Synthesis of the Marine Bromotyrosine Psammaplin F and Crystal Structure of a Psammaplin A Analogue

Abstract
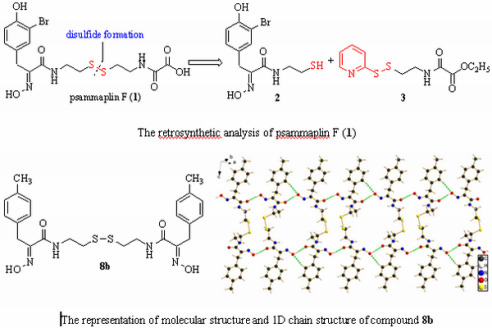
:
1. Introduction
2. Results and Discussion
2.1. Chemistry





{kind=link}
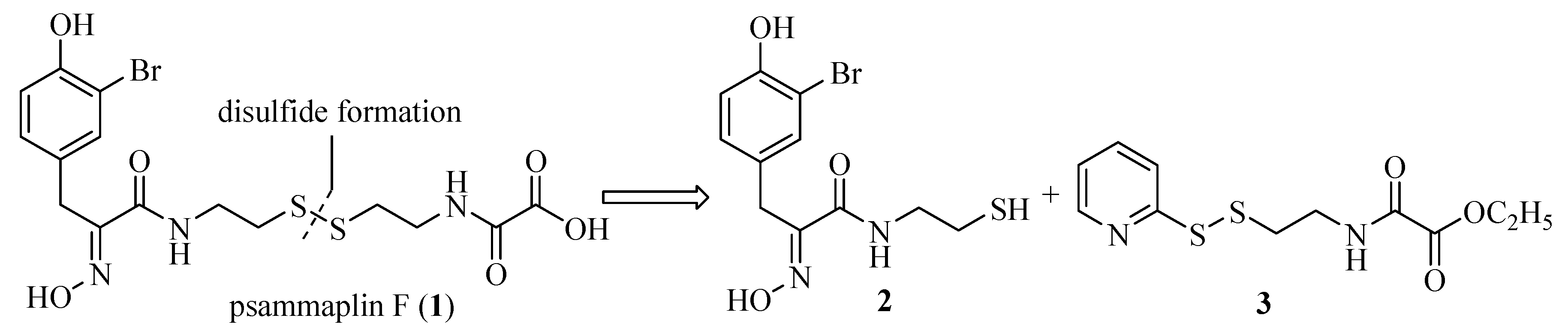{kind=link}
{kind=link}
{kind=link}
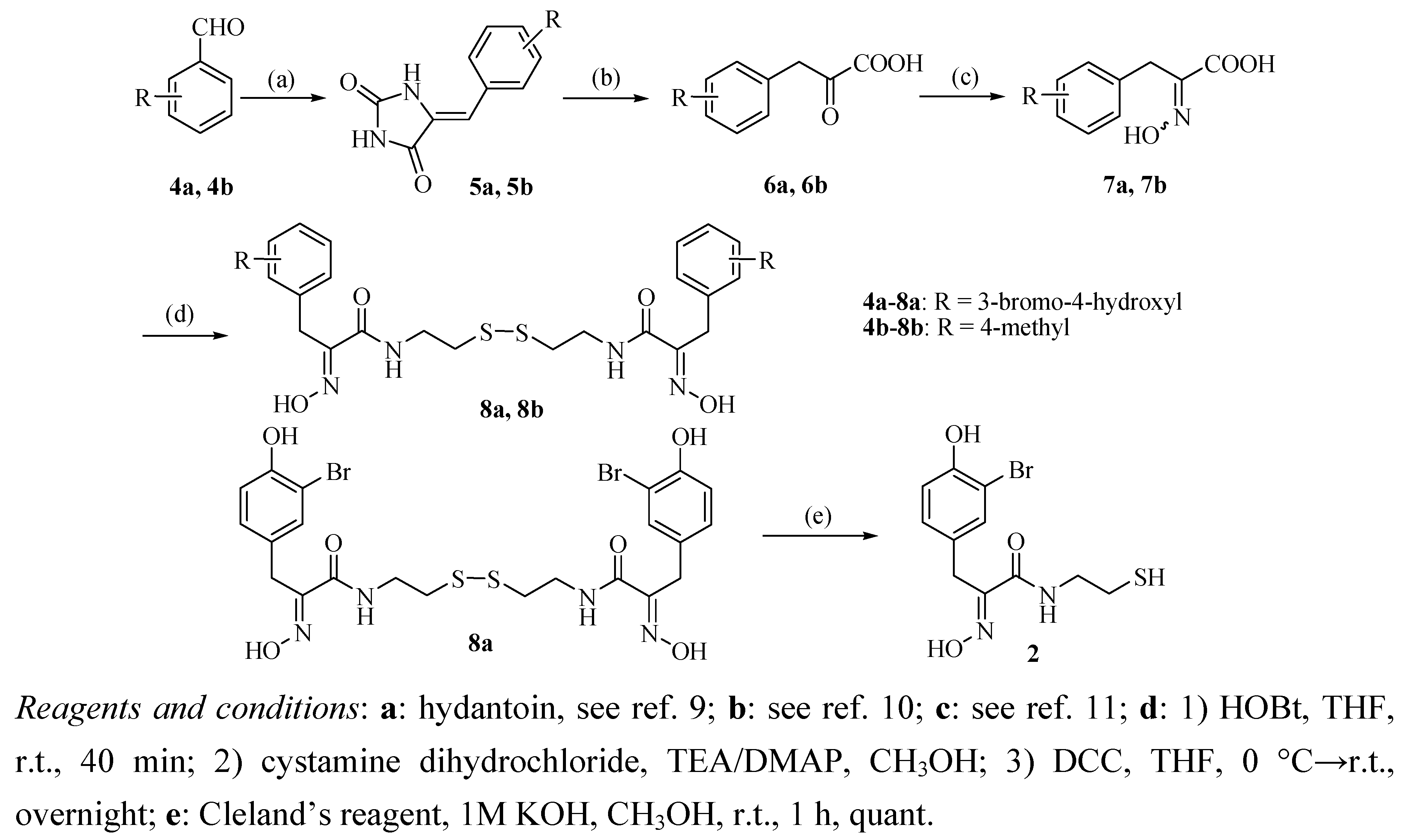{kind=link}
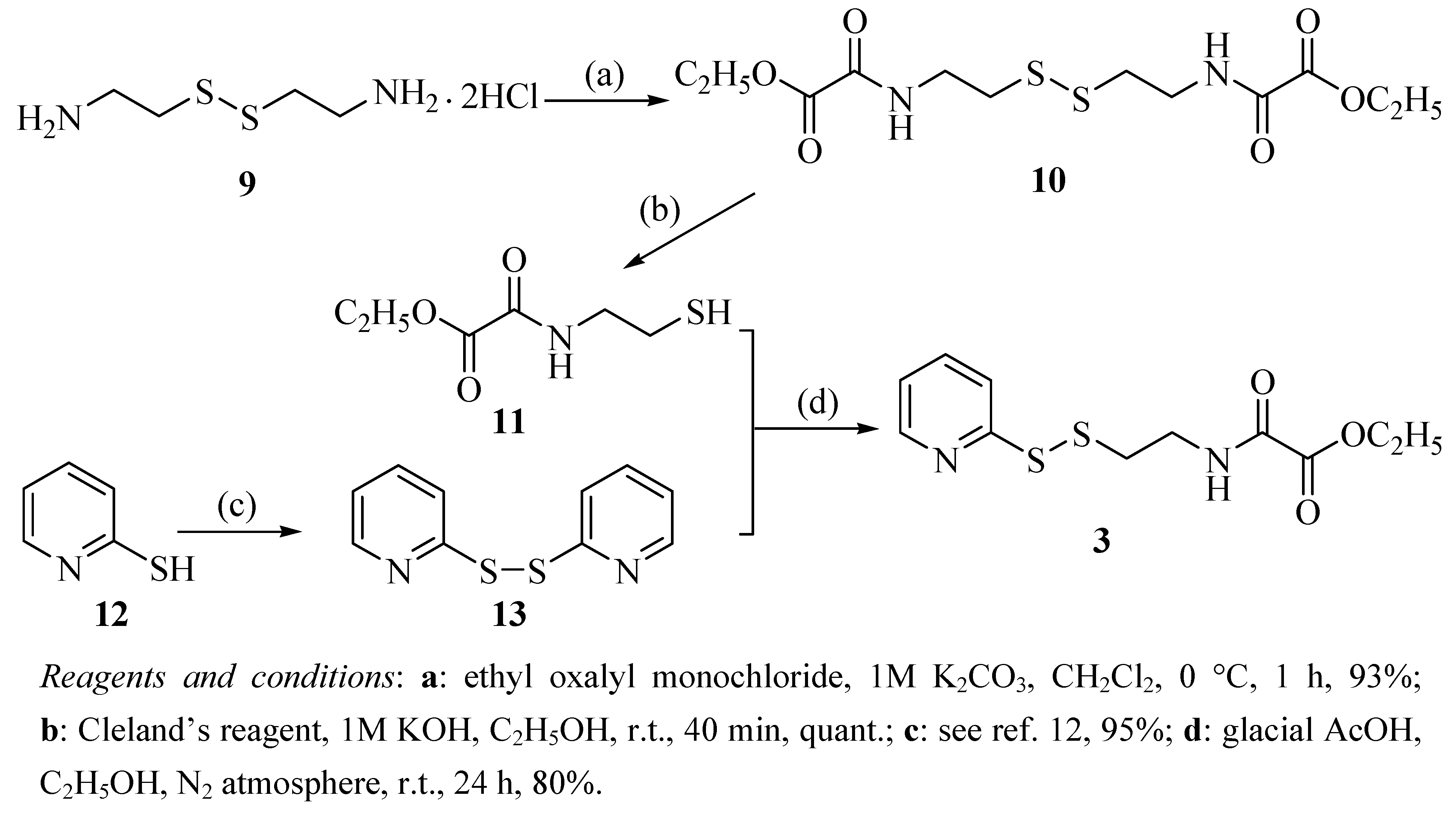{kind=link}
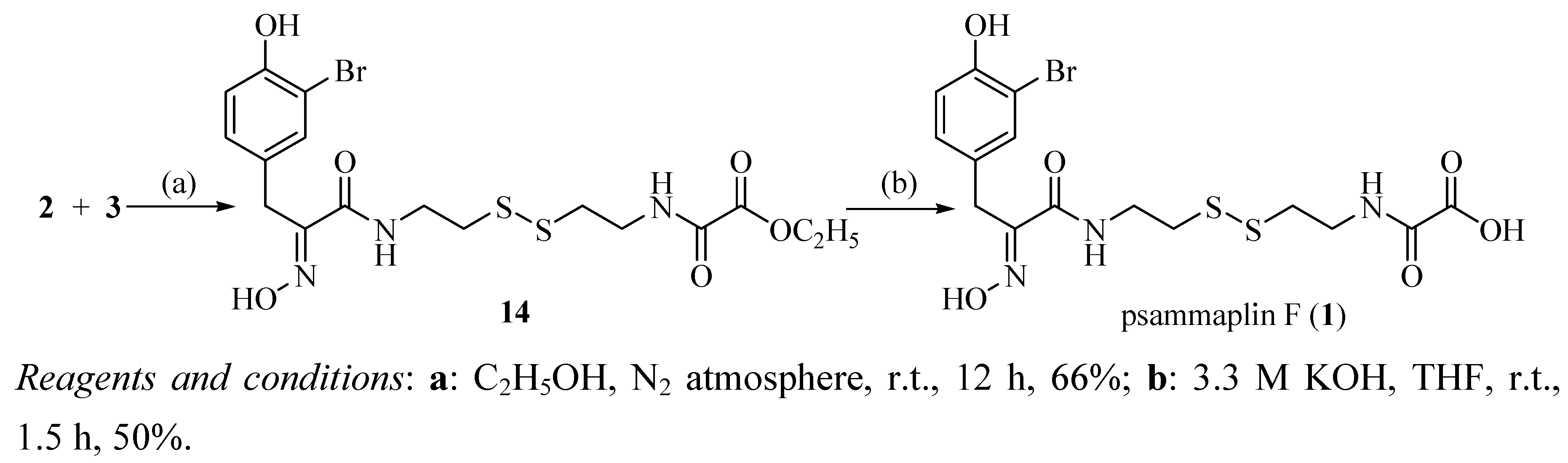{kind=link}
| Position | Synthetic psammaplin F | Natural psammaplin F[1] | ||
|---|---|---|---|---|
| H (δ) | C (δ, mult, J in Hz) | H (δ) | C (δ, mult, J in Hz) | |
| 2 | 2.85 (t, 6.0) | 38.7 | 2.84 (t, 6.0) | 38.6 |
| 2’ | 2.85 (t, 6.0) | 38.2 | 2.84 (t, 6.0) | 37.9 |
| 3 | 3.55 (t, 6.0) | 39.8 | 3.54 (t, 6.0) | 39.8 |
| 3’ | 3.55 (t, 6.0) | 40.0 | 3.54 (t, 6.0) | 40.1 |
