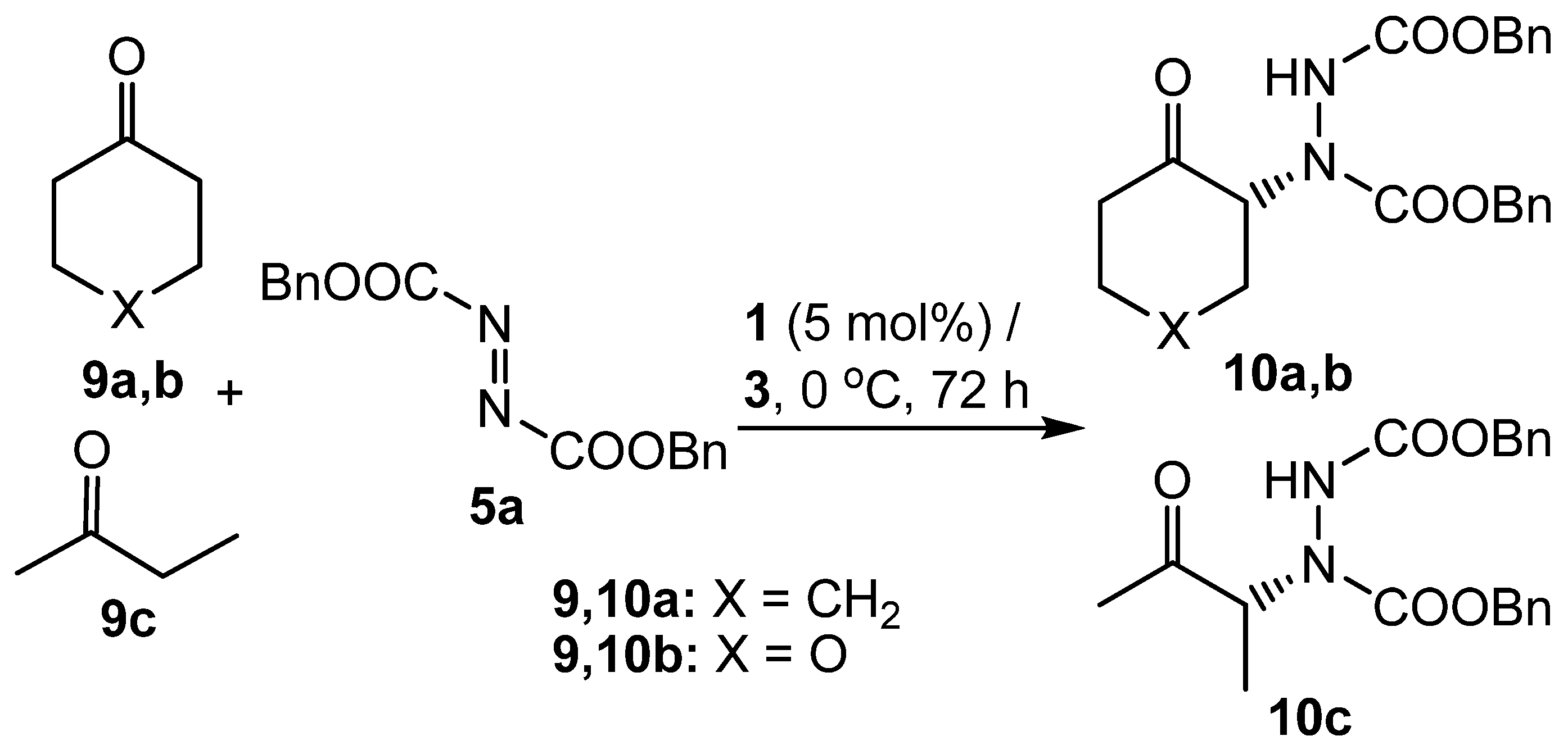

{kind=link}
{kind=link}
{kind=link}
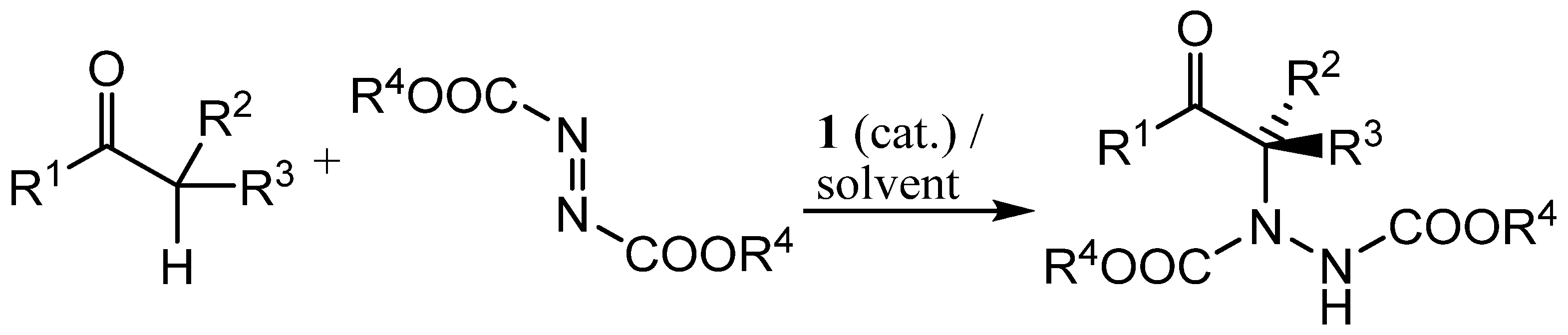{kind=link}
{kind=link}
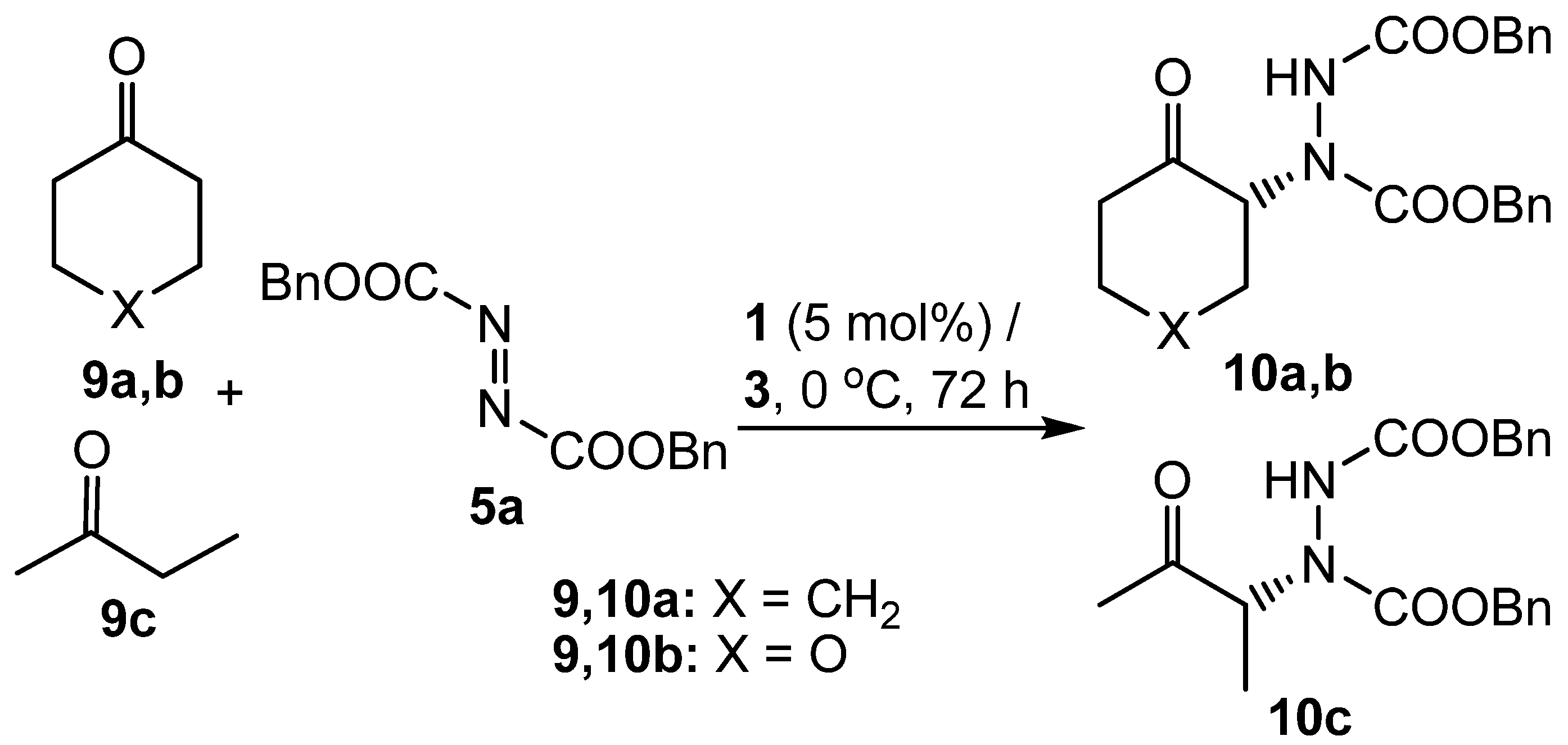{kind=link}
Table 1.
Effect of solvent and temperature on proline-catalysed α-hydrazinations of aldehydes.
| Entry | Solvent | Aldehyde | Product | Time (h) | T (°C) | Yield (%) | ee (%) |
|---|---|---|---|---|---|---|---|
| 1 | CH2Cl2 | 4a | 7a | 2 | RT | 86 | 98 (R)a |
| 2 | 2 | 4a | 7a | 2 | RT | 74 | 69 (R)a |
| 3 | 3 | 4a | 7a | 2 | RT | 81 | 80 (R)a |
| 4 | 3 | 4a | 7a | 2 | 0 | 18 | 99 (R)a |
| 5 | 3 | 4a | 7a | 24 | 0 | 69 | 97 (R)a |
