Electronic Structure of the Azide Group in 3¢-Azido-3¢-deoxythymidine (AZT) Compared to Small Azide Compounds

Abstract
:1. Introduction
2. Results and Discussion
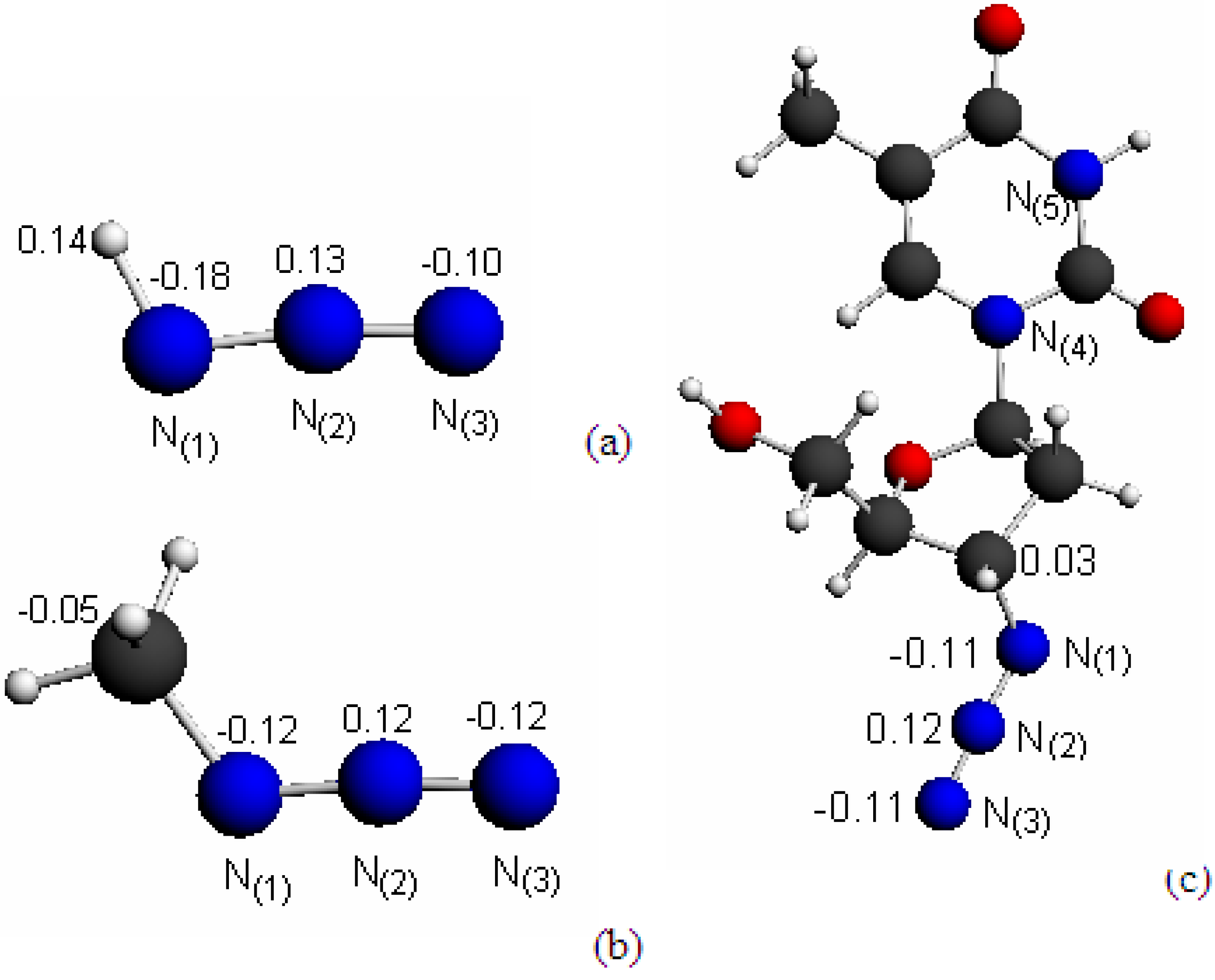
2.1. Structures and Hirshfeld Charge Distributions

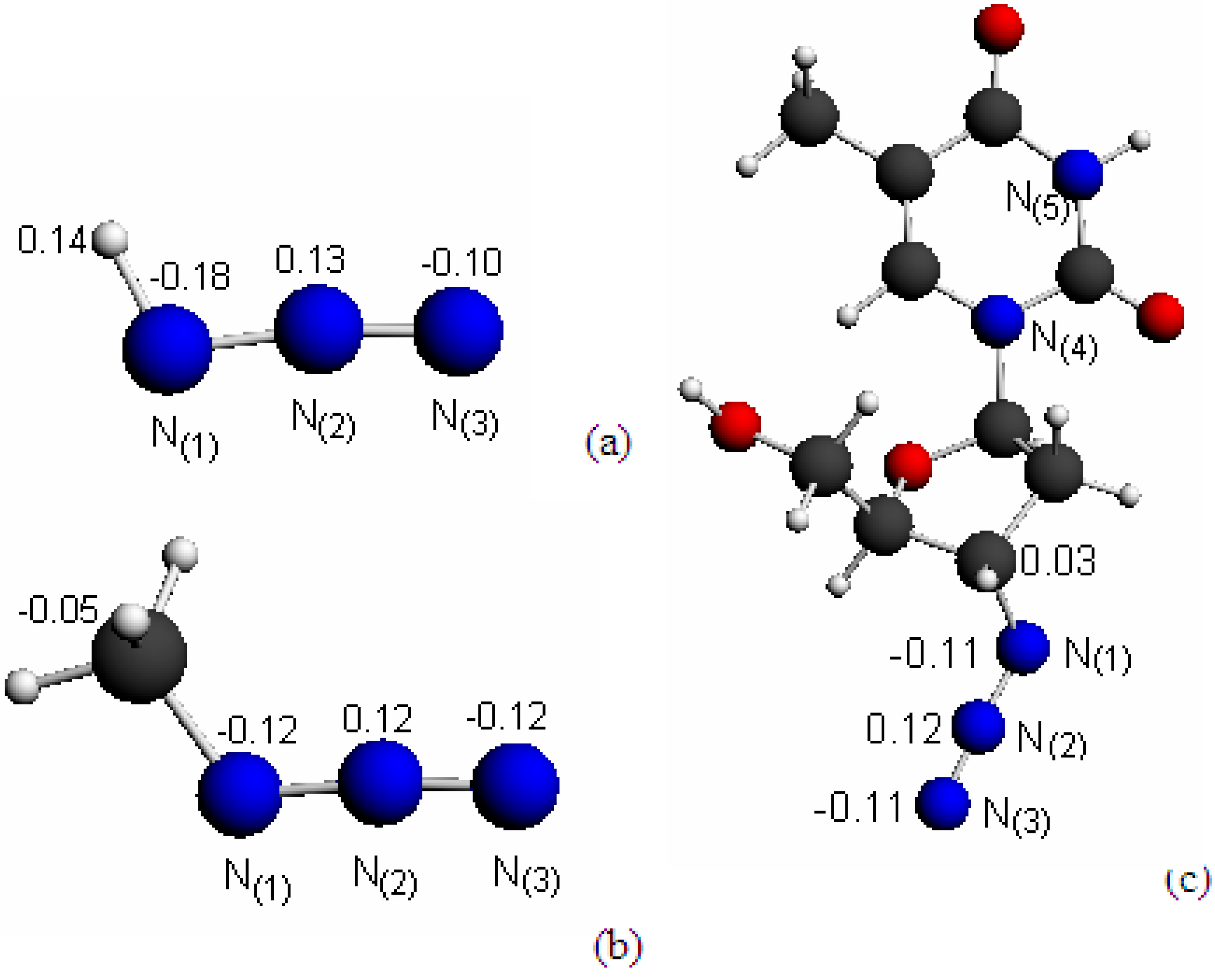{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure parameters | AZT | HN3 | CH3N3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| This Work | BLYP/6-31+G (d, p) [14] | Expt. [15] | This Work | MP2/TZ2P [13] | Expt. [13] | This Work | B3LYP/6-311++G** [16] | Expt. MW [17] | |
| N(1)-N(2)(Å) | 1.23 | 1.25 | 1.25 | 1.24 | 1.24 | 1.24 | 1.23 | 1.23 | 1.24 |
| N(2)-N(3)(Å) | 1.13 | 1.16 | 1.12 | 1.13 | 1.14 | 1.13 | 1.14 | 1.14 | 1.13 |
| R- N(1) (Å) | 1.48 | 1.49 | 1.49 | 1.02 | 1.02 | 1.02 | 1.47 | 1.47 | 1.46 |
| ∠N(1)-N(2)-N(3)(°) | 173.6 | 171.6 | 173.2 | 171.5 | 171.6 | 171.3 | 173.2 | 173.3 | |
| ∠R-N(1)-N(2)(°) | 115.9 | 116.8 | 115.0 | 110.1 | 109.5 | 108.8 | 116.1 | 116.1 | 117.0 |
| ∠R-N(1)-N(2) –N(3) (°) | -177.4 | 180.0 | 180.0 | 180.0 | |||||

2.2. IR Spectroscopy
| HN3 | CH3N3 | AZT | |||||||
|---|---|---|---|---|---|---|---|---|---|
| This Work a | B3LYP/6-31G** [22] | Expt. [23] | This Work* | B3LYP/6-31G** [22] | Expt. [23] | This Work a | BLYP/DZVP [24] | Expt. [25] | Assigned Vibration Mode |
| 2278 | 2294 | 2129 | 2247 | 2264 | 2106 | 2242 | 2103 | 2102 | ν1, Asym. NNN str. |
| 1300 | 1313 | 1264 | 1343 | 1474 | 1272 | 1328 | ν2, Symm. NNN str. | ||
