Diastereoselective Spiroannulation of Phenolic Substrates: Advances Towards the Asymmetric Formation of the Manumycin m-C7N Core Skeleton

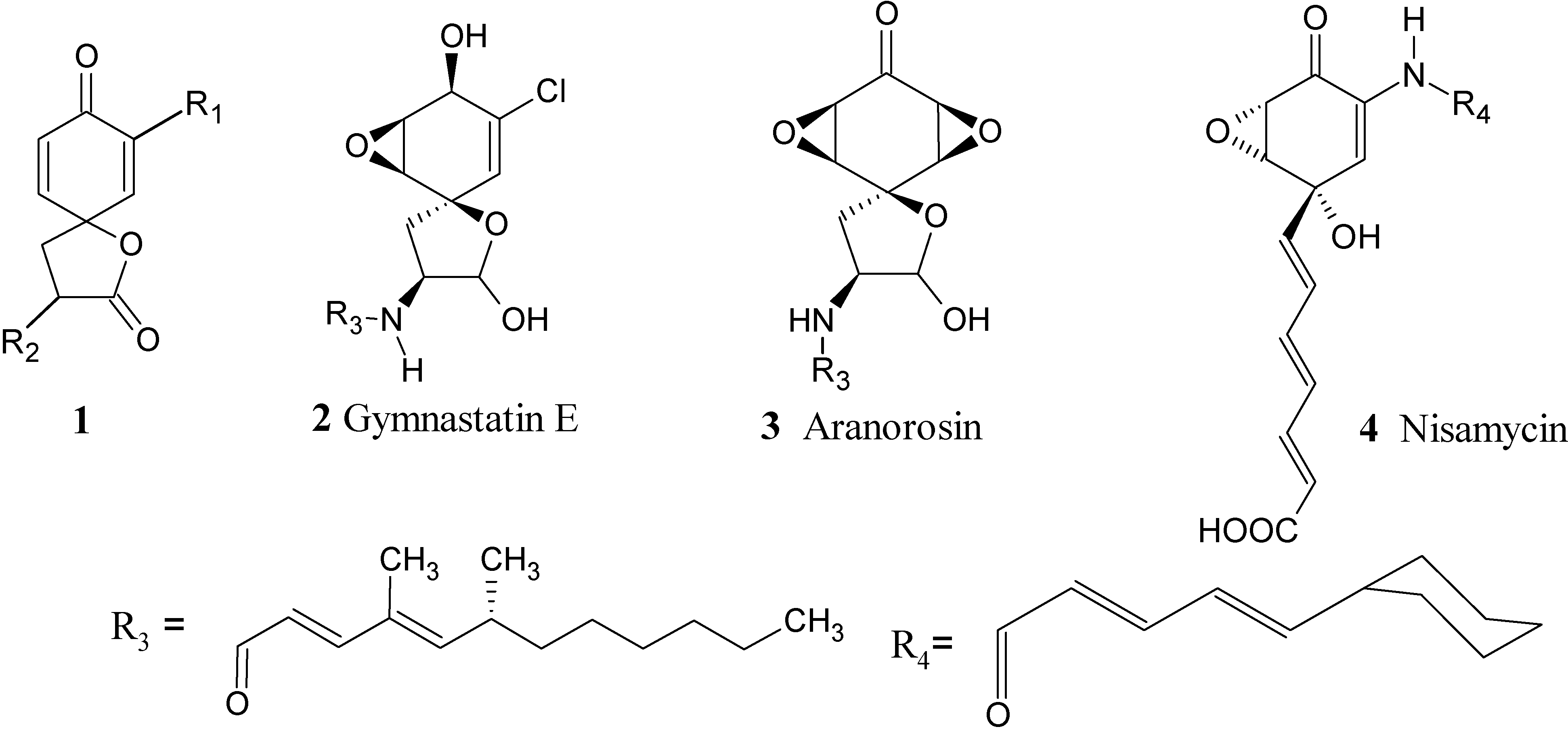
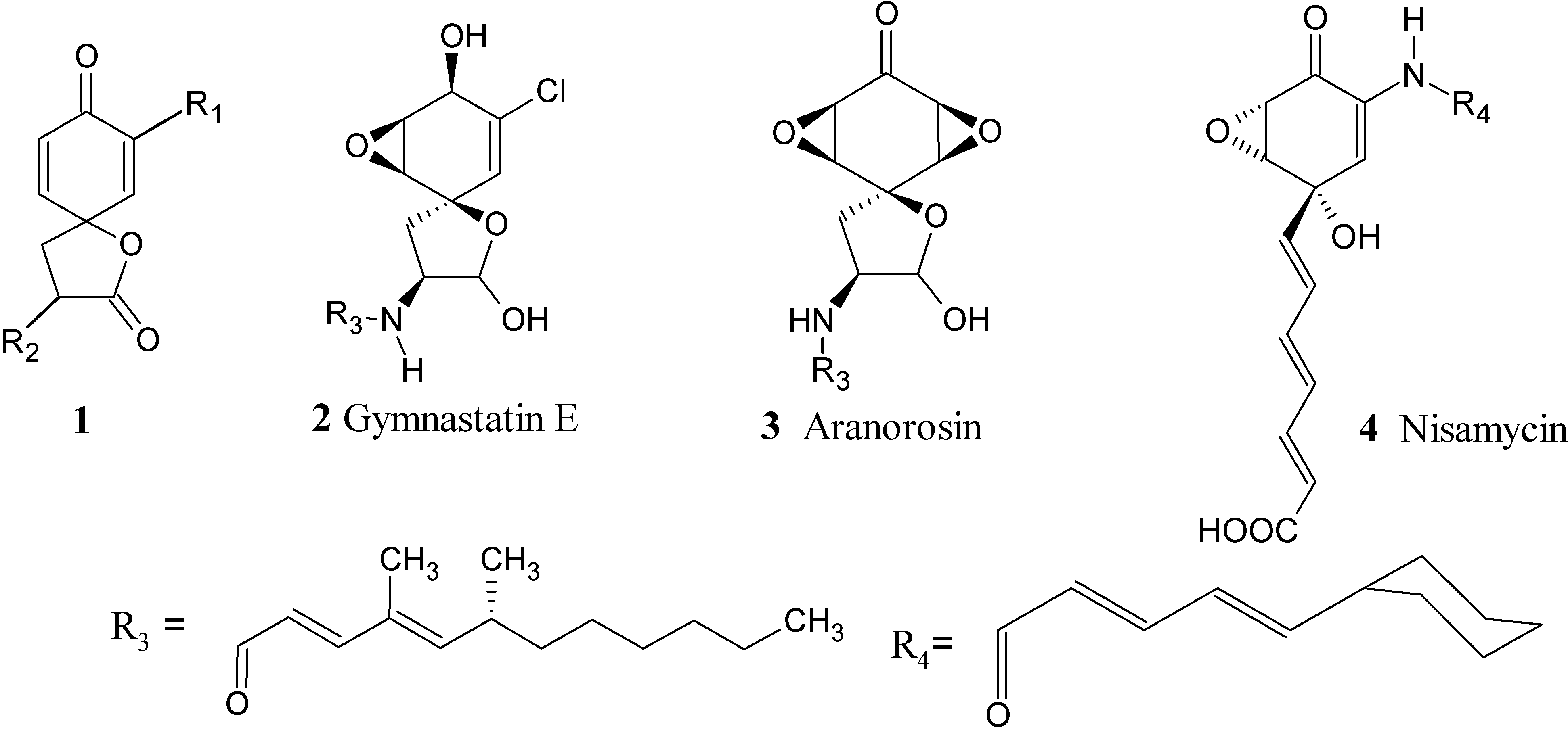{kind=link}
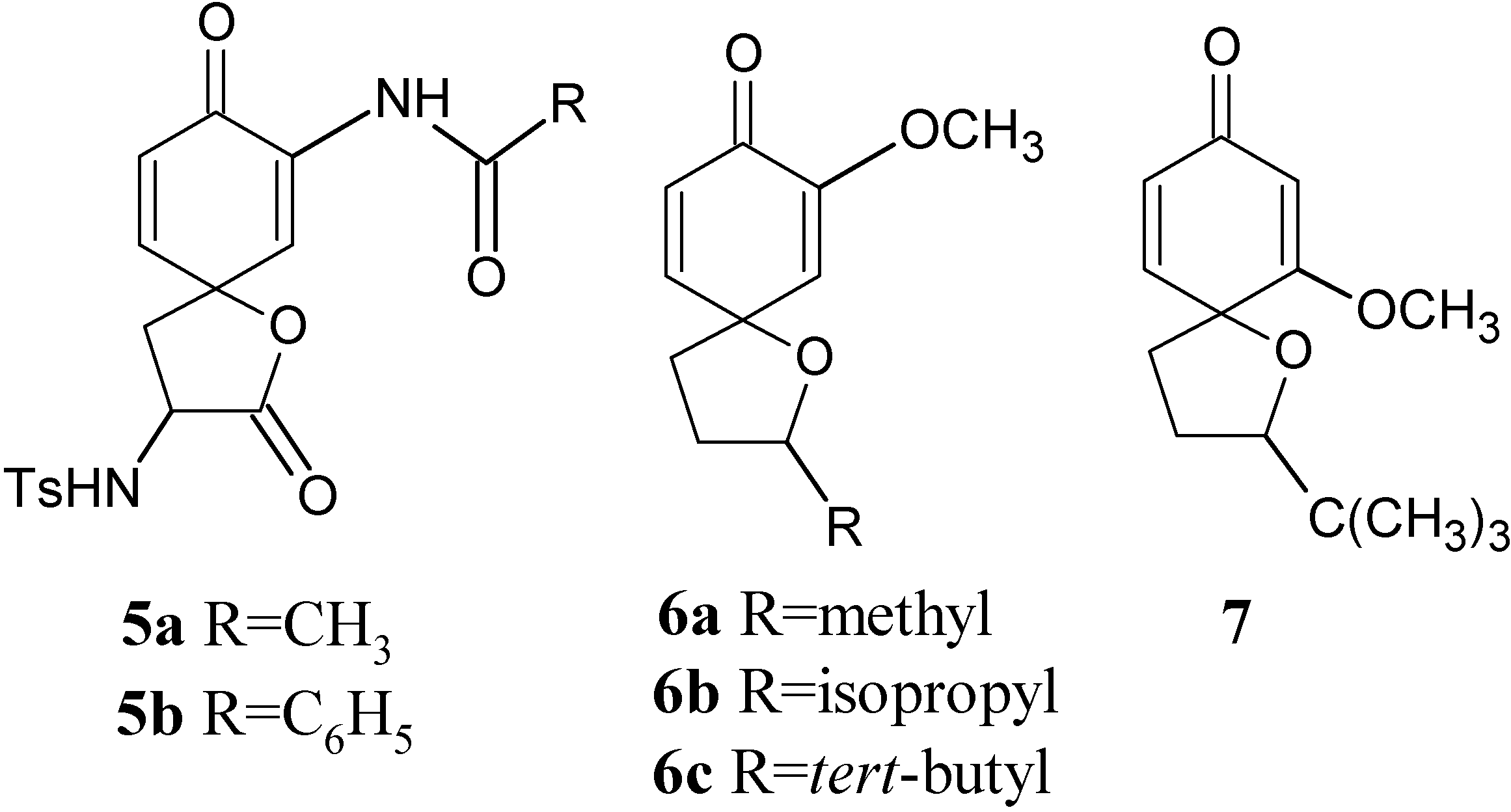{kind=link}
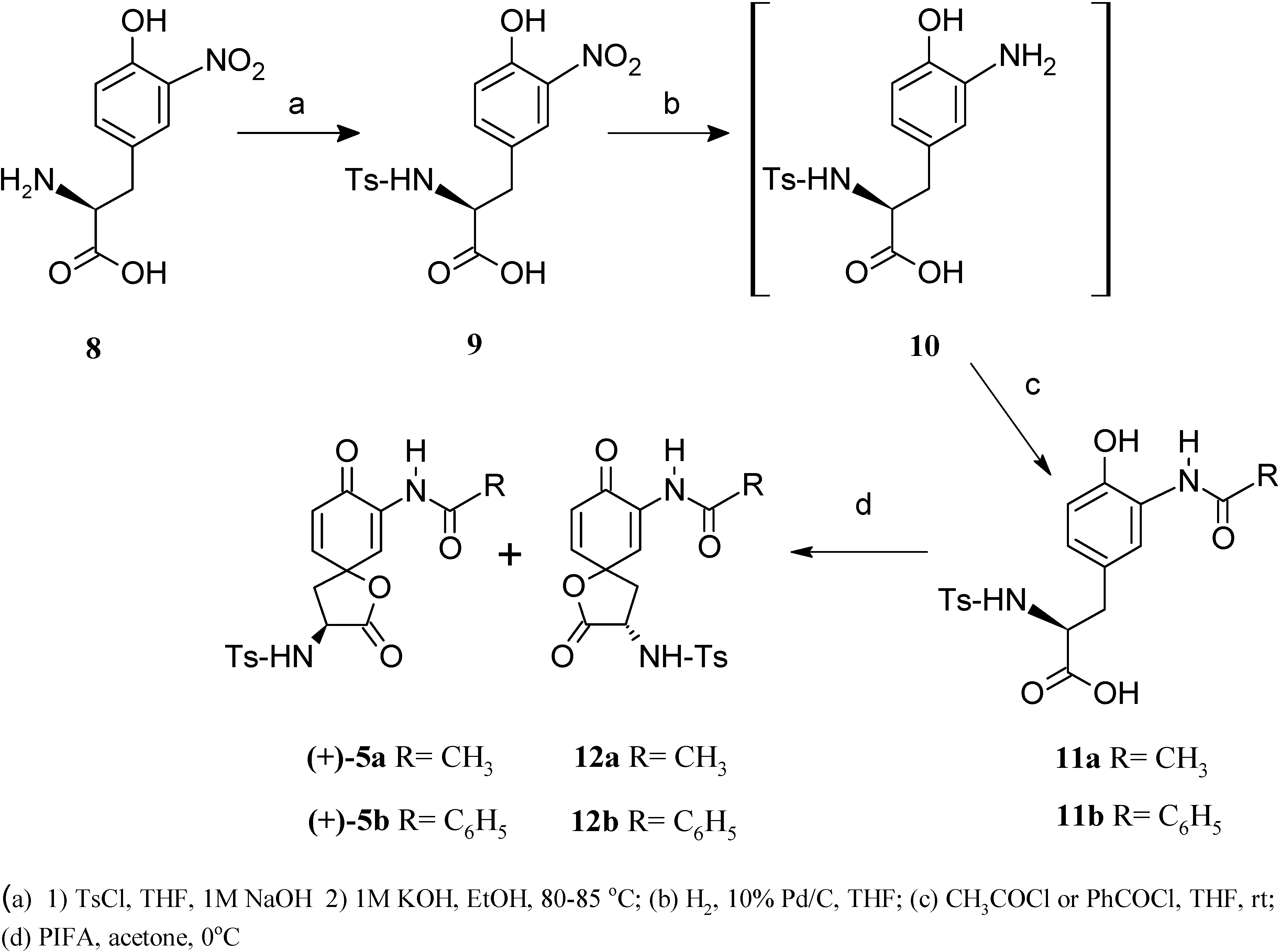{kind=link}
Abstract
:Introduction
Results and Discussion
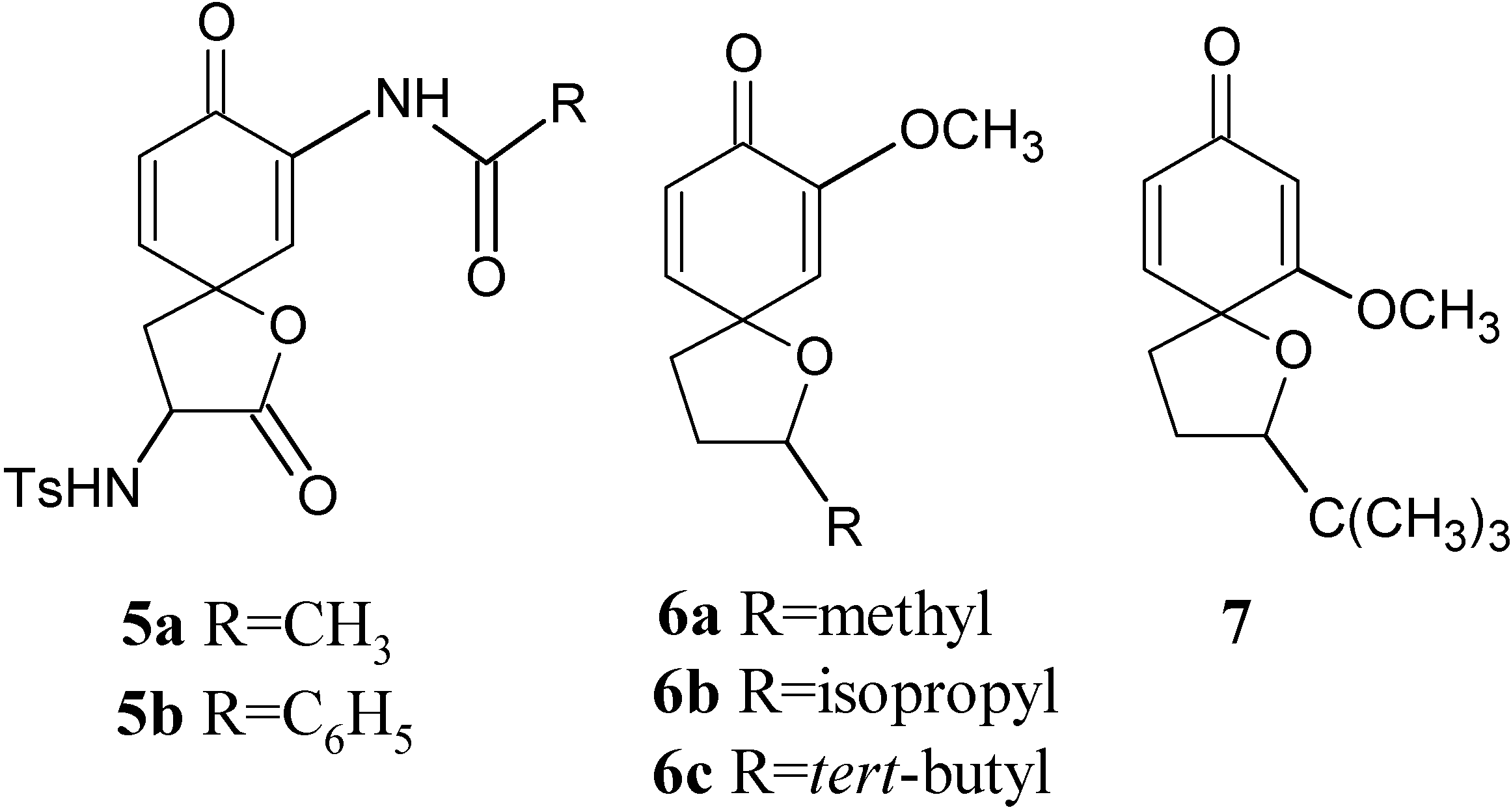
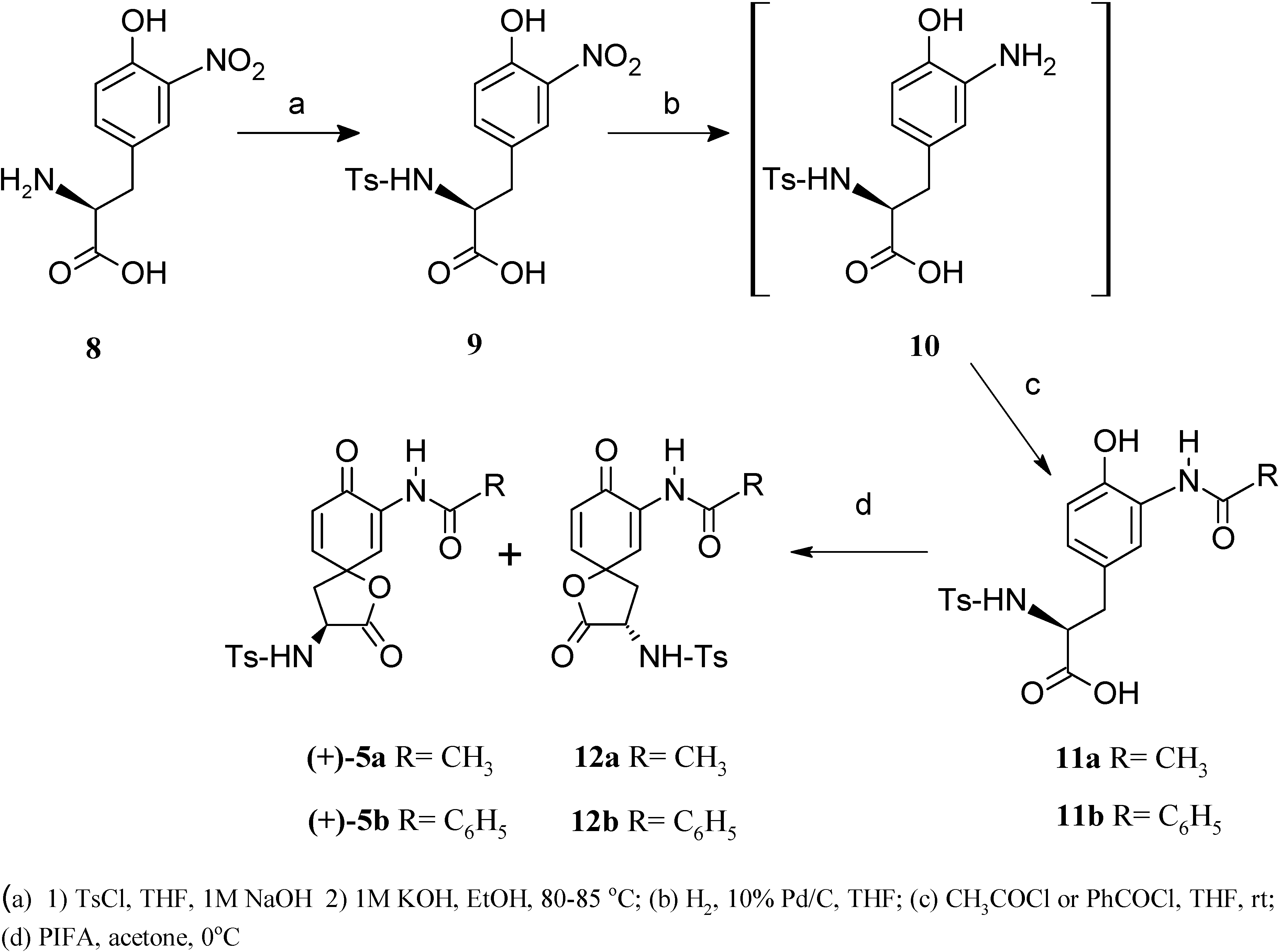
Conclusions
Acknowledgments
Experimental
General
(2S)-3-(4-Hydroxy-3-nitrophenyl)-2-{[(4-methylphenyl)sulfonyl]amino} propanoic acid [(-)-9]
(2S)-3-(3-Amino-4-hydroxylphenyl)-2-{[(4-methylphenyl)sulfonyl]amino} propanoic acid [(+)-10]
(2S)-3-[3(Acetylamino)-4-hydroxyphenyl]-2-{[(4-methylphenyl)sulfonyl]amino} propanoic acid [(-)-11a]
