Multiscale Modeling of Dendrimers and Their Interactions with Bilayers and Polyelectrolytes


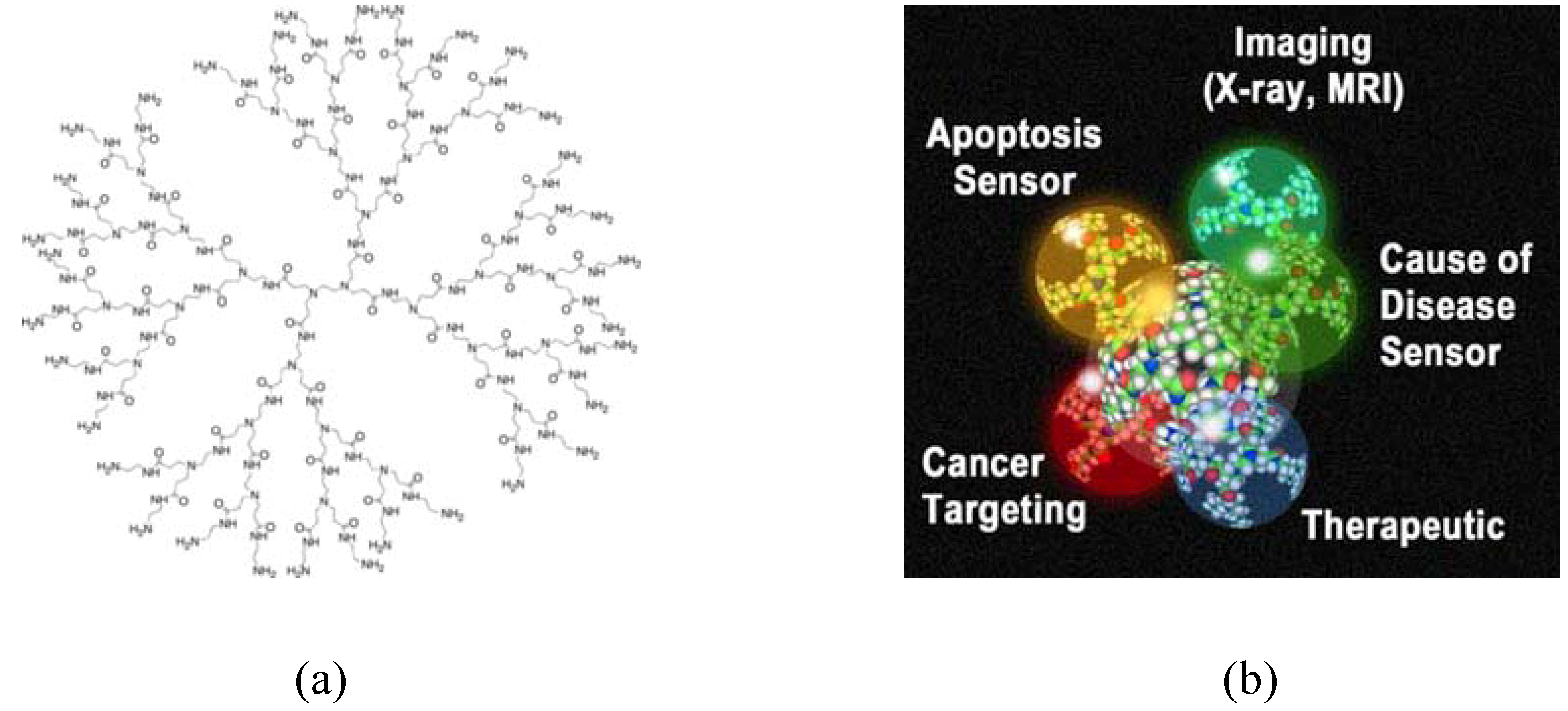{kind=link}
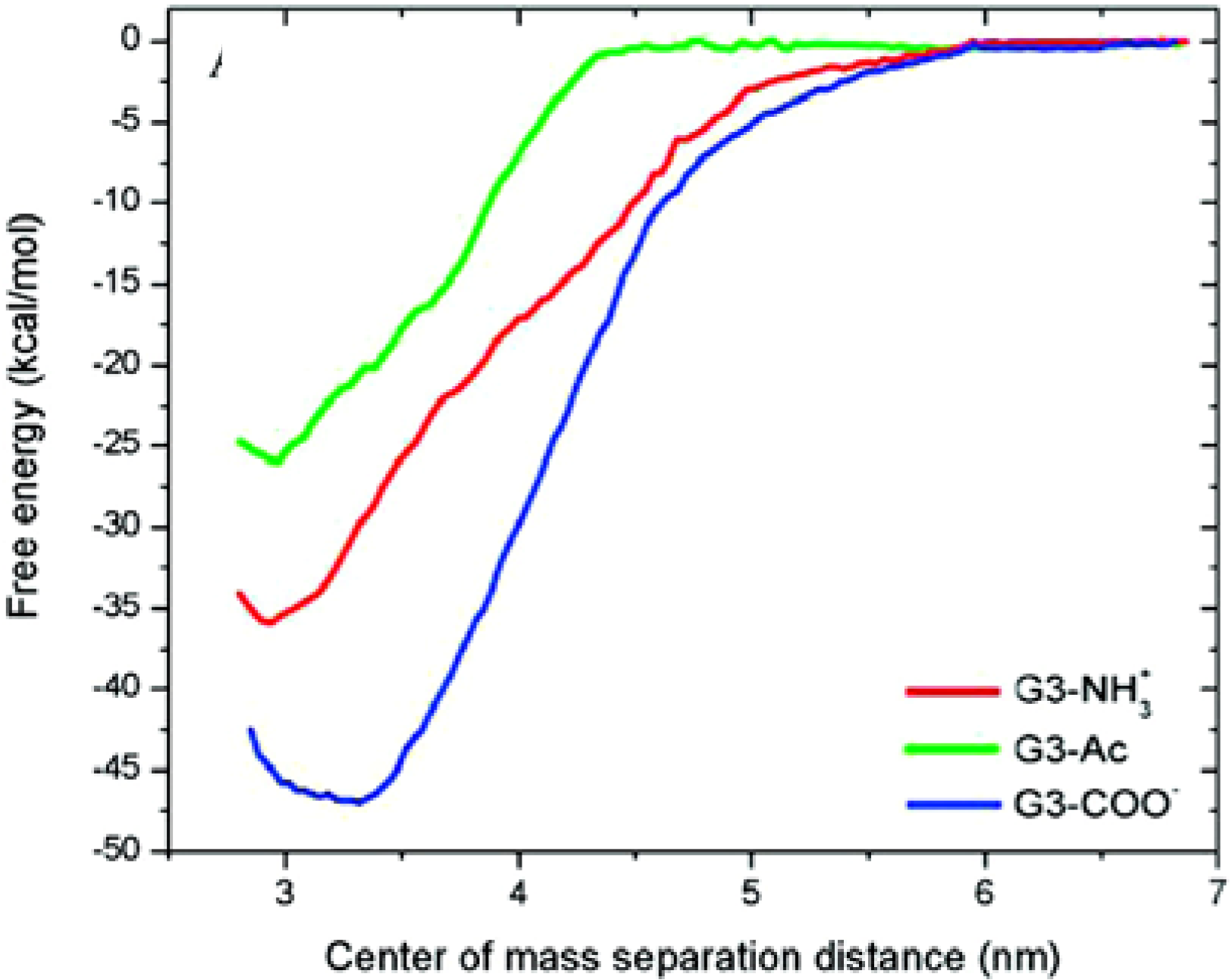{kind=link}
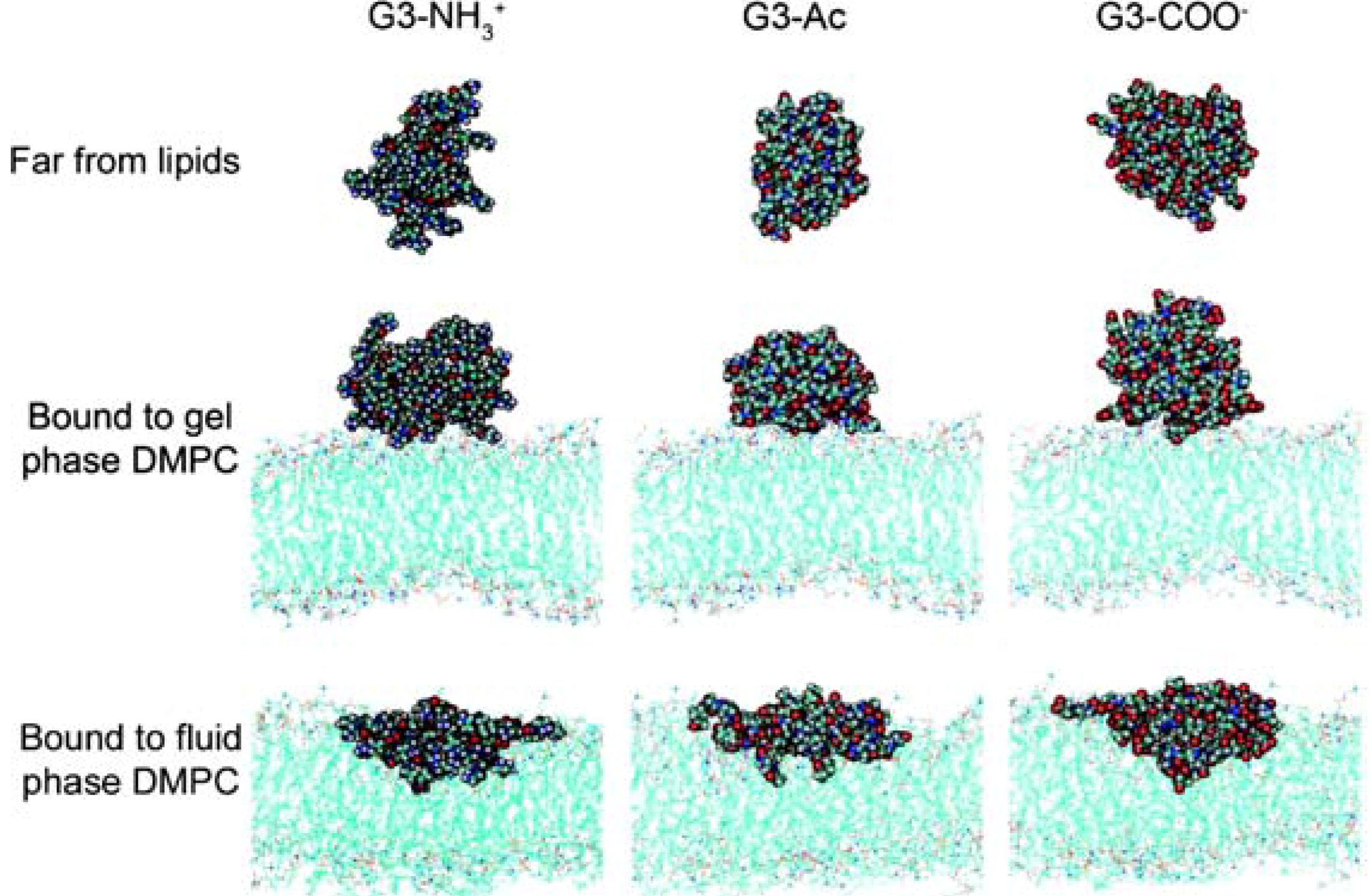{kind=link}
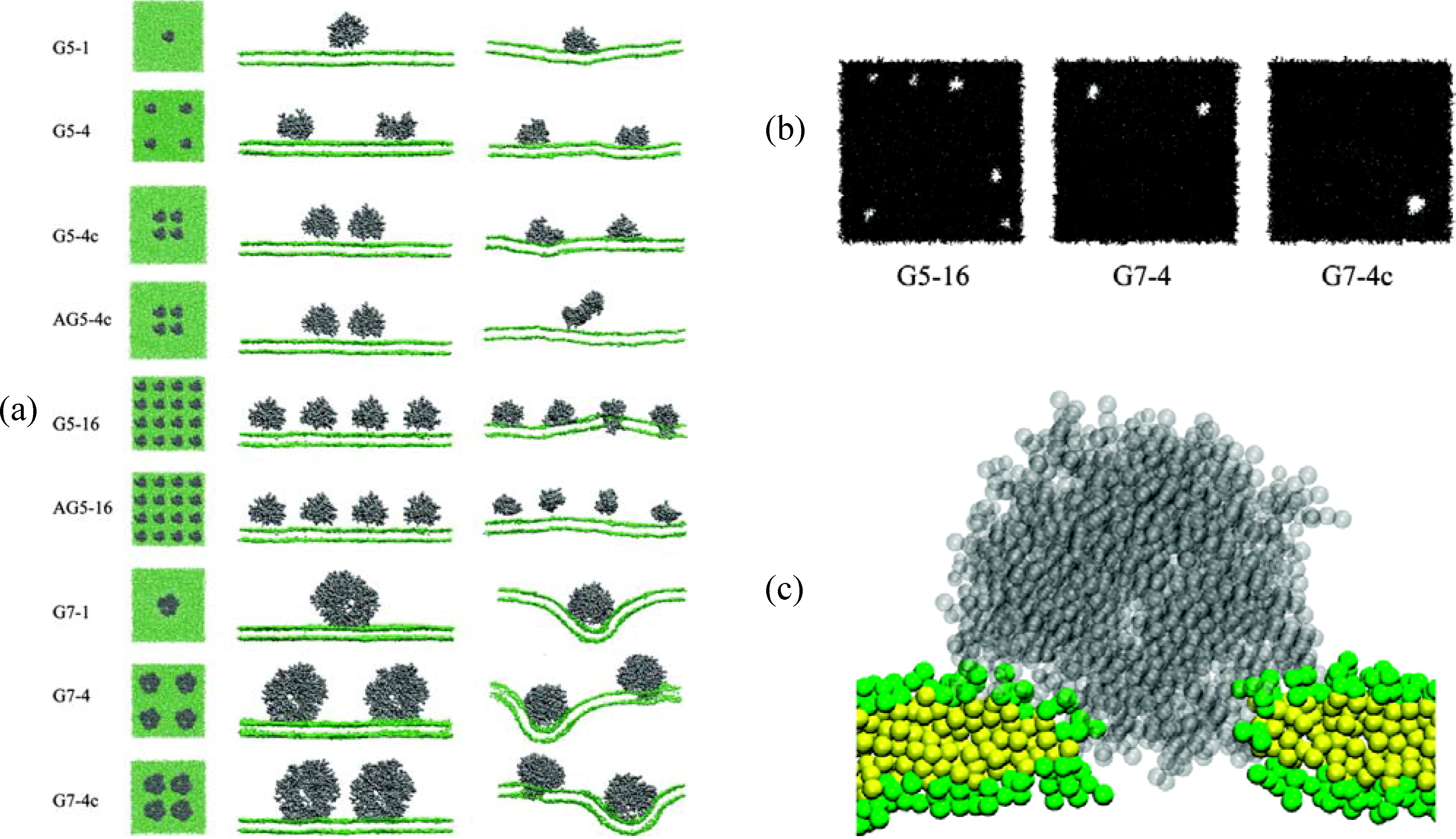{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Atomistic molecular dynamics simulations of dendrimers in explicit solvents
3. Simulations of dendrimers with lipid bilayers
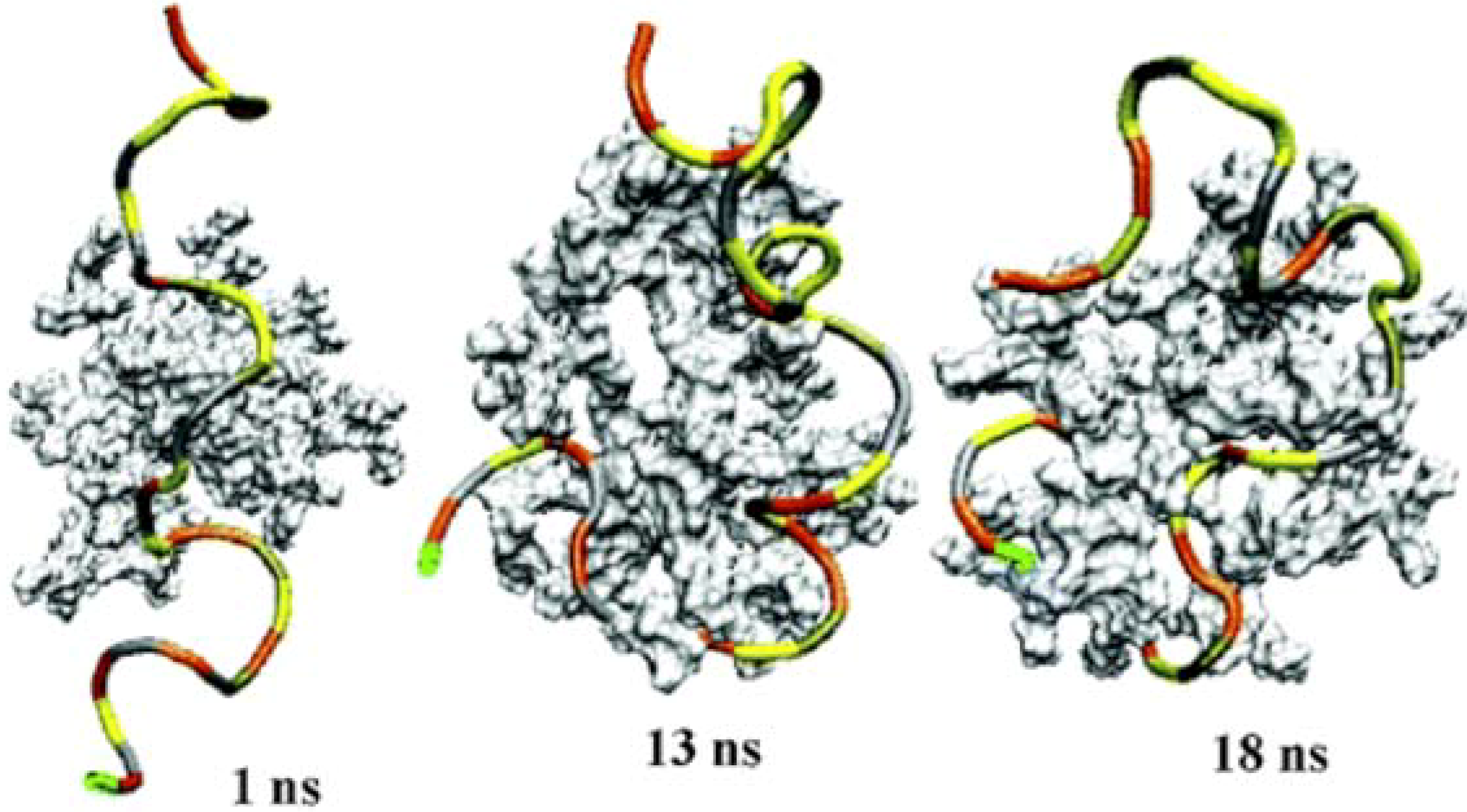
3.1 Atomistic molecular dynamics simulations

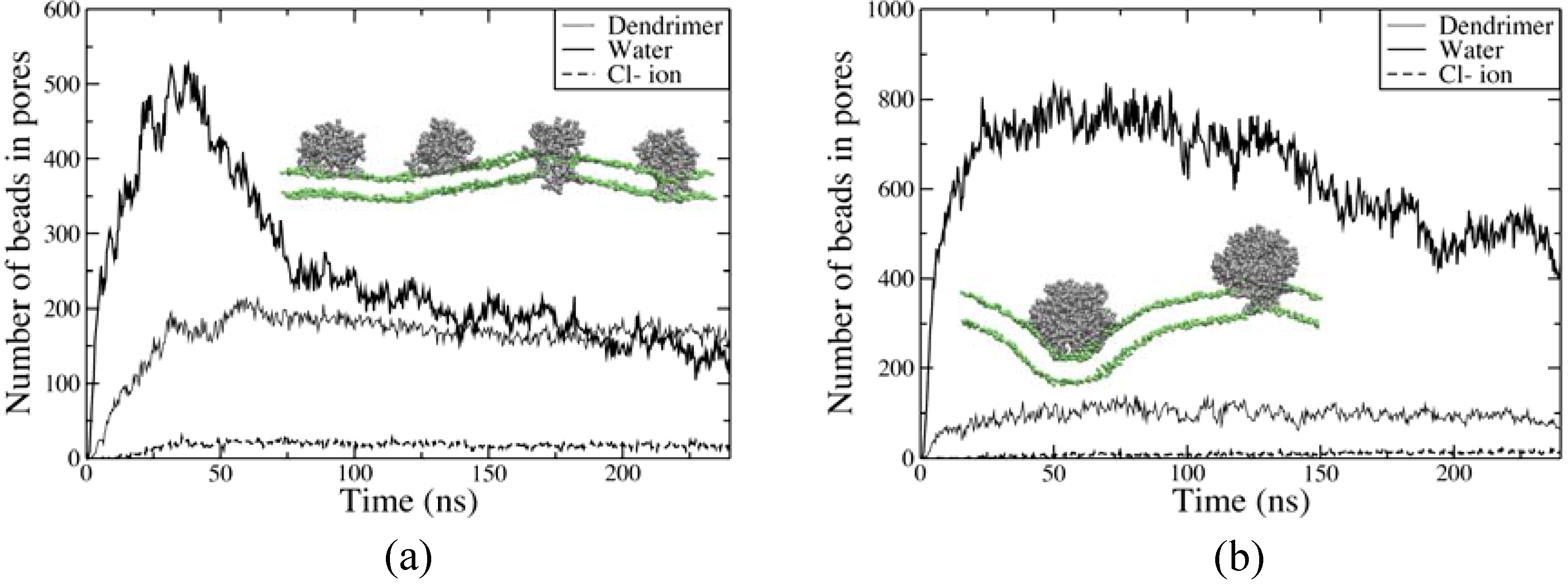
3.2 Coarse-grained molecular dynamics simulations


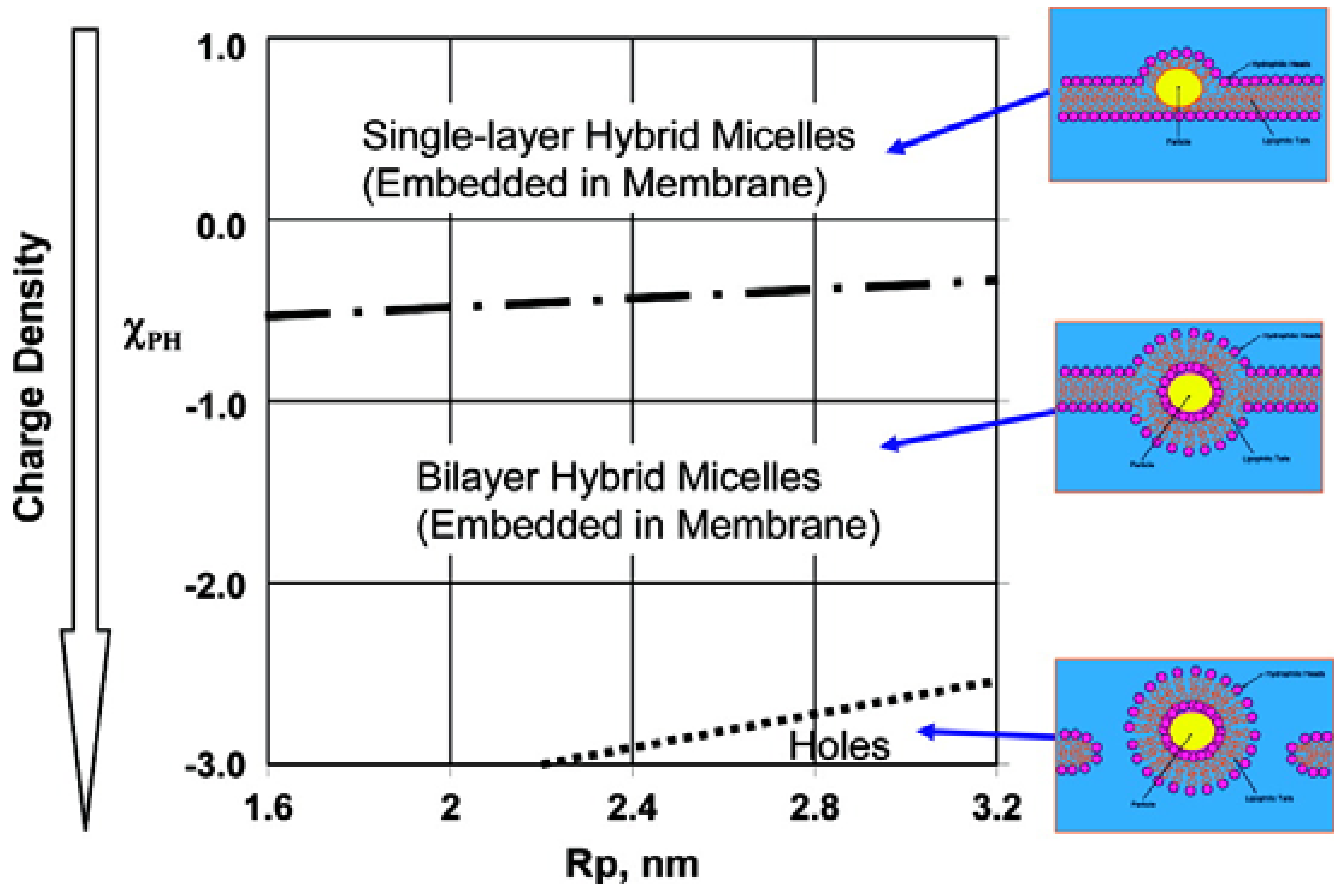
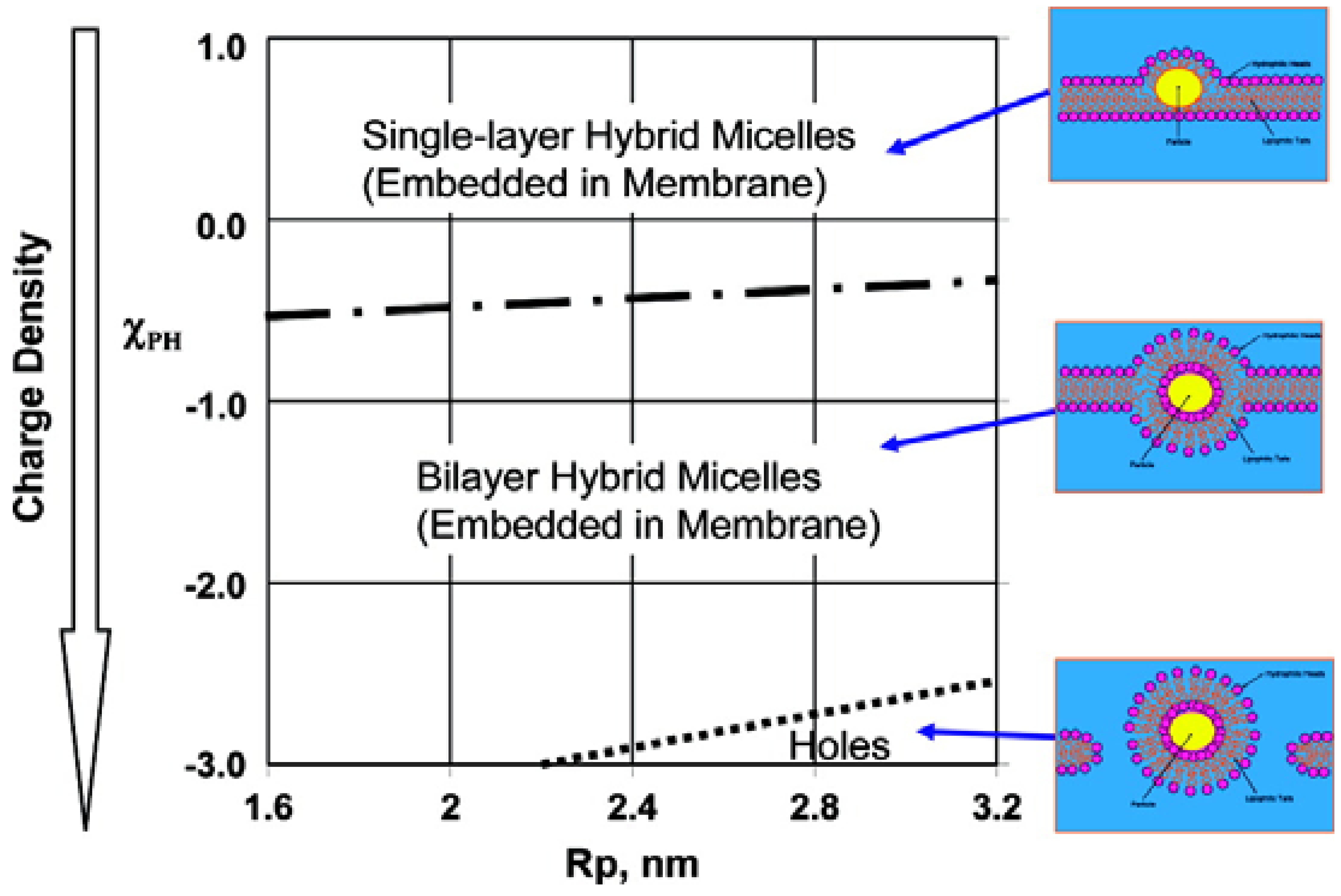
3.3 Mesoscale simulations
4. Simulations of dendrimers with polyelectrolytes
4.1 Monte Carlo and Brownian dynamics simulations
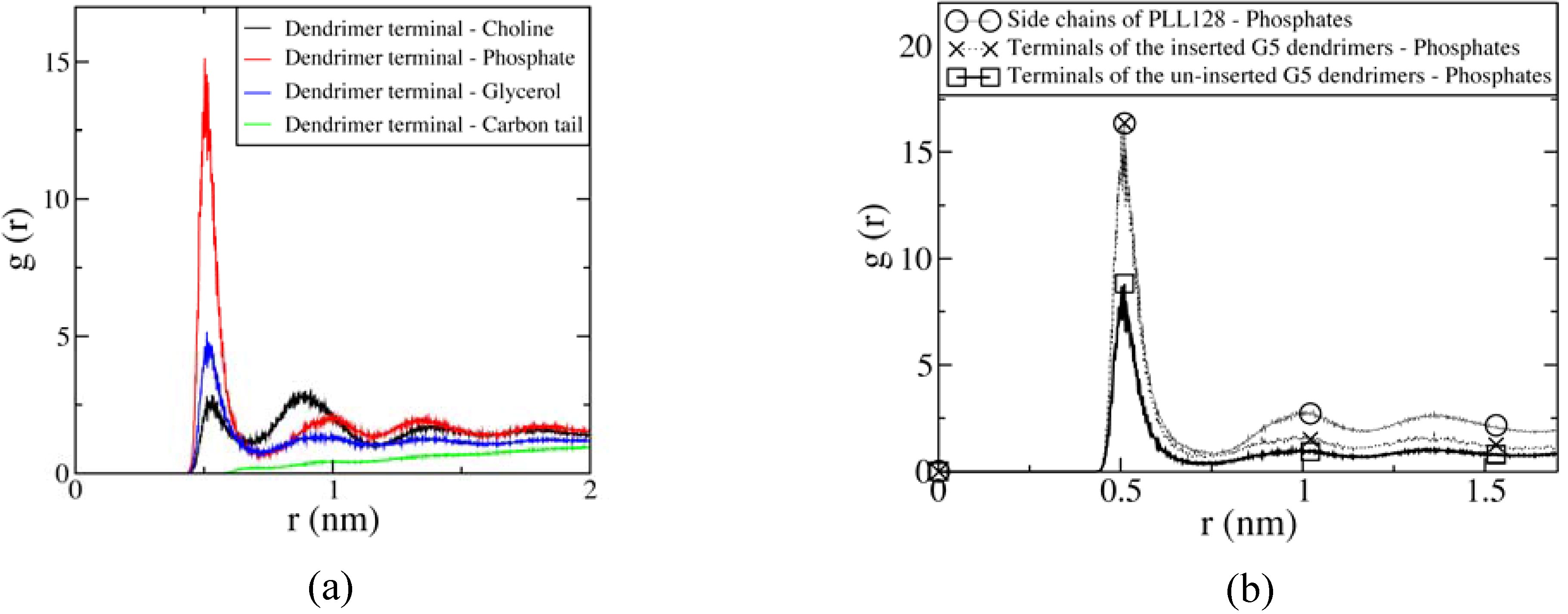
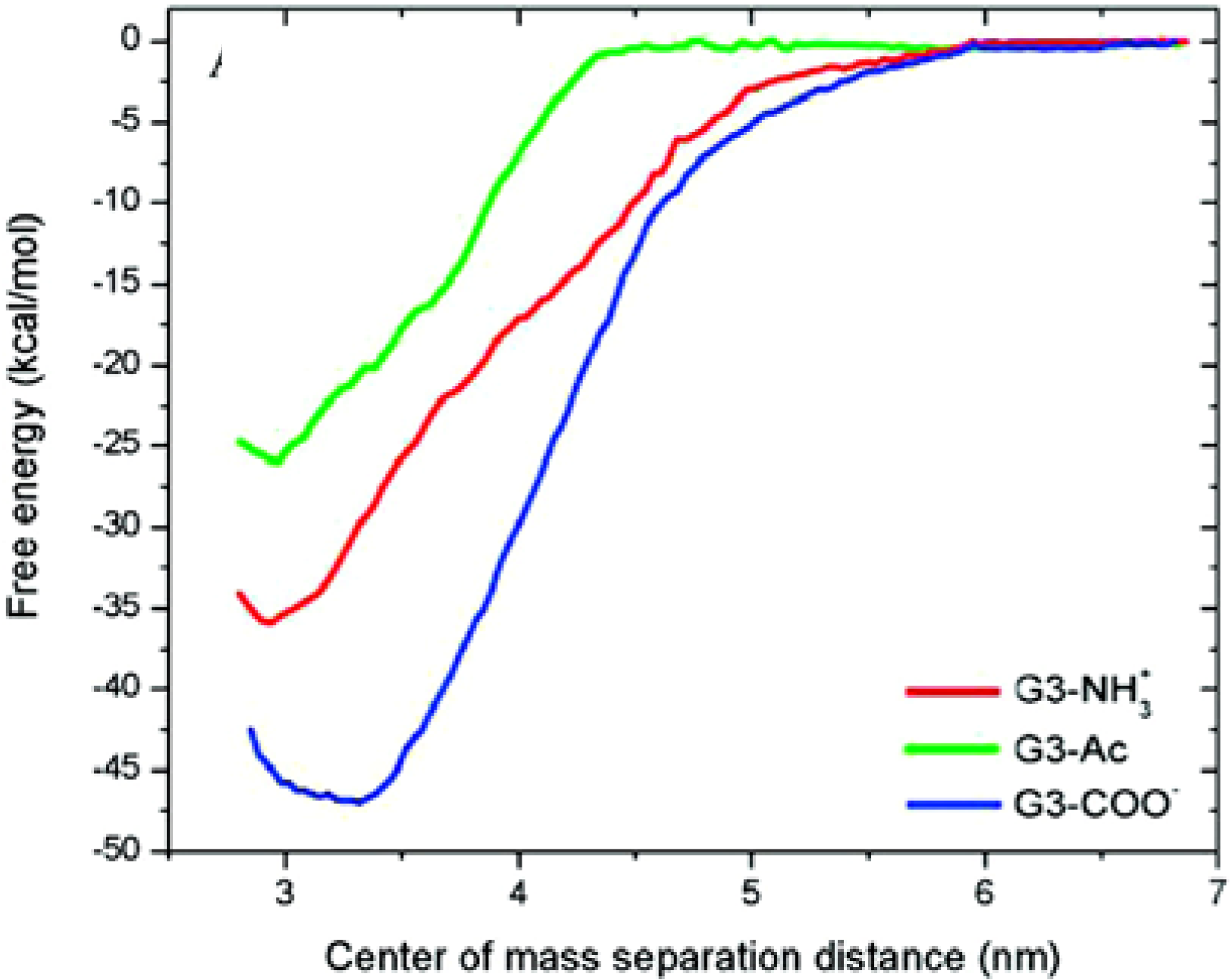
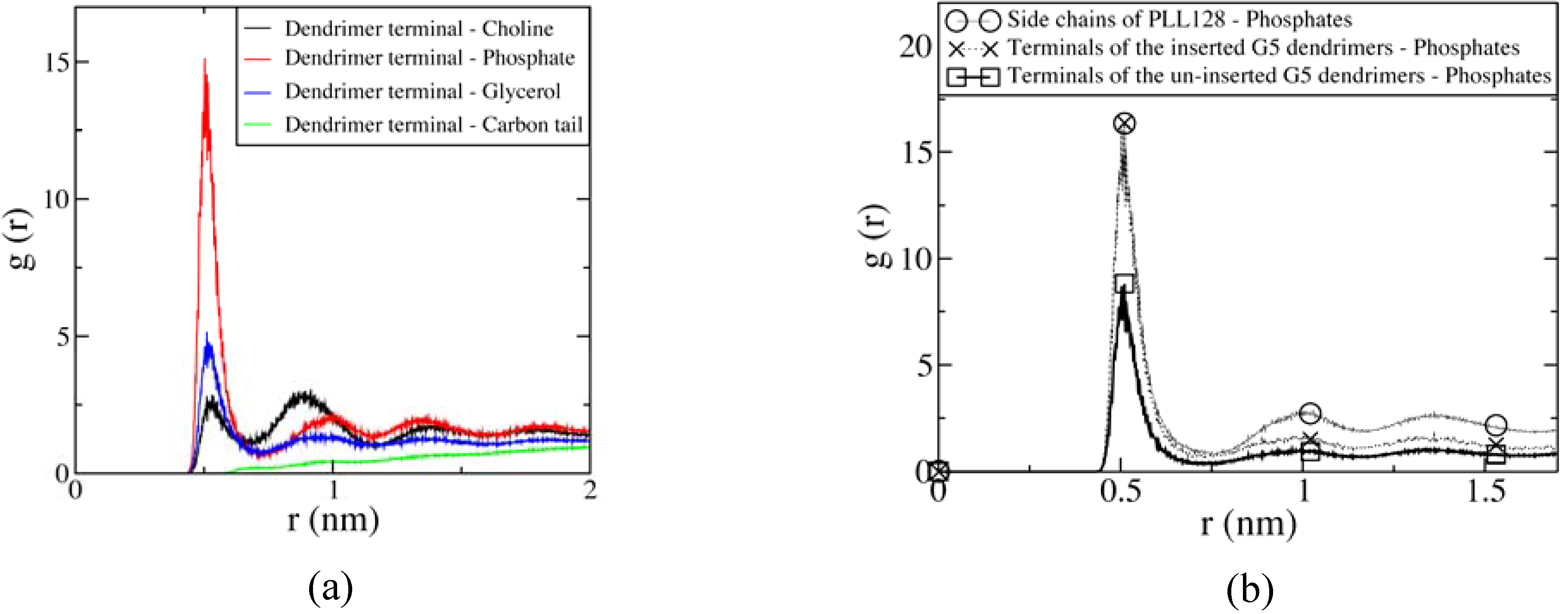
4.2 Atomistic and coarse-grained molecular dynamics simulations
5. Conclusions
Acknowledgments
References
- Malik, N.; Evagorou, E.G.; Duncan, R. Dendrimer-platinate: a novel approach to cancer chemotherapy. AntiCancer Drugs 1999, 10, 767–776. [Google Scholar] [CrossRef]
