Synthesis, Molecular Structure and Characterization of Allylic Derivatives of 6-Amino-3-methyl-1,2,4-triazolo[3,4-f][1,2,4]-triazin-8(7H)-one

Introduction

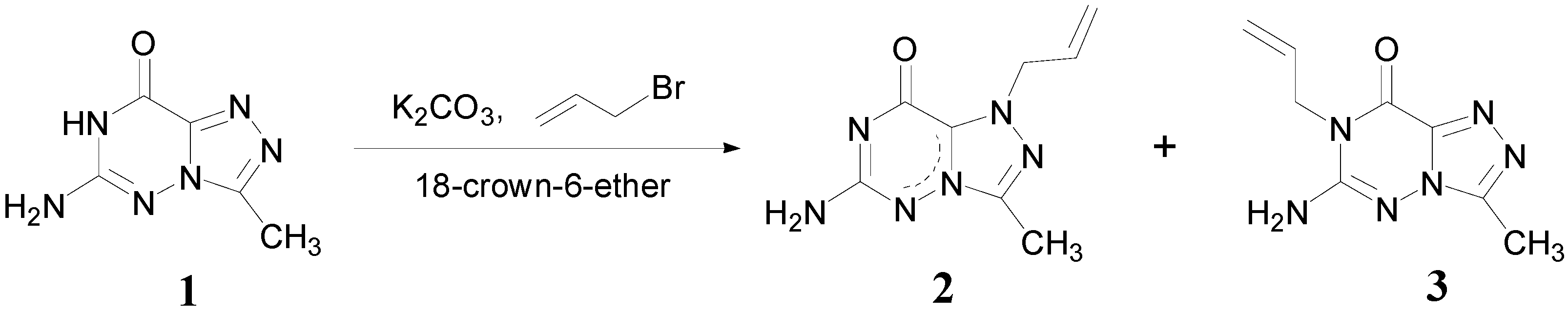
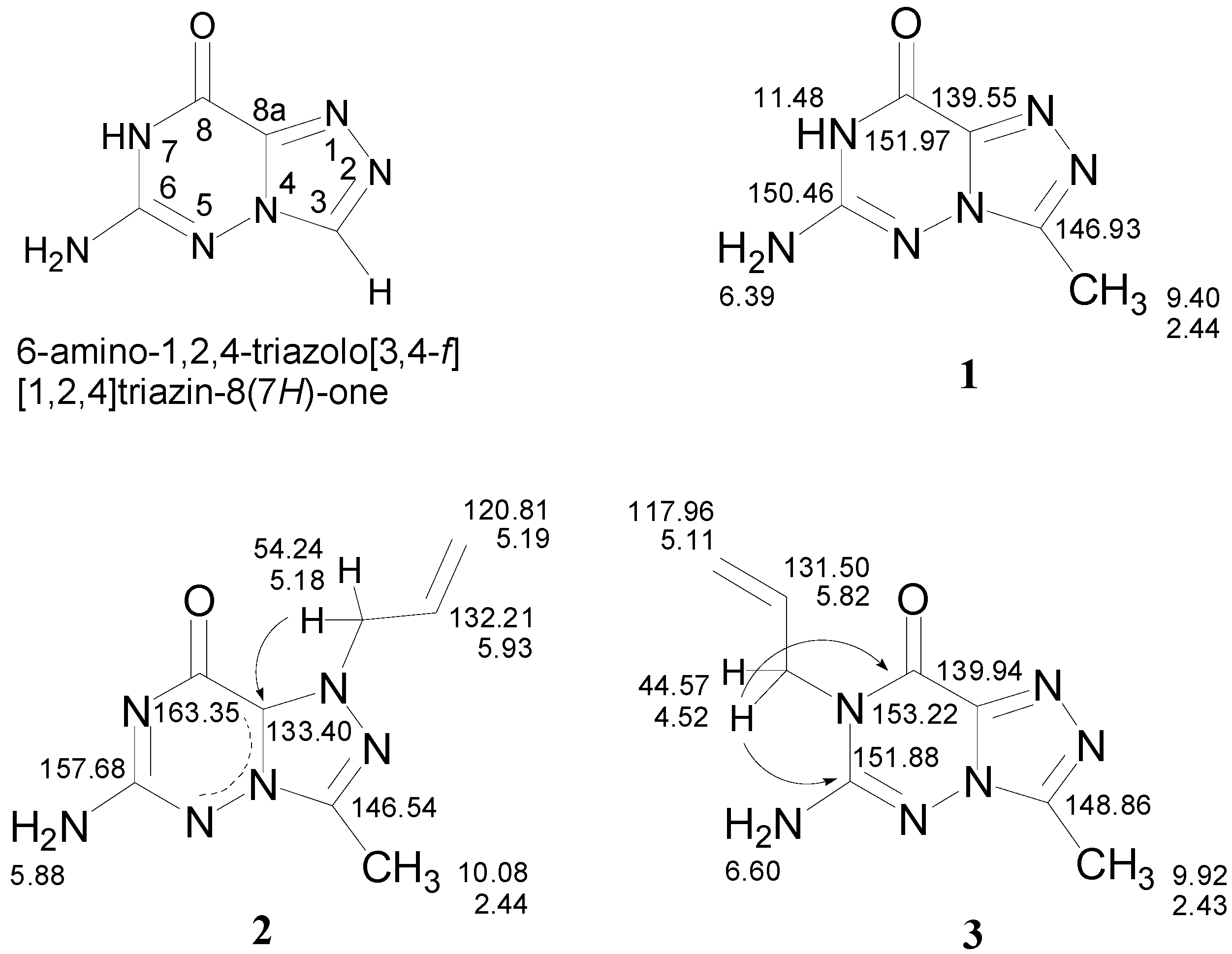
Results and Discussion

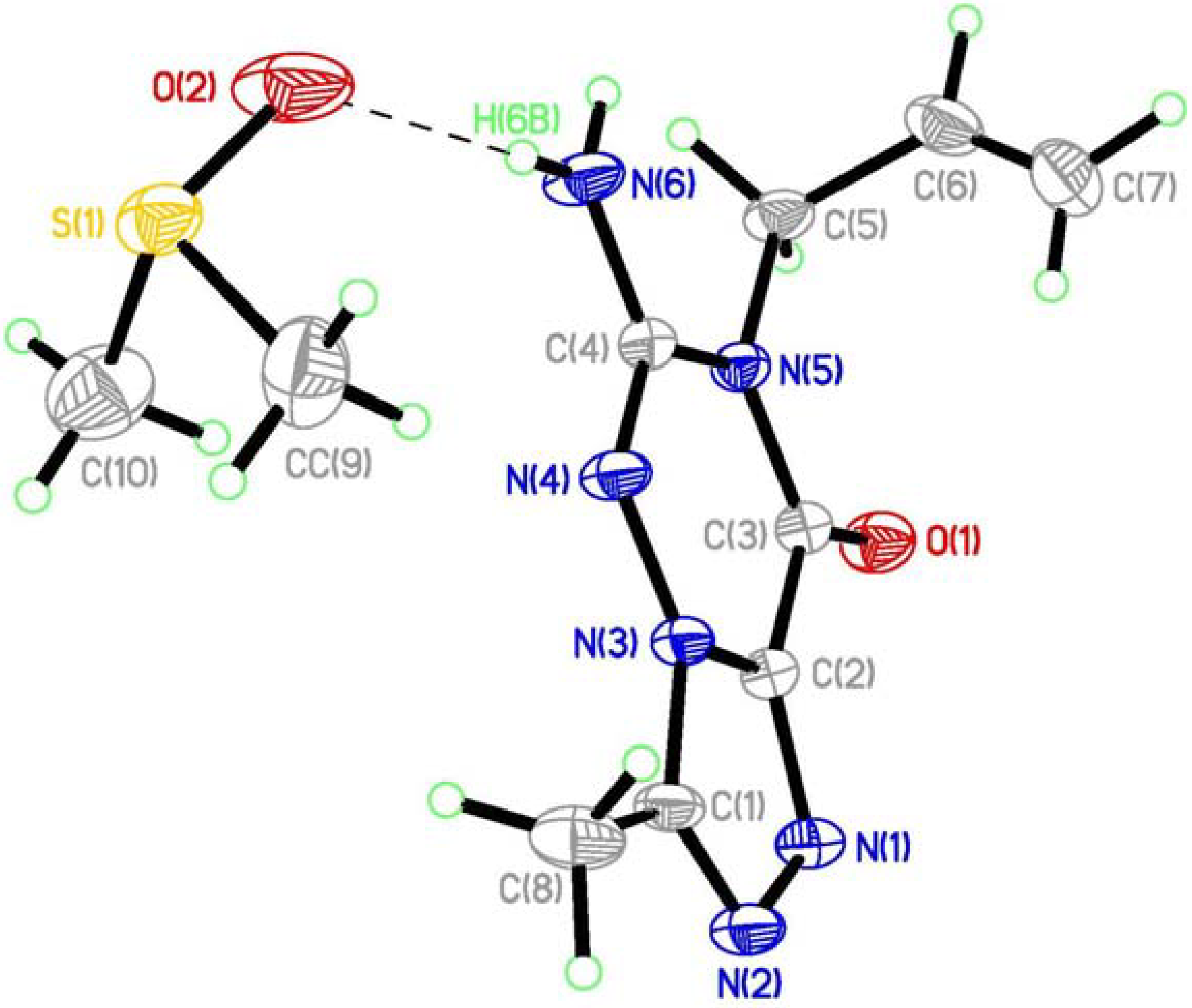
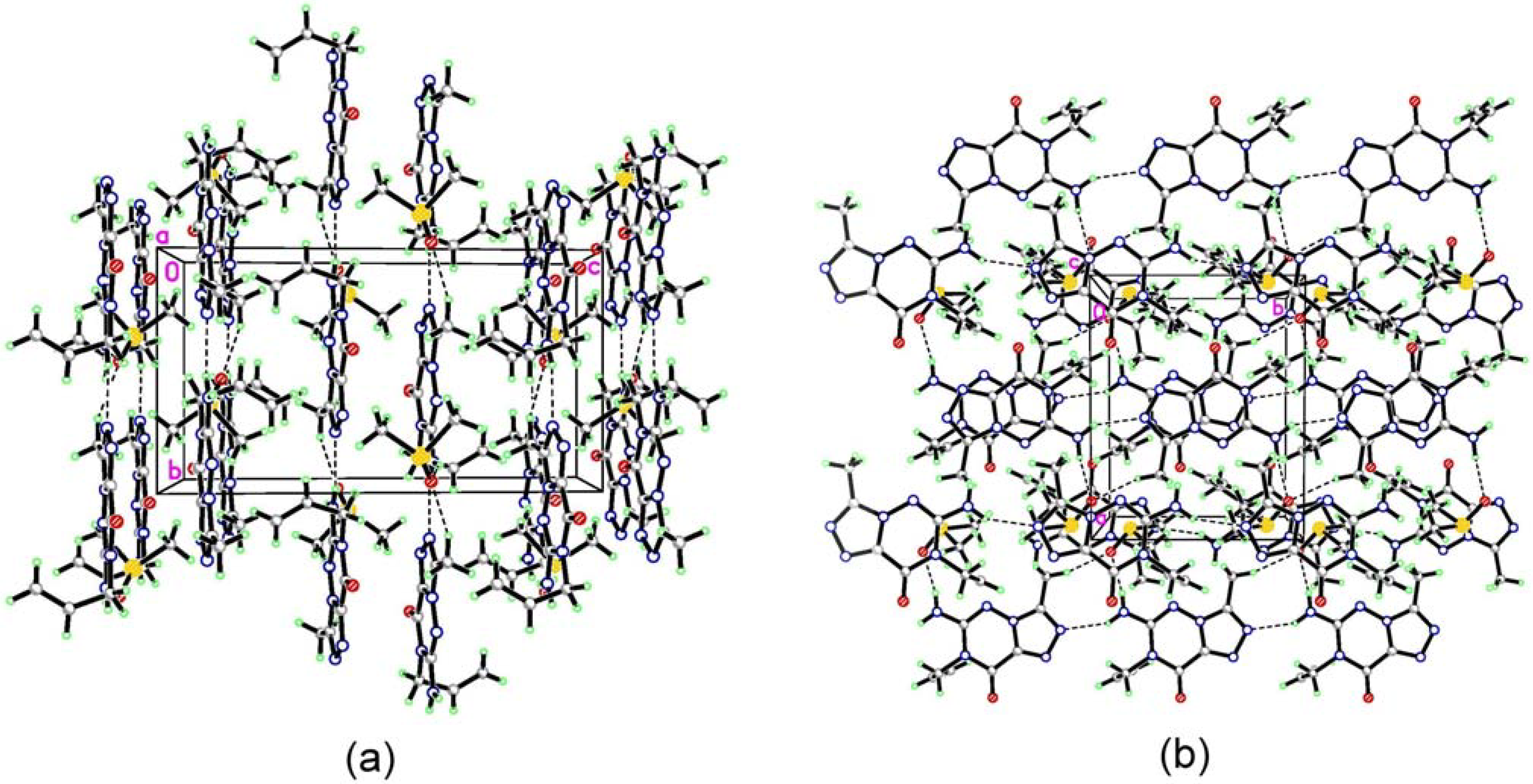
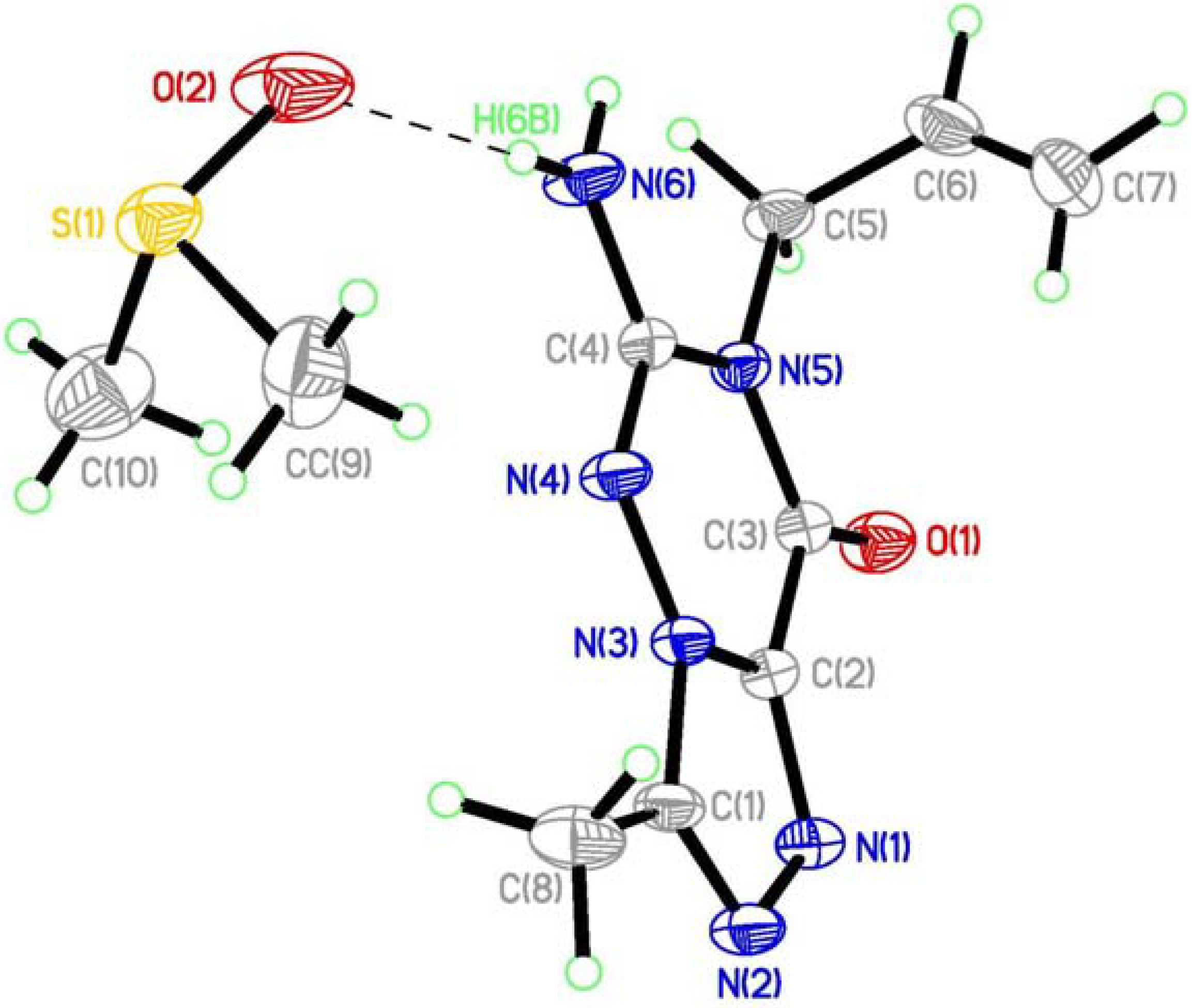
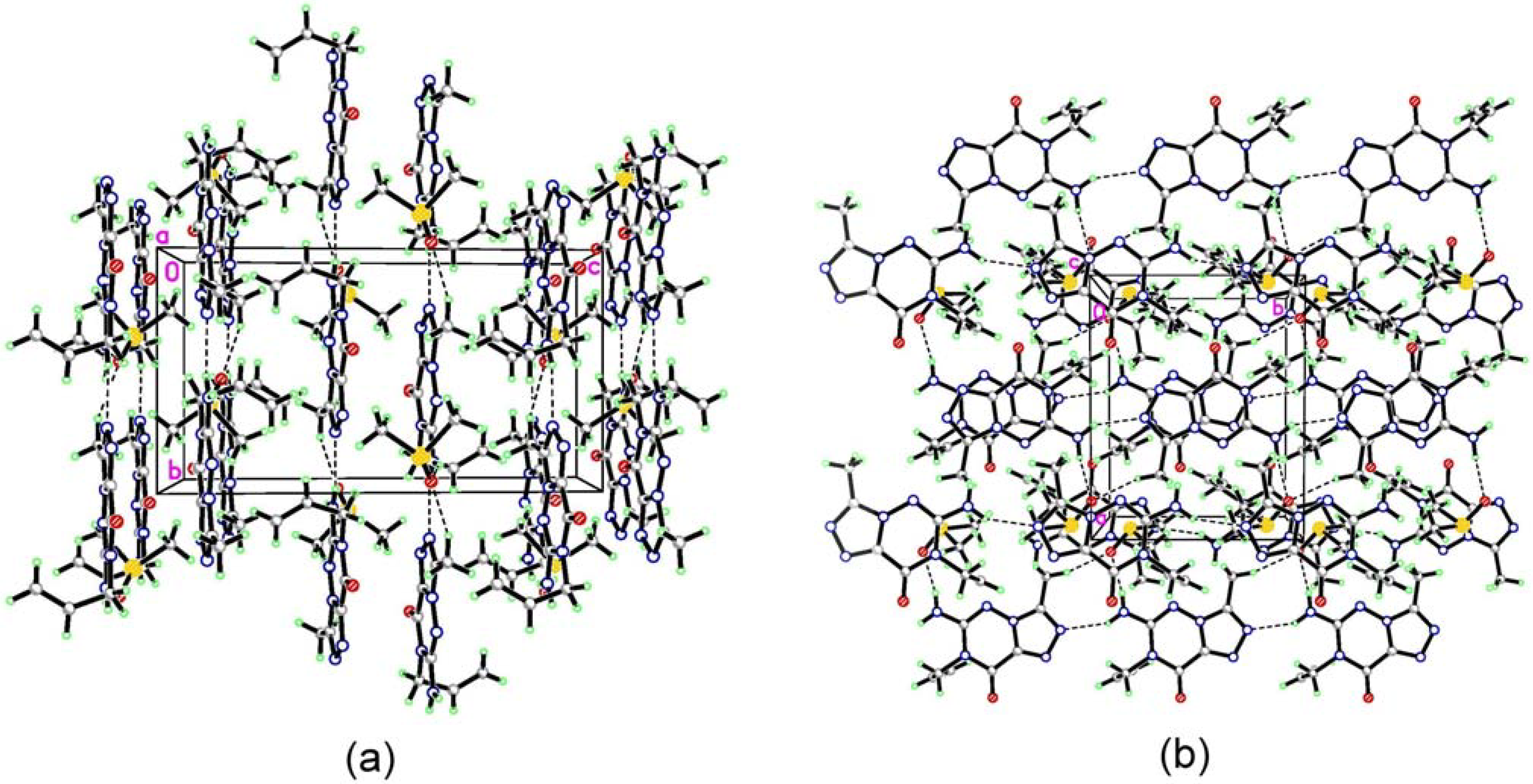
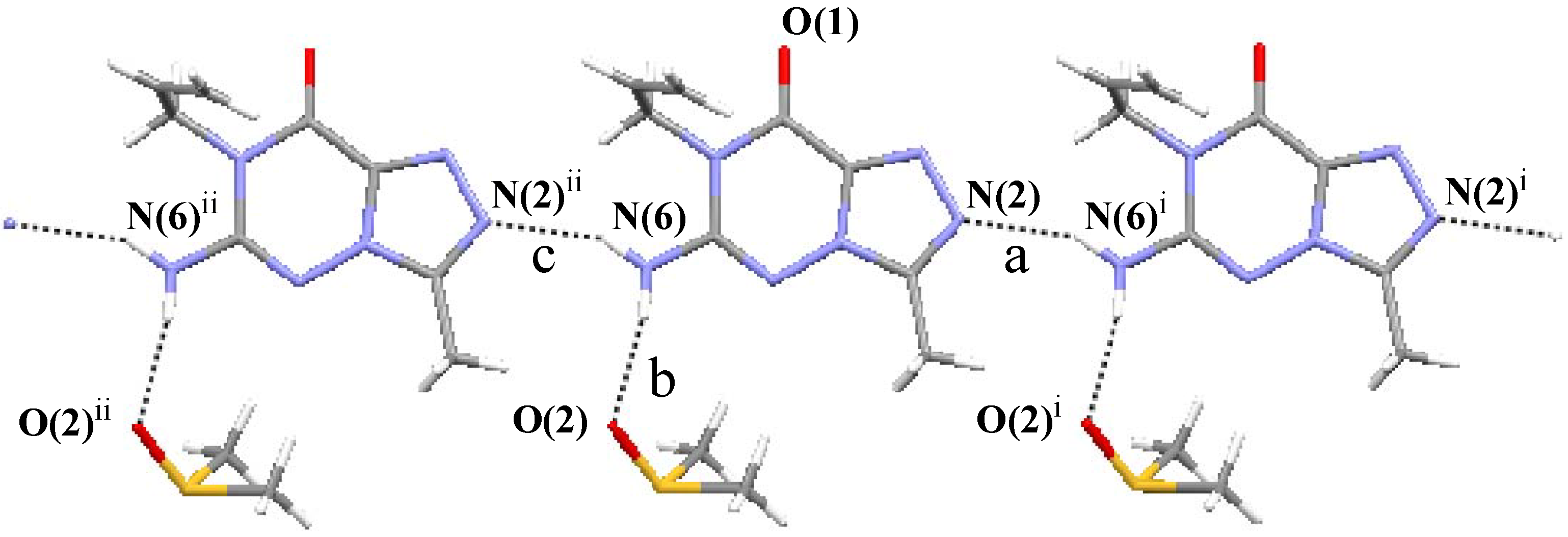
 (7) graph set association via N-H···N hydrogen bond interactions (Figure 3 notation [a] and [c]). Assignment of the H-bond descriptors is based on the graph-set theory [25]. The molecular graphic was obtained using the Mercury program (version 1.4, CCDC, Cambridge, UK). Meanwhile, each molecule links with