| 533 | 529 | 537 | 666 | 665 | 666 | 658 | ν4, NNN torsion | ||
| 603 | 603 | 606 | 574 | 574 | 560 | 572 | ν3, RNNN bend | ||
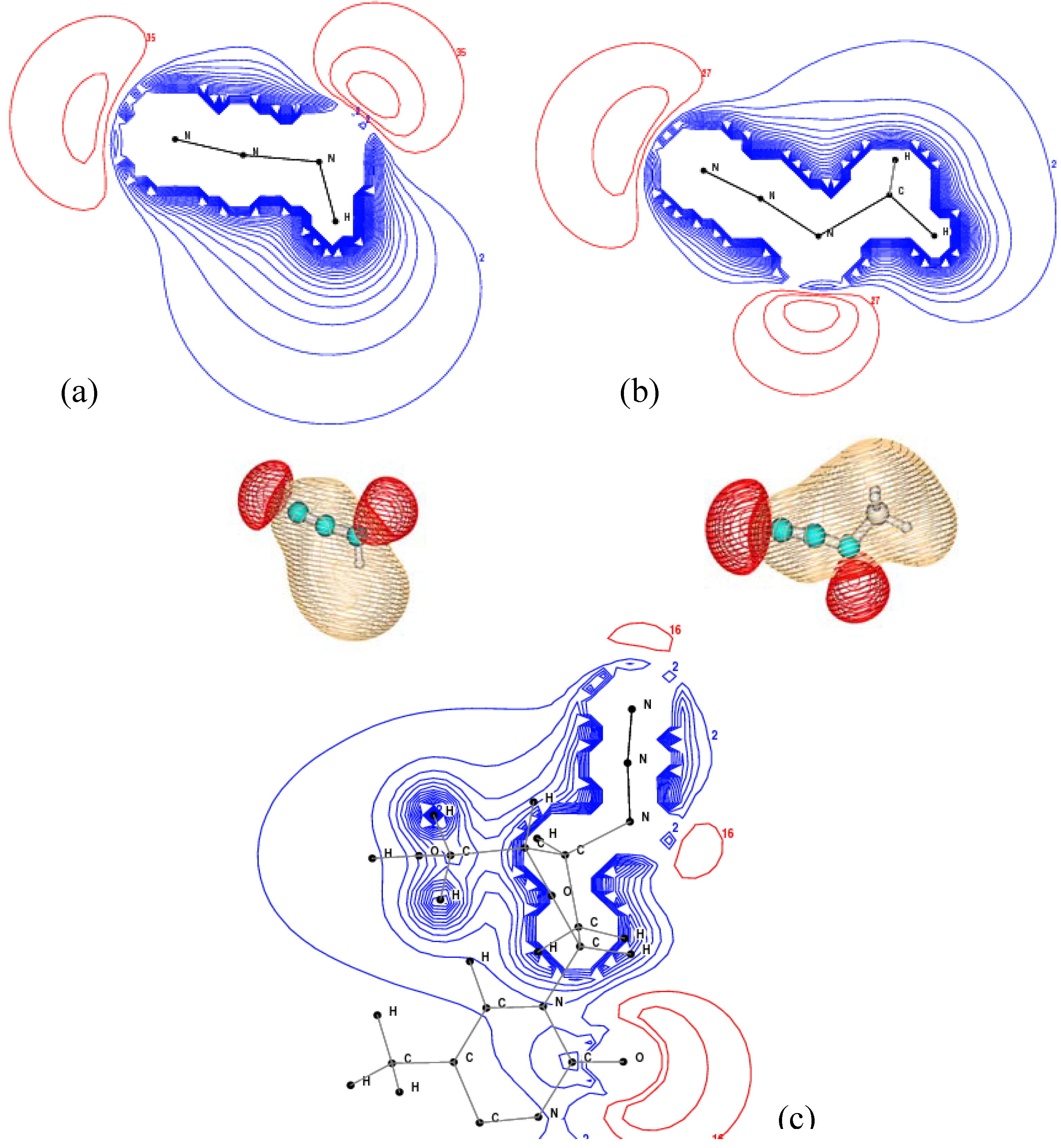
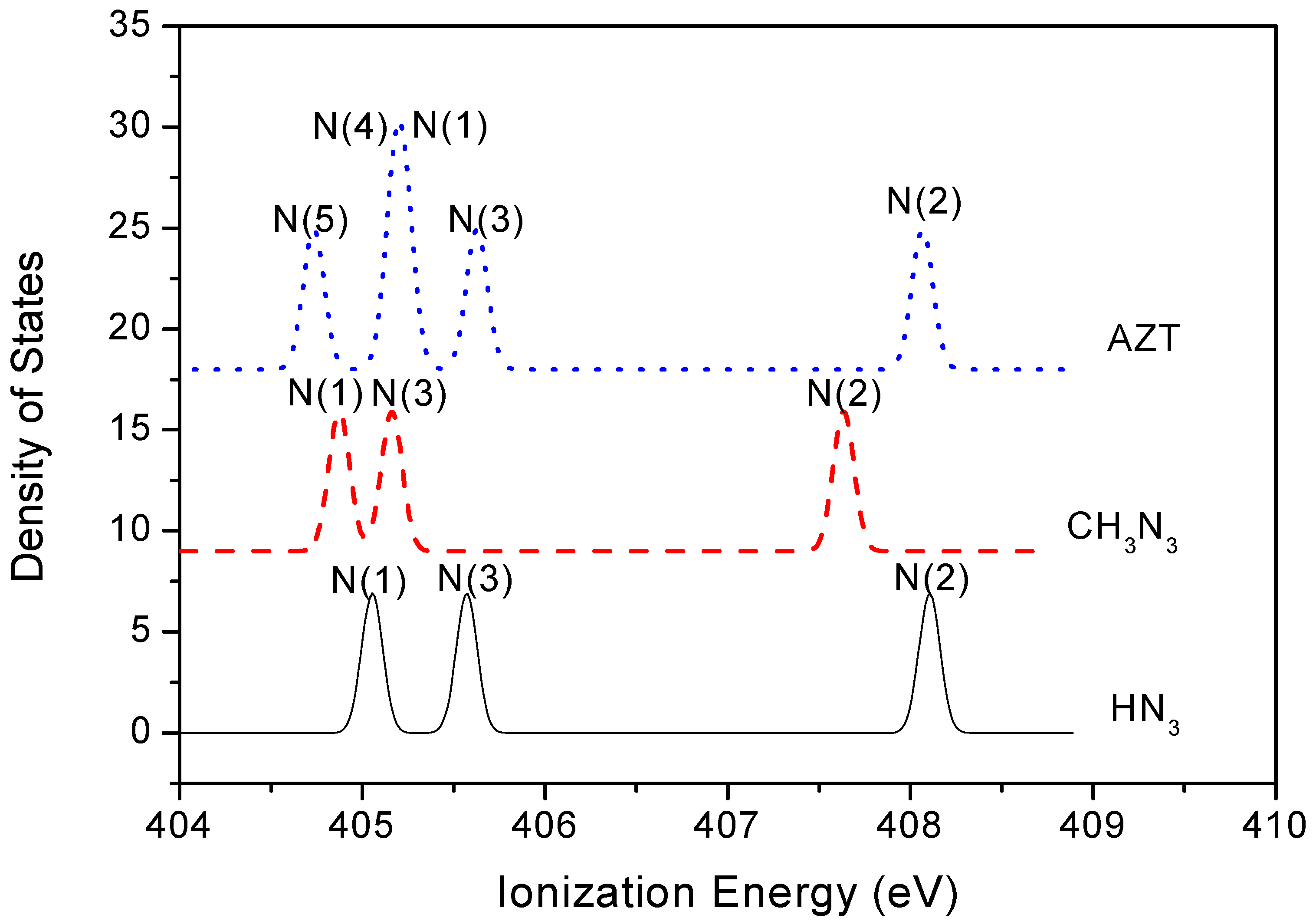
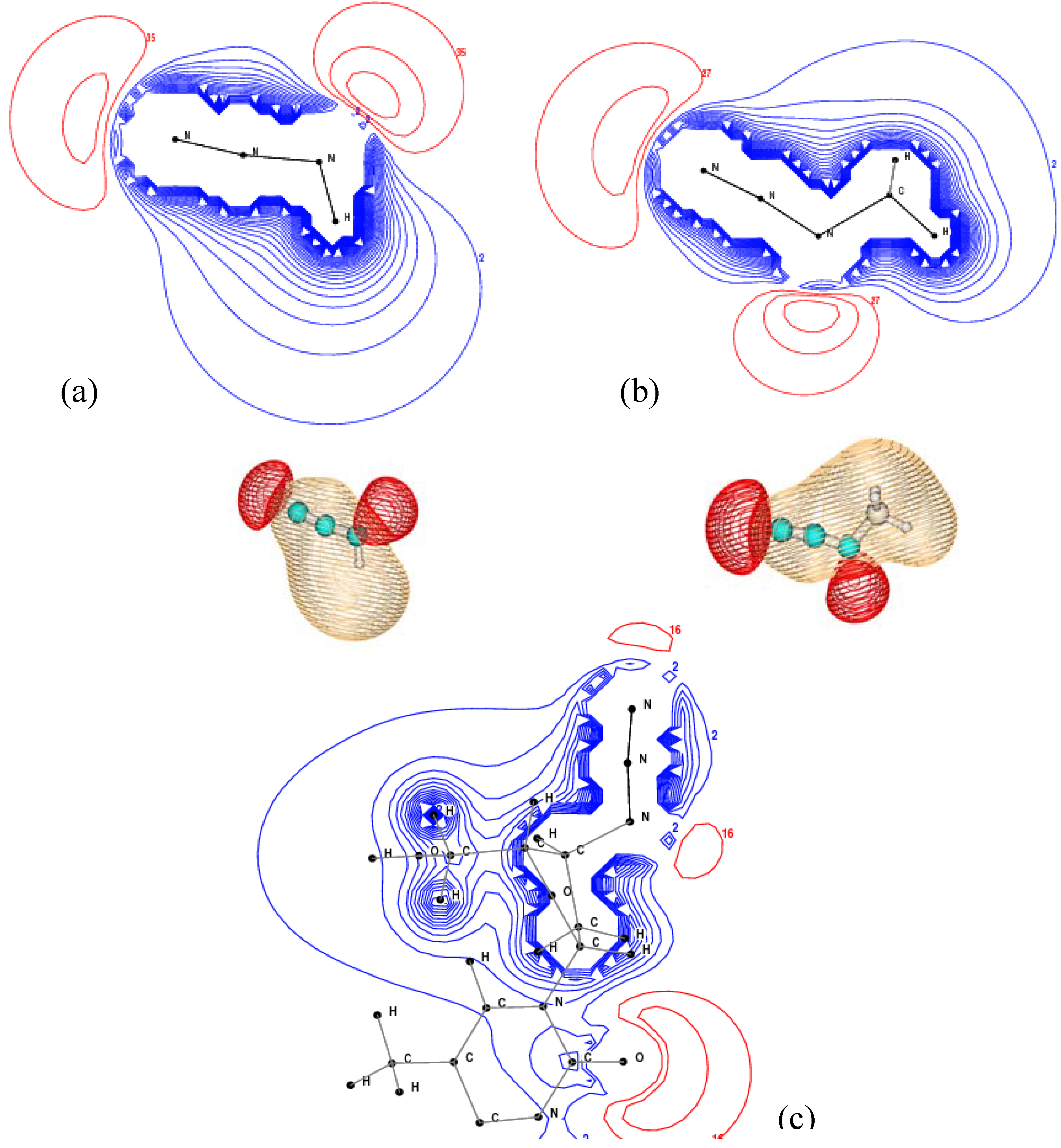
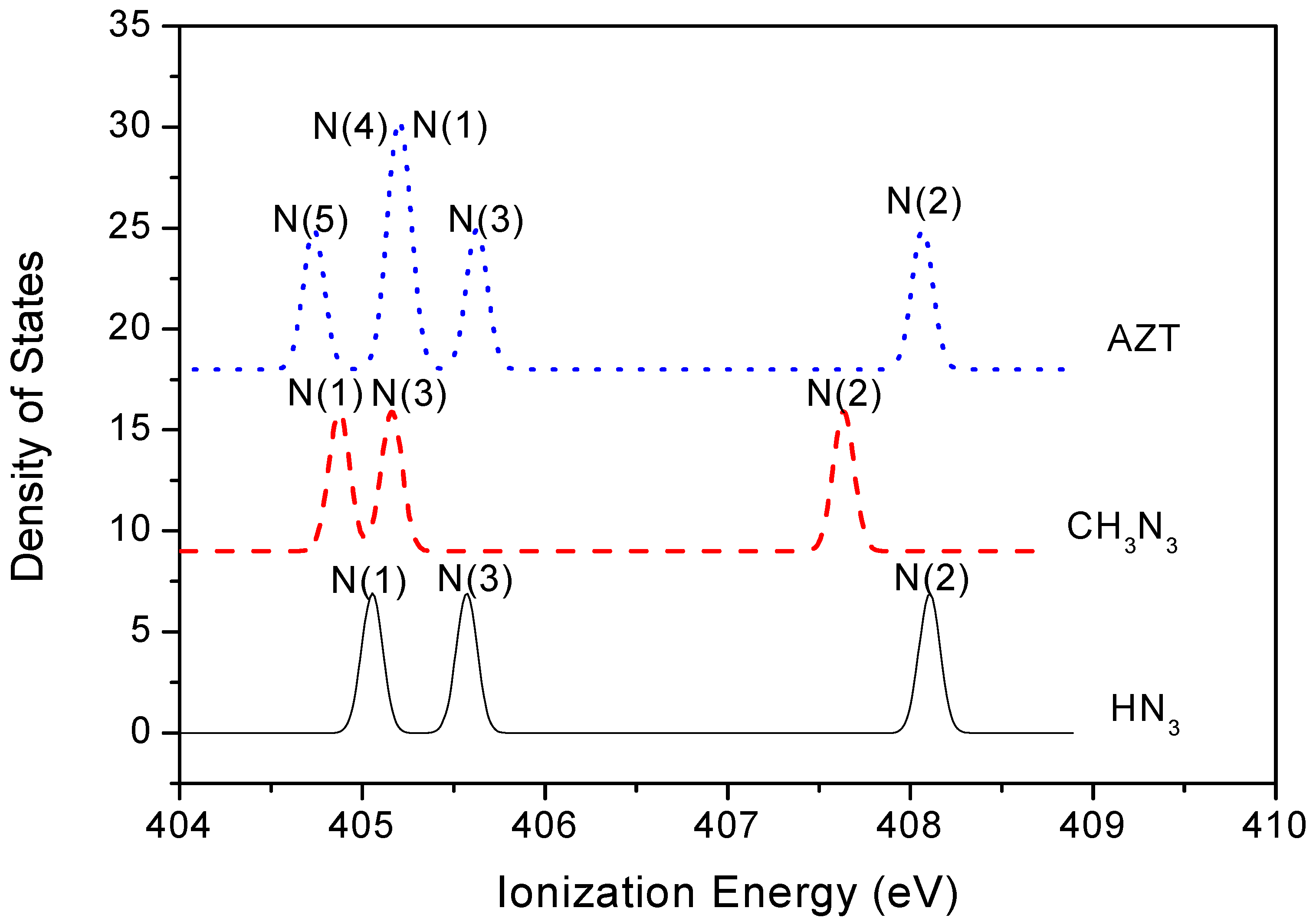
2.3. Core Shell Photoelectron Spectroscopy
| N1s sites | AZT | CH3N3 | HN3 (This work) | HN3 (Expt [10]) |
|---|---|---|---|---|
| N(1) | 405.23 | 404.87 | 405.05 | 399.5 |
| N(2) | 408.07 | 407.63 | 408.10 | 403.6 |
| N(3) | 405.62 | 405.16 | 405.57 | 400.4 |
| N(5) (base) | 404.73 | |||
| N(4) (base) | 405.18 |
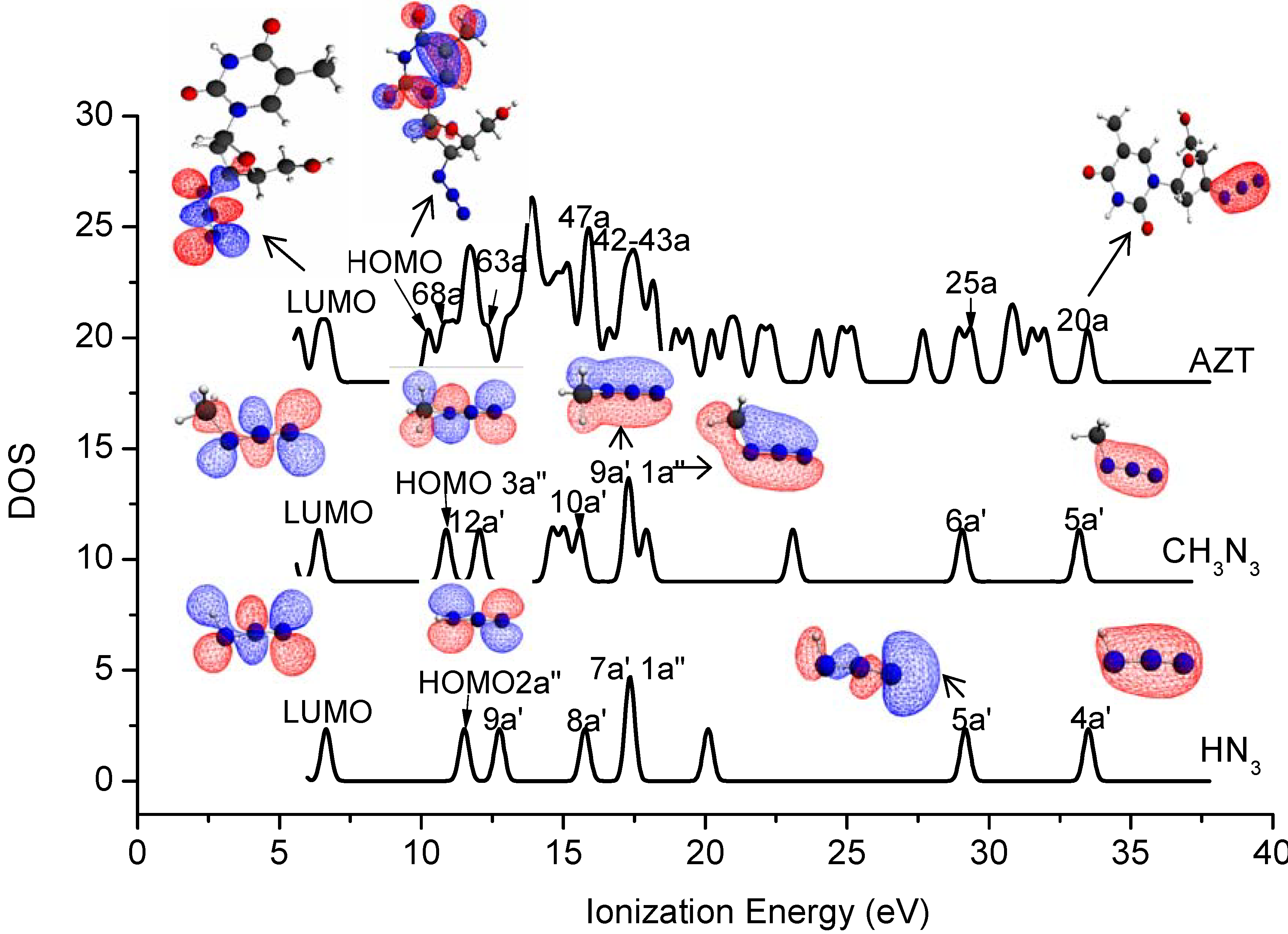
2.4. Valence Shell Photoelectron Spectroscopy
| Orbitals | SAOP/et-pVQZ | OVGF/TZVP | Expt. [10] |
|---|---|---|---|
| [HN3] | |||
| 2a″ | 11.51 | 10.13 (0.91) | 10.7 |
| 9a′ | 12.75 | 11.87 (0.92) | 12.2 |
| 8a′ | 15.76 | 15.88 (0.88) | 15.5 |
| 1a″ | 17.34 | 17.04* (0.81) | 16.7 |
| 7a′ | 17.35 | 17.17* (0.86) | 17.4 |
| 5a′ | 29.16 | ||
| 4a′ | 33.47 | ||
| [CH3N3] | |||
| 3a″ | 10.87 | 9.35 (0.91) | |
| 12a′ | 12.04 | 10.96 (0.92) | |
| 10a′ | 15.58 | 15.54 (0.88) | |
| 9a′ | 17.28 | 17.26 (0.85) | |
| 1a″ | 17.32 | ||
| 6a′ | 29.05 | ||
| 5a′ | 33.18 | ||
| [AZT] | |||
| 69a | 10.78 | ||
| 63a | 12.34 | ||
| 47a | 15.98 | ||
| 43a | 17.23 | ||
| 42a | 17.43 | ||
| 25a | 29.36 | ||
| 20a | 33.48 |
3. Theoretical Methods and Computational Details
4. Conclusions
Acknowledgements
References and Notes
