Stereochemistry of 16a-Hydroxyfriedelin and 3-Oxo-16-methylfriedel-16-ene Established by 2D NMR Spectroscopy

Abstract
:Introduction
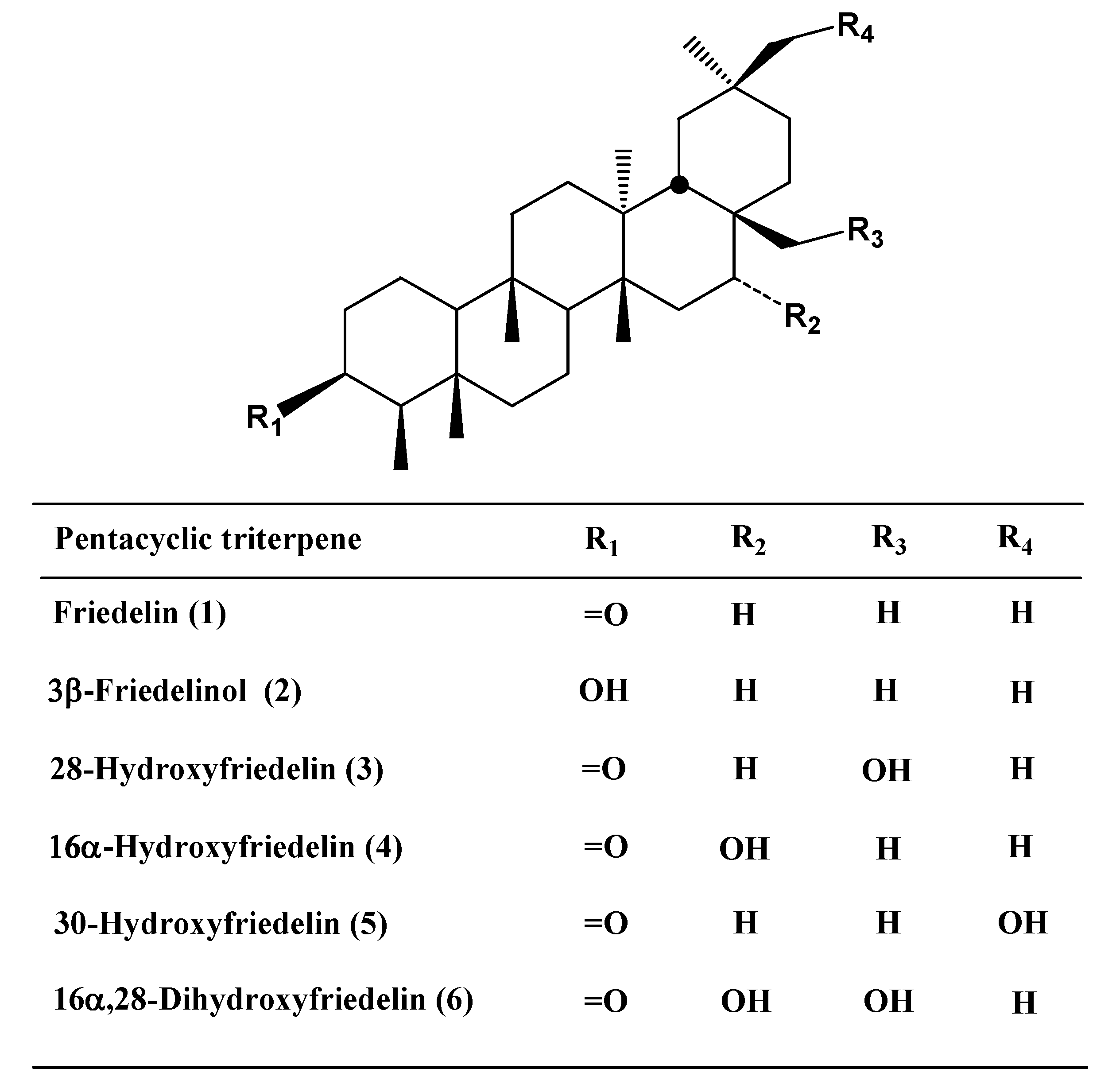
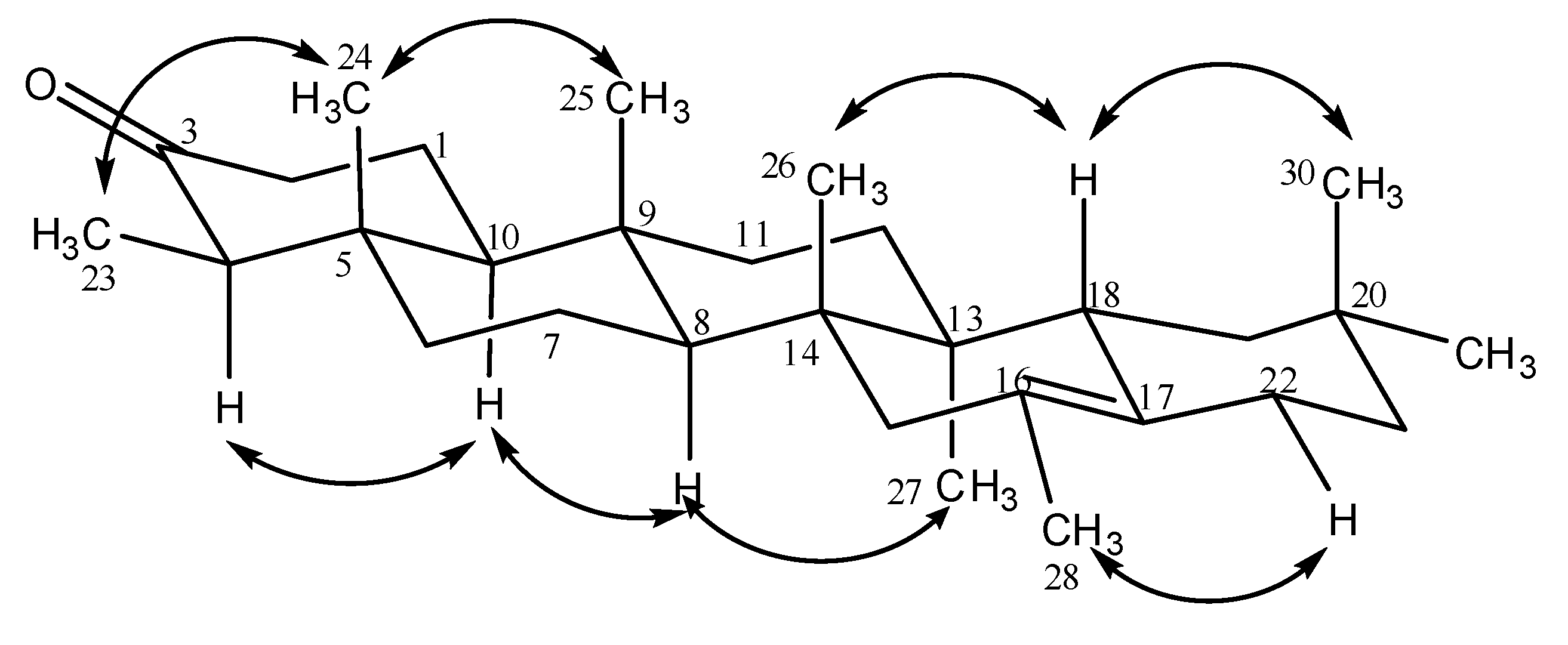
Results and Discussion
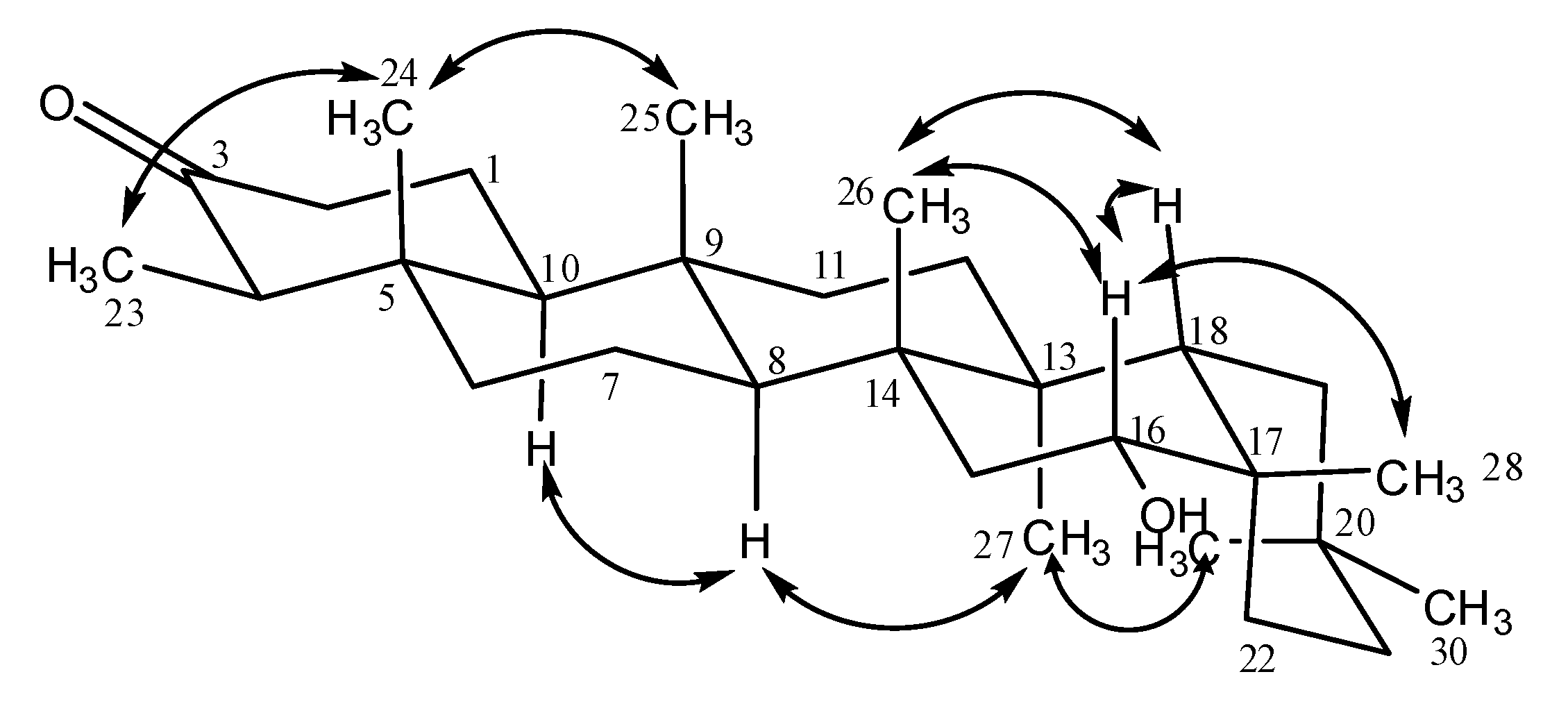

{kind=link}
{kind=link}
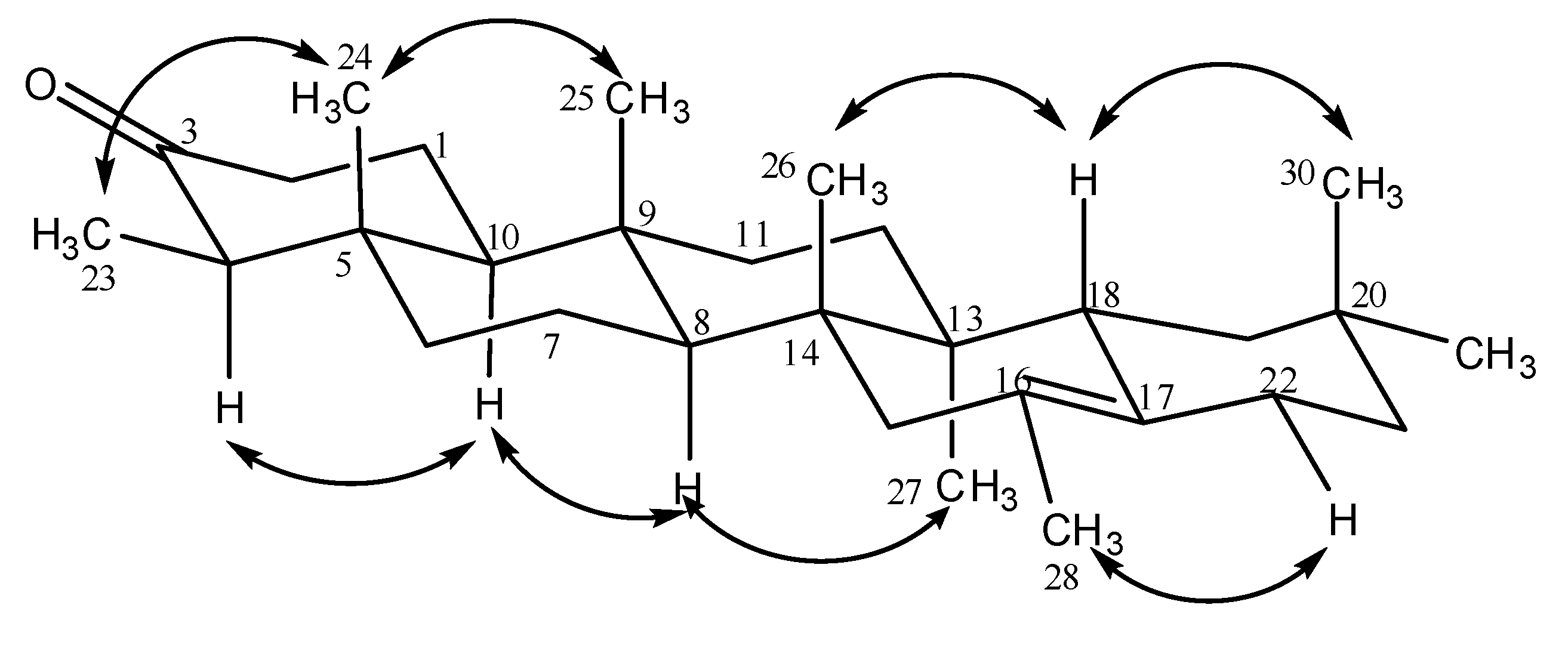{kind=link}
| δC (lit) * | δC (4) | δH (4) | HMBC | δC (7) | δH (7) |
|---|---|---|---|---|---|
| 22.3 | 22.77 | 1.62 ax 1.86 eq | 22.32 | 1.68 ax 1.98 eq | |
| 41.6 | 41.90 | 2.34 eq 2.44 ax | 41.55 | 2.39 ax 2.45 eq | |
| 212.5 | 212.12 | - | 1, 2, 4, 23 | 213.21 | - |
| 58.3 | 58.28 | 2.20, m | 23, 24, 5, 10 | 58.25 | 2.27, m |
| 42.3 | 42.37 | - | 42.07 | - | |
| 41.4 | 41.54 | 1.54, m | 41.09 | 1.23 eq 1.74 ax | |
| 18.6 | 18.95 | 1.64, m | 18.23 | 1.39 eq 1.48 ax | |
| 53.5 | 50.82 | 1.51, m | 12, 15, 25, 26 | 50.18 | 1.38, m |
| 37.6 | 37.97 | - | 37.53 | - | |
| 59.7 | 59.77 | 1.56, m | 2, 4, 8 | 59.25 | 1.55, m |
| 35.8 | 35.64 | 1.32, m | 35.50 | 1.27 ax 1.51 eq | |
| 30.8 | 30.42 | 1.29 ax 1.40 eq | 28.14 | 1.35, m | |
| 39.3 | 39.95 | - | 37.29 | - | |
| 40.1 | 40.25 | - | 37.65 | - | |
| 44.4 | 40.25 | 1.66 ax, m 2.08 eq, m | 43.07 | 1.52 eq 1.58 ax | |