the oxygen of DMSO via N-H···O hydrogen bonding ([b]). The structure is further stabilized by π-π stacking interactions (Figure 2: (b)), which results in the centroid···centroid distance (3.351 Å) being that between the layer of [1,2,4]triazolo[3,4-f][1,2,4]triazine ring.
(7) graph set association via N-H···N hydrogen bond interactions (Figure 3 notation [a] and [c]). Assignment of the H-bond descriptors is based on the graph-set theory [25]. The molecular graphic was obtained using the Mercury program (version 1.4, CCDC, Cambridge, UK). Meanwhile, each molecule links with the oxygen of DMSO via N-H···O hydrogen bonding ([b]). The structure is further stabilized by π-π stacking interactions (Figure 2: (b)), which results in the centroid···centroid distance (3.351 Å) being that between the layer of [1,2,4]triazolo[3,4-f][1,2,4]triazine ring.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Notation | D-H···A | D-H (Å) | H···A (Å) | D···A (Å) | Length-VdW | D-H···A(°) |
|---|---|---|---|---|---|---|
| a b c | N(2)···H(6C)−N(6)i N(6)−H(6B)···O(2) N(6)−H(6C)···N(2)ii | 0.860 0.860 0.860 | 2.190 2.033 2.190 | 2.978 2.848 2.978 | –0.122 –0.222 –0.122 | 152.27 157.57 152.27 |
Conclusions
Experimental Section
General
Syntheses: 6-Amino-3-methyl-1,2,4-triazolo[3,4-f][1,2,4]triazin-8(7H)-one (1)
1-Allyl-6-amino-3-methyl-1,2,4-triazolo[3,4-f][1,2,4]triazin-8(7H)-one (2) and 7-Allyl-6-amino-3-methyl-1,2,4-triazolo[3,4-f][1,2,4]triazin-8(7H)-one (3)
| Formula | C10H16N6O2S |
| Formula weight | 284.35 |
| Crystal system | Monoclinic |