- Tchertanov, L. Structural metrics relationships in covalently bonded organic azides. Acta Cryst. Sect. B 1999, 55, 807–809. [Google Scholar] [CrossRef]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic azides: an exploding diversity of a unique class of compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef]
- Bohn, M.A. Determination of the kinetic data of the thermal decomposition of energetic plasticizers and binders by adiabatic self heating. Thermochim. Acta 1999, 337, 121–139. [Google Scholar] [CrossRef]
- Tokumoto, M.; Tanaka, Y.; Kinoshita, N.; Kinoshita, T.; Ishibashi, S.; Ihara, H. Characterization of superconducting alkali and alkaline-earth fullerides prepared by thermal decomposition of azides. J. Phys. Chem. Sol. 1993, 54, 1667–1673. [Google Scholar] [CrossRef]
- Jönsson, B.M.; Håkansson, K.; Liljas, A. The structure of human carbonic anhydrase II in complex with bromide and azide. FEBS Lett. 1993, 322, 186–190. [Google Scholar] [CrossRef]
- Winnewisser, B.P. The substitution structure of hydrazoic acid, HNNN. J. Mol. Spectrosc. 1980, 82, 220–223. [Google Scholar] [CrossRef]
- Christe, K.O.; Wilson, W.W.; Dixon, D.A.; Khan, S.I.; Bau, R.; Metzenthin, T.; Lu, R. The aminodiazonium cation, H2N3+. J. Am. Chem. Soc. 1993, 115, 1836–1842. [Google Scholar] [CrossRef]
- Hooper, N.; Beeching, L.J.; Dyke, J.M.; Morris, A.; Ogden, J.S.; Dias, A.A.; Costa, M.L.; Barros, M.T.; Cabral, M.H.; Moutinho, A.M.C. A Study of the Thermal Decomposition of 2-Azidoethanol and 2-Azidoethyl Acetate by Ultraviolet Photoelectron Spectroscopy and Matrix Isolation Infrared Spectroscopy. J. Phys. Chem. A 2002, 106, 9968–9975. [Google Scholar] [CrossRef]
