Shortcut Access to Peptidosteroid Conjugates: Building Blocks for Solid-Phase Bile Acid Scaffold Decoration by Convergent Ligation

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
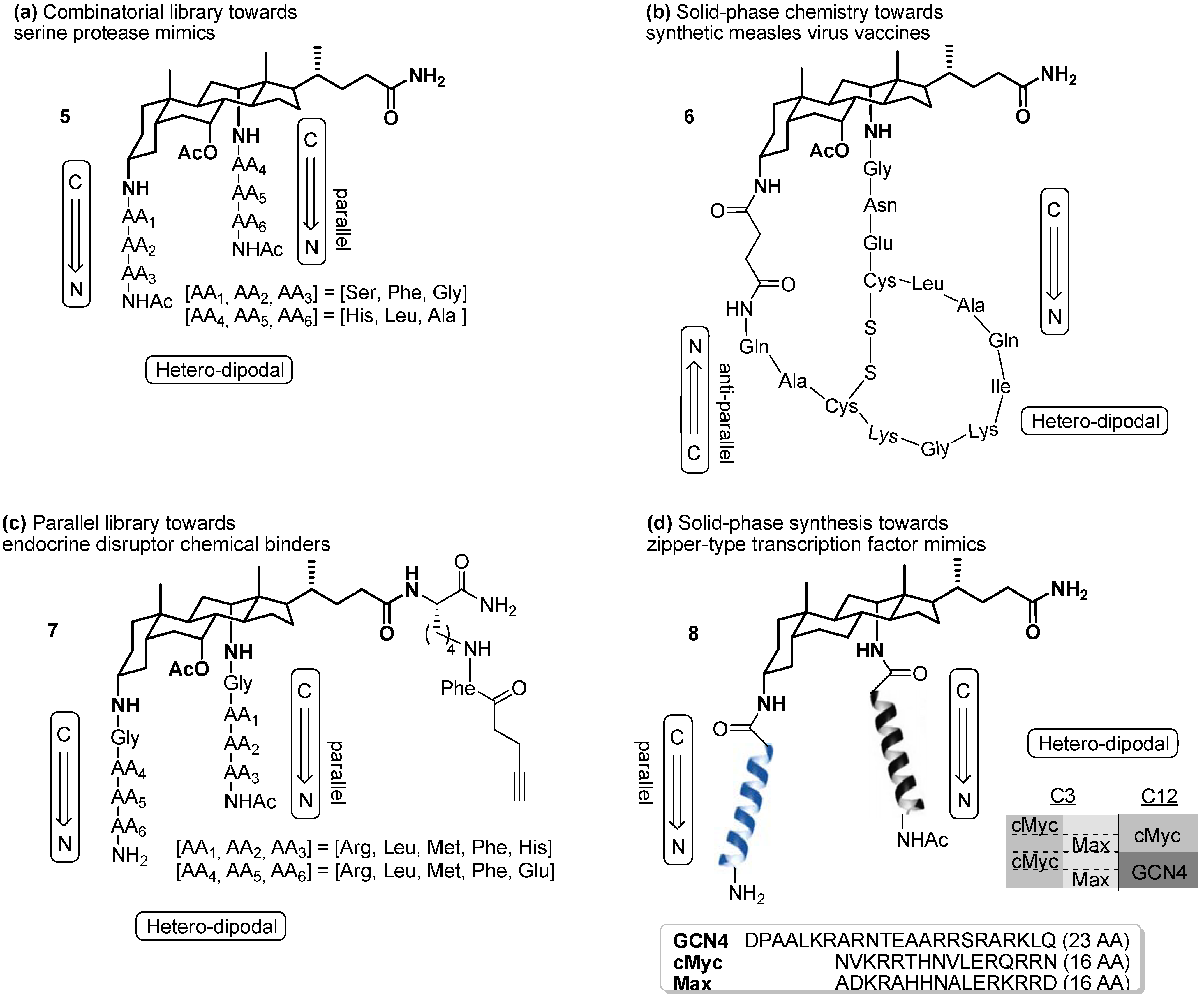
2. Results and Discussion
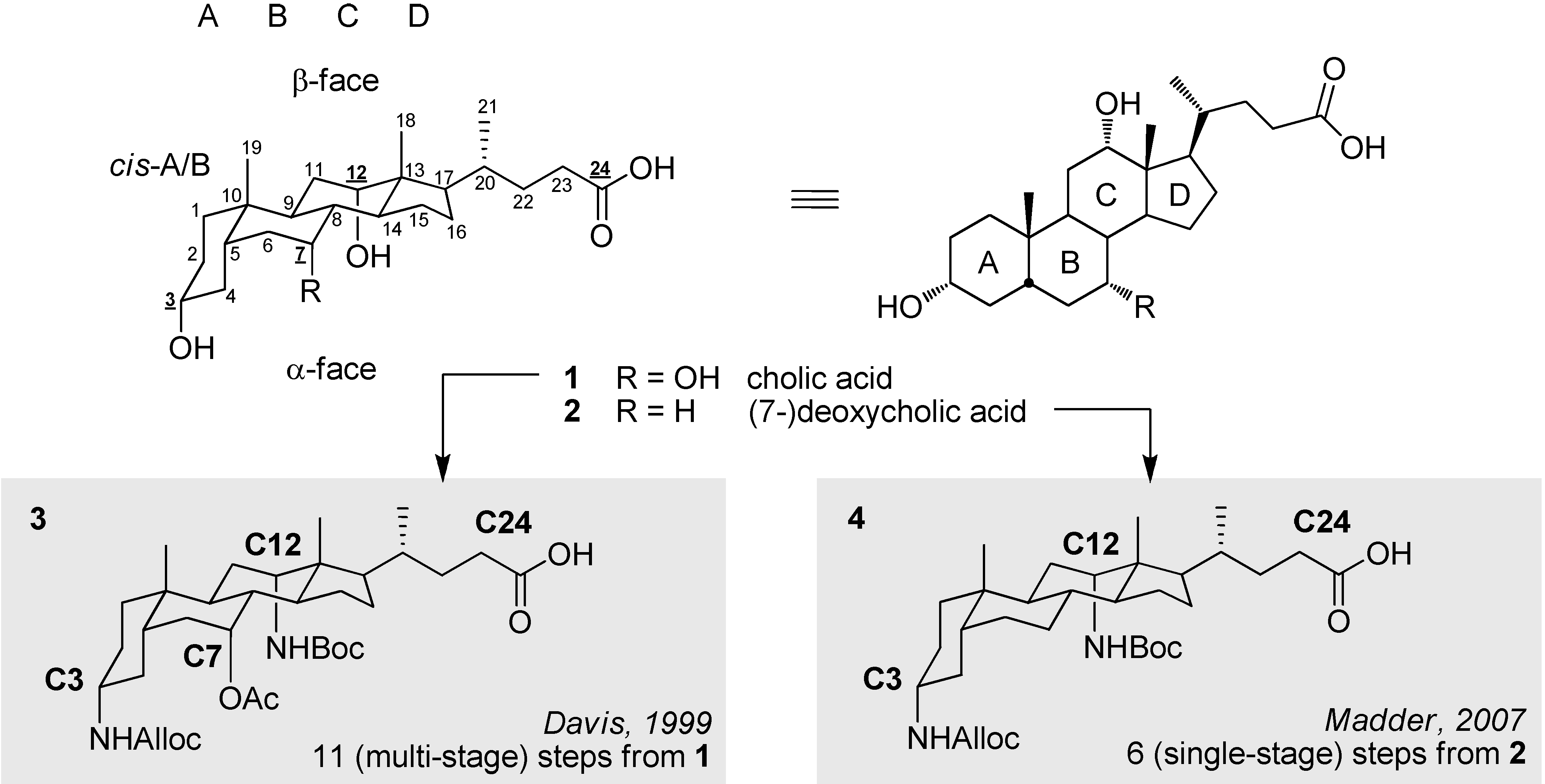
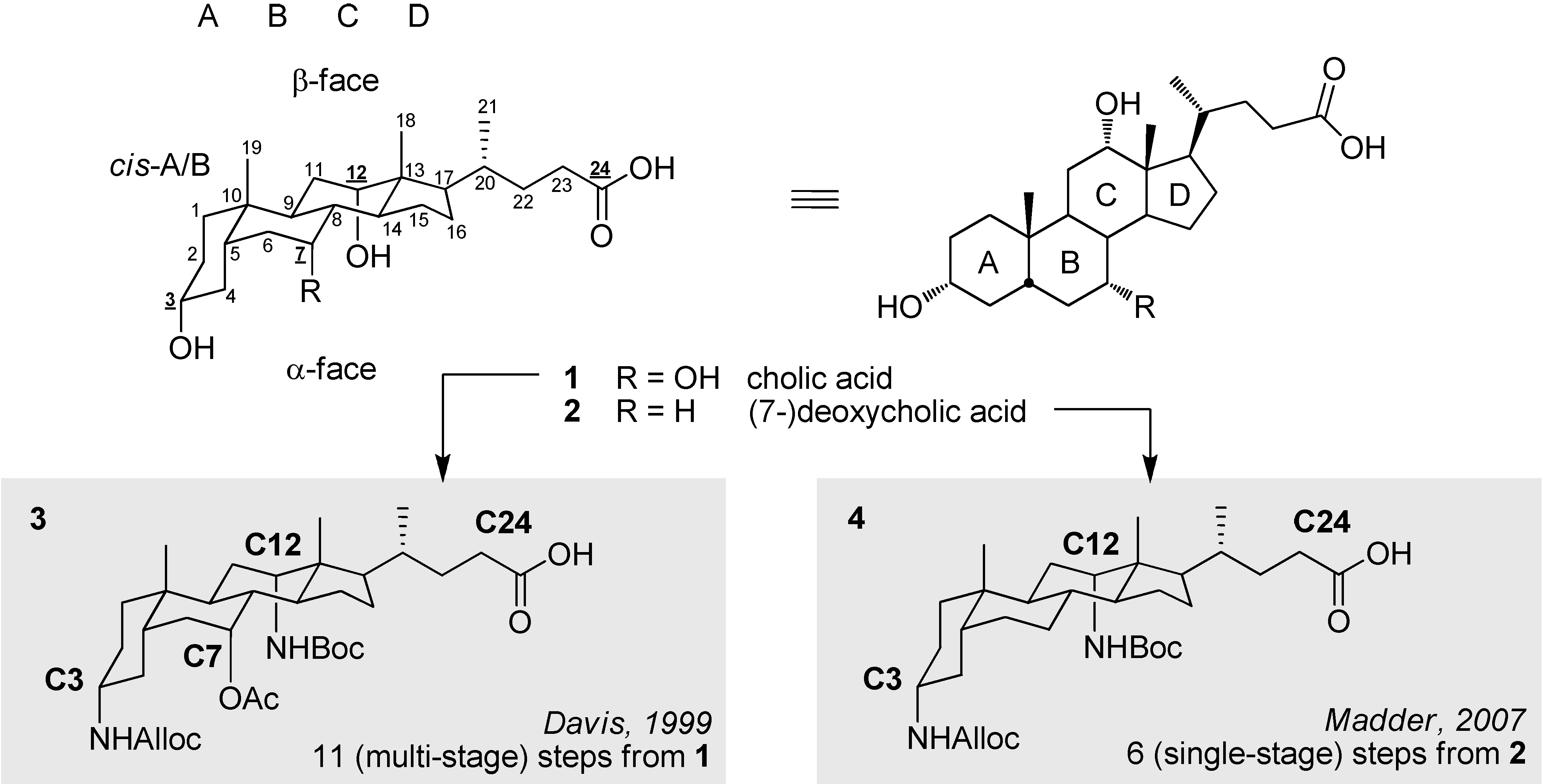
2.1. Ultrashort Access towards a Template with (Limited) Ligation Properties
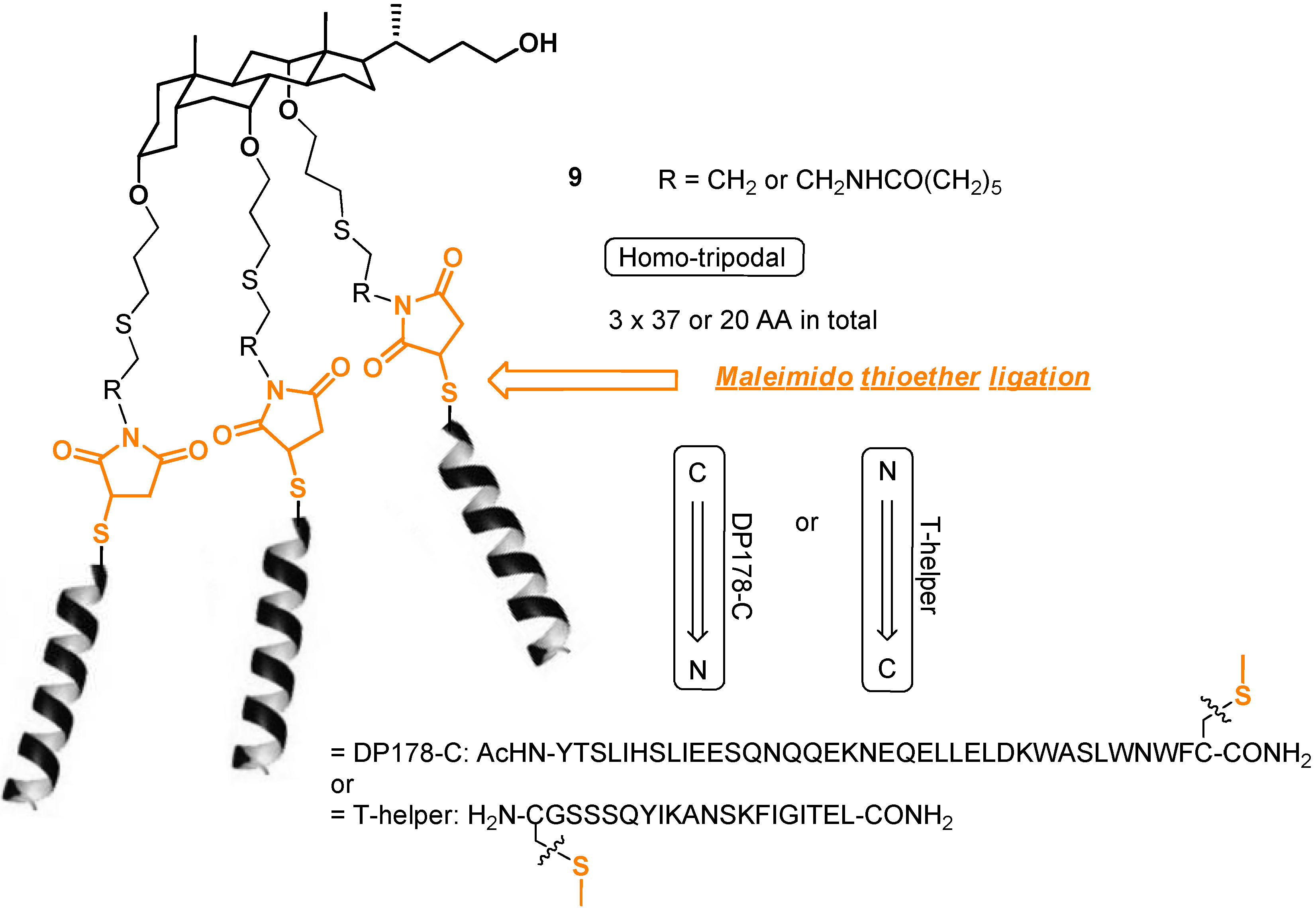
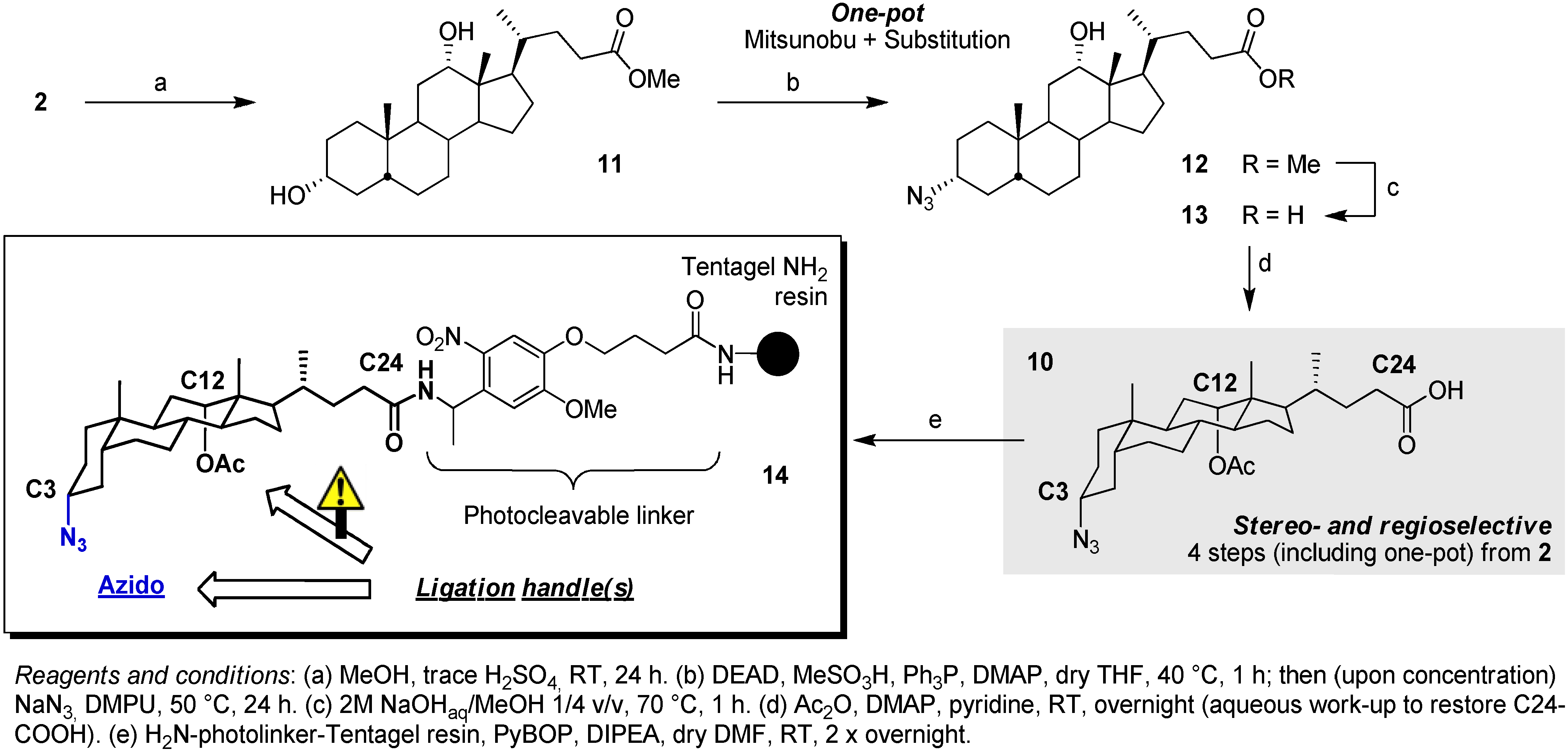
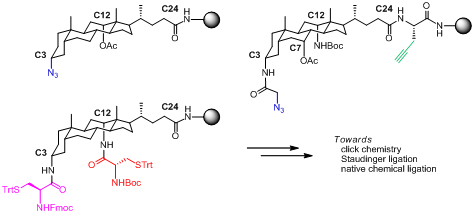
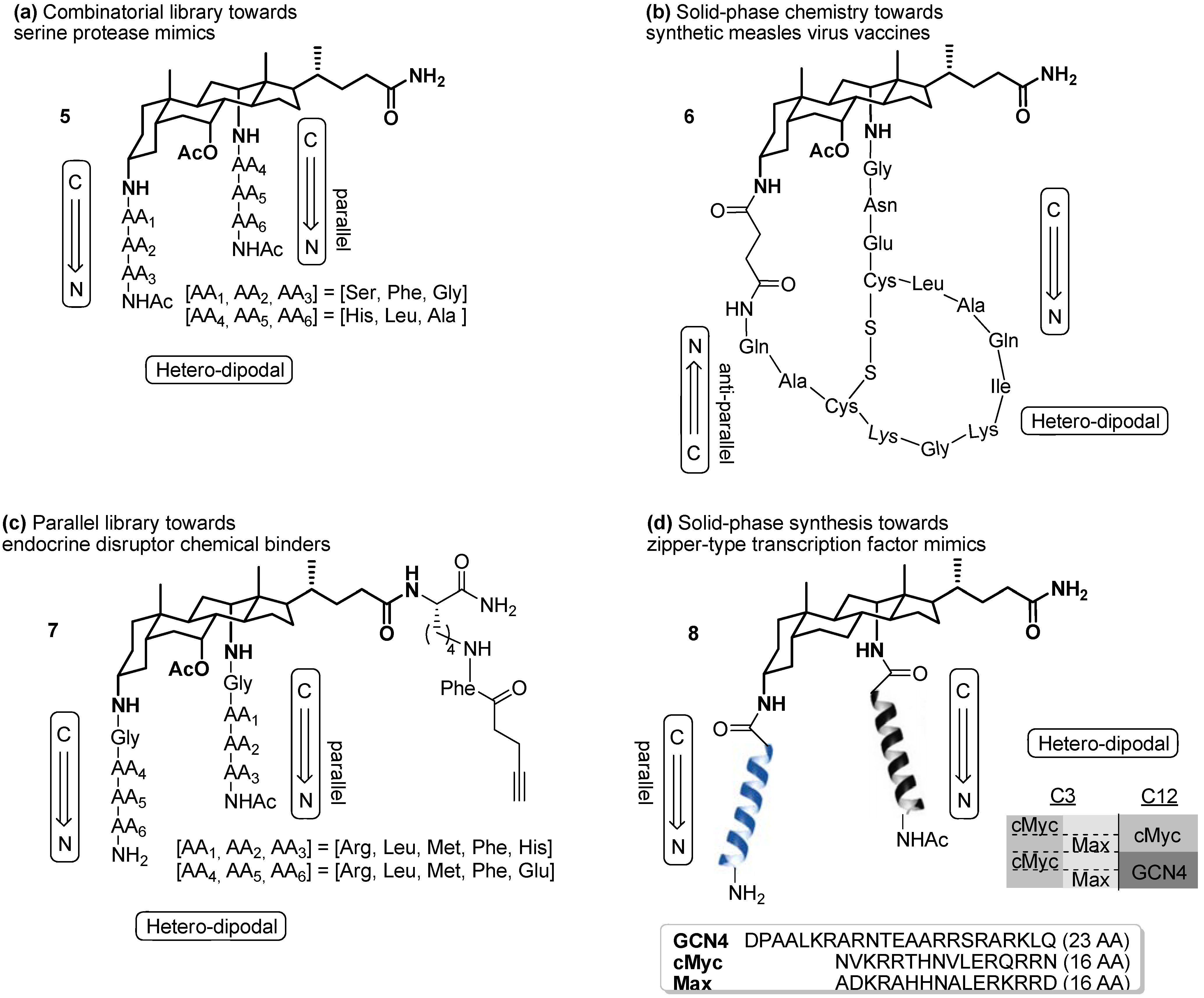
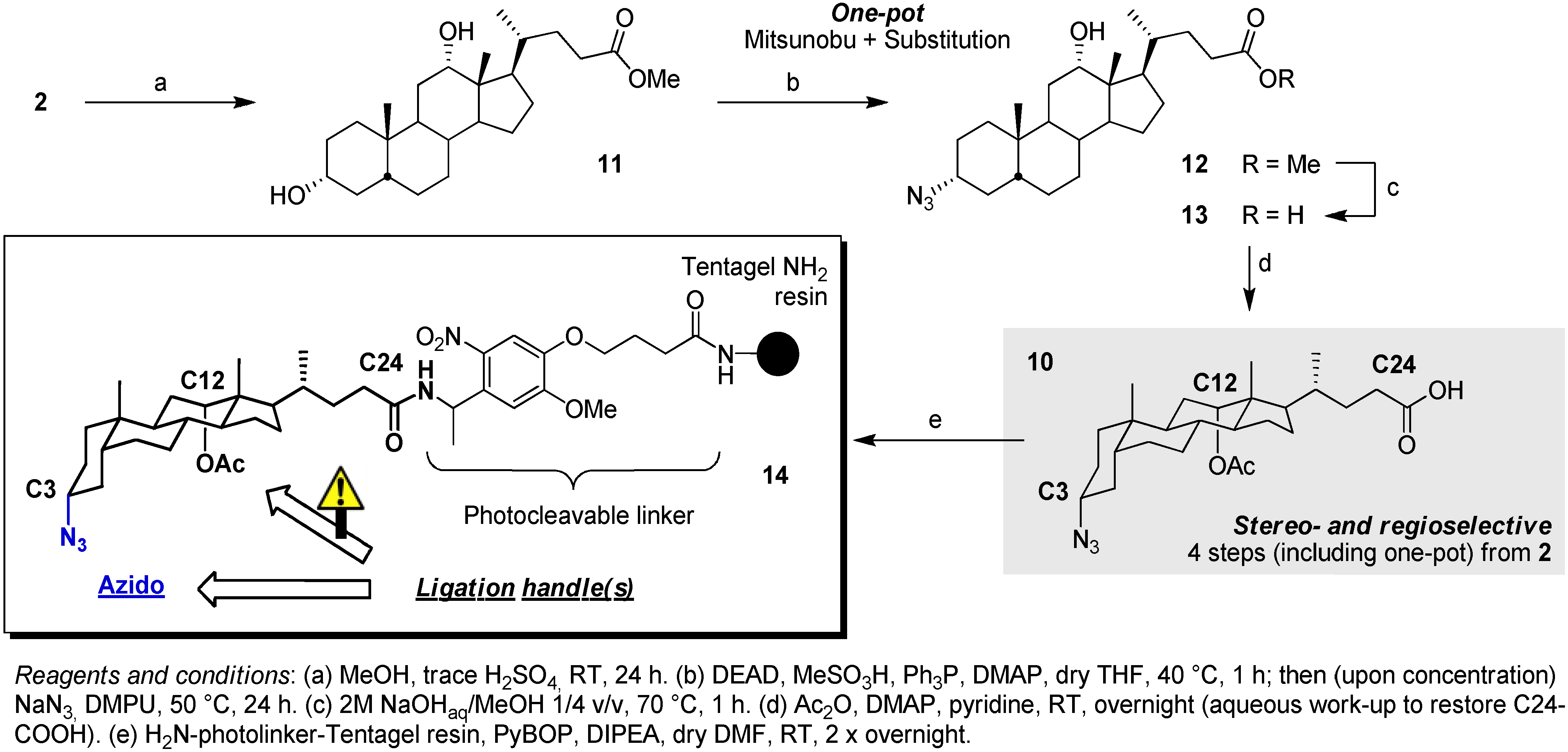
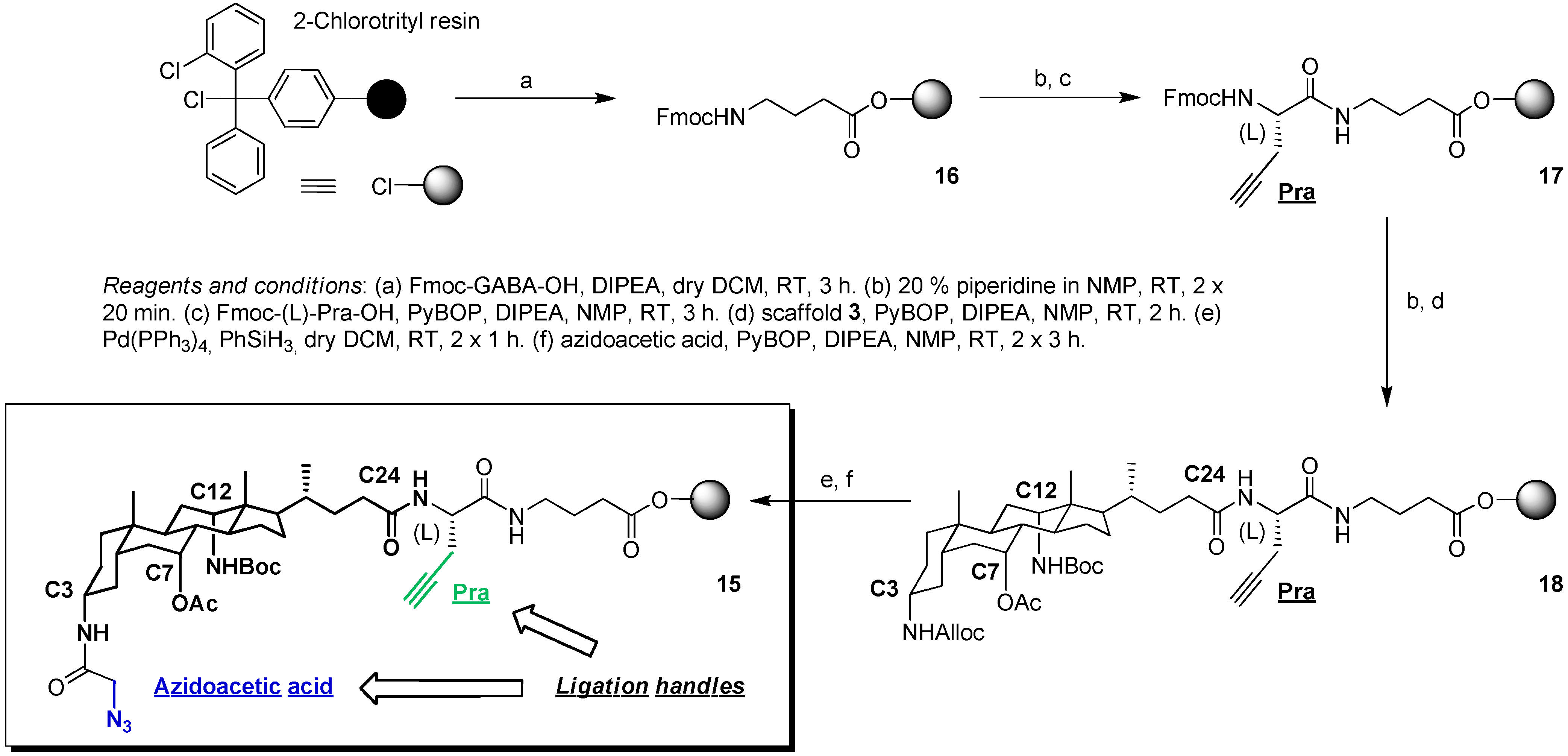
2.2. From EDC Receptors to a Simple Azide/Alkyne-Decorated Building Block for Click and Staudinger Ligation
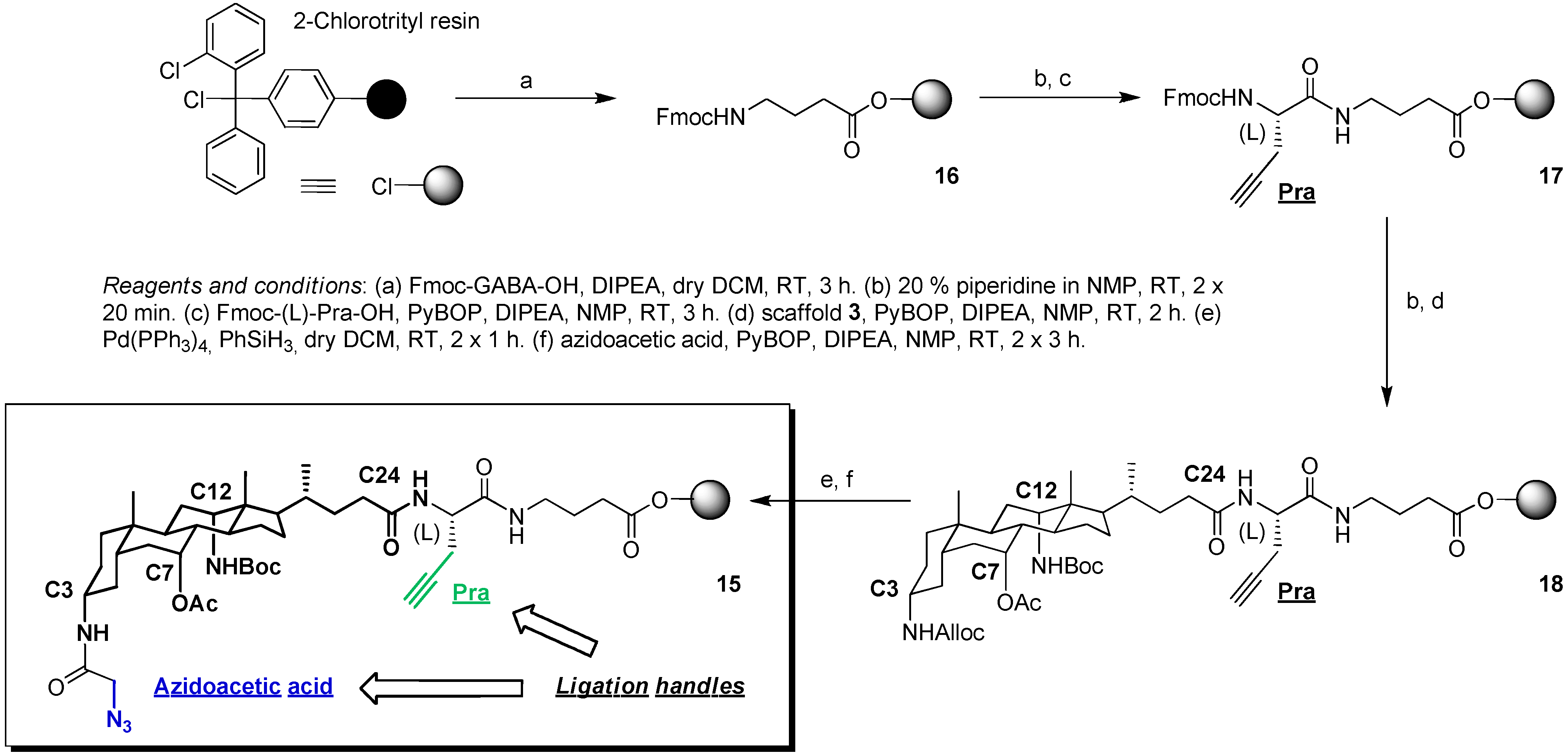
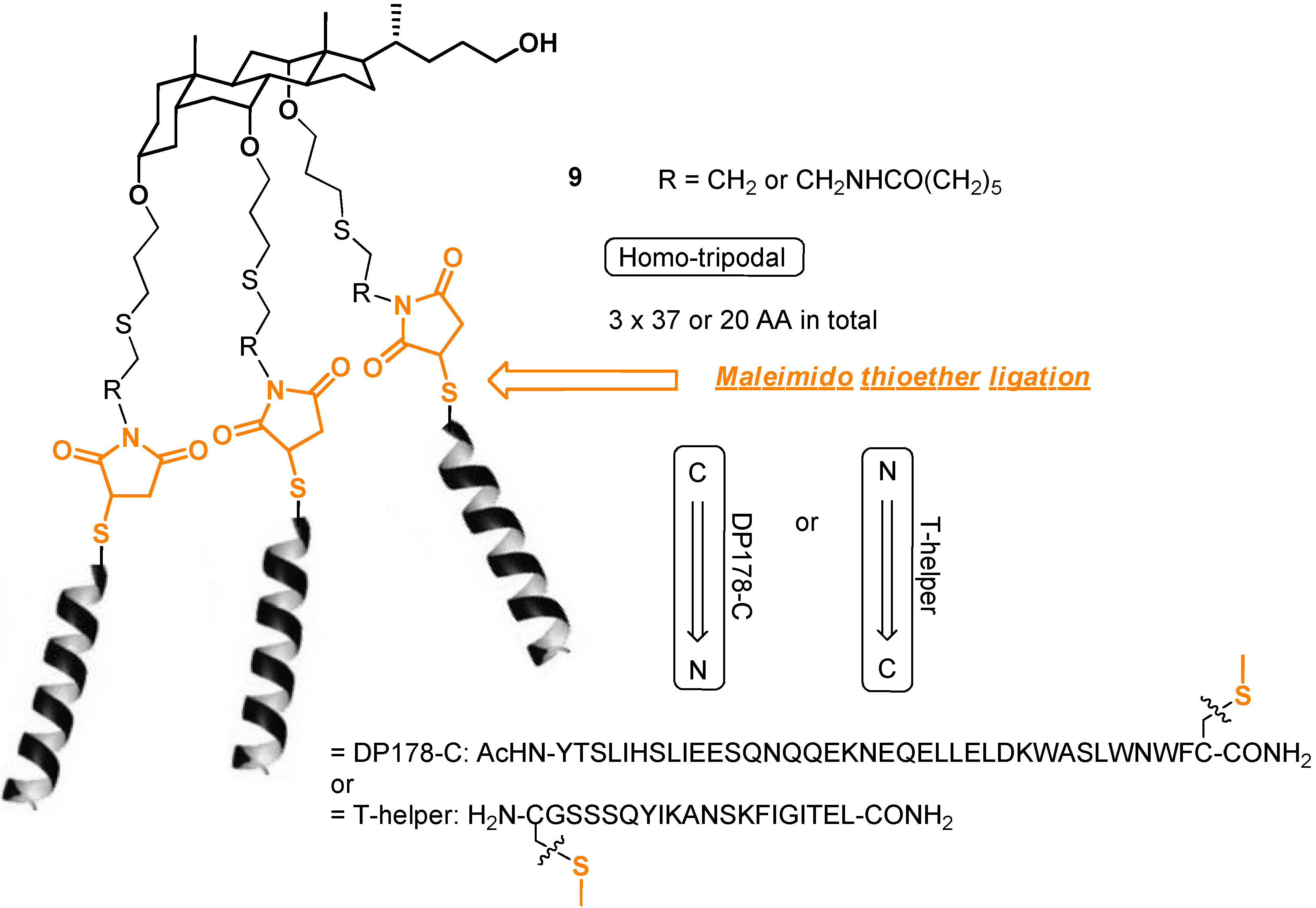
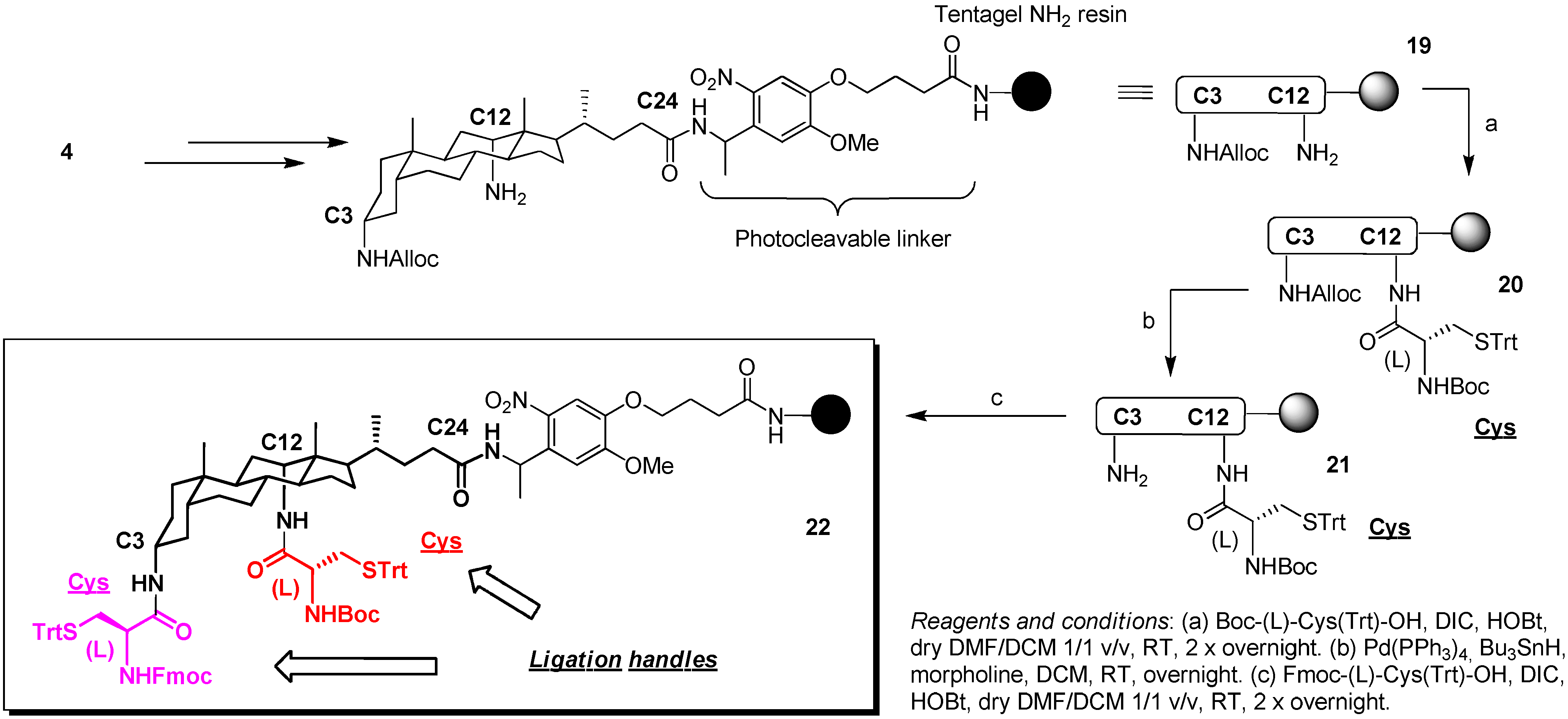
2.3. From Zipper-Type Protein Miniatures to Cys-Decorated Building Block for Double Orthogonal, Interthiol Assisted Native Chemical Ligation
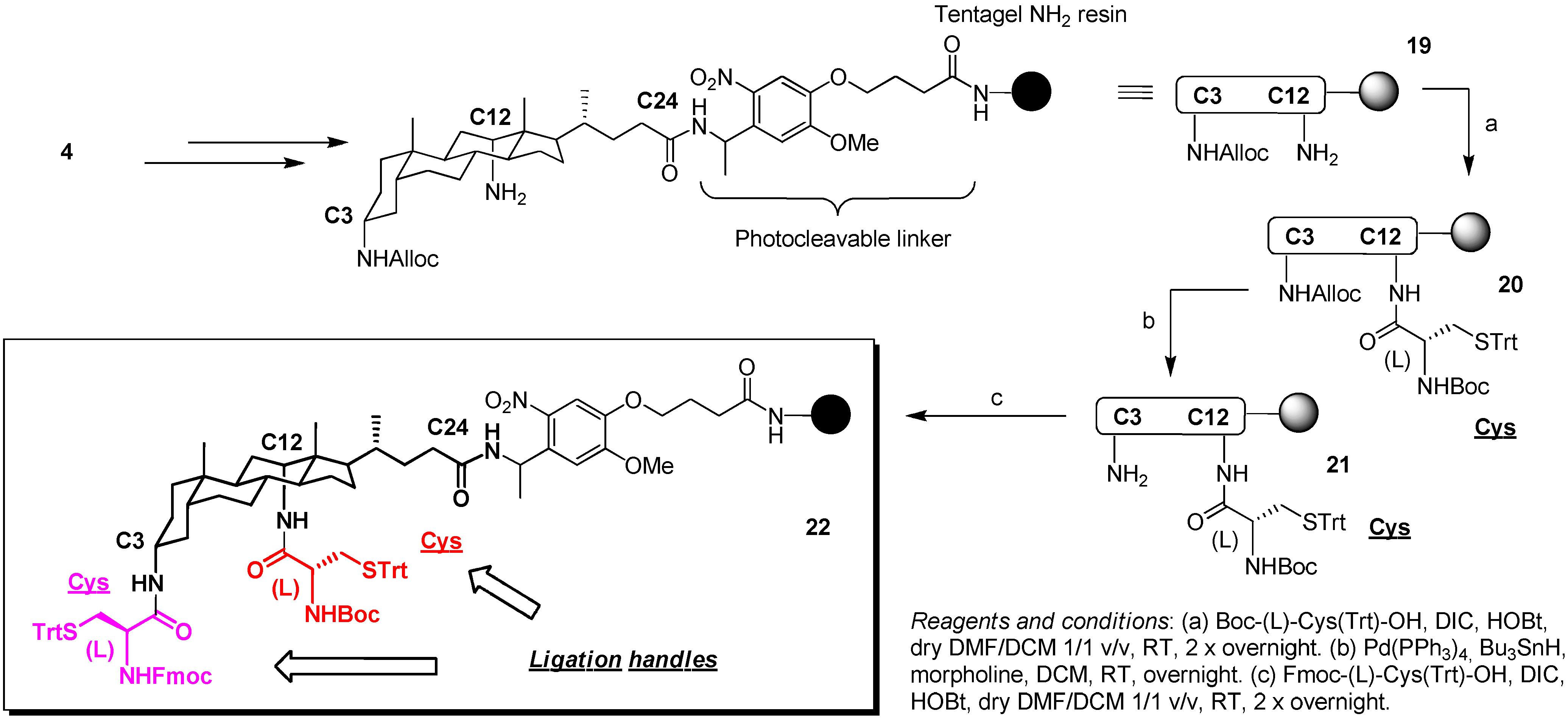
3. Experimental
3.1. General Information
3.2. Synthesis of Scaffold 14
3.3. Synthesis of Scaffold 15
3.4. Synthesis of Scaffold 22
4. Conclusions
Supplementary Materials
Acknowledgments
References and Notes