| Space group | P21/n |
| Unit-cell dimensions (Å) | a = 10.4850(6) |
| b = 8.4409(5) | |
| c = 15.5152(9) | |
| β = 95.963(1)° | |
| Unit-cell volume, V (Å3) | 1365.71(14) |
| Formula per unit cell, Z | 4 |
| Dcalcd (g/cm3) | 1.383 |
| Absorption coefficient, μ (mm-1) | 0.246 |
| F(000) | 600 |
| Crystal size (mm) | 0.36 × 0.25 × 0.08 |
| Index ranges | – 13 ≤ h ≤ 13 |
| – 10 ≤ k ≤ 10 | |
| – 20 ≤ l ≤ 20 | |
| Max. and min. transmission | 0.9806 and 0.9167 |
| Independent reflections | 3133 (Rint = 0.0480) |
| Reflections/restraints/parameters | 3133 /0 /175 |
| Final R indices [I > 2σ(I)] | R1 = 0.0590, wR2 = 0.1371 |
| R indices (all data) | R1 = 0.0736, wR2 = 0.1454 |
| Goodness-of-fit on F2 | 1.078 |
| Max. shift/error | 0.000 |
X-ray techniques
 ) + (0.0584P)2 + 0.7097P], where P = ( + 2
) + (0.0584P)2 + 0.7097P], where P = ( + 2  )/3). S = 1.078 and (Δ/σ)max= 0.000. The maximum peak on the final difference Fourier map is 0.321 and the minimum peak −0.319 eÅ–3.
)/3). S = 1.078 and (Δ/σ)max= 0.000. The maximum peak on the final difference Fourier map is 0.321 and the minimum peak −0.319 eÅ–3.Acknowledgements
References and Notes
- Pomarnacka, E. Acta Pol. Pharm. 1998, 55, 481.
- Abdel-Rahman, R. M.; Morsy, J. M.; Hanafy, F.; Amene, H. A. Pharmazie 1999, 54, 347.
- Abdel-Rahman, R. M.; Morsy, J. M.; El-Edfawy, S.; Amene, H. A. Pharmazie 1999, 54, 667.
- Abdel-Rahman, R. M. Pharmazie 2001, 56, 18.
- Abdel-Rahman, R. M. Pharmazie 2001, 56, 275.
- El-Gendy, Z.; Morsy, J. M.; Allimony, H. A.; Ali, W. R.; Abdel-Rahman, R. M. Pharmazie 2001, 56, 376.