- Naylor, A.M.; Goddard, W.A.; Kiefer, G.E.; Tomalia, D.A. Starburst Dendrimers .5. Molecular Shape Control. J. Am. Chem. Soc. 1989, 111, 2339–2341. [Google Scholar] [CrossRef]
- Patri, A.K.; Kukowska-Latallo, J.F.; Baker, J.R. Targeted drug delivery with dendrimers: Comparison of the release kinetics of covalently conjugated drug and non-covalent drug inclusion complex. Adv. Drug Delivery Rev. 2005, 57, 2203–2214. [Google Scholar] [CrossRef]
- Patri, A.K.; Majoros, I.J.; Baker, J.R. Dendritic polymer macromolecular carriers for drug delivery. Curr. Opin. Chem. Biol. 2002, 6, 466–471. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Naylor, A.M.; Goddard, W.A. Starburst Dendrimers - Molecular-Level Control of Size, Shape, Surface-Chemistry, Topology, and Flexibility from Atoms to Macroscopic Matter. Angew. Chem. Int. Ed. 1990, 29, 138–175. [Google Scholar] [CrossRef]
- Choi, Y.; Thomas, T.; Kotlyar, A.; Islam, M.T.; Baker, J.R. Synthesis and functional evaluation of DNA-assembled polyamidoamine dendrimer clusters for cancer cell-specific targeting. Chem. Biol. 2005, 12, 35–43. [Google Scholar] [CrossRef]
- Choi, Y.S.; Mecke, A.; Orr, B.G.; Holl, M.M.B.; Baker, J.R. DNA-directed synthesis of generation 7 and 5 PAMAM dendrimer nanoclusters. Nano Lett. 2004, 4, 391–397. [Google Scholar] [CrossRef]