- Wallimann, P.; Marti, T.; Furer, A.; Diederich, F. Steroids in molecular recognition. Chem. Rev. 1997, 97, 1567–1608. [Google Scholar] [CrossRef]
- Singh, Y.; Dolphin, G.T.; Razkin, J.; Dumy, P. Synthetic peptide templates for molecular recognition: Recent advances and applications. ChemBioChem 2006, 7, 1298–1314. [Google Scholar] [CrossRef]
- Martos, V.; Castreño, P.; Valero, J.; de Mendoza, J. Binding to protein surfaces by supramolecular multivalent scaffolds. Curr. Opin. Chem. Biol. 2008, 12, 698–706. [Google Scholar] [CrossRef]
- Maltais, R.; Tremblay, M.R.; Ciobanu, L.C.; Poirier, D. Steroids and combinatorial chemistry. J. Comb. Chem. 2004, 6, 443–456. [Google Scholar] [CrossRef]
- Tamminen, J.; Kolehmainen, E. Bile acids as building blocks of supramolecular hosts. Molecules 2001, 6, 21–46. [Google Scholar] [CrossRef]
- Virtanen, E.; Kolehmainen, E. Use of bile acids in pharmacological and supramolecular applications. Eur. J. Org. Chem. 2004, 3385–3399. [Google Scholar] [CrossRef]
- Davis, A.P. Bile acid scaffolds in supramolecular chemistry: The interplay of design and synthesis. Molecules 2007, 12, 2106–2122. [Google Scholar] [CrossRef]
- Nonappa; Maitra, U. Unlocking the potential of bile acids in synthesis, supramolecular/materials chemistry and nanoscience. Org. Biomol. Chem. 2008, 6, 657–669. [Google Scholar] [CrossRef]