- Holla, B. S.; Rao, B. S.; Gonsalves, R.; Sarojini, B. K.; Shridhara, K. Farmaco 2002, 57, 693.
- Habib, N. S.; Soliman, R.; Ismail, K.; Hassan, A. M.; Sarg, M. T. Boll. Chim. Farm. 2003, 142, 396.
- Iwashita, A.; Maemoto, T.; Nakada, H.; Shima, I.; Matsuoka, N.; Hisajima, H. J. Pharmacol. Exp. Ther. 2003, 307, 961. [CrossRef]
- Guertin, K. R.; Setti, L. Qi. L.; Dunsdon, R. M.; Dymock, B. W.; Jones, P. S.; Overton, H.; Taylor, M.; Williams, G.; Sergi, J. A.; Wang, K.; Peng, Y.; Renzetti, M.; Boyce, R.; Falcioni, F.; Garippa, R.; Olivier, A. R. Bioorg. Med. Chem. Lett. 2003, 13, 2895.
- Hunt, J. T.; Mitt, T.; Borzilleri, R.; Gullo-Brown, J.; Fargnoli, J.; Fink, B.; Han, W. C.; Mortillo, S.; Vite, G.; Wautlet, B.; Wong, T.; Yu, C.; Zheng, X.; Bhide, R. J. Med. Chem. 2004, 47, 4054.
- Chambers, M. S.; Atack, J. R.; Carling, R. W; Collinson, N.; Cook, S. M.; Dawson, G. R.; Ferris, P.; Hobbs, S. C.; O'Connor, D.; Marshall, G.; Rycroft, W.; Macleod, A. M. J. Med. Chem. 2004, 47, 5829. [CrossRef]
- Borzilleri, R. M.; Cai, Z. W.; Ellis, C.; Fargnoli, J.; Fura, A.; Gerhardt, T.; Goyal, B.; Hunt, J. T.; Mortillo, S.; Qian, L.; Tokarski, J.; Vyas, V.; Wautlet, B.; Zheng, X.; Bhide, R. S. Bioorg. Med. Chem. Lett. 2005, 15, 1429. [CrossRef]
- Borzilleri, R. M.; Zheng, X.; Qian, L.; Ellis, C.; Cai, Z. W.; Wautlet, B. S.; Mortillo, S.; Jeyaseelan, R., Sr.; Kukral, D. W.; Fura, A.; Kamath, A.; Vyas, V.; Tokarski, J. S.; Barrish, J. C.; Hunt, J. T.; Lombardo, L. J.; Fargnoli, J.; Bhide, R. S. J. Med. Chem. 2005, 48, 3991.
