Bohlmann-Rahtz Cyclodehydration of Aminodienones to Pyridines Using N-Iodosuccinimide

School of Chemistry, Main Building, Cardiff University, Park Place, Cardiff, CF10 3AT, UK
*
Author to whom correspondence should be addressed.
Molecules 2010, 15(5), 3211-3227; https://doi.org/10.3390/molecules15053211
Submission received: 22 March 2010
/
Accepted: 20 April 2010
/
Published: 30 April 2010
(This article belongs to the Special Issue Organic Iodine Chemistry)
Abstract
:Cyclodehydration of Bohlmann-Rahtz aminodienone intermediates using N‑iodosuccinimide as a Lewis acid proceeds at low temperature under very mild conditions to give the corresponding 2,3,6-trisubstituted pyridines in high yield and with total regiocontrol.
1. Introduction
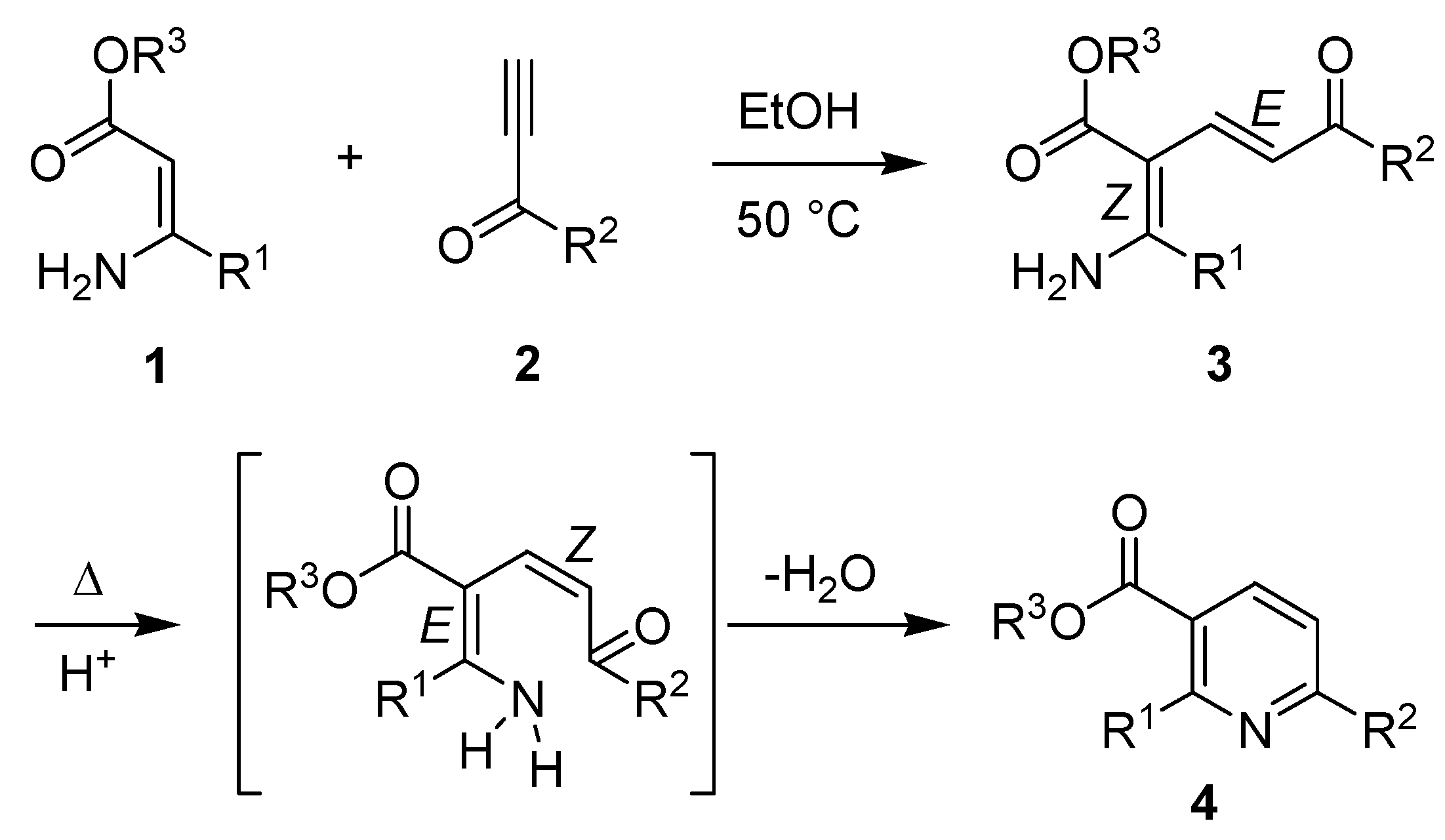
The synthesis, reactions and properties of pyridine-containing derivatives is an important component of modern heterocyclic chemistry. This heterocyclic motif is found in a large number of pharmaceutical agents [1], as a pharmacophore of considerable importance, and a valuable synthetic building block in drug discovery, heterocyclic chemistry and natural product synthesis [2]. Thus, new and facile methods for the synthesis of polysubstituted pyridines are of considerable current interest, in particular if they have a predictable and reliable regiochemical outcome and provide the heterocycle in high yield from readily-available precursors. The Bohlmann-Rahtz pyridine synthesis bears close relation to many well-established approaches to pyridines based upon heterocyclocondensation processes, such as the Hantzsch dihydropyridine synthesis [3], and was first reported back in 1957 [4]. This robust two-step method provides an efficient route to 2,3,6-trisubstituted pyridines 4 from stabilized enamines 1 and ethynyl ketones 2. It proceeds by an initial Michael addition to give an aminodienone intermediate 3, which is isolated and purified, and subsequent cyclodehydration under forcing conditions (up to 200 ºC) in order to facilitate E/Z-isomerization and subsequent spontaneous heterocyclization to the pyridine product 4 (Scheme 1). Since its discovery, this heterocyclocondensation reaction has received very little attention until recently [5], when it has found application in the synthesis of pyridine libraries [6,7], pyrido[2,3-d]pyrimidines [8,9,10,11], α-helix mimetics [12], nicotinonitrile-derived chromophores with tunable photophysical properties [13,14], heterocyclic amino acids [15,16,17], and in the preparation of pyridine-containing natural products such as the thiopeptide antibiotics [18,19,20,21,22] and chemical derivatives thereof [5,23,24,25,26,27].
Scheme 1.
Two-step Bohlmann-Rahtz synthesis of 2,3,6-trisubstituted pyridines 4.

Given its wide range of potential applications, many efforts have been made to improve the Bohlmann-Rahtz pyridine synthesis, with particular emphasis on catalyzing the E/Z-isomerization and subsequent cyclodehydration in order to avoid the use of very high temperatures and broaden its substrate specificity. Recent findings have shown that the heterocyclization can be accelerated through the use of a Brønsted or Lewis acid [5,9,28,29] and can be carried out in just one operation through the use of a protic solvent [30] or at high temperature under microwave dielectric heating [31]. Alternative processing methods have also been reported for the cyclodehydration reaction, such the use of continuous flow reactors [32,33], and this transformation can be incorporated into a one-pot multistep process by combining with reactions such as tandem oxidation [34] or enamine formation [35] which improves the overall chemical yield. However, despite the synthetic utility of Bohlmann-Rahtz aminodienones 3, as evidenced by their widespread application [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27], no studies had been undertaken to divert these intermediates down an alternative reaction path in order to expand the scope of possible products.
In 2002, Dechoux and co-workers described that the δ-dienaminoester 5 reacted with N‑iodosuccinimide (NIS) in methanol in the presence of sodium methoxide to give the corresponding 3-iodo-2(1H)-pyridinone 6 in 84% yield (Scheme 2) [36].
Scheme 2.
Synthesis of 3-iodo-2(1H)-pyridinone 6 from δ-dienaminoester 5 by Dechoux and co-workers [36].
Scheme 2.
Synthesis of 3-iodo-2(1H)-pyridinone 6 from δ-dienaminoester 5 by Dechoux and co-workers [36].

The reaction was found to be highly chemoselective and yet could be diverted to provide the polysubstituted pyrrole as an alternative product simply by changing the reagent and altering the conditions [37]. The use of NIS as an electrophilic iodinating agent for aldehydes, ketones and alkenes is well reported but recent developments have seen a host of further applications, such as the use of Brønsted or Lewis acids to improve its reactivity in electrophilic aromatic substitution reactions [38,39,40], asymmetric iodination of aldehydes using an axially chiral catalyst [41] and 1,3-dicarbonyl compounds under mild conditions [42], synthesis of haloalkenes and haloalkynes by catalytic Hunsdiecker reaction [43], the reaction of alkynes and NIS/water to give α-diketones [44], highly enantioselective iodocyclization of polyprenoids [45] and Au-catalyzed formation of 2-iodoenones from propargylic alcohol derivatives [46,47], amongst others. Interestingly, there have also been a few reports on the use of NIS as a mild and selective Lewis acid, such as in the catalytic deprotection of TBDMS ethers to alcohols in methanol under ambient conditions [48] and the N-debenzylation of benzylamino alcohols [49]. Given this spectrum of reactivity and the precedent offered by Dechoux [36,37], we set out to examine the reaction of the Bohlmann-Rahtz aminodienone intermediate 3 with NIS to establish the mechanistic course (Scheme 3). It was anticipated that reaction could proceed by iodination followed by heteroannulation to give the 5-iodopyridine 7 in a similar fashion to Dechoux’s pyridinone synthesis [36] (path a), iodination followed by displacement of iodide to give the acylpyrrole 8 following Dechoux’s pyrrole-5-carboxylate synthesis [37] (path b), or whether it would follow a new reaction course as a Lewis acid to give the Bohlmann-Rahtz pyridine 4 (path c).
