Unexplored Nucleophilic Ring Opening of Aziridines †


1
Instituto de Reconocimiento Molecular y Desarrollo Tecnológico (IDM), Centro Mixto Universidad Politécnica de Valencia, Universidad de Valencia, 46100 Burjassot, Spain
2
Department de Organic Chemistry, Universitat de València, Dr. Moliner 50, 46100 Burjassot, Spain
*
Author to whom correspondence should be addressed.
†
Dedicated to Professor Carmen Nájera on occasion of her 60th birthday.
Molecules 2010, 15(12), 9135-9144; https://doi.org/10.3390/molecules15129135
Submission received: 24 November 2010
/
Revised: 1 December 2010
/
Accepted: 6 December 2010
/
Published: 10 December 2010
Abstract
:The reactivity of dianions of carboxylic acids towards aziridines has been studied. Although, a similar reactivity to that of enolates from ketones, esters or amides has been observed, the method directly yields γ-aminoacids in one step. The method is complementary of previous results of enenediolate reactivity with other electrophiles. A comparative study with the reactivity of this enediolates with epoxides is included.
Introduction
Aziridines have been recognized as an attractive building block for the synthesis of a variety of nitrogen-containing biologically active compounds [1,2] Their high ring strain energy promotes high reactivity and many nucleophiles are able to cleave the ring [3,4]. The ring opening of aziridines is a widely investigated reaction and has been used to generate a large number of functionalized organic compounds that are not easily accessible by other means. However, the efficiency of the ring opening reactions of aziridines are heavily dependent upon the nature of the substituents on the three-member ring amines, the nucleophile and the reaction conditions employed [1,2,3,4,5,6,7].
The presence of electron-withdrawing substituents on the nitrogen atom activates the ring that then reacts easily with nucleophiles to form ring-opened products. In contrast, non-activated aziridines are relatively inert towards nucleophiles. Enolates derived from ketones, esters and amides have been used as effective nucleophiles to undergo addition to aziridines. The application of the enolate addition to aziridines has largely occurred in steroselective ring opening to form γ-amino carbonyl difunctionalized derivatives [3].
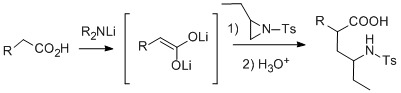
In order to extend our methodology using enediolates from carboxylic acids we wished to complete the study of nucleophilic ring opening with these nucleophiles, which permits direct acces to γ-aminoacids, as a full complement to the efficient synthesis of these compounds from enediolates of carboxylic acids and bromoacetonitrile, described previously by us [8,9].
Results and Discussion
Carboxylic acids are synthetically useful building blocks because, after double deprotonation, they afford enediolates (or dienediolates when starting from α,β-unsaturated carboxylic acids) that react with various electrophiles under adequate conditions [10,11,12]. Lithium dialkylamides are commonly used as bases to generate the lithium dianions [10,11,12,13], due to their strength and low nucleophilicity, specially when derived from sterically hindered amines, combined with their solubility in non-polar solvents [13,14]. It is well established that, in these solvents, lithium enolates exist as complex ion pair aggregates, whose metal center may be coordinated to solvent molecules or other chelating ligands, such as the amines resulting from deprotonation of the acid by the lithium amide. The available data confirm the complexity present in those aggregated reactive species, whose reactivity and selectivity products can be influenced by many factors [10,11,12,13,14,15,16,17,18].
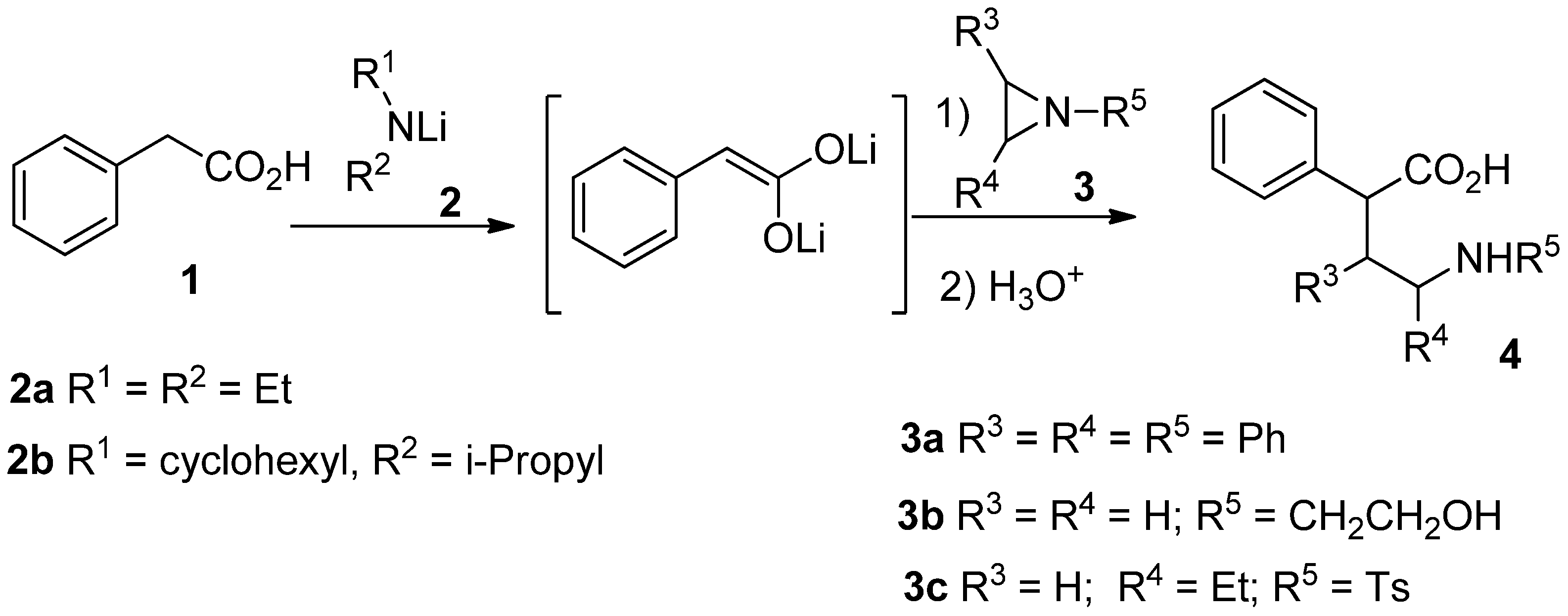
We began describing the optimization of the addition reaction of enediolate of phenylacetic acid (1) with aziridines (Scheme 1). We have used in a first experience the commercially available aziridines 3a and 3b and lithium diethylamide as base to generate the dianion under standard condition [19] (Table 1, entries 1 and 2). In both reactions, only starting material was recovered. Although this was expected from the reactivity of non-activated aziridines with nucleophiles, it was worth testing as enediolates have shown a distinctive reactivity from other nucleophiles [9,10,11]. Thus, the more reactive 2-ethyl-1-tosylaziridine (3c), synthesized as described [20], was used in the rest of the experiments.