- Fink, B. E.; Vite, G. D.; Mastalerz, H.; Kadow, J. F.; Kim, S. H.; Leavitt, K. J.; Du, K.; Crews, D.; Mitt, T.; Wong, T. W.; Hunt, J. T.; Vyas, D. M.; Tokarski, J. S. Bioorg. Med. Chem. Lett. 2005, 15, 4774. [CrossRef]
- Sztanke, K. Acta Pol. Pharm. 2005, 62, 221.
- Jones, P.; Atack, J. R.; Braun, M. P.; Cato, B. P.; Chambers, M. S.; O'Connor, D.; Cook, S. M.; Hobbs, S. C.; Maxey, R.; Szekeres, H. J.; Szeto, N.; Wafford, K. A.; MacLeod, A. M. Bioorg. Med. Chem. Lett. 2006, 16, 872. [CrossRef]
- Gupta, R.; Gupta, A. K.; Paul, S.; Kachroo, P. L. Indian J. Chem. 1998, 37B, 1211.
- Hiremath, S. P.; Ullagaddi, A.; Shivaramayya, K.; Purohit, M.G. Indian J. Heterocycl. Chem. 1999, 3, 145.
- Hwang, L. C.; Wu, R. R.; Tu, C. H. J. Heterocycl. Chem. 2005, 42, 851.
- Hwang, L. C.; Tu, C. H.; Wang, J. H. J. Heterocycl. Chem. 2006, in press.
- Hwang, L. C.; Tu, C. H.; Wang, J. H.; Lee, G. H.; Wang, Y. Anal. Sci. 2002, 18, 853.
- Hwang, L. C.; Tu, C. H.; Wang, J. H.; Lee, G. H. Molecules 2006, 11, 169.
- Lide, D. R. CRC Handbook of Chemistry and Physics, 80th ed.; CRC Press LLC: Boca Raton, FL, 1999-2000; Section 9; pp. 1–14. [Google Scholar]
- Bernstein, J.; Davis, R. E.; Shimoni, L.; Chang, N. L. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555. [CrossRef]
- Lovelette, C. A. J. Heterocycl. Chem. 1979, 16, 555.
- CCDC 608157 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033; e-mail: [email protected]).
- Sheldrick, G. M. SHELXS-97, Program for the Solution of Crystal Structure; Univ. of Göttingen: Göttingen, Germany, 1990. [Google Scholar]
- Sheldrick, G. M. SHELXL-97, Program for the Refinement of Crystal Structure; Univ. of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sample Availability: Samples of the compounds mentioned are available from the authors.
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Hwang, L.-C.; Jane, S.-Y.; Lai, H.-Y.; Tu, C.-H.; Lee, G.-H. Synthesis, Molecular Structure and Characterization of Allylic Derivatives of 6-Amino-3-methyl-1,2,4-triazolo[3,4-f][1,2,4]-triazin-8(7H)-one. Molecules 2006, 11, 444-452. https://doi.org/10.3390/11060444
Hwang L-C, Jane S-Y, Lai H-Y, Tu C-H, Lee G-H. Synthesis, Molecular Structure and Characterization of Allylic Derivatives of 6-Amino-3-methyl-1,2,4-triazolo[3,4-f][1,2,4]-triazin-8(7H)-one. Molecules. 2006; 11(6):444-452. https://doi.org/10.3390/11060444
Chicago/Turabian StyleHwang, Long-Chih, Shin-Yi Jane, Hsing-Yi Lai, Chun-Hsien Tu, and Gene-Hsiang Lee. 2006. "Synthesis, Molecular Structure and Characterization of Allylic Derivatives of 6-Amino-3-methyl-1,2,4-triazolo[3,4-f][1,2,4]-triazin-8(7H)-one" Molecules 11, no. 6: 444-452. https://doi.org/10.3390/11060444