Scheme 3.
Possible mechanistic course (path a, b or c) of the reaction between Bohlmann-Rahtzaminodienones 3 and NIS.
Scheme 3.
Possible mechanistic course (path a, b or c) of the reaction between Bohlmann-Rahtzaminodienones 3 and NIS.

2. Results and Discussion
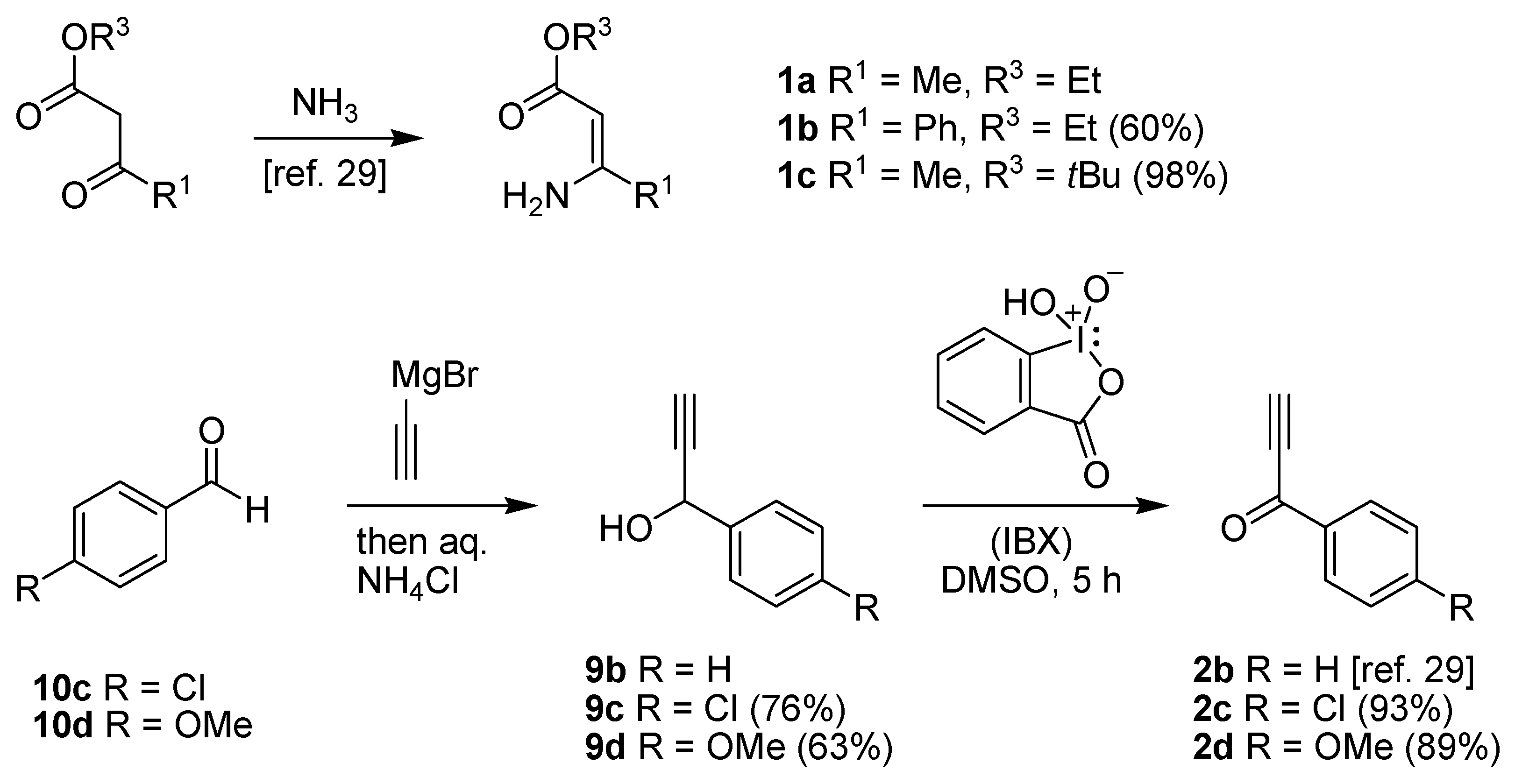
In order to test the behaviour of the Bohlmann-Rahtz intermediates on treatment with NIS, a sample library of aminodienones 3 was prepared from the corresponding enamines 1 and ethynyl ketones 2. The enamine subset 1a-c was generated according to our previously reported procedure (Scheme 4) [29] whereas the ethynyl ketone subset employed 4-(trimethylsilyl)but-3-yn-2-one 2a as a but-3-yn-2-one surrogate [29] and was complemented by additional aryl ethynyl ketone subset members 2b-d prepared by the o-iodoxybenzoic acid (IBX)-mediated oxidation [51] of propargylic alcohols 9b-d, which in turn could be generated by addition of ethynylmagnesium bromide to the corresponding benzaldehyde 10c,d [13,29,50] (Figure 4). This small library of ethynyl ketones 2a-d was chosen to probe if electronic effects had a major influence upon the mechanistic course and, while certainly not exhaustive in scope, was felt to be representative.
Scheme 4.
Enamine and aryl ethynylketone subsets, 1a-c and 2b-d, respectively.

The Michael addition of the enamine subset 1a-c and ethynyl ketones 2a-d was carried out under traditional Bohlmann-Rahtz conditions in ethanol at 50 ºC to give pure aminodienones 3 after purification by column chromatography on silica, in order to ensure no contamination from the corresponding pyridines 4 was evident prior to treatment with NIS. For the most part, the Michael addition reactions were highly efficient, apart from the synthesis of phenyldienone 3ba, which was prepared in low yield as significant cyclodehydration to 4ba (which was removed on purification) occurred spontaneously under the reaction conditions (Table 1).
With pure samples of the Bohlmann-Rahtz aminodienone intermediates 3 in hand, each was treated in turn with a stoichiometric quantity of NIS in ethanol at 0 ºC. In all cases, including both electron-poor and electron-rich aryldienones, only one reaction course was seen to operate: NIS was found to behave as a Lewis acid under these conditions, promoting spontaneous cyclodehydration of the normally kinetically-stable aminodienones to give the corresponding pyridine 4 in excellent yield (Table 1). In all cases only a single pyridine regioisomer was obtained, in accordance with the course of a Bohlmann-Rahtz cyclodehydration reaction [4,5]. The only substrate that was not efficient in this process was aminoheptadienone 3aa (entry 1), but by extending the reaction time to 4 h, pyridine 4aa was isolated in an acceptable 84% yield. All of the other substrates gave a near quantitative yield of the corresponding pyridine products 4. Furthermore, in order to establish the role of the NIS, one of these cyclodehydration reactions were carried out in the presence of a catalytic quantity (20 mol % NIS) of reagent (entry 3) and, for this substrate, the final yield of pyridine 4ab was found to be unchanged (>98% isolated yield).