- Hong, S.P.; Bielinska, A.U.; Mecke, A.; Keszler, B.; Beals, J.L.; Shi, X.Y.; Balogh, L.; Orr, B.G.; Baker, J.R.; Holl, M.M.B. Interaction of poly(amidoamine) dendrimers with supported lipid bilayers and cells: Hole formation and the relation to transport. Bioconjugate Chem. 2004, 15, 774–782. [Google Scholar] [CrossRef]
- Hong, S.P.; Leroueil, P.R.; Janus, E.K.; Peters, J.L.; Kober, M.M.; Islam, M.; Orr, B.G.; Baker, J.R.; Holl, M.M.B. Interaction of polycationic polymers with supported lipid bilayers and cells: Nanoscale hole formation and enhanced membrane permeability. Bioconj. Chem. 2006, 17, 728–734. [Google Scholar] [CrossRef]
- Leroueil, P.R.; Berry, S.A.; Duthie, K.; Han, G.; Rotello, V.M.; McNerny, D.Q.; Baker, J.R.; Orr, B.G.; Holl, M.M.B. Wide varieties of cationic nanoparticles induce defects in supported lipid bilayers. Nano Lett. 2008, 8, 420–424. [Google Scholar] [CrossRef]
- Leroueil, P.R.; Hong, S.Y.; Mecke, A.; Baker, J.R.; Orr, B.G.; Holl, M.M.B. Nanoparticle interaction with biological membranes: Does nanotechnology present a janus face? Acc. Chem. Res. 2007, 40, 335–342. [Google Scholar] [CrossRef]
- Mecke, A.; Lee, I.; Baker, J.R.; Holl, M.M.B.; Orr, B.G. Deformability of poly(amidoamine) dendrimers. Eur. Phys. J. E 2004, 14, 7–16. [Google Scholar] [CrossRef]