Two amines were tested as a base to generate the dienediolates, being cyclohexylisopropilamine 2b the most efficient in these reactions. Previous studies lead us to develop sub-stoichiometric lithium amide conditions for the generation of dianions of carboxylic acids, which, in some cases improve the yield and selectivity of the reaction [19]. These conditions are especially adequate when lithium amide attacks faster the electrophile than the dianion. In this case, sub-stoichiometric amount of the amide did not led to any improvement (Table 1, entry 6). The last two entries in Table 1 reproduce the best conditions that we obtained in the addition of dianions of carboxylic acids to epoxides [21,22] by using LiCl as a disaggregating agent in the enolate solution (entry 8) or as a Lewis acid activating the epoxide (entry 9) where the dianion solution was added (inverse addition) to a mixture of the electrophile with LiCl in THF.
In spite of the slight increase of yield when compared to entry 7, use of the latter procedure was discarded as it is more complex. The optimized conditions for reaction with aziridine 3c, namely lithium cyclohexylisopropylamide in equimolecular amount to generate the dianion and 1h reaction time at room temperature was extended to the rest of carboxylic acids (Table 2).
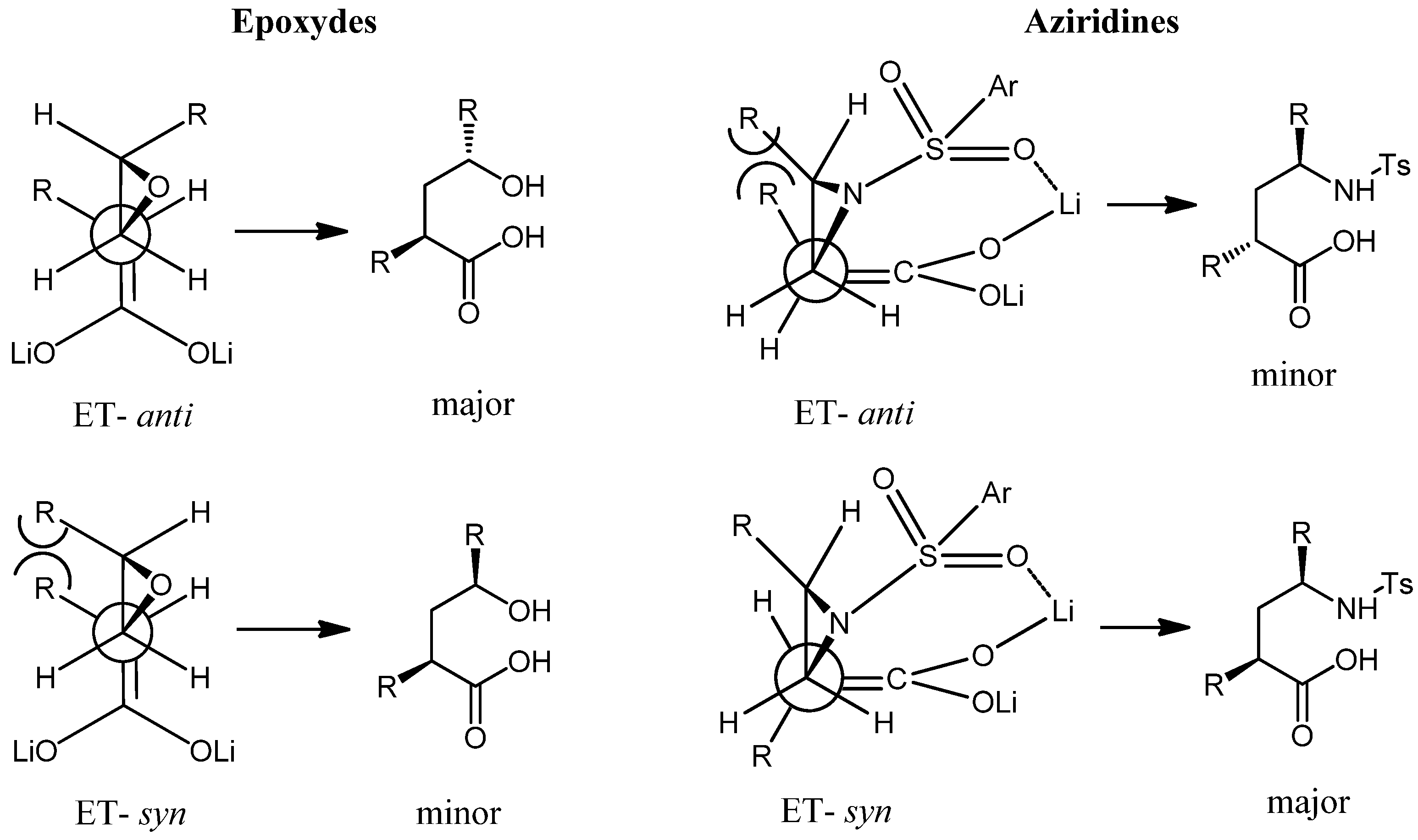
From non-conjugated carboxylic acids (i.e. 1, 5 and 6) γ-aminoacids were obtained straight away in moderate yield. As usual, the less hindered position of the aziridine was attacked to give compounds 4, 13, and 14, with the syn:anti diasteroselectivity shown in Table 2. Despite the low diasteroselectivity, the syn:anti ratio was determined by NOESY studies. It is worth mention that the major syn selectivity contrast with the results obtained in the addition of carboxylic acid dianion to epoxides [22]. In cases like this, Taylor [23] consider the pre-transition state for SN2 type reaction, where the bulky groups appear to be too far away from each other to show a significant effect (Figure 1). In the reaction of the dianion with aziridine the sulfoniloxy group play an important role in the pre-transition state by means of their coordination with the lithium ions. This can overcome steric hindrance and change the diastereoselective ratio. Similar effect has been observed by us in the regioselective alkylation of dienediolates of carboxylic acids with tosylates [24].