{kind=link}
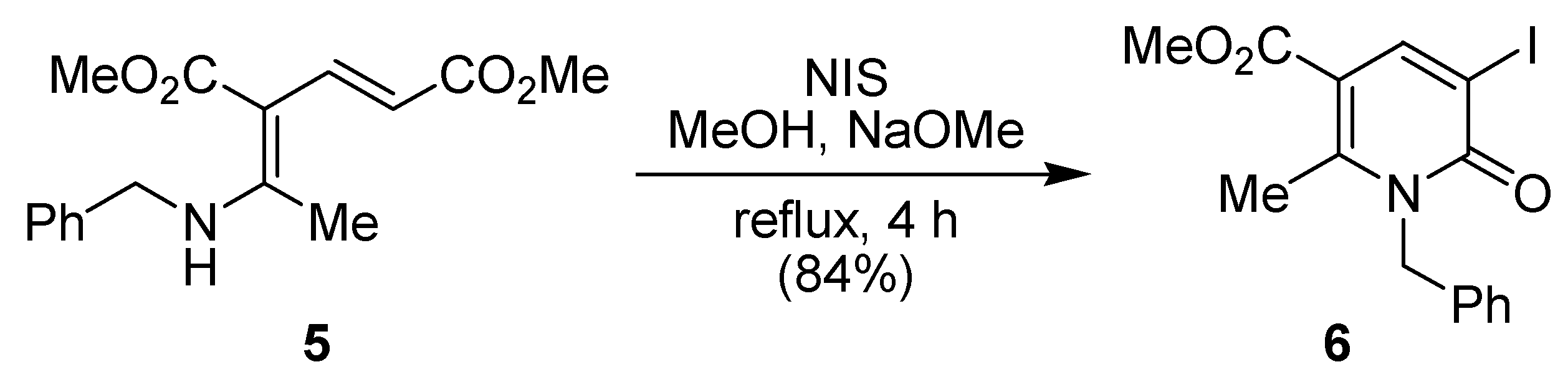{kind=link}
{kind=link}
{kind=link}
| Entry | 1 | 2 | 3 | Yield% a | 4 | Yield% b |
|---|---|---|---|---|---|---|
| 1 | 1a | 2a |  | 3aa (82) |  | 4aa (66 or 84c) |
| 2 | 1b | 2a |  | 3ba (23) |  | 4ba (>98) |
| 3 | 1a | 2b |  | 3ab (85) |  | 4ab (>98)d |
| 4 | 1a | 2c |  | 3ac (86) |  | 4ac (97) |
| 5 | 1a | 2d |  | 3ad (95) |  | 4ad (>98) |
| 6 | 1c | 2a |  | 3ca (68) |  | 4ca (>98)e |
a Isolated yield of 3, obtained after heating a solution of the corresponding subset members 1 and 2 in EtOH at 50 ºC followed by chromatographic purification on silica gel, is given in parentheses; b Isolated yield of 4, obtained after treatment with NIS (1 equiv.) in EtOH at 0 ºC followed by chromatographic purification on silica gel, is given in parentheses; c Reaction with NIS was carried out over 4 h at 0 ºC; d Isolated yield of 4ab was unchanged using either a stoichiometric (1 equiv.) or catalytic (20 mol%) amount of NIS; e Isolated yield of 4ca was unchanged if the reaction was carried out in the presence of NaHCO3.
The reaction course followed by Bohlmann-Rahtz intermediates on treatment with NIS (Scheme 3, path c) is in contrast to the behaviour of aminodienoates such as 5 as reported by Dechoux, which give the corresponding pyrrole derivatives under comparable reaction conditions (in analogy to Scheme 3, path b). This contrasting behaviour could be attributed to the increased reactivity of the ketone towards cyclodehydration but nonetheless is a remarkable switch in mechanism. The surprising facility of this process is also worthy of note. Typically, the Bohlmann-Rahtz cyclodehydration requires high temperatures (up to 200 ºC) or the action of a Brønsted or Lewis acid under heating (microwave or conventional), but, for most of the substrates examined, a near quantitative yield was obtained after only 1 h at 0 ºC. In order to establish if traces of a Brønsted acid (such as HI) had been responsible for the extremely facile cyclodehydration, the reaction of aminodienone 3ca with NIS (1 equiv.) was repeated in the presence of NaHCO3 base (Table 1, entry 6), but the yield of pyridine 4ca was found to be unchanged (>98% isolated yield). Purification of the NIS by recrystallization immediately prior to use was also found to have no effect upon the reaction course or efficiency. Furthermore, in the absence of NIS, when aminodienone 3aa was stirred in EtOH at 0 ºC, no cyclodehydration occurred at all and only unreacted starting material was obtained, as identified by 1H-NMR spectroscopic analysis, confirming a catalytic role for the reagent in the Z/E isomerisation and subsequent spontaneous cyclodehydration.
3. Experimental Section
3.1. General
Commercially available reagents were used without further purification; solvents were dried by standard procedures. Light petroleum refers to the fraction with bp 40–60 ºC, ether (Et2O) refers to diethyl ether and EtOAc refers to ethyl acetate. Column chromatography was carried out using Merck Kieselgel 60 H silica or Matrex silica 60. Analytical thin layer chromatography was carried out using aluminium-backed plates coated with Merck Kieselgel 60 GF254 that were visualised under UV light (at 254 and/or 360 nm). Melting points (mp) were determined on a Kofler hot stage apparatus and are uncorrected. Infra–red (IR) spectra were recorded in the range 4,000–600 cm–1 on a Perkin-Elmer 1600 series FTIR spectrometer using KBr disks for solid samples and thin films between NaCl plates for liquid samples or as a nujol mull and are reported in cm–1. Nuclear magnetic resonance (NMR) spectra were recorded in CDCl3 at 25 ºC unless stated otherwise using a Bruker DPX 400 or 500 Avance instrument operating at 400 or 500 MHz for 1H spectra and 100 or 125 MHz for 13C spectra and were reported in ppm; J values were recorded in Hz and multiplicities were expressed by the usual conventions (s = singlet, d = doublet, t = triplet, app = apparent, m = multiplet). Low-resolution mass spectra (MS) were determined using a Fisons VG Platform II Quadrupole instrument using atmospheric pressure chemical ionization (APcI) unless stated otherwise. ES refers to electrospray ionization, CI refers to chemical ionization (ammonia) and EI refers to electron impact. High-resolution mass spectra were obtained courtesy of the EPSRC Mass Spectrometry Service at Swansea, UK using the ionisation methods specified. Microanalyses were recorded using a Perkin-Elmer 240C Elemental Analyzer. In vacuo refers to evaporation at reduced pressure using a rotary evaporator and diaphragm pump, followed by the removal of trace volatiles using a vacuum (oil) pump.
3.2. Typical experimental procedures
3.2.1. Typical procedure for Michael addition of an enamine 1 and ethynyl ketone 2
A solution of the enamine 1 (0.36 mmol, 1 equiv.) and alkynone 2 (0.56 mmol, 1.5 equiv.) in EtOH (5 mL) was stirred at 50 ºC for 1–7 h, cooled and evaporated in vacuo to give the crude product. Purification by column chromatography on silica gel, eluting with EtOAc–light petroleum, gave the aminodienone 3.
3.2.2. Typical procedure for cyclodehydration of a Bohlmann-Rahtz aminodienone 3 using NIS
A solution of the aminodienone 3 (0.2 mmol, 1 equiv.) and N-iodosuccinimide (NIS) (0.25 mmol, 1.2 equiv.) in EtOH (4 mL) was stirred at 0 ºC for 1 h and then evaporated in vacuo to give the crude product. Purification by column chromatography on silica gel, eluting with EtOAc–light petroleum, gave the pyridine 4.
3.2.3. Typical procedure for the synthesis of propargylic alcohols 9 from aldehydes 10
A solution of the aldehyde 10 (5.0 mmol) in dry THF (10 mL) was added to a stirred solution of ethynylmagnesium bromide in THF (0.5 M; 15 mL, 7.5 mmol) at 0 ºC. The mixture was stirred at 0 ºC for 2 h, warmed to room temperature and stirred overnight. Saturated aqueous NH4Cl solution (2 mL) was added. The mixture was evaporated in vacuo and partitioned between ether (30 mL) and saturated aqueous ammonium chloride solution (30 mL). The ethereal layer was washed with brine (30 mL), dried (Na2SO4) and evaporated in vacuo.
3.3. Experimental procedures
3.3.1. Ethyl 3-amino-3-phenylpropenoate (1b)
Ammonium acetate (13.4 g, 0.17 mol) was added to a solution of ethyl benzoylacetate (5 mL, 29.0 mmol) and the mixture was heated at reflux in toluene–glacial acetic acid (5:1; 40 mL) for 20 h. After partitioning between H2O (100 mL) and ether (60 mL), the aqueous layer was further extracted with ether (2 × 25 mL) and the combined organic extracts were washed sequentially with saturated aqueous NaHCO3 solution (50 mL) and brine (25 mL), dried (MgSO4) and evaporated in vacuo. Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (3:1) (Rf = 0.11), gave the title compound [29] as a pale yellow oil (3.32 g, 60%) (Found: MNH4+, 209.1289. C11H17N2O2 [MNH4+] requires 209.1285); IR (film)/cm-1 νmax 3,441, 3,326, 2,979, 2,936, 1,663, 1,617, 1,555, 1,492, 1,364, 1,176, 1,095, 1,025, 796, 772, 699; 1H-NMR (400 MHz; CDCl3) δH 8.35 (1H, bs, NH), 7.33–7.12 (5H), 7.00 (1H, bs, NH), 4.75 (1H, s, CH), 3.95 (2H, q, J = 7.1 Hz, OCH2Me), 1.05 (3H, t, J = 7.1 Hz, CH2Me); 13C-NMR (100 MHz, CDCl3) δC 170.5 (C), 160.5 (C), 137.7 (C), 130.3 (CH), 128.9 (CH), 126.2 (CH), 84.7 (CH), 59.0 (CH2), 14.6 (Me); m/z (APcI) 192 (MH+, 100%) and 146 (13).