- Mecke, A.; Majoros, I.J.; Patri, A.K.; Baker, J.R.; Holl, M.M.B.; Orr, B.G. Lipid bilayer disruption by polycationic polymers: The roles of size and chemical functional group. Langmuir 2005, 21, 10348–10354. [Google Scholar] [CrossRef]
- Mecke, A.; Uppuluri, S.; Sassanella, T.M.; Lee, D.K.; Ramamoorthy, A.; Baker, J.R.; Orr, B.G.; Holl, M.M.B. Direct observation of lipid bilayer disruption by poly(amidoamine) dendrimers. Chem. Phys. Lipids 2004, 132, 3–14. [Google Scholar] [CrossRef]
- Lescanec, R.L.; Muthukumar, M. Configurational Characteristics and scaling behavior of starburst molecules - a computational study. Macromolecules 1990, 23, 2280–2288. [Google Scholar] [CrossRef]
- Boris, D.; Rubinstein, M. A self-consistent mean field model of a starburst dendrimer: Dense core vs dense shell. Macromolecules 1996, 29, 7251–7260. [Google Scholar] [CrossRef]
- Ballauff, M.; Likos, C.N. Dendrimers in solution: Insight from theory and simulation. Angew. Chem., Int. Ed. 2004, 43, 2998–3020. [Google Scholar] [CrossRef]
- Kandasamy, S.K.; Lee, H.; Larson, R.G. Computer simulations of dendrimers. In Dendrimer-based nanomedicine; Majoros, I.J., Baker, J.R.J., Eds.; Pan Stanford Publishing: Singapore, 2008; pp. 331–354. [Google Scholar]
- Maiti, P.K.; Cagin, T.; Lin, S.T.; Goddard, W.A. Effect of solvent and pH on the structure of PAMAM dendrimers. Macromolecules 2005, 38, 979–991. [Google Scholar] [CrossRef]