On the other hand, we have extended this methodology to α,β-unsaturated carboxylic acids, whose double deprotonation lead to dienediolates that behave as ambident nucleophiles through their α or γ carbon atoms [10,11,12]. Although α attack predominates for irreversible reactions, strong deviations are observed in alkylation reactions [25,26]. Steric and electronic effects and the aggregation states in the middle of reaction determine the regioselectivity of the reaction with each electrophile.
Reactions of dimethylacrilic acid (8) and crotonic acid (9) with aziridines showed no regioselectivity, in adition products from crotonic acid (9) showed to be unstable and decomposed on purification. Only for tiglic acid (7) with a methyl group in the α position, is the γ-adduct the only regioisomer observed.
The diasteroselectivity of α−products from crotonic acid was not determined, but in the case of α-adducts 16, the ratio was similar to that found for α-adducts from saturated acids, and as before, the corresponding γ-aminoacids were obtained. The method can be extended to o-methyl aromatic acids 10, 11, and 12 leading to aminoacids 20, 21 and 22 in 40–60% yield.
Conclusions
In summary, we have checked that the dianions of carboxylic acids react with aziridines in a similar way than enolates from ketones, esters or amides. In this case though, γ-aminoacids are directly obtained in one step. In those reactions leading to diastereosimers a moderate syn selectivity is observed. Thus, highly functionalized small molecules are obtained which are interesting building blocks to other transformations.
Experimental
General
IR spectral data were obtained for liquid films between KBr discs, and the measurements were carried out by the SCSIE (Servei Central de Suport a la Investigació Experimental de la Universitat de Valencia) on a Matteson Satellite FTIR 3000 model Spectrophotometer. NMR spectra were recorded at 25 °C for solutions in the stated solvent on Bruker Avance 300 or 400 spectrometers. High resolution mass spectra were determined with a Fison VG Autospec spectrometer. Flash Column Silica Gel (230–400 mesh, Scharlau) was used for flash column chromatography, with hexane/ethyl acetate mixtures for elution. All reactions were carried out under argon atmospheres, in oven dried glassware, using standard conditions for exclusion of moisture. THF was freshly distilled from blue benzophenone ketyl and amines were distilled from CaH2 and stored over molecular sieves and kept under Ar. The BuLi used was a 1.6 M hexane solution. This solution’s concentration was periodically checked before use. The −78 °C reaction temperature was achieved by cooling with a CO2/acetone bath and 0 °C achieved by an ice/water bath. Organic extracts were dried over anhydrous MgSO4, and solutions were evaporated under reduced pressure with a rotatory evaporator and a bath set at 40 °C.
General procedure for the reaction of lithium enediolates with N-tosylaziridine
n-BuLi (1.6 M in hexane, 3.1 mL, 5 mmol) was introduced into a previously purged reaction flask. The hexane was evaporated under vacuum and THF (2 mL), followed by cyclohexylisopropylamine (0.83 mL, 5 mmol) were added at -78 °C. The mixture was stirred for 15 min at 0 °C. The acid (2.25 mmol) in THF (2 mL) was added slowly at −78 °C and the mixture was kept at 0 °C for 30 min. N-tosyl-2-ethylaziridine (506 mg, 2.25 mmol) in THF (2 mL) was added slowly at −78 °C. The solution was stirred at room temperature for 1 h and quenched with H2O (15 mL). The reaction mixture was extracted with Et2O (3 × 15 mL). The aqueous phase at 0 °C, was acidified to pH 1 with conc. HCl and then extracted with EtOAc (3 × 15 mL) and the combined extracts were dried over anh. MgSO4. After evaporation of the solvent, the corresponding aminoacid was obtained.