- Bock, H.; Dammel, R. Gas-phase reactions. 66. Gas-phase pyrolyses of alkyl azides: experimental evidence for chemical activation. J. Am. Chem. Soc. 1988, 110, 5261–5269. [Google Scholar] [CrossRef]
- Lee, T.H.; Colton, R.J.; White, M.G.; Rabalais, J.W. Electronic structure of hydrazoic acid and the azide ion from x-ray and ultraviolet electron spectroscopy. J. Am. Chem. Soc. 1975, 97, 4845–4851. [Google Scholar] [CrossRef]
- Cabral, B.J.; Costa, M.L. Density functional study of molecular properties of hydrazoic acid and methyl azide. J. Mol. Struct. (THEOCHEM) 1995, 343, 31–41. [Google Scholar] [CrossRef]
- Klapötke, T.M. Recent developments in the chemistry of covalent azides. Chem. Ber. 1997, 130, 443–452. [Google Scholar] [CrossRef]
- Sklenák, S.; Gatial, A.; Biskupic, S. Ab initio study of small organic azides. J. Mol. Struct. (THEOCHEM) 1997, 397, 249–262. [Google Scholar] [CrossRef]
- Hernández, J.; Soscún, H.; Hinchliffe, A. Density functional studies of molecular polarizabilities. Part 11. The Dipole Polarizability of the Most Stable Conformation of 3'-Azido-3'-deoxythymidine AZT. Internet Electron. J. Mol. Des. 2003, 2, 589–598. [Google Scholar]
- Roey, P.V.; Salerno, J.M.; Duax, W.L.; Chu, C.K.; Ahn, M.K.; Schinazi, R.F. Solid-state conformation of anti-human immunodeficiency virus type-1 agents: Crystal structures of three 3'-azido-3'-deoxythymidine analogs. J. Am. Chem. Soc. 1988, 110, 2277–2282. [Google Scholar] [CrossRef]