(2S)-3-[3(Benzoylamino)-4-hydroxyphenyl]-2-{[(4-methylphenyl)sulfonyl]amino} propanoic acid [(-)-11b]
N-((3S)-3-{[(4-Methylphenyl)sulfonyl]amino}-1-oxaspiro[4.5]deca-6,9-dion-2,8-dien-7-yl) acetamide [(+)-5a]
N-((3S)-3-{[(4-Methylphenyl)sulfonyl]amino}-1-oxaspiro[4.5]deca-6,9-dion-2,8-dien-7-yl) benzamide [(+)-5b]
References
- Plourde, G.L. Studies Towards the Diastereoselective Spiroannulation of Phenolic Derivatives. Tetrahedron Lett. 2002, 43, 3597–3599. [Google Scholar] [CrossRef]
- Plourde, G.L. Molbank 2003, M315–M322. [CrossRef]
- Plourde, G.L.; Fisher, B.B. Synthesis of 6-Methoxy-1-oxaspiro[4,5]deca-6,9-diene-8-one. Molecules 2002, 7, 315–319. [Google Scholar] [CrossRef]
- Plourde, G.L.; English, N.J. Diastereoselective Spiroannulation of Phenolic Substrates: Synthesis of (±)-2-tert-Butyl-6-methoxy-1-oxaspiro[4,5]deca-6,9-diene-8-one. Molecules 2005, 10, 1335–1339. [Google Scholar] [CrossRef]
- MacDonald, G.; Alcaraz, L.; Lewis, N.J.; Taylor, R.J.K. Asymmetric Synthesis of the mC7N Core of the Manumycin Family: Preparation of (+)-MT35214 and a Formal Total Synthesis of (-)-Alisamycin. Tetrahedron Lett. 1998, 39, 5433–5436. [Google Scholar] [CrossRef]
- Alcaraz, L.; Macdonald, G.; Kapfer, I.; Lewis, N.J.; Taylor, R.J.K. The First Total Synthesis of a Member of the Manumycin Family of Antibiotics: Alisamycin. Tetrahedron Lett. 1996, 37, 6619–6622. [Google Scholar] [CrossRef]
- Alcaraz, L.; Taylor, R.J.K. The First Synthesis of the Streptomyces Derived Antibiotic U-62162. Chem. Comm. 1998, 1157–1158. [Google Scholar] [CrossRef]