| 5 | 166.0 | 166.0 | ||
| 5’ | 161.8 | 161.9 | ||
| 6 | 153.2 | 153.3 | ||
| 6’ | 159.0 | 159.2 | ||
| 7 | 3.80 (s) | 28.7 | 3.78 (s) | 28.8 |
| 8 | 130.6 | 130.7 | ||
| 9 | 7.34 (d, 1.8) | 134.5 | 7.35 (d, 2.0) | 134.6 |
| 10 | 110.5 | 110.6 | ||
| 11 | 153.7 | 153.9 | ||
| 12 | 6.77(d, 8.4) | 117.0 | 6.75 (d, 8.0) | 117.2 |
| 13 | 7.07 (dd, 1.8, 8.4) | 130.4 | 7.05 (dd, 2.0, 8.0) | 130.5 |
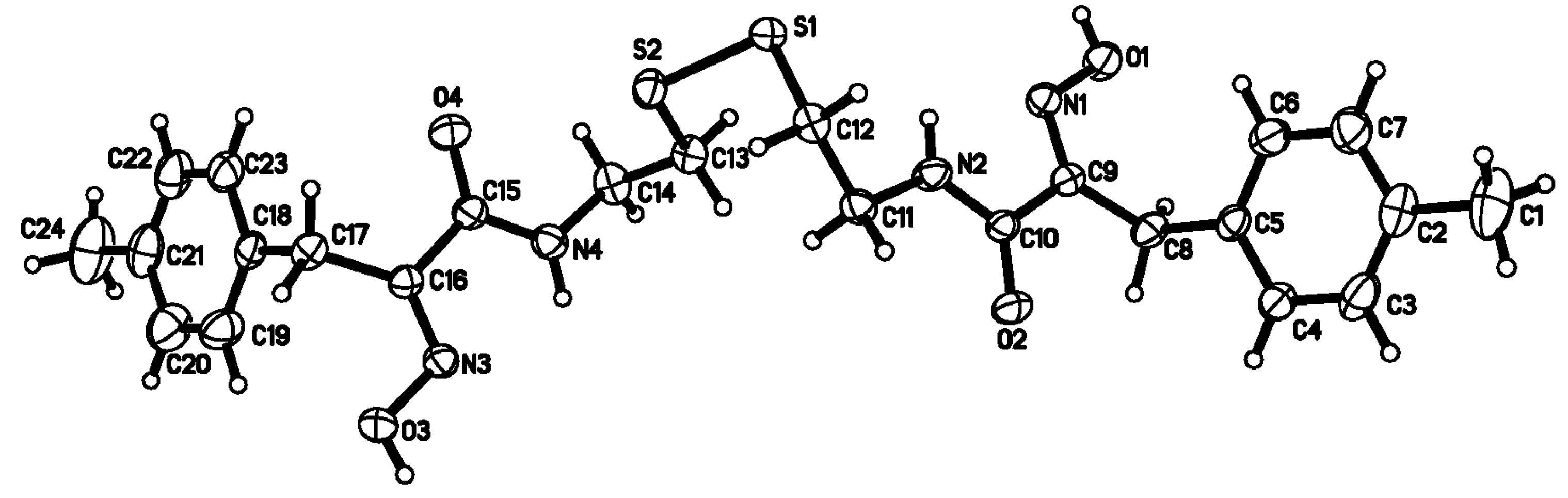
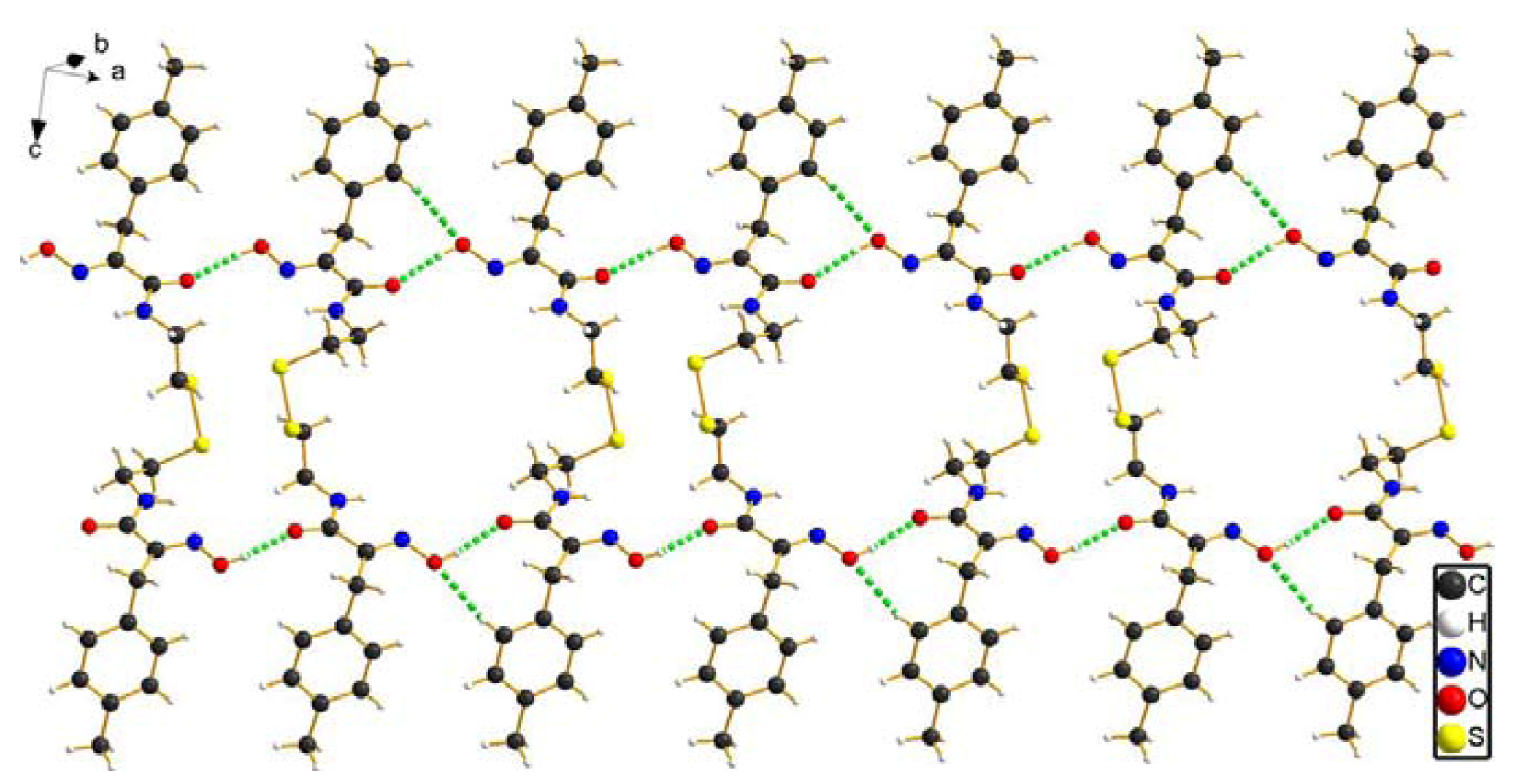
2.2. Crystal structure analysis of compound 8b

| Formula | C24 H30 N4 O4 S2 |
| Fw | 502.64 |
| Temp (K) | 295(2) |
| Cryst system | Triclinic |
| Space group | P-1 |
| a (Å) | 5.7159(9) |
| b (Å) | 9.6170(15) |
| c (Å) | 23.585(4) |
| α (°) | 94.030(2) |
| β (°) | 95.455(2) |
| γ (°) | 102.532(2) |
| V (Å3) | 1254.3(3) |
| Z | 2 |