2-Phenyl-4-(4-methylphenylsulfonamido)hexanoic acid (4). From phenylacetic acid (1, 306 mg); yield: 544 mg (67%); yellow oil; IR (KBr): υ = 3500–2700, 3258, 2974, 1710, 1600, 1496, 1367, 1164, 978, 712 cm−1; HRMS: m/z calcd. for C19H23NO4S [M+]: 361.1348; found: 361.1358. MS: m/z (%) = 361 [M+, 1%]; 343 [M+-H2O]; 332 [M+-CH3CH2, 34%]; 279 [C16H23O4+, 46%]; 212 [C13H10NO2+, 43%]. (2R*, 4R*) 1H-NMR (400 MHz, CDCl3): δ = 0.69 (t, J = 7.5 Hz, 3H, CH3CH2); 1.35 (m, 2H, CH3CH2); 1.92 (m, 1H, CHCHHCH); 2.23 (m, 1H, CHCHHCH); 2.41 (s, 3H, PhCH3); 3.17 (m, 1H, CHNH); 3.74 (t, J = 6.9 Hz, 1H, CHCOOH); 5.58 (d, J = 9.3 Hz, 1H, NH); 7.26 (m, 7H, CHAr); 7.75 (d, J = 8.4 Hz, 2H, CHAr) ppm; 13C-NMR (100 MHz, CDCl3): δ = 9.3 (CH3CH2); 21.5 (PhCH3); 28.2 (CH3CH2); 37.6 (CHCH2CH); 48.1 (CHCOOH); 53.7 (CHNH); 127.0 (CHAr); 128.2 (CHAr); 128.7 (CHAr); 128.8 (CHAr); 129.7 (CHAr); 137.8 (CAr); 138.4 (CAr); 143.2 (CAr); 179.2 (COOH) ppm. (2R*, 4S*) 1H-NMR (400 MHz, CDCl3): δ = 0.73 (t, J = 7.2 Hz, 3H, CH3CH2); 1.35 (m, 2H, CH3CH2); 1.70 (m, 1H, CHCHHCH); 2.23 (m, 1H, CHCHHCH); 2.38 (s, 3H, PhCH3); 3.18 (m, 1H, CHNH); 3.71 (t, J = 6.9 Hz, 1H, CHCOOH); 5.46 (d, J = 9.7 Hz, 1H, NH); 7.27 (m, 7H, CHAr); 7.72 (d, J = 8.4 Hz, 2H, CHAr) ppm; 13C-NMR (100 MHz, CDCl3): δ = 9.4 (CH3CH2); 21.0 (PhCH3); 27.9 (CH3CH2); 38.1 (CHCH2CH); 47.6 (CHCOOH); 53.5 (CHNH); 127.1 (CHAr); 127.5 (CHAr); 128.2 (CHAr); 128.8 (CHAr); 129.6 (CHAr); 137.8 (CAr); 138.5 (CAr); 143.3 (CAr); 178.6 (COOH) ppm.
(E)-2-methyl-6-(4-methylphenylsulfonamido)oct-2-enoic acid (15). From 2-methyl-2-butenoic acid (1, 225 mg); yield: 415 mg (68%); brown oil; IR (KBr): υ = 3400–2700, 2923, 2850, 1698, 1417, 1325, 1159, 1093, 815, 667 cm−1; HRMS: m/z calcd. for C16H23NO4S: 325,1348; found: 325.1422; MS: m/z (%):325 [M+, 97%]; 308 [M+-OH, 100%]; 212 [C10H14NO2S+, 23%]; 155 [C7H7O2S+, 48%]. 1H-NMR (300 MHz, CDCl3): δ = 0.75 (t, J = 7.3 Hz, 3H, CH3CH2); 1.30-2.00 (m, 4H, CH2CHCH2); 2.17 (s, 3H, CH3CCOOH); 2.32 (m, 2H, CHCH2CH2); 2.41 (s, 3H, PhCH3); 3.12 (m, 1H, CHNH); 6.96 (m, 1H, CH=CCOOH); 7.21 (d, J = 7.9 Hz, 2H, CHAr); 7.73 (d, J = 8.1 Hz, 2H, CHAr); 13C-NMR (75 MHz, CDCl3): δ = 9.81 (CH3CH2); 21.9 (PhCH3); 28.7 (CH3CCOOH); 36.6 (CH3CH2); 41.2 (=CHCH2CH2); 46.9 (=CHCH2CH2); 53.7 (CHNH); 119.2 (CH=CCOOH); 127.4 (CHAr); 130.1 (CHAr); 135.4 (CAr); 138.7 (CAr); 143.8 (CCOOH); 171.8 (COOH).
Reaction with 3-methyl-2-butenoic acid: From 8 (225 mg); yield: 366 mg (60%) as a 59:41 mixture of 16 and 17
4-(4-Methylphenylsulfonamido)-2-(prop-1-en-2-yl)hexanoic acid (16): brown oil; IR (KBr): υ = 3300–2900, 3272, 2925, 1696, 1644, 1455, 1321, 1158, 1092, 814, 666 cm−1; HRMS: m/z calcd. for C16H23NO4S: 325.1348; found: 325.1335; MS: m/z (%) = 325 [M+, 1%]; 308 [M+-OH, 100%]; 212 [C10H14NO2S+, 19%]; 155 [C7H7O2S+, 25%]. 1H-NMR (400 MHz, CDCl3): δ = 0.74 (t, J = 7.4 Hz, 3H, CH3CH2); 1.27 (m, 4H, CH2CHCH2); 1.85 (s, 3H,); 2.43 (s, 3H, PhCH3); 3.15 (m, 2H, CHNH, CHCOOH); 4.83 (s, 1H, CHH=CCH3), 4.93 (s, 1H, CHH=CCH3); 7.34(d, J = 8.0 Hz, 2H, CHAr); 7.81 (d, J = 8.1 Hz, 2H, CHAr) ppm; 13C-NMR (100 MHz, CDCl3): δ = 9.3 (CH3CH2); 20.3 (CH3C=CH2); 25.2 (PhCH3); 27.7 (CH3CH2); 34.4 (CHCH2CH); 50.7 (CHNH); 54.0 (CHCOOH); 114.9 (CH3C=CH2); 127.1 (CHAr); 129.6 (CHAr); 138.5 (CAr); 143.2 (CAr); 163.3 (CH3C=CH2); 178.8 (COOH) ppm.