- Badawi, H.M. Theoretical study of the structure and vibrational spectra of formyl and methyl azides. J. Mol. Struct. (THEOCHEM) 2002, 583, 89–97. [Google Scholar] [CrossRef]
- Salathiel, W.M.; Curl, J.R.F. Microwave Spectrum of Methylazide. J. Chem. Phys. 1966, 44, 1288–1290. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, G.; Tylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part1. Bond lengths in organic compounds. J. Chem. Soc., Perkin Trans. 1987, 2, S1–S19. [Google Scholar]
- Janoschek, R. Pentazole and Other Nitrogen Rings. Angew. Chem. Int. Ed. Engl. 1992, 32, 230–232. [Google Scholar] [CrossRef]
- Harcourt, R.D. On the "pentavalent" nitrogen atom and nitrogen pentacoordination. J. Mol. Struct. 1993, 300, 245–256. [Google Scholar] [CrossRef]
- Brockway, L.O.; Pauling, L. The electron-diffraction investigation of the structure of molecules of methyl azide and carbon suboxide. Proc. Natl. Acad. Sci. 1933, 19, 860–867. [Google Scholar] [CrossRef]
- Fu, A.; Du, D.; Zhou, Z. Density functional study on the vibrational frequencies of hydrazoic acid and methyl azide. Chinese Chem. Letts. 1999, 10, 619–622. [Google Scholar]
- Nielsen, C.J.; Sjøgfogren, C.E. The vibrational spectra, molecular structure and conformation of organic azides : Part IV. An ab initio study of hydrazoic acid, azidomethane, azidoethane, azidoethene and azidomethanal. J. Mol. Struct. (THEOCHEM) 1987, 150, 361–379. [Google Scholar] [CrossRef]