- Hirschmann, R.; Sprengeler, P.A.; Kawasaki, T.; Leahy, J.W.; Shakespeare, W.C.; Smith, A.B., III. The first design and synthesis of a steroidal peptidomimetic. The potential value of peptidomimetics in elucidating the bioactive conformation of peptide ligands. J. Am. Chem. Soc. 1992, 114, 9699–9701. [Google Scholar] [CrossRef]
- Hirschmann, R.; Sprengeler, P.A.; Kawasaki, T.; Leahy, J.W.; Shakespeare, W.C.; Smith, A.B., III. The versatile steroid nucleus: Design and synthesis of a peptidomimetic employing this novel scaffold. Tetrahedron 1993, 49, 3665–3676. [Google Scholar] [CrossRef]
- Swaan, P.; Hillgren, K.M.; Szoka, F.C., Jr.; Øie, S. Enhanced transepithelial transport of peptides by conjugation to cholic acid. Bioconjugate Chem. 1997, 8, 520–525. [Google Scholar] [CrossRef]
- Jing, B.W.; Janout, V.; Regen, S.L. Fully detachable molecular umbrellas as peptide delivery agents. Bioconjugate Chem. 2003, 14, 1191–1196. [Google Scholar] [CrossRef]
- Lee, S.; Kim, K.; Kumar, T.S.; Lee, J.; Kim, S.K.; Lee, D.Y.; Lee, Y.; Byun, Y. Synthesis and biological properties of insulin-deoxycholic acid chemical conjugates. Bioconjugate Chem. 2005, 16, 615–620. [Google Scholar] [CrossRef]
- Enhsen, A.; Kramer, W.; Wess, G. Bile acids in drug discovery. Drug Discov. Today 1998, 3, 409–418. [Google Scholar] [CrossRef]