| 75.6 | 75.70 | 4.25 J = 7.0; 10.4 Hz | 14, 15, 17, 18, 22, 28 | 122.57 | - |
| 32.1 | 37.75 | - | 129.50 | - | |
| 44.8 | 46.66 | 1.63, m | 19, 27, 28 | 40.43 | 1.87, m |
| 35.8 | 34.28 | 1.35, m | 37.69 | 1.03 ax 1.29 eq | |
| 28.0 | 28.62 | - | 30.00 | - | |
| 32.1 | 34.97 | 1.62, m | 38.33 | 1.19 eq 1.36 ax | |
| 36.0 | 27.76 | 1.97, m 2.05, m | 24.60 | 1.91 ax 2.48 eq | |
| 6.8 | 7.55 | 0.95, d J = 6.7 Hz | 3, 4, 5 | 6.84 | 0.88, d J = 6.8 |
| 14.7 | 15.08 | 0.69, s | 4, 5, 10 | 14.64 | 0.72, s |
| 18.2 | 19.43 | 0.84, s | 8, 9, 10 | 17.20 | 0.86, s |
| 20.1 | 17.70 | 0.93, s | 8, 13 14 15 | 16.43 | 0.75, s |
| 21.5 | 20.02 | 1.34, s | 12, 13, 14, 18 | 16.59 | 0.84, s |
| 24.9 | 31.03 | 1.37, s | 17, 22 | 19.62 | 1.59, s |
| 30.8 | 32.67 | 1.09, s | 19, 20, 21 | 33.07 | 0.92, s |
| 35.5 | 36.95 | 1.05, s | 19, 20, 21 | 24.45 | 0.94, s |
Experimental
General
NMR spectra
Plant Material and Compound Isolation
Acknowledgements
References
- Wolf, B.M.; Weisbrode, S.E. Safety evaluation of an extract from Salacia oblonga. Food Chem. Toxicol. 2003, 4, 867–870. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Murakami, T.; Shimada, H.; Matsuda, H.; Yamahara, J.; Tanabe, G.; Muraoka, O. Salacinol, potent antidiabetic principle with unique thiosugar sulfonium sulfate structure from the ayurvedic traditional medicine Salacia reticulata in Sri Lanka and India. Tetrahedron Lett. 1997, 38, 8367–8370. [Google Scholar] [CrossRef]