- Essa, A.H.; Ibrahim, M.; Hameed, A.J.; AI-Masoudi, N.A. Theoretical investigation of 3’-substituted -2’-3’-dideoxythymidines related to AZT. QSAR infrared and substituent electronic effect studies. ARKIVOC 2008, viii, 255–265. [Google Scholar]
- Araújo, A.A.S.; Storpirtis, S.; Mercuri, L.P.; Carvalho, F.M.S.; Filho, M.D.S.; Matos, J.R. Thermal analysis of the antiretroviral zidovudine (AZT) and evaluation of the compatibility with excipients used in solid dosage forms. Intel. J. Pharm. 2003, 260, 303–314. [Google Scholar] [CrossRef]
- Gritsenko, O.V.; Braida, B.; Baerends, E.J. Physical interpretation and evaluation of the Kohn–Sham and Dyson components of the epsilon–I relations between the Kohn--Sham orbital energies and the ionization potentials. J. Chem. Phys. 2003, 119, 1937–1950. [Google Scholar] [CrossRef]
- Plekan, O.; Feyer, V.; Richter, R.; Coreno, M.; de Simone, M.; Prince, K.C.; Trofimov, A.B.; Gromov, E.V.; Zaytseva, I.L.; Schirmer, J. A theoretical and experimental study of the near edge X-ray absorption fine structure (NEXAFS) and X-ray photoelectron spectra (XPS) of nucleobases: Thymine and adenine. Chem. Phys. 2008, 347, 360–375. [Google Scholar] [CrossRef]
- Wang, F.; Pang, W. Valence orbital response to conformers of n-butane. Mol. Simul. 2007, 33, 1173–1185. [Google Scholar] [CrossRef]
- Selvam, L.; Vasilyev, V.; Wang, F. Methylation of zebularine: a quantum mechanical study incorporating interactive 3D PDF graphs. J. Phys. Chem. B 2009, in press. [Google Scholar]