3.3.2. tert-Butyl β-aminocrotonate (1c)
Ammonium hydroxide solution (35%, 40 mL) was added to tert-butyl acetoacetate (4 mL, 24.2 mmol) in MeOH (40 mL) and the mixture was stirred at 50 ºC for 18 h. After cooling, the solution was evaporated in vacuo and partitioned between H2O (40 mL) and ether (40 mL). The aqueous layer was further extracted with EtOAc (2 × 35 mL) and the combined organic extracts were washed with brine (25 mL), dried (Na2SO4) and evaporated in vacuo to give the title compound [52] as a colourless oil (3.72 g, 98%) (Found: MH+, 158.1178. C8H16NO2 [MH+] requires 158.1176); IR (film)/cm-1 νmax 3,554, 3,341, 2,980, 2,919, 1,666, 1,622, 1,567, 1,454, 1,390, 1,366, 1,296, 1,150, 983, 790; 1H-NMR (400 MHz; CDCl3) δH 8.20 (1H, bs, NH), 4.20 (1H, bs, NH), 4.35 (1H, s, CH), 1.80 (3H, s, Me), 1.38 (9H, s, CMe3); 13C-NMR (100 MHz, CDCl3) δC 170.3 (C), 158.8 (C), 85.9 (CH), 78.2 (C), 28.6 (Me), 22.4 (Me); m/z (APcI) 158 (MH+, 77%).
3.3.3. 1-Phenylprop-2-yn-1-one (2b)
A solution of o-iodoxybenzoic acid (IBX) [51] (3.65 g, 13.0 mmol) in DMSO (110 mL) was stirred for 15 min at room temperature until homogeneous. A solution of 1-phenyl-2-propyn-1-ol 9b (1.32 g, 10.0 mmol) in DMSO (10 mL) was added and the mixture was stirred for 5 h. H2O (30 mL) was added and the mixture was stirred at room temperature for 10 min, cooled in ice and partitioned between H2O (120 mL) and ether (90 mL). The mixture was filtered through Celite® and the aqueous layer was further extracted with ether (50 mL). The organic extracts were combined, washed sequentially with H2O (3 × 50 mL), saturated aqueous NaHCO3 solution (70 mL) and brine (70 mL), dried (Na2SO4) and evaporated in vacuo to give the title compound as a pale yellow solid (1.0 g, 77%), m.p. 49–50 ºC (MeOH) (literature [29] m.p. 47–48 ºC) (Found: M+, 130.0414. C9H6O [M+] requires 130.0413); IR (KBr)/cm-1 νmax 3,231, 2,094, 1,645, 1,593, 1,578, 1,452, 1,317, 1,261, 1,173, 1,005, 695; 1H-NMR (400 MHz; CDCl3) δH 8.12 (2H, m, o-PhH), 7.55 (1H, m, p-PhH), 7.45 (2H, m, m-PhH), 3.36 (1H, s, CH); 13C-NMR (100 MHz; CDCl3) δC 177.5 (C), 136.1 (C), 134.6 (CH), 129.7 (CH), 128.7 (CH), 80.9 (C), 80.3 (CH); m/z (EI) 130 (M•+, 16%), 77 (32), 53 (100).
3.3.4. 1-(4-Chlorophenyl)prop-2-yn-1-one (2c)
Propargylic alcohol 9c was prepared using aldehyde 10c according to the general procedure 3.2.3. Purification by column chromatography on SiO2 gel, eluting with CH2Cl2 (Rf 0.27), gave 1-(4-chlorophenyl)prop-2-yn-1-ol (9c) [50] as a pale yellow oil (635 mg, 76%) (Found: M+, 166.0181. C9H7ClO [M+] requires 166.0180); IR (film)/cm–1νmax 3418, 3296, 2884, 2119, 1904, 1645, 1597, 1490, 1406, 1257, 1192, 1092, 1015, 950, 909, 835, 791, 734, 650; 1H NMR (400 MHz; CDCl3) δH 7.48 (2H, app d, J = 8.4 Hz, 2’,6’-PhH), 7.33 (2H, app d, J = 8.4 Hz, 3’,5’-PhH), 5.45 (1H, s, 1-H), 2.68 (1H, s, 3-H), 2.28 (1H, s, OH); 13C NMR (100 MHz; CDCl3) δC 138.4 (C), 134.4 (C), 128.8 (CH), 128.0 (CH), 83.1 (CH), 75.3 (C), 63.7 (CH); m/z (EI) 166 (M•+, 9%), 164 (27), 113 (5), 111 (14), 53 (100). A solution of IBX (5.84 g, 20.8 mmol) in DMSO (120 mL) was stirred for 15 min at room temperature until homogeneous. A solution of 1-(4-chlorophenyl)prop-2-yn-1-ol (9c) (2.72 g, 16.3 mmol) in DMSO (20 mL) was added and the mixture was stirred for 5 h. H2O (40 mL) was added and the mixture was stirred at room temperature for 10 min, cooled in ice and partitioned between H2O (120 mL) and ether (90 mL). The mixture was filtered through Celite® and the aqueous layer was further extracted with ether (50 mL). The organic extracts were combined, washed sequentially with H2O (3 × 50 mL), saturated aqueous NaHCO3 solution (70 mL) and brine (70 mL), dried (Na2SO4) and evaporated in vacuo. Purification by column chromatography on SiO2 gel, eluting with CHCl3 (Rf = 0.45), gave the title compound as a pale yellow solid (2.50 g, 93%), m.p. 68–69 ºC (MeOH) (literature [6] m.p. 68–69 ºC) (Found: M+, 165.9991. C9H5Cl37O [M+] requires 165.9994); IR (KBr)/cm-1 νmax 2,921, 2,853, 2,360, 1,662, 1,462, 1,377, 1,248, 1,094, 1,003, 722; 1H-NMR (400 MHz; CDCl3) δH 7.98 (2H, app d, J = 8.6 Hz, 2’,6’-PhH), 7.41 (2H, app d, J = 8.6 Hz, 3’,5’-PhH), 3.38 (1H, s, CH); 13C-NMR (100 MHz; CDCl3) δC 176.1 (C), 141.3 (C), 134.5 (C), 131.0 (CH), 129.1 (CH), 81.4 (C), 79.9 (CH); m/z (EI) 166 (M•+, 7%), 164 (M•+, 21), 138 (11), 136 (33), 113 (3), 111 (12), 53 (100).
3.3.5. 1-(4-Methoxyphenyl)prop-2-yn-1-one (2d)
Propargylic alcohol 9d was prepared using aldehyde 10d according to the general procedure 3.2.3. Purification by column chromatography on SiO2 gel, eluting with CH2Cl2 (Rf 0.10), gave 1-(4-methoxyphenyl)prop-2-yn-1-ol (9c) [6,53] as a pale yellow oil (513 mg, 63%) (Found: MH+, 161.0596. C10H10O2 [MH+] requires 161.0597); IR (film)/cm-1 νmax 3,438, 3,284, 3,003, 2,935, 2,837, 1,892, 1,611, 1,512, 1,464, 1,442, 1,304, 1,249, 1,174, 1,112, 1,032, 948, 833, 768; 1H-NMR (400 MHz; CDCl3) δH 7.50 (2H, app d, J 8.6, 2’,6’-PhH), 6.90 (2H, app d, J 8.6, 3’,5’-PhH), 5.42 (1H, s, 1-H), 3.85 (3H, s, OMe), 2.65 (1H, s, 3-H); 13C-NMR (100 MHz; CDCl3) δC 159.8 (C), 132.4 (C), 128.1 (CH), 114.0 (CH), 83.7 (CH), 74.7 (C), 64.0 (CH), 55.4 (Me); m/z (EI) 162 (M•+, 100%), 161 (54), 145 (35), 131 (38), 89 (57), 53 (43). A solution of IBX (5.14 g, 18.3 mmol) in DMSO (120 mL) was stirred for 15 min at room temperature until homogeneous. A solution of 1-(4-methoxyphenyl)prop-2-yn-1-ol (9d) (2.27 g, 14.0 mmol) in DMSO (20 mL) was added and the mixture was stirred for 5 h. H2O (40 mL) was added and the mixture was stirred at room temperature for 10 min, cooled in ice and partitioned between H2O (120 mL) and ether (90 mL). The mixture was filtered through Celite® and the aqueous layer was further extracted with ether (80 mL). The organic extracts were combined, washed sequentially with H2O (3 × 50 mL), saturated aqueous NaHCO3 solution (70 mL) and brine (70 mL), dried (Na2SO4) and evaporated in vacuo. Purification by column chromatography on SiO2 gel, eluting with CHCl3 (Rf = 0.29), gave the title compound as a pale yellow solid (2.50 g, 93%), m.p. 86–87 ºC (MeOH) (literature [6] m.p. 85–87 ºC) (Found: MH+, 161.0597. C10H9O2 [MH+] requires 161.0597); IR (KBr)/cm-1 νmax 3,297, 2,092, 1,641, 1,597, 1,572, 1,511, 1,423, 1,252, 1,170, 1,116, 1,023, 841, 758, 710, 685; 1H-NMR (400 MHz; CDCl3) δH 8.05 (2H, app d, J = 8.7 Hz, 2’,6-PhH), 6.88 (2H, app d, J = 8.7 Hz, 3’,5’-PhH), 3.88 (3H, s, OMe), 3.29 (1H, s, CH); 13C-NMR (100 MHz; CDCl3) δC 176.0 (C), 164.8 (C), 132.2 (CH), 129.2 (CH), 113.9 (CH), 80.4 (C), 80.1 (C), 55.6 (Me); m/z (APcI) 161 (MH+, 100%).