- Salunke, D.B.; Hazra, B.G.; Pore, V.S. Steroidal conjugates and their pharmacological applications. Curr. Med. Chem. 2006, 13, 813–847. [Google Scholar] [CrossRef]
- Kramer, W. Transporters, trojan horses and therapeutics: Suitability of bile acid and peptide transporters for drug delivery. Biol. Chem. 2011, 392, 77–94. [Google Scholar]
- Chen, Y.; Suenaga, T.; Still, W.C. Sequence-selective peptide binding with a peptido-A,B-trans-steroidal receptor selected from an encoded combinatorial receptor library. J. Am. Chem. Soc. 1996, 118, 1813–1814. [Google Scholar] [CrossRef]
- Boyce, R.; Li, G.; Nestler, H.P.; Suenaga, T.; Still, W.C. Peptidosteroid receptors for opioid peptides. Sequence-selective binding using a synthetic receptor library. J. Am. Chem. Soc. 1994, 116, 7955–7956. [Google Scholar] [CrossRef]
- Still, W.C.; Li, G.; Wennemers, H. Synthetic receptors, libraries and uses thereof. EP Patent 0739486, 1996. [Google Scholar]
- Wess, G.; Bock, K.; Kleine, H.; Kurz, M.; Guba, W.; Hemmerle, H.; Lopez-Calle, E.; Baringhaus, K.; Glombik, H.; Enhsen, A.; Kramer, W. The design and synthesis of a scaffold for combinatorial chemistry based on bile acid. Angew. Chem. Int. Ed. 1996, 35, 2222–2224. [Google Scholar] [CrossRef]