- Jeller, A.H.; Silva, D.H.S.; Lião., L.M.; Bolzani, V.S.;; Furlan, M. Antioxidant phenolic and quinonemethide triterpenes from Cheiloclinium cognatum. Phytochemistry 2004, 65, 1977–1982. [Google Scholar] [CrossRef]
- Deepa, M.A.; Bai, V.N. Antibacterial activity of Salacia beddomei. Fitoterapia 2004, 75, 589–591. [Google Scholar] [CrossRef]
- Kuo, Y.H.; Kuo, L.M.Y. Antitumour and anti-AIDS triterpenes from Celastrus hindsii. Phytochemistry 1997, 44, 1275–1281. [Google Scholar] [CrossRef]
- Oliveira, M. L. G.; Duarte, L. P.; Silva, G. D. F.; Vieira Filho, S.A.; Knupp, V.F.; Alves, F.G.P. 3-Oxo-12α-hydroxyfriedelane from Maytenus gonoclada: structure elucidation by (1)H and (13)C chemical shifts assignments and 2D-NMR spectroscopy. Magn. Reson. Chem. 2007, 45, 895–898. [Google Scholar] [CrossRef]
- Oliveira, D.M.; Silva, G.D.F.; Duarte, L.P.; Vieira Filho, S.A. Chemical constituents isolated from roots of Maytenus acanthophylla Reissek (Celastraceae). Bioch. Syst. Ecol. 2006, 34, 661–665. [Google Scholar] [CrossRef]