3.3.6. (2Z,4E)-2-Amino-3-ethoxycarbonylheptadien-6-one (3aa)
Aminodienone 3aa was prepared according to the general procedure 3.2.1 using ethyl β-aminocrotonate (1a) and 4-(trimethylsilyl)but-3-yn-2-one (2a). Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (1:1) (Rf 0.23), gave the title compound as a yellow solid (58 mg, 82%), m.p. 125–126 ºC(light petroleum–EtOAc) (literature [29] m.p. 125.5–126.4 ºC) (Found: C, 60.6; H, 7.7; N, 7.0. Calc. for C10H15NO3: C, 60.1; H, 7.7; N, 7.1%) (Found: MH+, 198.1125. C10H16NO3 [MH+] requires 198.1125); IR (KBr)/cm-1 νmax 3,334, 3,193, 2,977, 1,647, 1,546, 1,488, 1,459, 1,362, 1,319, 1,286, 1,205, 1,180, 1,112, 1,024, 970, 950, 844; 1H- NMR (400 MHz; CDCl3) δH 9.65 (1H, bs, NH), 7.55 (1H, d, J = 15.6 Hz, 4-H), 6.50 (1H, d, J = 15.6 Hz, 5-H), 5.5 (1H, bs, NH), 4.22 (2H, q, J = 7.1 Hz, OCH2Me), 2.22 (3H, s, Me), 2.15 (3H, s, Me), 1.28 (3H, t, J = 7.1 Hz, CH2Me); 13C-NMR (100 MHz; CDCl3) δC 199.0 (C), 169.7 (C), 165.7 (C), 139.5 (CH), 121.1 (CH), 94.4 (C), 60.0 (CH2), 28.4 (Me), 22.6 (Me), 14.4 (Me); m/z (APcI) 198 (MH+, 100%), 181 (48).
3.3.7. (2Z,4E)-2-Amino-3-ethoxycarbonyl-1-phenylhexadien-6-one (3ba)
Aminodienone 3ba was prepared according to the general procedure 3.2.1 using ethyl 3-amino-3-phenylpropenoate (1b) and 4-(trimethylsilyl)but-3-yn-2-one (2a). Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (1:1) (Rf 0.24), gave the title compound as a yellow solid (22 mg, 23%), m.p. 104–105 ºC (light petroleum–EtOAc) (Found: C, 69.2; H, 6.6; N, 5.2. Calc. for C15H17NO3: C, 69.5; H, 6.6; N, 5.4%); IR (KBr)/cm-1 νmax 3,317, 3,120, 2,980, 1,654, 1,586, 1,507, 1,461, 1,345, 1,285, 1,257, 1,210, 1,119, 1,023, 1,002, 979, 927, 852, 768, 702, 632, 358; 1H-NMR (400 MHz; CDCl3) δH 9.40 (1H, bs, NH), 7.50–7.30 (5H), 7.08 (1H, d, J = 15.9 Hz, 4-H), 6.45 (1H, d, J = 15.9 Hz, 5-H), 5.45 (1H, bs, NH), 4.25 (2H, q, J = 7.1 Hz, OCH2Me), 1.88 (3H, s, Me), 1.35 (3H, t, J = 7.1 Hz, CH2Me); 13C-NMR (100 MHz; CDCl3) δC 190.2 (C), 169.8 (C), 167.4 (C), 142.1 (CH), 136.8 (CH), 130.5 (CH), 128.9 (CH), 128.4 (CH), 122.6 (CH), 95.5 (C), 60.3 (CH2), 26.8 (Me), 14.5 (CH2); m/z (APcI) 260 (MH+, 100%), 243 (12).
3.3.8. (2Z,4E)-2-Amino-3-ethoxycarbonyl-6-phenylhexadien-6-one (3ab)
Aminodienone 3ab was prepared according to the general procedure 3.2.1 using ethyl β-aminocrotonate (1a) and 1-phenylprop-2-yn-1-one (2b). Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (1:1) (Rf 0.24), gave the title compound as a yellow solid (79 mg, 85%), mp 156–157 ºC (light petroleum–EtOAc) (lit. [4] mp 164 °C) (Found: C, 69.4; H, 6.6; N, 5.2. Calc. for C15H18NO3: C, 69.5; H, 6.6; N, 5.4%) (Found: MH+, 260.1282. C15H17NO3 [MH+] requires 260.1281); IR (KBr)/cm-1 νmax 3,342, 3,203, 2,976, 1,623, 1,580, 1,539, 1,497, 1,378, 1,354, 1,320, 1,286, 1,223, 1,205, 1,178, 1,110, 1,055, 1,036, 1,023, 976, 847, 705, 626; 1H-NMR (400 MHz; CDCl3) δH 9.67 (1H, bs, NH), 7.94 (2H, m, o-PhH), 7.86 (1H, d, J = 15.0 Hz, 4-H), 7.43 (3H, m,p-PhH), 7.39 (1H, d, J = 15.0 Hz, 5-H), 5.70 (1H, bs, NH), 4.24 (2H, q, J = 7.1 Hz, OCH2Me), 2.33 (3H, s, Me), 1.35 (3H, t, J = 7.1 Hz, CH2Me); 13C-NMR (100 MHz; CDCl3) δC 190.9 (C), 169.8 (C), 166.7 (C), 141.1 (CH), 139.7 (C), 131.8 (CH), 128.4 (CH), 128.1 (CH), 115.8 (CH), 95.6 (C), 60.0 (CH2), 22.6 (Me), 14.5 (Me); m/z (APcI) 260 (MH+, 100%).
3.3.9. (2Z,4E)-2-Amino-3-ethoxycarbonyl-6-(4-chlorophenyl)hexadien-6-one (3ac)
Aminodienone 3ac was prepared according to the general procedure 3.2.1 using ethyl β-aminocrotonate (1a) and 1-(4-chlorophenyl)prop-2-yn-1-one (2c). Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (1:1) (Rf 0.18), gave the title compound as a yellow solid (91 mg, 86%), m.p. 163–164 ºC (light petroleum–EtOAc) (literature [14] m.p. 164–165 ºC) (Found: MH+, 294.0893. C15H1735ClNO3 [MH+] requires 294.0891); IR (KBr)/cm-1νmax 3,315, 3,168, 2,964, 1,653, 1,630, 1,591, 1,571, 1,539, 1,485, 1,350, 1,323, 1,288, 1,224, 1,177, 1,120, 1,089, 1,057, 1,037, 1,012, 975, 856, 824, 743, 713, 642, 589, 538, 502; 1H-NMR (400 MHz; CDCl3) δH 9.71 (1H, bs, NH), 7.88 (1H, d, J = 15.0 Hz, 4-H), 7.84 (2H, app d, J = 8.5 Hz, 2’,6’-PhH), 7.36 (2H, app d, J = 8.5 Hz, 3’,5’-PhH), 7.33 (1H, d, J = 15.0 Hz, 5-H), 5.78 (1H, bs, NH), 4.20 (2H, q, J = 7.1 Hz, OCH2Me), 2.27 (3H, s Me), 1.35 (3H, t, J = 7.1 Hz, CH2Me); 13C-NMR (100 MHz; CDCl3) δC 189.5 (C), 169.7 (C), 167.0 (C), 141.6 (CH), 138.0 (C), 138.0 (C), 129.5 (CH), 128.6 (CH), 115.1 (CH), 95.7 (C), 60.1 (CH2), 22.6 (Me), 14.5 (Me); m/z (APcI) 296 (MH+, 32%), 294 (MH+, 100).
3.3.10. (2Z,4E)-2-Amino-3-ethoxycarbonyl-6-(4-methoxyphenyl)hexadien-6-one (3ad)
Aminodienone 3ad was prepared according to the general procedure 3.2.1 using ethyl β-aminocrotonate (1a) and 1-(4-methoxyphenyl)prop-2-yn-1-one (2d). Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (1:1) (Rf 0.50), gave the title compound as a yellow solid (99 mg, 95%), mp 155–156 ºC (light petroleum–EtOAc) (literature [54] m.p. 159 ºC) (Found: MH+, 290.1386. C16H20NO4 [MH+] requires 290.1387); IR (KBr)/cm-1 νmax 3,297, 3,145, 2,974, 1,717, 1,627, 1,598, 1,576, 1,539, 1,511, 1,475, 1,362, 1,321, 1,300, 1,263, 1,231, 1,168, 1,126 1,021, 834, 586; 1H-NMR (400 MHz; CDCl3) δH 9.63 (1H, bs, NH), 7.91 (2H, app d, J = 8.8 Hz, 2’,6’-PhH), 7.82 (1H, d, J = 15.0 Hz, 4-H), 7.38 (1H, d, J = 15.0 Hz, 5-H), 6.85 (1H, app d, J = 8.8 Hz, 3’,5’-PhH), 5.63 (1H, bs, NH), 4.23 (2H, q, J = 7.2 Hz, OCH2Me), 3.83 (3H, s, OMe), 2.25 (3H, s Me), 1.35 (3H, t, J = 7.2 Hz, CH2Me); 13C-NMR (100 MHz; CDCl3) δC 189.3 (C), 169.9 (C), 166.3 (C), 162.7 (C), 140.2 (CH), 132.4 (C), 130.2 (CH), 115.7 (CH), 113.6 (CH), 95.5 (C), 60.0 (CH2), 55.4 (Me), 22.6 (Me), 14.5 (Me); m/z (APcI) 290 (MH+, 100%).