- Nestler, H.P. Sequence-selective nonmacrocyclic two-armed receptors for peptides. Mol. Divers. 1996, 2, 35–40. [Google Scholar] [CrossRef]
- HØeg-Jensen, T. Cholic acid-based scaffolds for multidimensional molecular presentation of peptides. WO Patent 1999031124, 1999. [Google Scholar]
- Ding, B.; Taotofa, U.; Orsak, T.; Chadwell, M.; Savage, P.B. Synthesis and characterization of peptide-cationic steroid antibiotic conjugates. Org. Lett. 2004, 6, 3433–3436. [Google Scholar] [CrossRef]
- Barry, J.F.; Davis, A.P.; Pérez-Payán, M.N.; Elsegood, M.R.J.; Jackson, R.F.W.; Gennari, C.; Piarulli, U.; Gude, M. A trifunctional steroid-based scaffold for combinatorial chemistry. Tetrahedron Lett. 1999, 40, 2849–2852. [Google Scholar]
- Verzele, D.; Madder, A. Short synthesis of orthogonally protected 3α,12α-diamino-5β-cholan-24-oicacid, a dipodal steroid scaffold for combinatorial chemistry. Eur. J. Org. Chem. 2007, 11, 1793–1797. [Google Scholar] [CrossRef]
- Verzele, D.; Goeman, J.L.; Madder, A. LC-(TIC/EIC)-MS as tool in the analysis of diastereomeric 3,12-aza-analogues of deoxycholic acid. ARKIVOC 2007, 10, 325–336. [Google Scholar]