- Rizvi, S.H.; Shoeb, A.; Kapil, R.S.; Popli, S.P. Antidesmanol - New Pentacyclic Triterpenoid From Antidesma menasu Miq Ex Tul. Experientia 1980, 36, 146–147. [Google Scholar] [CrossRef]
- lves, J.S.; Castro, J.C.M.; Freire, M.O.; da-Cunha, E.V.M.; Barbosa-Filho, J.M.; Silva, M.S. Complete assignment of the 1H and 13C NMR spectra of four triterpenes of the ursane, artane, lupane and friedelane groups. Magn. Reson.Chem. 2000, 38, 201–206. [Google Scholar] [CrossRef]
- Vieira, H.S.; Takahashi, J.A.; Gunatilaka, A.A.L.; Boaventura, M.A.D. 1H and 13C NMR signal assignments of a novel Baeyer-Villiger originated diterpene lactone. Magn. Reson. Chem. 2006, 44, 146–150. [Google Scholar] [CrossRef]
- Fernandez, A.H.; Cerero, S.M.; Jimenez, F.M. About the Timing of Wagner-Meerwein and Nametkin Rearrangements, 6,2-Hydride Shift, Proton Elimination and Cation Trapping in 2-Norbornyl Carbocations. Tetrahedron 1998, 54, 4607–4614. [Google Scholar] [CrossRef]
- Starling, S.M.; Vonwiller, S.C.; Reek, J.N.H. Effect of Ortho Substituents on the Direction of 1,2-Migrations in the Rearrangement of 2-exo-Arylfenchyl Alcohols. J. Org. Chem. 1998, 63, 2262–2272. [Google Scholar] [CrossRef]