- Van Leeuwen, R.; Baerends, E.J. Exchange-correlation potential with correct asymptotic behavior. Phys. Rev. A 1994, 49, 2421. [Google Scholar] [CrossRef]
- Schipper, P.R.T.; Gritsenko, O.V.; van Gisbergen, S.J.A.; Baerends, E.J. Molecular calculations of excitation energies and (hyper)polarizabilities with a statistical average of orbital model exchange-correlation potentials. J. Chem. Phys. 2000, 112, 1344–1352. [Google Scholar] [CrossRef]
- Chong, D.P.; Lenthe, E.V.; Gisbergen, S.V.; Evert, J.B. Even-tempered slater-type orbitals revisited: From hydrogen to krypton. J. Comput. Chem. 2004, 25, 1030–1036. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Electron Density Distributions in Molecules. Crystallogr. Rev. 1991, 2, 169–200. [Google Scholar] [CrossRef]
- Thompson, A.; Saha, S.; Wang, F.; Tsuchimochi, T.; Nakata, A.; Imamura, Y.; Nakai, H. Density functional study on core ionization spectra of cytidine and its fragments. Bull. Chem. Soc. Jpn. 2009, 82, 187–195. [Google Scholar] [CrossRef]
- Nakata, A.; Imamura, Y.; Otsuka, T.; Nakai, H. Time-dependent density functional theory calculations for core-excited states: Assessment of standard exchange-correlation functionals and development of a novel hybrid functional. J. Chem. Phys. 2006, 124, 094105:1–094105:9. [Google Scholar]
- Duffy, P.; Sordo, J.A.; Wang, F. Valence orbital response to pseudorotation of tetrahydrofuran: A snapshot using dual space analysis. J. Chem. Phys 2008, 128, 125102. [Google Scholar] [CrossRef]