(E)-3-Methyl-6-(4-methylphenylsulfonamido)oct-2-enoic acid (17): brown oil; 1H-NMR (400 MHz, CDCl3): δ = 0.76 (t, J = 7.5 Hz, 3H, CH3CH2); 1.43 (m, 4H, CH3CH2, CCH2); 2.33 (dt, J1 = 5.1 Hz, J2 = 7.4 Hz, 1H, CCH2CHH); 2.42 (s, 3H, PhCH3); 2.52 (dt, J1 = 5.2 Hz, J2 = 7.4 Hz, 1H, CCH2CHH); 3.19 (m, 1H, CHNH); 5.66 (s, 1H, CHCOOH); 7.29 (d, J = 7.8 Hz, 2H, CHAr); 7.78 (d, J = 8.3 Hz, 2H, CHAr) ppm; 13C-NMR (100 MHz, CDCl3): δ = 9.8 (CH3CH2); 21.5 (CH3CCH2); 28.4 (PhCH3); 29.5 (CH3CH2); 32.8 (CHCH2CH2); 34.4 (CHCH2CH2); 55.5 (CHNH); 115.7 (CHCOOH); 127.0 (CHAr); 129.6 (CHAr); 138.3 (CAr); 143.2 (CAr); 163.3 (C=CHCOOH); 171.3 (COOH) ppm.
4-(4-Methylphenylsulfonamido)-2-propylhexanoic acid (13). From pentenoic acid (5, 230 mg); yield: 198 mg (31%); yellow oil; IR (KBr): υ = 3500–2700, 3235, 2917, 1712, 1591, 1465, 1382, 1131, 1021, 732 cm−1; HRMS: m/z calcd. for C16H25NO4S: 327,1504, found: 327.1979; MS: m/z (%): 327 [M+, 16%]; 311 [C16H25NO3S+, 20%]; 310 [M+-OH, 100%]; (2R*, 4R*) 1H-NMR (300 MHz, CDCl3): δ = 0.77 (t, J = 7.5 Hz, 3H, CH3CH2CH2); 0.96 (t, J = 7.6 Hz, 3H, CH3CH2CH); 1.30-2.00 (m, 9H, CH3CH2CH2CHCH2CHCH2); 2.51 (s, 3H, PhCH3); 3.31 (bs, 1H, CHNH); 7.37 (d, J = 8.1 Hz, 2H, CHAr); 8.20 (d, J = 8.4 Hz, 2H, CHAr) ppm; 13C-NMR (75 MHz, CDCl3): δ = 9.0 (CH3CH2CH); 14.1 (CH3CH2CH2); 21.9 (CH3CH2CH2); 27.6 (CH3CH2CH); 30.3 (CH3CH2CH2); 35.1 (CHCH2CH); 41.5 (CHCOOH); 54.3 (CHNH); 127.4 (CHAr); 129.8 (CHAr); 138.8 (CAr); 143.7 (CAr); 181.6 (COOH) ppm. (2R*, 4S*) 1H-NMR (300 MHz, CDCl3): δ = 0.79 (t, J = 7.5 Hz, 3H, CH3CH2CH2); 0.98 (t, J = 7.7 Hz, 3H, CH3CH2CH); 1.30-2.00 (m, 9H, CH3CH2CH2CHCH2CHCH2); 2.50 (s, 3H, PhCH3); 3.31 (bs, 1H, CHNH); 7.38 (d, J = 8.0 Hz, 2H, CHAr); 7.86 (d, J = 8.3 Hz, 2H, CHAr) ppm; 13C-NMR (75 MHz, CDCl3): δ = 9.1 (CH3CH2CH); 13.8 (CH3CH2CH2); 22.0 (CH3CH2CH2); 28.3 (CH3CH2CH); 30.3 (CH3CH2CH2); 35.0 (CHCH2CH); 41.4 (CHCOOH); 54.6 (CHNH); 127.5 (CHAr); 130.0 (CHAr); 138.3 (CAr); 143.6 (CAr); 182.2 (COOH) ppm.