| 6 | 3 | 4a | 7b | 24 | 0 | 39c | 66 (R)b |
| 7 | 3 | 4b | 7c | 24 | 0 | 41 | 90 (R)d |
| 8 | 3 | 4c | 7d | 24 | 0 | 70 | 36 (R)d |
| 9 | 3 | 4d | 7e | 24 | 0 | 76 | 99 (R)a |
| 10 | 3 | 4e | 7f | 24 | 0 | 87 | 92 (R)a |
Table 2.
Proline-catalysed reaction of ketones with dibenzyl azodicarboxylate.
| Entry | Solvent | Ketone | Product | Yield (%) | ee (%) |
|---|---|---|---|---|---|
| 1 | Racemic-3 | 9a | 10a | 71 | 77 (R)a |
| 2 | Racemic-3 | 9b | 10b | 51 | 72 (R)b |
| 3 | Racemic-3 | 9c | 10c | 31 | 52 (R)a |
| 4 | (R)-3 | 9a | 10a | 81 | 74 (R)a |
| 5c | (R)-3 | 9a | 10a | 78 | 75 (S)a |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Beattie, C.; North, M.; Villuendas, P. Proline-Catalysed Amination Reactions in Cyclic Carbonate Solvents. Molecules 2011, 16, 3420-3432. https://doi.org/10.3390/molecules16043420
AMA Style
Beattie C, North M, Villuendas P. Proline-Catalysed Amination Reactions in Cyclic Carbonate Solvents. Molecules. 2011; 16(4):3420-3432. https://doi.org/10.3390/molecules16043420
Chicago/Turabian StyleBeattie, Christopher, Michael North, and Pedro Villuendas. 2011. "Proline-Catalysed Amination Reactions in Cyclic Carbonate Solvents" Molecules 16, no. 4: 3420-3432. https://doi.org/10.3390/molecules16043420