- Lin, S.T.; Maiti, P.K.; Goddard, W.A. Dynamics and thermodynamics of water in PAMAM dendrimers at subnanosecond time scales. J. Phys. Chem. B 2005, 109, 8663–8672. [Google Scholar] [CrossRef]
- Maiti, P.K.; Goddard, W.A. Solvent quality changes the structure of G8 PAMAM dendrimer, a disagreement with some experimental interpretations. J. Phys. Chem. B 2006, 110, 25628–25632. [Google Scholar] [CrossRef]
- Nisato, G.; Ivkov, R.; Amis, E.J. Size invariance of polyelectrolyte dendrimers. Macromolecules 2000, 33, 4172–4176. [Google Scholar] [CrossRef]
- Han, M.; Chen, P.Q.; Yang, X.Z. Molecular dynamics simulation of PAMAM dendrimer in aqueous solution. Polymer 2005, 46, 3481–3488. [Google Scholar] [CrossRef]
- Lee, H.; Baker, J.R.; Larson, R.G. Molecular dynamics studies of the size, shape, and internal structure of 0% and 90% acetylated fifth-generation polyamidoamine dendrimers in water and methanol. J. Phys. Chem. B 2006, 110, 4014–4019. [Google Scholar] [CrossRef]
- Maiti, P.K.; Messina, R. Counterion distribution and zeta-potential in PAMAM dendrimer. Macromolecules 2008, 41, 5002–5006. [Google Scholar] [CrossRef]