2-Benzyl-4-(4-methylphenylsulfonamido)hexanoic acid (14). From 3-phenylpropinoic acid (6, 338 mg); yield: 498 mg (59%); yellow oil; IR (KBr): υ = 3500–2700, 3270, 2930, 1708, 1599, 1495, 1322, 1159, 1092, 700 cm−1; HRMS: m/z calcd. for C20H25NO4S: 375.1504; found: 375.1525; MS: m/z (%): 375 [M+, 1%]; 357 [M+-H2O]; 293 [C20H23NO+, 22%]; 155 [C7H7O2S+, 32%]; 91 [C7H7+, 100%]; (2R*, 4R*) 1H-NMR (300 MHz, CDCl3): δ = 0.73 (t, J = 7.2 Hz, 3H, CH3CH2); 1.37 (m, 2H, CH3CH2); 1.42 (m, 1H, CHCHHCH); 1.92 (m, 1H, CHCHHCH); 2.50 (s, 3H, PhCH3); 2.84 (m, 2H, CHCOOH, PhCHH); 3.30 (m, 1H, PhCHH); 3.32 (bs, 1H, CHNH); 5.52 (d, J = 9.0 Hz, 1H, NH); 7.34 (m, 5H, CHAr); 7.35 (d, J = 7.8 Hz, 2H, CHAr); 7.83 (d, J = 8.1 Hz, 2H, CHAr) ppm; 13C-NMR (75 MHz, CDCl3): δ = 9.7 (CH3CH2); 22.1 (PhCH3); 28.4 (CH3CH2); 36.2 (CHCH2CH); 38.6 (PhCH2); 44.6 (CHCOOH); 54.4 (CHNH); 126.9 (CHAr); 127.4 (CHAr); 128.7 (CHAr); 129.0 (CHAr); 129.4 (CHAr); 138.6 (CAr); 140.5 (CAr); 143.7 (CAr); 181.1 (COOH) ppm. (2R*, 4S*) 1H-NMR (300 MHz, CDCl3): δ = 0.73 (t, J = 7.2 Hz, 3H, CH3CH2); 1.37 (m, 2H, CH3CH2); 1.42 (m, 1H, CHCHHCH); 1.96 (m, 1H, CHCHHCH); 2.50 (s, 3H, PhCH3); 2.84 (m, 2H, CHCOOH, PhCHH); 3.30 (m, 1H, PhCHH); 3.32 (bs, 1H, CHNH); 5.23 (d, J = 9.0 Hz, 1H, NH); 7.34 (m, 5H, CHAr); 7.35 (d, J = 7.8 Hz, 2H, CHAr); 7.84 (d, J = 8.1 Hz, 2H, CHAr) ppm; 13C-NMR (75 MHz, CDCl3): δ = 10.1 (CH3CH2); 22.0 (PhCH3); 28.2 (CH3CH2); 36.0 (CHCH2CH); 38.4 (PhCH2); 43.4 (CHCOOH); 54.2 (CHNH); 126.8 (CHAr); 127.4 (CHAr); 128.6 (CHAr); 128.9 (CHAr); 129.4 (CHAr); 138.3 (CAr); 140.5 (CAr); 143.7 (CAr); 179.2 (COOH) ppm.
2-(3-(4-Methylphenylsulfonamido)pentyl)benzoic acid (20). From 2-methylbenzoic acid (10, 306 mg); yield: 325 mg (40%); yellow oil; IR (KBr): υ = 3300–2900, 3272, 2926, 1696, 1600, 1455, 1321, 1158, 1093, 666 cm−1; HRMS: m/z calcd. for C19H23NO4S: 361.1348; found: 361.1443; MS: m/z (%): 361 [M+, 0.1%]; 343 [M+-H2O, 1%]; 332 [M+-CH3CH2, 30%]; 314 [C17H16NOS+, 63%]; 212 [C13H10NO2+, 100%]; 155 [C7H7O2S+, 85%]; 91 [C7H7+, 93%]. 1H-NMR (300 MHz, CDCl3): δ = 0.91 (t, J = 7.5 Hz, 3H, CH3CH2); 1.59 (m, 2H, CH3CH2); 1.77 (m, 2H, PhCH2CH2); 2.29 (s, 3H, PhCH3); 2.81 (dt, J1 = 10.8, J2 = 5.0, 1H, PhCHH); 3.06 (dt, J1 = 10.6, J2 = 5.3 Hz, 1H, PhCHH); 3.32 (m, 1H, CHNH); 5.64 (d, J = 7.5 Hz, 1H, NH); 7.37 (m, 4H, CHAr); 7.55 (t, J = 6.0 Hz, 1H, CHAr); 7.91 (d, J = 10.1 Hz, 1H, CHAr); 8.16 (d, J = 6.6 Hz, 1H, CHAr) ppm; 13C-NMR (75 MHz, CDCl3): δ = 10.2 (CH3CH2); 21.9 (PhCH3); 28.6 (CH3CH2); 30.9 (PhCH2); 36.9 (PhCH2CH2); 55.9 (CHNH); 126.6 (CHAr); 127.5 (CHAr); 129.9 (CHAr); 130.1 (CHAr); 131.6 (CHAr); 132.4 (CHAr); 133.4 (CAr); 136.7 (CAr); 143.4 (CAr); 145.3 (CAr); 173.2 (COOH) ppm.
5-Methyl-2-(3-(4-methylphenylsulfonamido)pentyl)furan-3-carboxylic acid (21). From 2,5-dimethyl-3-furanoic acid (11, 315 mg); yield: 435 mg (53%); brown oil; IR (KBr): υ = 3300–2900, 3271, 2927, 1682, 1583, 1435, 1323, 1235, 1159, 815 cm−1; HRMS: m/z calcd. for C18H23NO5S: 365,1297; found: 365.1292; MS: m/z (%): 365 [M+, 0.2%]; 347 [M+-H2O]; 212 [C10H14NO2S+, 49%]; 155 [C7H7O2S+, 50%]; 91 [C7H7+, 100%]; 1H-NMR (300 MHz, CDCl3): δ = 0.79 (t, J = 7.2 Hz, 3H, CH3CH2); 1.37 (m, 2H, CH3CH2); 1.64 (m, 2H, CHCH2CH2); 2.21 (s, 3H, CH3C-O); 2.51 (s, 3H, PhCH3); 2.70 (m, 1H, CHCH2CHH); 2.84 (m, 1H, CHCH2CHH); 3.15 (m, 1H, CHNH); 5.03 (d, J = 8.4 Hz, 1H, NH); 6.18 (s, 1H, CHCCOOH); 7.22 (d, J = 8.1 Hz, 2H, CHAr); 7.70 (d, J = 8.1 Hz, 2H, CHAr) ppm; 13C-NMR (75 MHz, CDCl3): δ = 10.1 (CH3CH2); 13.6 (CH3C-O); 21.9 (CCH2); 24.1 (PhCH3); 28.1 (CH3CH2); 33.0 (CCH2CH2); 55.4 (CHNH); 106.6 (CHCOOH); 113.6 (CCOOH); 127.4 (CHAr); 130.1 (CHAr); 138.6 (CAr); 143.5 (CAr); 151.0 (CH3C); 162.7 (CCH2); 169.8 (COOH) ppm.