| ρcalc (g cm-3) | 1.331 |
| Μ (mm-1) | 0.250 |
| F(000) | 532 |
| Limiting indices | -6<=h<=6, -11<=k<=5, -28<=l<=27 |
| Data / restraints / parameters | 4589 / 0 / 311 |
| θ range for data collection (°) | 1.74 to 25.66 |
| Completeness to theta = 25.66 | 97.2 % |
| Reflnscollected/unique | 6718 / 4589 |
| GOF | 1.049 |
| Final R indices [I>2σ(I)] | R1a = 0.0484, ωR2b = 0.1271 |
| R indices (all data) | R1 = 0.0595, ωR2 = 0.1364 |


| C(1)-C(2) | 1.516(4) | C(14)-N(4) | 1.446(3) |
| C(2)-C(3) | 1.361(4) | C(15)-O(4) | 1.222(3) |
| C(2)-C(7) | 1.380(4) | C(15)-N(4) | 1.337(3) |
| C(3)-C(4) | 1.393(4) | C(15)-C(16) | 1.500(3) |
| C(4)-C(5) | 1.375(3) | C(16)-N(3) | 1.271(3) |
| C(5)-C(6) | 1.379(3) | C(16)-C(17) | 1.501(3) |
| C(5)-C(8) | 1.518(3) | C(17)-C(18) | 1.510(3) |
| C(6)-C(7) | 1.382(4) | C(18)-C(23) | 1.372(3) |