- Kelly, C.V.; Leroueil, P.R.; Nett, E.K.; Wereszczynski, J.M.; Baker, J.R.; Orr, B.G.; Holl, M.M.B.; Andricioaei, I. Poly(amidoamine) dendrimers on lipid bilayers I: Free energy and conformation of binding. J. Phys. Chem. B 2008, 112, 9337–9345. [Google Scholar] [CrossRef]
- Kelly, C.V.; Leroueil, P.R.; Orr, B.G.; Holl, M.M.B.; Andricioaei, I. Poly(amidoamine) dendrimers on lipid bilayers II: Effects of bilayer phase and dendrimer termination. J. Phys. Chem. B 2008, 112, 9346–9353. [Google Scholar] [CrossRef]
- Lee, H.; Larson, R.G. Molecular dynamics simulations of PAMAM dendrimer-induced pore formation in DPPC bilayers with a coarse-grained model. J. Phys. Chem. B 2006, 110, 18204–18211. [Google Scholar] [CrossRef]
- Marrink, S.J.; de Vries, A.H.; Mark, A.E. Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. B 2004, 108, 750–760. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef]
- Lee, H.; Larson, R.G. Coarse-grained molecular dynamics studies of the concentration and size dependence of fifth- and seventh-generation PAMAM dendrimers on pore formation in DMPC bilayer. J. Phys. Chem. B 2008, 112, 7778–7784. [Google Scholar] [CrossRef]
- Lee, H.; Larson, R.G. Lipid bilayer curvature and pore formation induced by charged linear polymers and dendrimers: The effect of molecular shape. J. Phys. Chem. B 2008, 112, 12279–12285. [Google Scholar] [CrossRef]