- Del Amo, V.; Siracusa, L.; Markidis, T.; Baragana, B.; Bhattarai, K.; Galobardes, M.; Naredo, G.; Pérez-Payán, M.N.; Davis, A.P. ifferentially-protected steroidal triamines; scaffolds with potential for medicinal, supramolecular, and combinatorial chemistry. Org. Biomol. Chem. 2004, 2, 3320–3328. [Google Scholar] [CrossRef]
- de Muynck, H.; Madder, A.; Farcy, N.; de Clercq, P.J.; Pérez-Payán, M.N.; Öhnberg, L.M.; Davis, A.P. Application of combinatorial procedures in the search for serine-protease-like activity with focus on the acyl transfer step. Angew. Chem. Int. Ed. 2000, 39, 145–148. [Google Scholar] [CrossRef]
- Madder, A.; Li, L.; de Muynck, H.; Farcy, N.; van Haver, D.; Fant, F.; Vanhoenacker, G.; Sandra, P.; Davis, A.P.; de Clercq, P.J. Evaluation of a two-stage screening procedure in the combinatorial search for serine protease-like activity. J. Comb. Chem. 2002, 4, 552–562. [Google Scholar] [CrossRef]
- Bodé, C.A.; Muller, C.P.; Madder, A. Validation of a solid-phase-bound steroid scaffold for the synthesis of novel cyclic peptidosteroids. J. Pept. Sci. 2007, 13, 702–708. [Google Scholar] [CrossRef]
- Bodé, C.A.; Bechet, T.; Prodhomme, E.; Gheysen, K.; Gregoir, P.; Martins, J.C.; Muller, C.P.; Madder, A. Towards the conformational mimicry of the measles virus HNE loop: Design, synthesis and biological evaluation of a cyclic bile acid-peptide conjugate. Org. Biomol. Chem. 2009, 7, 3391–3399. [Google Scholar] [CrossRef]