- Kikuchi, T.; Niwa, M.; Yokoi, T.; Kadota, S. Studies on the Neutral Constituents of Pachysandra terminalis SIEB. et ZUCC. VIII. Methyl Migration in the Dehydration Reaction of Pachysonol and Pachysandiol-B Derivatives. Chem. Pharm. Bull. 1981, 29, 1819–1826. [Google Scholar] [CrossRef]
- Hao, X.; Xuehui, L.; Yuxin, C.; Min, Z.; Jiaxiang, S. NMR study on the reaction of rearrangement of 17β-hydroxy-7α-methyl-19-nor-17-α-pregn-5(10)-en-20-yn-3-one. Bopuxue Zazhi 2000, 17, 17–22. [Google Scholar]
- Lebedeva, T.L.; Vointseva, I.I.; Gilman, L.M.; Petrovskii, P.V.; Larina, T.A.; Topchiev, A.V. Solvent-induced allylic rearrangements in poly(trichlorobutadiene) chains. Russ. Chem. Bull. 1997, 46, 732–738. [Google Scholar] [CrossRef]
- Mitsuo, K.; Zhang, L.C.; Kabuto, C.; Sakurai, H. Synthesis and Reactions of Neutral Hypercoordinate Allylsilicon Compounds Having a Tropolonato Ligand Organometallics. 1996, 15, 5335–5341. [Google Scholar]
- Mahato, S.B.; Kundu, A.P. 13C NMR Spectra of pentacyclic triterpenoids - a compilation and some salient features. Phytochemistry 1994, 37, 1517–1575. [Google Scholar] [CrossRef]
- Courtney, J.L.; Shannon, J.S. Studies in mass spectrometry triterpenoids: structure assignment to some friedelane derivatives. Tetrahedron Lett. 1963, 1, 13–20. [Google Scholar] [CrossRef]