| C(8)-C(9) | 1.507(3) | C(18)-C(19) | 1.376(4) |
| C(9)-N(1) | 1.274(3) | C(19)-C(20) | 1.387(4) |
| C(9)-C(10) | 1.502(3) | C(20)-C(21) | 1.370(5) |
| C(10)-O(2) | 1.223(3) | C(21)-C(22) | 1.360(5) |
| C(10)-N(2) | 1.327(3) | C(21)-C(24) | 1.518(4) |
| C(11)-N(2) | 1.445(3) | C(22)-C(23) | 1.395(4) |
| C(11)-C(12) | 1.509(3) | N(1)-O(1) | 1.384(2) |
| C(12)-S(1) | 1.817(2) | N(3)-O(3) | 1.388(2) |
| C(13)-C(14) | 1.518(3) | S(1)-S(2) | 2.0376(8) |
| C(13)-S(2) | 1.830(2) | ||
| C(3)-C(2)-C(7) | 117.0(3) | O(4)-C(15)-C(16) | 120.5(2) |
| C(3)-C(2)-C(1) | 121.2(3) | N(4)-C(15)-C(16) | 117.6(2) |
| C(7)-C(2)-C(1) | 121.9(3) | N(3)-C(16)-C(15) | 114.33(19) |
| C(2)-C(3)-C(4) | 121.9(3) | N(3)-C(16)-C(17) | 127.5(2) |
| C(5)-C(4)-C(3) | 120.7(3) | C(15)-C(16)-C(17) | 118.11(19) |