3.3.11. (2Z,4E)-2-Amino-3-tert-butoxycarbonylheptadien-6-one (3ca)
Aminodienone 3ca was prepared according to the general procedure 3.2.1 using tert-butyl β-aminocrotonate (1c) and 4-(trimethylsilyl)but-3-yn-2-one (2a). Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (1:1) (Rf 0.26), gave the title compound as a yellow solid (55 mg, 68%), m.p. 142–143 ºC(light petroleum–EtOAc) (literature [14] m.p. 142–144 ºC) (Found: C, 63.8; H, 8.5; N, 6.1. Calc. for C12H19NO3: C, 64.0; H, 8.5; N, 6.2%) (Found: MH+, 226.1437. C12H20NO3 [MH+] requires 226.1438); IR (KBr)/cm-1 νmax 3,341, 3,198, 2,975, 1,654, 1,539, 1,485, 1,454, 1,362, 1,323, 1,295, 1,218, 1,160, 1,111, 1,034, 970, 840, 692, 572; 1H-NMR (400 MHz; CDCl3) δH 9.60 (1H, bs, NH), 7.48 (1H, d, J = 15.4 Hz, 4-H), 6.45 (1H, d, J = 15.4 Hz, 5-H), 5.39 (1H, bs, NH), 2.20 (3H, s, Me), 2.18 (3H, s, Me), 1.48 (9H, s, CMe3); 13C-NMR (100 MHz; CDCl3) δC 198.8 (C), 169.1 (C), 165.2 (C), 139.9 (CH), 120.8 (CH), 95.7 (C), 80.8 (C), 28.5 (Me), 28.4 (Me), 22.7 (Me); m/z (APcI) 226 (MH+, 98%), 208 (54), and 152 (100).
3.3.12. Ethyl 2,6-dimethylpyridine-3-carboxylate (4aa)
Pyridine 4aa was prepared according to the general procedure 3.2.2 using aminodienone 3aa. Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (1:1) (Rf 0.44), gave the title compound as a pale yellow oil (24 mg, 66%) (Found: MH+, 180.1017. C10H14NO2 [MH+] requires 180.1019); IR (film)/cm-1 νmax 2,924, 2,825, 1,730, 1,593, 1,462, 1,377, 1,272, 1,236, 1,148, 1,079, 770, 722; 1H-NMR (400 MHz; CDCl3) δH 8.03 (1H, d, J = 8.0 Hz, 4-H), 7.00 (1H, d, J = 8.0 Hz, 5-H), 4.27 (2H, q, J = 7.2 Hz, OCH2Me), 2.75 (3H, s, Me), 2.50 (3H, s, Me), 1.33 (3H, t, J = 7.2 Hz, CH2Me); 13C-NMR (100 MHz; CDCl3) δC 166.7 (C), 161.1 (C), 159.4 (C), 138.9 (CH), 122.8 (C), 120.5 (CH), 61.1 (CH2), 24.8 (Me), 24.6 (Me), 13.7 (Me); m/z (APcI) 180 (MH+, 100%).
3.3.13. Ethyl 6-methyl-2-phenylpyridine-3-carboxylate (4ba)
Pyridine 4ba was prepared according to the general procedure 3.2.2 using aminodienone 3ba. Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (1:1) (Rf 0.56), gave the title compound as a pale yellow solid (47 mg, 98%), m.p. 45–46 ºC (EtOH) (literature [55] m.p. 46 ºC) (Found: MH+, 242.1175. C15H16NO2 [MH+] requires 242.1176); IR (film)/cm-1 νmax 3,056, 2,963, 2,915, 2,855, 2,363, 1,715, 1,589, 1,440, 1,356, 1,278 1,211, 1,137, 1,106, 1,053, 797, 767, 740, 699, 650; 1H-NMR (400 MHz; CDCl3) δH 7.95 (1H, d, J = 8.0 Hz, 4-H), 7.33–7.25 (5H, PhH), 7.12 (1H, d, J = 8.0 Hz, 5-H), 4.05 (2H, q, J = 7.1 Hz, OCH2Me), 2.55 (3H, s, Me), 0.94 (3H, t, J = 7.1 Hz, CH2Me); 13C-NMR (100 MHz; CDCl3) δC 168.2 (C), 160.8 (C), 158.7 (C), 140.5 (C), 138.4 (CH), 128.5 (CH), 128.4 (CH), 128.1 (CH), 124.4 (C), 121.3 (CH), 61.3 (CH2), 24.8 (Me), 13.6 (Me); m/z (APcI) 242 (MH+, 100%).
3.3.14. Ethyl 2-methyl-6-phenylpyridine-3-carboxylate (4ab)
Pyridine 4ab was prepared according to the general procedure 3.2.2 using aminodienone 3ab. Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (1:1) (Rf 0.62), gave the title compound as a pale yellow solid (47 mg, 98%), m.p. 44–45 ºC (EtOH) (literature [4] m.p. 44 ºC) (Found: MH+, 242.1176. C15H16NO2 [MH+] requires 242.1179); IR (KBr)/cm-1 νmax 2,981, 1,715, 1,582, 1,496, 1,455, 1,382, 1,264, 1,185, 1,154, 1,091, 1,026, 922, 848, 798, 757, 692; 1H-NMR (400 MHz; CDCl3) δH 8.21 (1H, d, J = 8.2 Hz, 4-H), 8.02 (2H, m, o-PhH), 7.60 (1H, d, J = 8.2 Hz, 5-H), 7.42 (3H, m,p-PhH) 4.32 (2H, q, J = 7.2 Hz, OCH2Me), 2.85 (3H, s, Me), 2.45 (3H, t, J = 7.2 Hz, CH2Me); 13C- NMR (100 MHz; CDCl3) δC 166.9 (C), 160.0 (C), 159.1 (C), 139.3 (CH), 138.5 (C), 129.7 (CH), 128.8 (CH), 127.3 (CH), 123.7 (C), 117.4 (CH), 61.2 (CH2), 25.3 (Me), 14.3 (Me); m/z (APcI) 242 (MH+, 100%).
3.3.15. Ethyl 2-methyl-6-(4-chlorophenyl)pyridine-3-carboxylate (4ac)
Pyridine 4ac was prepared according to the general procedure 3.2.2 using aminodienone 3ac. Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (1:1) (Rf 0.68), gave the title compound as a pale yellow solid (59 mg, 97%), m.p. 48–49 ºC (aqueous EtOH) (literaute [6] m.p. 47–48 ºC) (Found: MH+, 276.0784. C15H1535ClNO2 [MH+] requires 276.0786); IR (KBr)/cm-1 νmax 2,985, 1,720, 1,585, 1,492, 1,455, 1,372, 1,265, 1,180, 1,155, 1,096, 1,013, 896, 833, 786, 742, 705; 1H-NMR (400 MHz; CDCl3) δH 8.23 (1H, d, J = 8.2 Hz, 4-H), 7.95 (2H, app d, J = 8.5 Hz, 2’,6’-PhH), 7.54 (1H, d, J = 8.2 Hz, 5-H), 7.40 (2H, app d, J = 8.5 Hz, 3’,5’-PhH), 4.38 (2H, q, J = 7.1 Hz, OCH2Me), 1.60 (3H, s, Me), 1.35 (3H, t, J = 7.1 Hz, CH2Me); 13C-NMR (100 MHz; CDCl3) δC 166.5 (C), 160.1 (C), 157.7 (C), 139.4 (CH), 136.9 (C), 135.9 (C), 129.0 (CH), 128.6 (CH), 123.9 (C), 117.4 (CH), 61.2 (CH2), 25.3 (Me), 14.3 (Me); m/z (APcI) 278 (MH+, 33%), 276 (MH+, 100).