- Taylor, R.J.K.; Alcaraz, L.; Kapfer-Eyer, I.; Macdonald, G.; Wei, X.; Lewis, N. The Synthesis of Alisamycin, Nisamycin, LL-C10037α and Novel Epoxyquinone Analogues of Manumycin A. Synthesis 1998, 775–790. [Google Scholar]
- Wei, X.; Cronje Grove, J.J.; Taylor, R.J.K. The First Total Synthesis of (±)-Colabomycin D. J. Chem. Soc. Perkin Trans. 1 1999, 1143–1145. [Google Scholar]
- Wipf, P.; Coish, P.D.G. Total Synthesis of (±)-Nisamycin. J. Org. Chem. 1999, 64, 5053–5061. [Google Scholar] [CrossRef]
- Alcaraz, L.; Macdonald, G.; Ragot, J.P.; Lewis, N.; Taylor, R.J.K. Manumycin A: Synthesis of the (+)-Enantiomer and Revision of Stereochemical Assignment. J. Org. Chem. 1998, 63, 3526–3527. [Google Scholar] [CrossRef]
- Alcaraz, L.; Macdonald, G.; Ragot, J.; Lewis, N.J.; Taylor, R.J.K. Synthetic Approaches to the Manumycin A, B and C Antibiotics: The First Total Synthesis of (+)-Manumycin A. Tetrahedron 1999, 55, 3707–3716. [Google Scholar] [CrossRef]
- Cronje Grove, J.J.; Wei, X.; Taylor, R.J.K. The First Total Synthesis of a Type I Manumycin Antibiotic, (+)-TMC-1A: The Total Synthesis of (-)-LL-C10037β and (+)-Manumycin B. Chem. Comm. 1999, 421–422. [Google Scholar] [CrossRef]
- Wipf, P.; Kim, Y. Stereoselective Synthesis of the Functionalized Spirocyclic Core of Aranorosin. J. Org. Chem. 1993, 58, 1649–1650. [Google Scholar] [CrossRef]
- Wipf, P.; Kim, Y.; Fritch, P.C. Total Synthesis and Structure Assignment of the Antitumor Antibiotic Aranorosin. J. Org. Chem. 1993, 58, 7195–7203. [Google Scholar] [CrossRef]