3-(3-(4-Methylphenylsulfonamido)pentyl)thiophene-2-carboxylic acid (22). From 3-methyl-2-tiophenoic acid (12, 320 mg); yield: 496 mg (60%); brown oil; IR (KBr): υ = 3400–2600, 2967, 2639, 1671, 1598, 1537, 1428, 1303, 1158, 1093 cm−1; HRMS: m/z calcd. for C17H21NO4S2: 367,0912; found: 367.0912; MS: m/z (%): 367 [M+, 1%]; 338 [M+-CH3CH2, 27%]; 320 [C15H14NO3S2+, 36%]; 212 [C10H14NO2S+, 100%]; 155 [C7H7O2S+, 77%]; 91 [C7H7+, 78%]; 1H-NMR (400 MHz, CDCl3): δ = 0.78 (t, J = 7.4 Hz, 3H, CH3CH2); 1.48 (m, 2H, CH3CH2); 1.71 (m, 2H, CH2CH2CH); 2.40 (s, 3H, PhCH3); 2.84 (m, 1H, CHHCH2CH); 2.95 (m, 1H, CHHCH2CH); 3.20 (m, 1H, CH2CH); 6.89 (d, J = 5.0 Hz, 1H, SCH=CH); 7.27 (d, J = 8.0 Hz, 2H, CHAr); 7.47 (d, J = 5.0 Hz, 1H; SCH=CH); 7.78 (d, J = 8.3 Hz, 2H, CHAr) ppm; 13C-NMR (100 MHz, CDCl3): δ = 9.5 (CH3CH2); 21.5 (PhCH3); 25.6 (CH2CH2CH); 27.5 (CH3CH2); 31.5 (CH2CH2CH); 34.5 (CH2CH); 127.0 (CHAr); 129.7 (CHAr); 131.7 (SCH=CH); 132.1 (SCH=CH); 138.2 (CAr); 143.2 (CAr); 147.9 (CCOOH); 151.8 (CCH2); 167.6 (COOH) ppm.
Acknowledgements
We thank the Spanish Government (projects MAT2009-14564-C04). SCSIE (Universidad de Valencia) is gratefully acknowledged for all the equipment employed.
References and Notes
- Lowden, P.A.S. Aziridines and Epoxides in Organic Synthesis; Yudin, A.K., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 399–442. [Google Scholar]
- Padwa, A.; Woolhouse, A.D. Comprehensive Heterocyclic Chemistry III; Lwowski, W., Ed.; Pergamon: Oxford, UK, 1984; pp. 47–93. [Google Scholar]
- Hu, X.E. Nucleophilic ring opening of aziridines. Tetrahedron 2004, 60, 2701–2743. [Google Scholar] [CrossRef]
- Pineschi, M. Asymmetric Ring-Opening of Epoxides and Aziridines with Carbon Nucleophiles. Eur. J. Org. Chem. 2006, 4979–4988. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Hu, Y.; Wang, D.-X.; Pan, J.; Huang, Z.-T.; Wang, M.-X. Unprecedented carbon-carbon bond cleavage in nucleophilic aziridine ring opening reaction, efficient ring transformation of aziridines to imidazolidin-4-ones. Chem. Commun. 2009, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Pandey, S.K.; Gandhi, S.; Singh, V.K. PPh3/halogenating agent-mediated highly efficient ring opening of activated and non-activated aziridines. Tetrahedron Lett. 2009, 50, 263–265. [Google Scholar] [CrossRef]
- Tarrade-Matha, A.; Valle, M.S.; Tercinier, P.; Dauban, P.; Dodd, R.H. Enantiospecific Synthesis of a Protected Equivalent of APTO, the beta-Amino Acid Fragment of Microsclerodermins C and D, by Aziridino-gamma-lactone Methodology. Eur. J. Org. Chem. 2009, 673–686. [Google Scholar] [CrossRef]
- Gil, S.; Parra, M.; Rodriguez, P. A simple synthesis of γ-aminoacids. Tetrahedron Lett. 2007, 48, 3451–3453. [Google Scholar] [CrossRef]
- Gil, S.; Parra, M.; Rodriguez, P. An efficient synthesis of γ-aminoacids. Attempts to drive it enantioselectively. Molecules 2008, 13, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Thomson, C.M. Dianion Chemistry in Organic Synthesis; CRC Press: Boca Raton, FL, USA, 1994; pp. 88–129. [Google Scholar]
- Gil, S.; Parra, M. Dienediolates of carboxylic acids in synthesis. Recent advances. Curr. Org. Chem. 2002, 6, 283–302. [Google Scholar] [CrossRef]
- Gil, S.; Parra, M. Reactivity control of dianions of carboxylic acids. Synthetic applications. Recent Res. Devel. Org. Chem. 2002, 6, 449–481. [Google Scholar] [CrossRef]
- Clayden, I. Organolithiums: Selectivity for Synthesis; Pergamon Press: Oxford, UK, 2002; p. 73. [Google Scholar]
- Juaristi, E.; Beck, A.K.; Hansen, J.; Matt, T.; Mukhopadhyay, T.; Simson, M.; Seebach, D. Enantioselective Aldol and Michael Additions of Achiral Enolates in the presence of chiral lithium amides and amines. Synthesis 1993, 1271–1290. [Google Scholar] [CrossRef]
- Streitwieser, A.; Husemann, M.; Kim, Y.-J. Aggregation and reactivity of the dilithium and dicesium N-diolates of 1-naphthyl acetic acid. J. Org. Chem. 2003, 68, 7937–7942. [Google Scholar] [CrossRef] [PubMed]