| C(4)-C(5)-C(6) | 117.8(2) | C(16)-C(17)-C(18) | 112.58(18) |
| C(4)-C(5)-C(8) | 120.6(2) | C(23)-C(18)-C(19) | 117.3(2) |
| C(6)-C(5)-C(8) | 121.6(2) | C(23)-C(18)-C(17) | 122.0(2) |
| C(5)-C(6)-C(7) | 120.6(3) | C(19)-C(18)-C(17) | 120.7(2) |
| C(2)-C(7)-C(6) | 122.0(3) | C(18)-C(19)-C(20) | 121.2(3) |
| C(9)-C(8)-C(5) | 112.76(18) | C(21)-C(20)-C(19) | 121.4(3) |
| N(1)-C(9)-C(10) | 113.78(18) | C(22)-C(21)-C(20) | 117.5(3) |
| N(1)-C(9)-C(8) | 126.7(2) | C(22)-C(21)-C(24) | 120.8(3) |
| C(10)-C(9)-C(8) | 119.52(18) | C(20)-C(21)-C(24) | 121.7(4) |
| O(2)-C(10)-N(2) | 122.1(2) | C(21)-C(22)-C(23) | 121.6(3) |
| O(2)-C(10)-C(9) | 121.86(19) | C(18)-C(23)-C(22) | 121.0(3) |
| N(2)-C(10)-C(9) | 116.09(18) | C(9)-N(1)-O(1) | 113.18(18) |
| N(2)-C(11)-C(12) | 110.54(18) | C(10)-N(2)-C(11) | 124.39(19) |
| C(11)-C(12)-S(1) | 114.69(15) | C(16)-N(3)-O(3) | 112.38(18) |
| C(14)-C(13)-S(2) | 110.52(16) | C(15)-N(4)-C(14) | 122.4(2) |
| N(4)-C(14)-C(13) | 113.57(19) | C(12)-S(1)-S(2) | 105.05(8) |
| O(4)-C(15)-N(4) | 121.9(2) | C(13)-S(2)-S(1) | 104.27(8) |
 C(9)-C(8)-C(5) and C(16)-C(17)-C(18) are 112.76(18) and 112.58(18)°, respectively. This suggests that the dihedral angles between the amide and oxime group plane and phenyl group are 75.02 and 71.79°, respectively. The two benzene rings are close to parallel, with a dihedral angle of 3.92°. The torsion angles of C(12)-S(1)-S(2)-C(13) and C(11)-C(12)-S(1)-S(2) are -88.50(11) and 77.45(16)°, respectively, which leads to the conclusion that the 2,2'-disulfandiyldiethanamino chain is seriously twisted. It is interesting that the overall shape of the compound 8b takes on zigzag-like chain.