3.3.16. Ethyl 2-methyl-6-(4-methoxyphenyl)pyridine-3-carboxylate (4ad)
Pyridine 4ad was prepared according to the general procedure 3.2.2 using aminodienone 3ad. Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (1:1) (Rf 0.63), gave the title compound as a pale yellow solid (53 mg, 98%), m.p. 68–69 ºC (aqueous EtOH) (literature [6] m.p. 68–69 ºC) (Found: MH+, 272.1284. C16H18NO3 [MH+] requires 272.1281); IR (KBr)/cm-1 νmax 2,989, 2,895, 1,717, 1,605, 1,579, 1,509, 1,452, 1,439, 1,389, 1,362, 1,311, 1,264, 1,172, 1,112, 1,088, 1,071, 1,031, 832, 785; 1H-NMR (500 MHz; CDCl3) δH 8.20 (1H, d, J = 8.2 Hz, 4-H), 7.95 (2H, app d, J = 8.7 Hz, 2’,6’-PhH), 7.53 (1H, d, J = 8.2 Hz, 5-H), 7.37 (2H, app d, J = 8.7 Hz, 3’,5’-PhH), 4.34 (2H, q, J = 7.1 Hz, OCH2Me), 3.82 (3H, s, OMe), 2.85 (3H, s, Me), 1.36 (3H, t, J = 7.1 Hz, CH2Me); 13C-NMR (125 MHz; CDCl3) δC 166.8 (C), 161.0 (C), 160.0 (C), 158.7 (C), 139.3 (CH), 131.7 (C), 128.7 (CH), 122.8 (C), 116.5 (CH), 114.2 (CH), 61.1 (CH2), 55.4 (Me), 25.4 (Me), 14.4 (Me); m/z (APcI) 272 (MH+, 100%).
3.3.17. tert-Butyl 2,6-dimethylpyridine-3-carboxylate (4ca)
Pyridine 4ca was prepared according to the general procedure 3.2.2 using aminodienone 3ca. Purification by column chromatography on SiO2 gel, eluting with light petroleum–EtOAc (1:1) (Rf 0.59), gave the title compound [29] as a pale yellow oil (41 mg, 98%) (Found: MH+, 208.1329. C12H18NO2 [MH+] requires 208.1332); IR (film)/cm-1 νmax 2,921, 2,852, 1,724, 1,592, 1,462, 1,377, 1,284, 1,175, 1,144, 1,080, 771, 722; 1H-NMR (400 MHz; CDCl3) δH 7.95 (1H, d, J = 8.0 Hz, 4-H), 6.95 (1H, d, J = 8.0 Hz, 5-H), 2.68 (3H, s, Me), 2.45 (3H, s, Me), 1.50 (9H, s, CMe3); 13C-NMR (100 MHz; CDCl3) δC 166.1 (C), 160.6 (C), 158.7 (C), 138.8 (CH), 124.5 (C), 120.5 (CH), 81.7 (C), 28.2 (Me), 24.8 (Me), 24.6 (Me); m/z (APcI) 208 (MH+, 100%).
4. Conclusions
The use of NIS in EtOH represents a new and rapid mild method for the low temperature cyclodehydration of Bohlmann-Rahtz aminodienone intermediates 3 to give the corresponding 2,3,6-trisubstituted pyridines 4 in excellent yield and with total regiocontrol. In this process, the NIS appears to be behaving as a remarkable Lewis acid, that catalyzes E/Z isomerisation and spontaneous cyclodehydration at 0 ºC, rather than an iodinating agent. This method for mild and efficient cyclodehydration is likely to find application, in particular for the synthesis of pyridines from acid-sensitive substrates, and will be reported [23] in due course.
Acknowledgements
We thank the EPSRC Mass Spectrometry Service, Swansea for high resolution spectra and the EPSRC for funding (studentship award to CG).
References and Notes
- Roth, H.J.; Kleemann, A. Pharmaceutical Chemistry. Drug Synthesis; John Wiley & Sons: New York, NY, USA, 1988; Volume 1. [Google Scholar]
- Henkel, T.; Brunne, R.M.; Müller, H.; Reichel, F. Statistical investigation into the structural complexity of natural products and synthetic compounds. Angew. Chem. Int. Ed. Engl. 1999, 38, 643–647. [Google Scholar]
- Hantzsch, A. Condensationprodukte aus Aldehydammoniak und Ketoniartigen Verbindungen. Berichte 1881, 14, 1637–1638. [Google Scholar]
- Bohlmann, F.; Rahtz, D. Űber eine neue pyridinsynthese. Chem. Ber. 1957, 90, 2265–2272. [Google Scholar] [CrossRef]
- Bagley, M.C.; Glover, C.; Merritt, E.A. The Bohlmann–Rahtz pyridine synthesis: from discovery to applications. Synlett 2007, 2459–2482. [Google Scholar]
- Bagley, M.C.; Dale, J.W.; Ohnesroge, M.; Xiong, X.; Bower, J. A facile solution phase combinatorial synthesis of tetrasubstituted pyridines using the Bohlmann–Rahtzheteroannulation reaction. J. Comb. Chem. 2003, 5, 41–44. [Google Scholar] [CrossRef]
- Bashford, K.E.; Burton, M.B.; Cameron, S.; Cooper, A.L.; Hogg, R.D.; Kane, P.D.; MacManus, D.A.; Matrunola, C.A.; Moody, C.J.; Robertson, A.A.B.; Warne, M.R. The Bohlmann–Rahtz route to functionalised pyridine scaffolds and their use in library synthesis. Tetrahedron Lett. 2003, 44, 1627–1629. [Google Scholar]
- Bagley, M.C.; Hughes, D.D.; Lloyd, R.; Powers, V.E.C. A new and highly expedient synthesis of pyrido[2,3–d]pyrimidines. Tetrahedron Lett. 2001, 42, 6585–6588. [Google Scholar]
- Bagley, M.C.; Dale, J.W.; Hughes, D.D.; Ohnesorge, M.; Phillips, N.G.; Bower, J. Synthesis of pyridines and pyrido[2,3–d]pyrimidines by the Lewis acid catalysed Bohlmann–Rahtz heteroannulation reaction. Synlett 2001, 1523–1526. [Google Scholar]
- Bagley, M.C.; Hughes, D.D.; Lloyd, R.; Powers, V.E.C. New approaches towards inhibitors of folate-dependent enzymes: rapid synthesis of 5-deazapterins from uracil derivatives. In Chem. Biol., Pteridines Folates 2001 (2002), Proc. Int. Symp. Pteridines Folates; Kluwar Academic Publishers: Norwell, Massachusetts, USA, 2002; pp. 415–420. [Google Scholar]
- Hughes, D.D.; Bagley, M.C. One or two-step Bohlmann–Rahtz heteroannulation of 6-aminouracil derivatives for the synthesis of pyrido[2,3–d]pyrimidines. Synlett 2002, 1332–1334. [Google Scholar]
- Davis, J.M.; Truong, A; Hamilton, A.D. Synthesis of a 2,3; 6,3-terpyridine scaffold as an α-helix mimetic. Org. Lett. 2005, 7, 5405–5408. [Google Scholar] [CrossRef]
- Bagley, M.C.; Lin, Z.; Pope, S.J.A. Rapid synthesis of 3-cyanopyridine-derived chromophores with two-dimensional tunability and solvatochromicphotophysical properties. Chem. Commun. 2009, 5165–5167. [Google Scholar]
- Bagley, M.C.; Lin, Z.; Pope, S.J.A. Microwave-assisted synthesis and complexation of luminescent cyanobipyridyl-zinc(II) bis(thiolate) complexes with intrinsic and ancillary photophysical tunability. Dalton Trans. 2010, 39, 3163–3166. [Google Scholar] [CrossRef]
- Adlington, R.M.; Baldwin, J.E.; Catterick, D.; Pritchard, G.J.; Tang, L.T. The synthesis of novel heterocyclic substituted α-amino acids; further exploitation of α-amino acid alkynyl ketones. J. Chem. Soc., Perkin Trans. 2000, 1, 303–305. [Google Scholar]
- Adlington, R.M.; Baldwin, J.E.; Catterick, D.; Pritchard, G.J.; Tang, L.T. The synthesis of novel heterocyclic substituted α-amino acids; further exploitation of α-amino acid alkynyl ketones as reactive substrates. J. Chem. Soc., Perkin Trans. 2000, 1, 2311–2316. [Google Scholar]
- Adamo, M.F.A.; Adlington, R.M.; Baldwin, J.E.; Pritchard, G.J.; Rathmell, R.E. Practical routes to diacetylenicketones and their application for the preparation of alkynyl substituted pyridines, pyrimidines and pyrazoles. Tetrahedron 2003, 59, 2197–2205. [Google Scholar] [CrossRef]
- Bagley, M.C.; Dale, J.W.; Merritt, E.A.; Xiong, X. Thiopeptide antibiotics. Chem. Rev. 2005, 105, 685–714. [Google Scholar]
- Hughes, R.A.; Moody, C.J. From amino acids to heteroaromatics–thiopeptide antibiotics, nature’s heterocyclic peptides. Angew. Chem. Int. Ed. 2007, 46, 7930–7954. [Google Scholar] [CrossRef]
- Moody, C.J.; Bagley, M.C. The first synthesis of promothiocin A. Chem. Commun. 1998, 2049–2050. [Google Scholar]
- Bagley, M.C.; Bashford, K.E.; Hesketh, C.L.; Moody, C.J. Total synthesis of the thiopeptidepromothiocin A. J. Am. Chem. Soc. 2000, 122, 3301–3313. [Google Scholar]