- McKillop, A.; McLaren, L.; Taylor, R.J.K.; Watson, R.J.; Lewis, N.J. The Total Synthesis of the Diepoxycyclohexanone Antibiotic Aranorosin and Novel Synthetic Analogues. J. Chem. Soc. Perkin Trans. 1 1996, 1385–1393. [Google Scholar]
- Phoon, C.W.; Somanadhan, B.; Heng, S.C.H.; Ngo, A.; Ng, S.B.; Butler, M.S.; Buss, A.D.; Sim, M.M. Isolation and Total Synthesis of Gymnastatin N, a POLO-like Kinase 1 Active Constituent from the Fungus Arachniotus Punctatus. Tetrahedron 2004, 60, 11619–11628. [Google Scholar] [CrossRef]
- Sample availability: Available from the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Plourde, G.L.; Spaetzel, R.R.; Kwasnitza, J.S.; Scully, T.W. Diastereoselective Spiroannulation of Phenolic Substrates: Advances Towards the Asymmetric Formation of the Manumycin m-C7N Core Skeleton. Molecules 2007, 12, 2215-2222. https://doi.org/10.3390/12092215
Plourde GL, Spaetzel RR, Kwasnitza JS, Scully TW. Diastereoselective Spiroannulation of Phenolic Substrates: Advances Towards the Asymmetric Formation of the Manumycin m-C7N Core Skeleton. Molecules. 2007; 12(9):2215-2222. https://doi.org/10.3390/12092215
Chicago/Turabian StylePlourde, Guy L., Randy R. Spaetzel, Jolene S. Kwasnitza, and Thomas W. Scully. 2007. "Diastereoselective Spiroannulation of Phenolic Substrates: Advances Towards the Asymmetric Formation of the Manumycin m-C7N Core Skeleton" Molecules 12, no. 9: 2215-2222. https://doi.org/10.3390/12092215