- Ginzburg, V.V.; Balijepailli, S. Modeling the thermodynamics of the interaction of nanoparticles with cell membranes. Nano Lett. 2007, 7, 3716–3722. [Google Scholar] [CrossRef]
- Smith, K.A.; Jasnow, D.; Balazs, A.C. Designing synthetic vesicles that engulf nanoscopic particles. J. Chem. Phys. 2007, 127, 10. [Google Scholar]
- Kabanov, V.A.; Sergeyev, V.G.; Pyshkina, O.A.; Zinchenko, A.A.; Zezin, A.B.; Joosten, J.G.H.; Brackman, J.; Yoshikawa, K. Interpolyelectrolyte complexes formed by DNA and astramol poly(propylene imine) dendrimers. Macromolecules 2000, 33, 9587–9593. [Google Scholar] [CrossRef]
- Kabanov, V.A.; Zezin, A.B.; Rogacheva, V.B.; Gulyaeva, Z.G.; Zansochova, M.F.; Joosten, J.G.H.; Brackman, J. Interaction of Astramol poly(propyleneimine) dendrimers with linear polyanions. Macromolecules 1999, 32, 1904–1909. [Google Scholar] [CrossRef]
- Gurtovenko, A.A.; Lyulin, S.V.; Karttunen, M.; Vattulainen, I. Molecular dynamics study of charged dendrimers in salt-free solution: Effect of counterions. J. Chem. Phys. 2006, 124, 8. [Google Scholar]
- Welch, P.; Muthukumar, M. Dendrimer-polyelectrolyte complexation: A model guest-host system. Macromolecules 2000, 33, 6159–6167. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Darinskii, A.A.; Lyulin, A.V. Computer simulation of complexes of dendrimers with linear polyelectrolytes. Macromolecules 2005, 38, 3990–3998. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Darinskii, A.A.; Lyulin, A.V. Energetic and conformational aspects of dendrimer overcharging by linear polyelectrolytes. Phys. Rev. E 2008, 78, 9. [Google Scholar]
- Maiti, P.K.; Bagchi, B. Structure and dynamics of DNA-dendrimer complexation: Role of counterions, water, and base pair sequence. Nano Lett. 2006, 6, 2478–2485. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Vattulainen, I.; Gurtovenko, A.A. Complexes comprised of charged dendrimers, linear polyelectrolytes, and counterions: Insight through coarse-grained molecular dynamics Simulations. Macromolecules 2008, 41, 4961–4968. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, H.; Larson, R.G. Multiscale Modeling of Dendrimers and Their Interactions with Bilayers and Polyelectrolytes. Molecules 2009, 14, 423-438. https://doi.org/10.3390/molecules14010423
Lee H, Larson RG. Multiscale Modeling of Dendrimers and Their Interactions with Bilayers and Polyelectrolytes. Molecules. 2009; 14(1):423-438. https://doi.org/10.3390/molecules14010423
Chicago/Turabian StyleLee, Hwankyu, and Ronald G. Larson. 2009. "Multiscale Modeling of Dendrimers and Their Interactions with Bilayers and Polyelectrolytes" Molecules 14, no. 1: 423-438. https://doi.org/10.3390/molecules14010423