- Albert, D.; Feigel, M. Beta-loop, gamma-loop, and helical peptide conformations in cyclopeptides containing a steroidal pseudo-amino acid. Helv. Chim. Acta 1997, 80, 2168–2181. [Google Scholar] [CrossRef]
- Albert, D.; Feigel, M.; Benet-Buchholz, J.; Boese, R. Crystal structure of a peptide-steroid macrocycle—intramolecular attraction between steroids and peptidic β(I) turns. Angew. Chem. Int. Ed. 1998, 37, 2727–2729. [Google Scholar] [CrossRef]
- Albert, D.; Feigel, M. Steroidal cyclopeptide, synthesis and shape of the cavity. Tetrahedron Lett. 1994, 35, 565–568. [Google Scholar] [CrossRef]
- Wessjohann, L.A.; Voigt, B.; Rivera, D.G. Diversity oriented one-pot synthesis of complex macrocycles: Very large steroid-peptoid hybrids from multiple multicomponent reactions including bifunctional building blocks. Angew. Chem. Int. Ed. 2005, 44, 4785–4790. [Google Scholar] [CrossRef]
- Figaroli, S.; Madder, A. Design and automated generation of artificial estrogen receptor as potential endocrine disruptor chemical binders. Tetrahedron 2010, 66, 6912–6918. [Google Scholar] [CrossRef]
- Verzele, D.; Madder, A. Towards the first miniature cMyc-Max model: Semi-on-line monitoring gives solid-phase access to hydrophobic b(-HLH-)ZIP peptidosteroid tweezers. ChemBioChem. Submitted.