C(9)-C(8)-C(5) and C(16)-C(17)-C(18) are 112.76(18) and 112.58(18)°, respectively. This suggests that the dihedral angles between the amide and oxime group plane and phenyl group are 75.02 and 71.79°, respectively. The two benzene rings are close to parallel, with a dihedral angle of 3.92°. The torsion angles of C(12)-S(1)-S(2)-C(13) and C(11)-C(12)-S(1)-S(2) are -88.50(11) and 77.45(16)°, respectively, which leads to the conclusion that the 2,2'-disulfandiyldiethanamino chain is seriously twisted. It is interesting that the overall shape of the compound 8b takes on zigzag-like chain. 
| D-H...A | d(D-H) | d(H-A) | d(D-A) | D-H...A |
|---|---|---|---|---|
| O(1)-H(1)...O(4)#1 | 0.82 | 1.87 | 2.671(2) | 167.0 |
| O(3)-H(3A)...O(2)#2 | 0.82 | 1.90 | 2.711(2) | 171.5 |
| C(4)-H(4)...O(3)#2 | 0.93 | 2.53 | 3.229(3) | 133 |
3. Experimental
3.1. General
3.2. Single crystal X-ray determination of compound 8b
4. Conclusions
Acknowledgements
- Sample Availability: Samples are available from the authors.
References and Notes
- Piña, I.C.; Gautschi, J.T.; Wang, G.Y.S.; Miranda, L.S.; Francis, J.S.; Dennis, F.; Susan, C.K.; Lidia, C.S.; Stacy, W.R.; Larry, B.P.; Kenneth, W.B.; Phillip, C. Psammaplins from the sponge Pseudoceratina purpurea: inhibition of both histone deacetylase and DNA methyltransferase. J. Org. Chem. 2003, 68, 3866–3873. [Google Scholar]
- Kim, D.; Lee, I.S.; Jung, J.H.; Lee, C.O.; Choi, S.U. Psammaplin A, a natural phenolic compound, has inhibitory effect on human topoisomerase II and is cytotoxic to cancer cells. Anticancer Res. 1999, 19, 4085–4090. [Google Scholar]
- Nicolaou, K.C.; Hughes, R.; Pfefferkorn, J.A.; Barluenga, S.; Roecker, A.J. Combinatorial synthesis through disulfide exchange: discovery of potent psammaplin A type antibacterial agents active against methicillin-resistant staphylococcus aureus (MRSA). Chem. Eur. J. 2001, 7, 4280–4295. [Google Scholar] [CrossRef]
- Tabudravu, J.N.; Eijsink, V.G.H.; Gooday, G.W.; Jaspars, M.; Komander, D.; Legg, M.; Synstad, B.; van Aalten, D.M.F. Psammaplin A, a chitinase inhibitor isolated from the Fijian marine sponge Aplysinella rhax. Bioorg. Med. Chem. 2002, 10, 1123–1128. [Google Scholar] [CrossRef]
- Shim, J.S.; Lee, H.S.; Shin, J.; Kwon, H.J. Psammaplin A, a marine natural product, inhibits aminopeptidase N and suppresses angiogenesis in vitro. Cancer Lett. 2004, 203, 163–169. [Google Scholar]
- Hoshino, O.; Murakata, M.; Yamada, K. A convenient synthesis of a bromotyrosine derived metabolite, psammaplin A, from psammaplysilla SP. Bioorg. Med. Chem. Lett. 1992, 2, 1561–1562. [Google Scholar] [CrossRef]
- Godert, A.M.; Angelino, N.; Woloszynska-Read, A.; Morey, S.R.; James, S.R.; Karpf, A.R.; Sufrin, J.R. An improved synthesis of psammaplin A. Bioorg. Med. Chem. Lett. 2006, 16, 3330–3333. [Google Scholar]
- Thomas, W.L.; Edel, O’T.; Orlagh, F. Solid oral dosage form containing an enhancer. U.S. Patent 20070292512 (A1), 20 December 2007. [Google Scholar]
- Thenmozhiyal, J.C.; Wong, P.T.H.; Chui, W.K. Anticonvulsant activity of phenylmethylenehydantoins: a structure-activity relationship study. J. Med. Chem. 2004, 47, 1527–1535. [Google Scholar] [CrossRef]
- Naoyuki, T.; Hiroyuki, M. Method for producing 4-hydroxyphenylpyruvic acid. Japan Patent 2003104932 (A), 9 April 2003. [Google Scholar]