- Aulakh, V.S.; Ciufolini, M.A. An improved synthesis of pyridine-thiazole cores of thiopeptide antibiotics. J. Org. Chem. 2009, 74, 5750–5753. [Google Scholar] [CrossRef]
- Bagley, M.C.; Xiong, X. An improved synthesis of pyridine-thiazole cores of thiopeptide antibiotics. Org. Lett. 2004, 6, 3401–3404. [Google Scholar] [CrossRef]
- Bagley, M.C.; Dale, J.W.; Jenkins, R.L.; Bower, J. First synthesis of an amythiamicin pyridine cluster. Chem. Commun. 2004, 102–103. [Google Scholar]
- Bagley, M.C.; Dale, J.W.; Xiong, X.; Bower, J. Synthesis of dimethyl sulfomycinamate. Org. Lett. 2003, 5, 4421–4424. [Google Scholar] [CrossRef]
- Bagley, M.C.; Chapaneri, K.; Dale, J.W.; Xiong, X.; Bower, J. One-pot multistep Bohlmann–Rahtz heteroannulation reactions: synthesis of dimethyl sulfomycinamate. J. Org. Chem. 2005, 70, 1389–1399. [Google Scholar]
- Merritt, E.A.; Bagley, M.C. Synthesis of the central heterocyclic domain of micrococcin P1. Synlett 2007, 954–958. [Google Scholar]
- Bagley, M.C.; Dale, J.W.; Bower, J. A new modification of the Bohlmann–Rahtz pyridine synthesis. Synlett 2001, 1149–1151. [Google Scholar]
- Bagley, M.C.; Brace, C.; Dale, J.W.; Ohnesorge, M.; Phillips, N.G.; Xiong, X.; Bower, J. Synthesis of tetrasubstituted pyridines by the acid-catalysed Bohlmann–Rahtz reaction. J. Chem. Soc., Perkin Trans. 2002, 1, 1663–1671. [Google Scholar]
- Xiong, X.; Bagley, M.C.; Chapaneri, K. A new mild method for the one-pot synthesis of pyridines. Tetrahedron Lett. 2004, 45, 6121–6124. [Google Scholar] [CrossRef]
- Bagley, M.C.; Lunn, R.; Xiong, X. A new one-step synthesis of pyridines under microwave-assisted conditions. Tetrahedron Lett. 2002, 43, 8331–8334. [Google Scholar] [CrossRef]
- Bagley, M.C.; Jenkins, R.L.; Lubinu, M.C.; Mason, C.; Wood, R. A simple continuous flow microwave reactor. J. Org. Chem. 2005, 70, 7003–7006. [Google Scholar]
- Bagley, M.C.; Fusillo, V.; Jenkins, R.L.; Lubinu, M.C.; Mason, C. Continuous flow processing from microreactors to mesoscale: the Bohlmann–Rahtzcyclodehydration reaction. Org. Biomol. Chem. 2010, 8, 2245–2251. [Google Scholar]
- Bagley, M.C.; Hughes, D.D.; Sabo, H.M.; Taylor, P.H.; Xiong, X. One pot synthesis of pyridines or pyrimidines by tandem oxidation-heteroannulation of propargylic alcohols. Synlett 2003, 1443–1446. [Google Scholar]
- Bagley, M.C.; Dale, J.W.; Bower, J. A new one-pot three-component condensation reaction for the synthesis of 2,3,4,6-tetrasubstituted pyridines. Chem. Commun. 2002, 1682–1683. [Google Scholar]
- Agami, C.; Dechoux, L.; Hebbe, S.; Moulinas, J. An efficient synthesis of polysubstituted 3-halo-2(1H)-pyridinones. Synthesis 2002, 79–82. [Google Scholar]
- Agami, C.; Dechoux, L.; Hebbe, S. Efficient and flexible access to polysubstituted pyrroles. Synlett 2001, 1440–1442. [Google Scholar]
- Thiebes, C.; Prakash, G.K.S.; Petasis, N.A.; Olah, G.A. Mild preparation of haloarenes by ipso-substitution of arylboronic acids with N-halosuccinimides. Synlett 1998, 141–142. [Google Scholar]
- Castanet, A.-S.; Colobert, F.; Broutin, P.-E. Mild and regioselective iodination of electron-rich aromatics with N-iodosuccinimide and catalytic trifluoroacetic acid. Tetrahedron Lett. 2002, 43, 5047–5048. [Google Scholar]
- Olah, G.A.; Wang, Q.; Sandford, G.; Prakash, G.K.S. Iodination of deactivated aromatics with N-iodosuccinimide in trifluoromethanesulfonic acid (NIS–CF3SO3H) via in situ generated superelectrophilic iodine(I) trifluoromethanesulfonate. J. Org. Chem. 1993, 58, 3194–3195. [Google Scholar] [CrossRef]
- Kano, T.; Ueda, M.; Maruoka, K. Direct asymmetric iodination of aldehydes using an axially chiralbifunctional amino alcohol catalyst. J. Am. Chem. Soc. 2008, 130, 3728–3729. [Google Scholar] [CrossRef]
- Yang, D.; Yan, Y.-L.; Lui, B. Mild α-halogenation reactions of 1,3-dicarbonyl compounds catalyzed by Lewis acids. J. Org. Chem. 2002, 67, 7429–7431. [Google Scholar]
- Das, J.P.; Roy, S. Catalytic Hunsdiecker reaction of α,β-unsaturated carboxylic acids: how efficient is the catalyst? J. Org. Chem. 2002, 67, 7861–7864. [Google Scholar] [CrossRef]
- Niu, M.; Fu, H.; Jiang, Y.; Zhao, Y. A convenient and practical approach to α-diketones via reactions of internal aryl alkynes with N-iodosuccinimide/water. Synthesis 2008, 2879–2882. [Google Scholar]
- Sakakura, A.; Ukai, A.; Ishihara, K. Enantioselective halocyclization of polyprenoids induced by nucleophilic phosphoramidites. Nature 2007, 445, 900–903. [Google Scholar]
- Crone, B.; Kirsch, S.F. Synthesis of 4-iodo-3-furanones utilizing electrophile-induced tandem cyclization/1,2-migration reactions. J. Org. Chem. 2007, 72, 5435–5438. [Google Scholar] [CrossRef]
- Yu, M.; Zhang, G.; Zhang, L. Gold-catalyzed efficient preparation of linear α-iodoenones from propargylic acetates. Org. Lett. 2007, 9, 2147–2150. [Google Scholar] [CrossRef]
- Karimi, B.; Zamani, A.; Zareyee, D. N-Iodosuccinimide (NIS) as as mild and highly chemoselective catalyst for deprotection of tert-butyldimethylsilyl ethers. Tetrahedron Lett. 2004, 45, 9139–9141. [Google Scholar] [CrossRef]
- Grayson, E.J.; Davis, B.G. A tuneable method for N-debenzylation of benzylamino alcohols. Org. Lett. 2005, 7, 2361–2364. [Google Scholar] [CrossRef]
- Bagley, M.C.; Hughes, D.D.; Lubinu, M.C.; Merritt, E.A.; Taylor, P.H.; Tomkinson, N.C.O. Microwave-assisted synthesis of pyrimidine libraries. QSAR Comb. Sci. 2004, 23, 859–867. [Google Scholar] [CrossRef]
- Frigerio, M.; Santagostino, M.; Sputore, S. A user-friendly entry to 2-iodoxybenzoic acid (IBX). J. Org. Chem. 1999, 64, 4537–4538. [Google Scholar] [CrossRef]
- Buckler, R.T.; Hartzler, H.E.; Phillips, B.M. Antiinflammatory β-arylamidoacrylic acids. J. Med. Chem. 1975, 18, 509–513. [Google Scholar] [CrossRef]
- Irving, F.; Johnson, A.W. Synthesis of 3-acyl- and 3-aroyl-meso benzanthrones. J. Chem. Soc. 1948, 2037–2038. [Google Scholar] [CrossRef]
- Bohlmann, F. Verfahren zur herstellung von pyridinderivaten. Ger. Pat. DE 1190464, 1965. [Chem. Abs.1965, 63, 3272].. [Google Scholar]
- Ohta, K.; Iwaoka, J.; Kamijo, Y.; Okada, M.; Nomura, Y. Formation of pyridines by the reaction of isoxazoles with enamines. Nippon Kagaku Kaishi 1989, 1593–1600. [Google Scholar]
- Sample Availability: Samples of the pyridines 4 are available from the authors.
© 2010 by the authors;
Share and Cite
MDPI and ACS Style
Bagley, M.C.; Glover, C. Bohlmann-Rahtz Cyclodehydration of Aminodienones to Pyridines Using N-Iodosuccinimide. Molecules 2010, 15, 3211-3227. https://doi.org/10.3390/molecules15053211
AMA Style
Bagley MC, Glover C. Bohlmann-Rahtz Cyclodehydration of Aminodienones to Pyridines Using N-Iodosuccinimide. Molecules. 2010; 15(5):3211-3227. https://doi.org/10.3390/molecules15053211
Chicago/Turabian StyleBagley, Mark C., and Christian Glover. 2010. "Bohlmann-Rahtz Cyclodehydration of Aminodienones to Pyridines Using N-Iodosuccinimide" Molecules 15, no. 5: 3211-3227. https://doi.org/10.3390/molecules15053211