- Matos, F.J.A. Introdução a Fitoquímica Experimental; Editora UFC: Fortaleza, Brazil, 1988; p. 126. [Google Scholar]
- Sample Availability: Samples of compounds 1, 2, 3 and 5 are available from the authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an-open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Duarte, L.P.; Silva de Miranda, R.R.; Rodrigues, S.B.V.R.; De Fátima Silva, G.D.; Filho, S.A.V.; Knupp, V.F. Stereochemistry of 16a-Hydroxyfriedelin and 3-Oxo-16-methylfriedel-16-ene Established by 2D NMR Spectroscopy. Molecules 2009, 14, 598-607. https://doi.org/10.3390/molecules14020598
Duarte LP, Silva de Miranda RR, Rodrigues SBVR, De Fátima Silva GD, Filho SAV, Knupp VF. Stereochemistry of 16a-Hydroxyfriedelin and 3-Oxo-16-methylfriedel-16-ene Established by 2D NMR Spectroscopy. Molecules. 2009; 14(2):598-607. https://doi.org/10.3390/molecules14020598
Chicago/Turabian StyleDuarte, Lucienir Pains, Roqueline Rodrigues Silva de Miranda, Salomão Bento Vasconcelos Rodrigues Rodrigues, Grácia Divina De Fátima Silva, Sidney Augusto Vieira Filho, and Vagner Fernandes Knupp. 2009. "Stereochemistry of 16a-Hydroxyfriedelin and 3-Oxo-16-methylfriedel-16-ene Established by 2D NMR Spectroscopy" Molecules 14, no. 2: 598-607. https://doi.org/10.3390/molecules14020598