- Falzon, C.T.; Wang, F.; Pang, W. Orbital signatures of methyl in l-Alanine. J. Chem. Phys. B 2006, 110, 9713–9719. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03, Revision C.02; Gaussian, Inc: Wallingford, CT, USA, 2004. [Google Scholar]
- Velde, G.T.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, C.F.; Gisbergen, S.J.A.V.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- ADF2008.01, SCM, Theoretical Chemistry. Vrije Universiteit: Amsterdam, the Netherlands, 2008.
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput.-Aided Mol. Design 2000, 14, 123–134. [Google Scholar] [CrossRef]
- Sample Availability: Calculations of the compunds are available from the authors.
© 2009 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, F.-F.; Wang, F. Electronic Structure of the Azide Group in 3¢-Azido-3¢-deoxythymidine (AZT) Compared to Small Azide Compounds. Molecules 2009, 14, 2656-2668. https://doi.org/10.3390/molecules14072656
Chen F-F, Wang F. Electronic Structure of the Azide Group in 3¢-Azido-3¢-deoxythymidine (AZT) Compared to Small Azide Compounds. Molecules. 2009; 14(7):2656-2668. https://doi.org/10.3390/molecules14072656
Chicago/Turabian StyleChen, Fang-Fang, and Feng Wang. 2009. "Electronic Structure of the Azide Group in 3¢-Azido-3¢-deoxythymidine (AZT) Compared to Small Azide Compounds" Molecules 14, no. 7: 2656-2668. https://doi.org/10.3390/molecules14072656