- Li, H.; Wang, L. Cholic acid as template for multivalent peptide assembly. Org. Biomol. Chem. 2003, 1, 3507–3513. [Google Scholar] [CrossRef]
- Cline, L.L.; Janout, V.; Fisher, M.; Juliano, R.L.; Regen, S.L. A molecular umbrella approach to the intracellular delivery of small interfering RNA. Bioconjugate Chem. 2011, 22, 2210–2216, For a recently disclosed related example using similar (maleimido-thioether and 2,2′-dipyridyl-disulfide) ligation chemistries, see. [Google Scholar] [CrossRef]
- Nilsson, B.L.; Kiessling, L.L.; Raines, R.T. Staudinger ligation: A peptide from a thioester and azide. Org. Lett. 2000, 2, 1939–1941. [Google Scholar] [CrossRef]
- Lawless, L.J.; Blackburn, A.G.; Ayling, A.J.; Pérez-Payán, M.N.; Davis, A.P. Steroidal guanidines as enantioselective receptors for N-acyl α-amino acids. Part 1. 3α-guanylated carbamates derived from cholic acid. J. Chem. Soc. Perkin Trans. 2001, 1, 1329–1341. [Google Scholar]
- Ayling, A.J.; Broderick, S.; Clare, J.P.; Davis, A.P.; Pérez-Payán, M.N.; Lahtinen, M.; Nissinen, M.J.; Rissanen, K. An extraction-based assay for neutral anionophores: The measurement of high binding constants to steroidal receptors in a nonpolar solvent. Chem. Eur. J. 2002, 8, 2197–2203. [Google Scholar] [CrossRef]
- Pore, V.S.; Aher, N.G.; Kumar, M.; Shukla, P.K. Design and synthesis of fluconazole/bile acid conjugates using click reaction. Tetrahedron 2006, 62, 11178–11186. [Google Scholar] [CrossRef]
- Aher, N.G.; Pore, V.S.; Patil, S.P. Design, synthesis, and micellar properties of bile acid dimers and oligomers linked with a 1,2,3-triazole ring. Tetrahedron 2007, 63, 12927–12934. [Google Scholar] [CrossRef]
- Gao, H.; Dias, J.R. Selective protection of the various hydroxy groups of cholic acid and derivatives. A review. Org. Prep. Proced. Int. 1999, 31, 145–166. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Vatmurge, N.S.; Hazra, B.G.; Pore, V.S.; Shirazi, F.; Deshpande, M.V.; Kadreppa, S.; Chattopadhyay, S.; Gonnade, R.G. Synthesis and biological evaluation of bile acid dimers linked with 1,2,3-triazole and bis-β-lactam. Org. Biomol. Chem. 2008, 6, 3823–3830. [Google Scholar] [CrossRef]
- Zhang, J.; Luo, J.; Zhu, X.X.; Junk, M.J.N.; Hinderberger, D. Molecular pockets derived from cholic acid as chemosensors for metal ions. Langmuir 2010, 26, 2958–2962. [Google Scholar] [CrossRef]
- Sokolova, N.V.; Latyshev, G.V.; Lukashev, N.V.; Nenajdenko, V.G. Design and synthesis of bile acid-peptide conjugates via triazole moiety. Org. Biomol. Chem. 2011, 9, 4921–4926. [Google Scholar] [CrossRef]
- Chhatra, R.K.; Kumar, A.; Pandey, P.S. Synthesis of a bile acid-based click-macrocycle and its application in selective recognition of chloride ion. J. Org. Chem. 2011, 76, 9086–9089. [Google Scholar]
- Avrutina, O.; Empting, M.; Fabritz, S.; Daneschdar, M.; Frauendorf, H.; Diederichsen, U.; Kolmar, H. Application of copper(I) catalyzed azide-alkyne [3+2] cycloaddition to the synthesis of template-assembled multivalent peptide conjugates. Org. Biomol. Chem. 2009, 7, 4177–4185. [Google Scholar] [CrossRef]
- Kent, S.B.H. Total chemical synthesis of proteins. Chem. Soc. Rev. 2009, 38, 338–351. [Google Scholar] [CrossRef]
- Han, Y.; Albericio, F.; Barany, G. Occurrence and minimization of cysteine racemization during stepwise solid-phase peptide synthesis. J. Org. Chem. 1997, 62, 4307–4312. [Google Scholar] [CrossRef]
- Banaszynski, L.A.; Liu, C.W.; Wandless, T.J. Characterization of the FKBP.rapamycin.FRB ternary complex. J. Am. Chem. Soc. 2005, 127, 4715–4721. [Google Scholar] [CrossRef]
- Bunin, B.A. The Combinatorial Index; Academic Press: Waltham, MA, USA, 1998; pp. 219–220. [Google Scholar]
- Sample Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Verzele, D.; Figaroli, S.; Madder, A. Shortcut Access to Peptidosteroid Conjugates: Building Blocks for Solid-Phase Bile Acid Scaffold Decoration by Convergent Ligation. Molecules 2011, 16, 10168-10186. https://doi.org/10.3390/molecules161210168
Verzele D, Figaroli S, Madder A. Shortcut Access to Peptidosteroid Conjugates: Building Blocks for Solid-Phase Bile Acid Scaffold Decoration by Convergent Ligation. Molecules. 2011; 16(12):10168-10186. https://doi.org/10.3390/molecules161210168
Chicago/Turabian StyleVerzele, Dieter, Sara Figaroli, and Annemieke Madder. 2011. "Shortcut Access to Peptidosteroid Conjugates: Building Blocks for Solid-Phase Bile Acid Scaffold Decoration by Convergent Ligation" Molecules 16, no. 12: 10168-10186. https://doi.org/10.3390/molecules161210168