- Eames, J.; Suggate, M.J. Recent developments in the transfer of chirality within enolate alkylation reactions. Angew. Chem. Int. Ed. 2005, 44, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Sott, R.; Granader, J.; Hilmersson, G. Solvent-dependent mixed complex formation–NMR studies and asymmetric addition reactions of lithiumacetonitrile to benzaldehyde mediated by chiral lithium amides. Chem. Eur. J. 2002, 8, 2081–2087. [Google Scholar] [CrossRef]
- Sotoca, E.; Bouillon, J.P.; Gil, S.; Parra, M.; Portella, C. Reaction of lithium enediolates with perfluoroketene dithioacetals. Synthesis of α-trifluoromethyl γ-dicarboxylic acid derivatives. Tetrahedron 2005, 61, 4395–4402. [Google Scholar] [CrossRef]
- Brun, E.M.; Casades, I.; Gil, S.; Mestres, R.; Parra, M. New conditions for the generation of dianions of carboxylic acids. Tetrahedron Lett. 1998, 39, 5443–5446. [Google Scholar] [CrossRef]
- Bieber, L.W.; de Araújo, M.C.F. Short and Efficient Synthesis of Optically Active N-Tosyl Aziridines from 2-Amino Alcohols. Molecules 2002, 7, 902–906. [Google Scholar] [CrossRef]
- Gil, S.; Torres, M.; Ortúzar, N.; Wincewicz, R.; Parra, M. Eficient addition of acid enediolates to epoxides. Eur. J. Org. Chem. 2004, 2160–2165. [Google Scholar] [CrossRef]
- Domingo, L.R.; Gil, S.; Parra, M.; Segura, J. Unusual Regioselectivity in the Opening of Epoxides by Carboxylic Acid Enediolates. Molecules 2008, 13, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.K. Reactions of epoxides with ester, ketone and amide enolates. Tetrahedron 2000, 56, 1149–1163. [Google Scholar] [CrossRef]
- Brun, E.M.; Gil, S.; Mestres, R.; Parra, M. Regioselective Alkylation of Lithium Dienediolates of α,β-unsaturated Carboxylic Acids. Synthesis-Stuttgart 2000, 1160–1165. [Google Scholar] [CrossRef]
- Brun, E.M.; Gil, S.; Mestres, R.; Parra, M. Lithium enediolates and dienediolates of carboxylic acids in synthesis: Alkylation with secondary halides. Tetrahedron 1998, 54, 15305–15320. [Google Scholar] [CrossRef]
- Aurell, M.J.; Gil, S.; Mestres, R.; Parra, M.; Parra, L. Alkylation of lithium dienediolates of butenoic acids. Regioselectivity effects of structure and leaving group of the alkylating agent. Tetrahedron 1998, 54, 4357–4366. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 4, 15, 16, 17, 20 are available from the authors. |
Scheme 1.
Reaction of aziridines with enediolate of phenylacetic acid.

Figure 1.
Transition States in the addition of enediolates to epoxydes and aziridines.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of the reaction conditions of enediolate of phenylacetic acid with aziridines.
Table 1.
Optimization of the reaction conditions of enediolate of phenylacetic acid with aziridines.
| Entry | Aziridine | Amine | Time at r.t. | Yield (%) |
|---|---|---|---|---|
| 1 | 3a | 2a | 1h | 0 |
| 2 | 3b | 2a | 1h | 0 |
| 3 | 3c | 2a | 1h | 50 |
| 4 | 3c | 2a | 3h | 44 |
| 5 | 3c | 2a | 24h | 35 |
| 6 | 3c | 2a* | 1h | 46 |
| 7 | 3c | 2b | 1h | 67 |
| 8 | 3c | 2b | 1h | 58** |
| 9 | 3c | 2b | 1h | 71** |
* 0.5 eq. of amine; ** Using LiCl as additive in normal (entry 8) or inverse addition (entry 9).
Table 2.
Addition of dianions of carboxylic acids to aziridine 3c.
| Entry | Acid | Product | Yield (%) | γ : α | Syn : Anti |
|---|---|---|---|---|---|
| 1 |  |  | 31 | 66 : 33 | |
| 2 |  |  | 67 | 64 :36 | |
| 3 |  |  | 59 | 67 : 33 | |
| 4 |  |  | 68 | 100 : 0 | |
| 5 |  |  | 60 | 41 : 59 | 69 : 31 |
| 6 |  |  | 25 | 50 : 50 | |
| 7 |  |  | 40 | ||
| 8 |  |  | 53 | ||
| 9 |  |  | 60 |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Costero, A.M.; Gil, S.; Parra, M.; Rodríguez, P. Unexplored Nucleophilic Ring Opening of Aziridines. Molecules 2010, 15, 9135-9144. https://doi.org/10.3390/molecules15129135
AMA Style
Costero AM, Gil S, Parra M, Rodríguez P. Unexplored Nucleophilic Ring Opening of Aziridines. Molecules. 2010; 15(12):9135-9144. https://doi.org/10.3390/molecules15129135
Chicago/Turabian StyleCostero, Ana María, Salvador Gil, Margarita Parra, and Pablo Rodríguez. 2010. "Unexplored Nucleophilic Ring Opening of Aziridines" Molecules 15, no. 12: 9135-9144. https://doi.org/10.3390/molecules15129135