- Knapp, S.; Amorelli, B.; Darout, E.; Ventocilla, C.C.; Goldman, L.M.; Huhn, R.A.; Minnihan, E.C. A family of mycothiol analogues. J. Carbohydr.Chem. 2005, 24, 103–130. [Google Scholar]
- Leino, R.; Lönnqvist, J.E. A very simple method for the preparation of symmetrical disulfides. Tetrahedron Lett. 2004, 45, 8489–8491. [Google Scholar] [CrossRef]
- Fournie-Zaluski, M.C.; Coric, P.; Turcaud, S.; Lucas, E.; Noble, F.; Maldonado, R.; Roques, B.P. Mixed-inhibitor-prodrug as a new approach toward systemically active inhibitors of enkephalin-degrading enzymes. J. Med. Chem. 1992, 35, 2473–2481. [Google Scholar] [CrossRef]
- Kemp, D.S.; Galakatos, N.G.; Dranginis, S.; Ashton, C.; Fotouhi, N.; Curran, T.P. Peptide synthesis by prior thiol capture. 4. Amide bond formation: the effect of a side-chain substituent on the rates of intramolecular O, N-acyl transfer. J. Org. Chem. 1986, 51, 3320–3324. [Google Scholar] [CrossRef]
- Zugates, G.T.; Anderson, D.G.; Little, S.R.; Lawhorn, I.E.B.; Langer, R. Synthesis of poly(β-amino esters) with thiol-reactive side chains for DNA delivery. J. Am. Chem. Soc. 2006, 128, 12726–12734. [Google Scholar] [CrossRef]
- Kotoku, N.; Tsujita, H.; Hiramatsu, A.; Mori, C.; Koizumi, N.; Kobayashi, M. Efficient total synthesis of bastadin 6, an anti-angiogenic brominated tyrosine-derived metabolite from marine sponge. Tetrahedron 2005, 61, 7211–7218. [Google Scholar]
- Dhar, S.; Nethajiand, M.; Chakravarty, A.R. Designing molecules for PDT: red light-induced DNA cleavage on disulfide bond activation in a dicopper (II) complex. Dalton Trans. 2005, 344–348. [Google Scholar]
- Katti, K.V.; Singh, P.R.; Barnes, C.L. Transition-metal chemistry of main-group hydrazides. Part 2. A new oxime thiosemicarbazide framework as a novel SN multifunctional tripodal ligand for palladium (II): synthetic and X-ray crystal structural investigations. J. Chem. Soc. Dalton Trans. 1993, 2153–2156. [Google Scholar]
- Arabshahi, L.; Schmitz, F.J. Brominated tyrosine metabolites from an unidentified sponge. J. Org. Chem. 1987, 52, 3584–3586. [Google Scholar] [CrossRef]
- Sheldrick, G. Program SADABS: Area-Detector Absorption Correction; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXL-97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL-97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. A short history of SHELXL. Acta Cryst. 2008, A64, 112–122. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, Q.; Liu, D.; Sun, D.; Yang, S.; Hu, G.; Wu, Z.; Zhao, L. Synthesis of the Marine Bromotyrosine Psammaplin F and Crystal Structure of a Psammaplin A Analogue. Molecules 2010, 15, 8784-8795. https://doi.org/10.3390/molecules15128784
Yang Q, Liu D, Sun D, Yang S, Hu G, Wu Z, Zhao L. Synthesis of the Marine Bromotyrosine Psammaplin F and Crystal Structure of a Psammaplin A Analogue. Molecules. 2010; 15(12):8784-8795. https://doi.org/10.3390/molecules15128784
Chicago/Turabian StyleYang, Qianjiao, Dan Liu, Deyang Sun, Sen Yang, Guodong Hu, Zuti Wu, and Linxiang Zhao. 2010. "Synthesis of the Marine Bromotyrosine Psammaplin F and Crystal Structure of a Psammaplin A Analogue" Molecules 15, no. 12: 8784-8795. https://doi.org/10.3390/molecules15128784