A Convenient Asymmetric Synthesis of a β-amino Ester with Additional Functionalization as a Precursor for Peptide Nucleic Acid (PNA) Monomers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
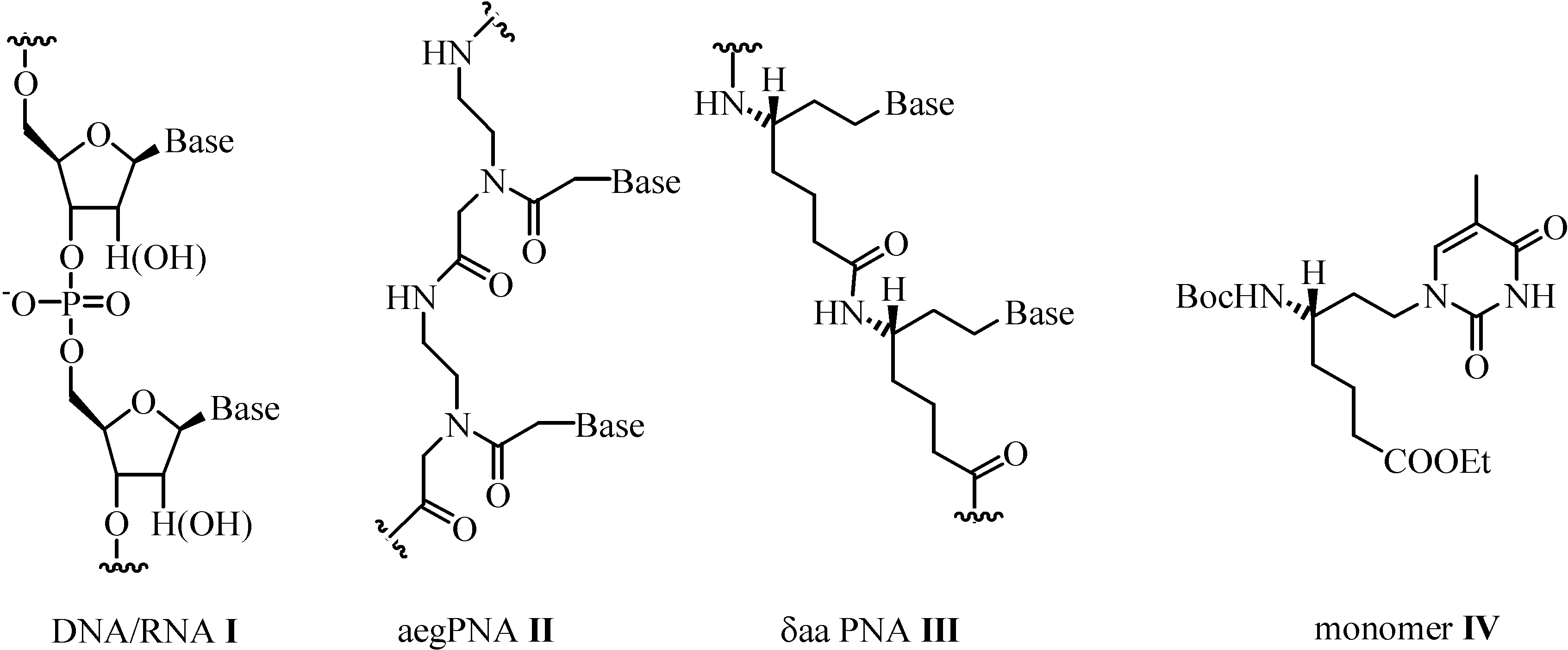
:Introduction
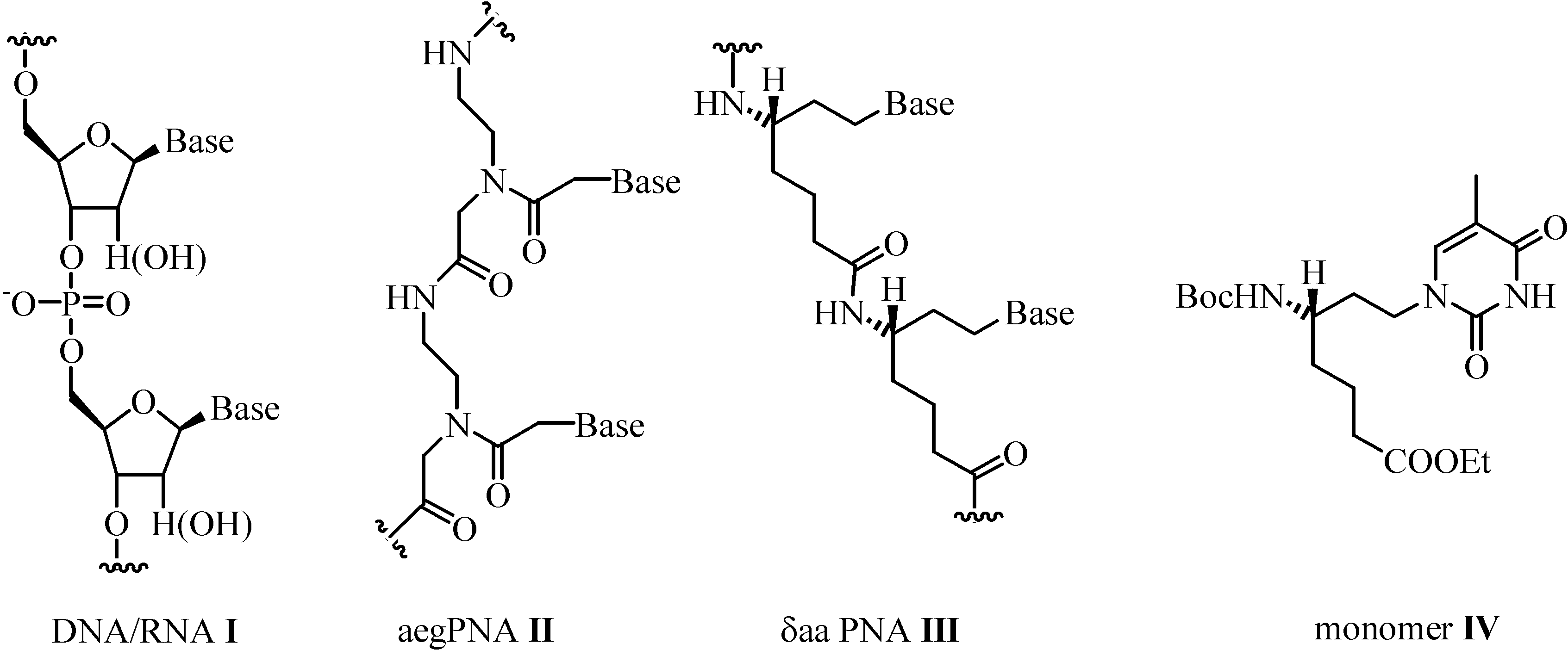
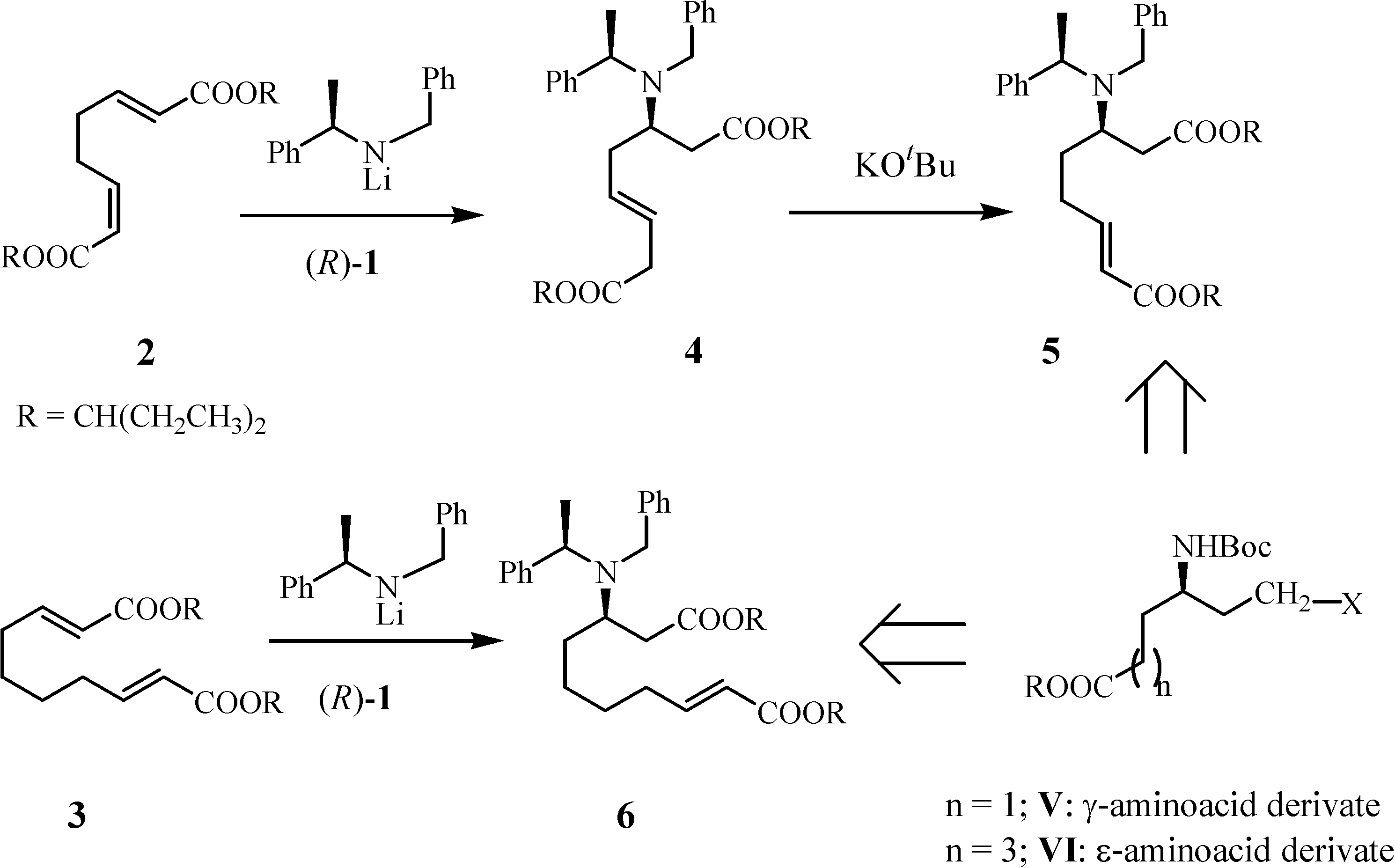
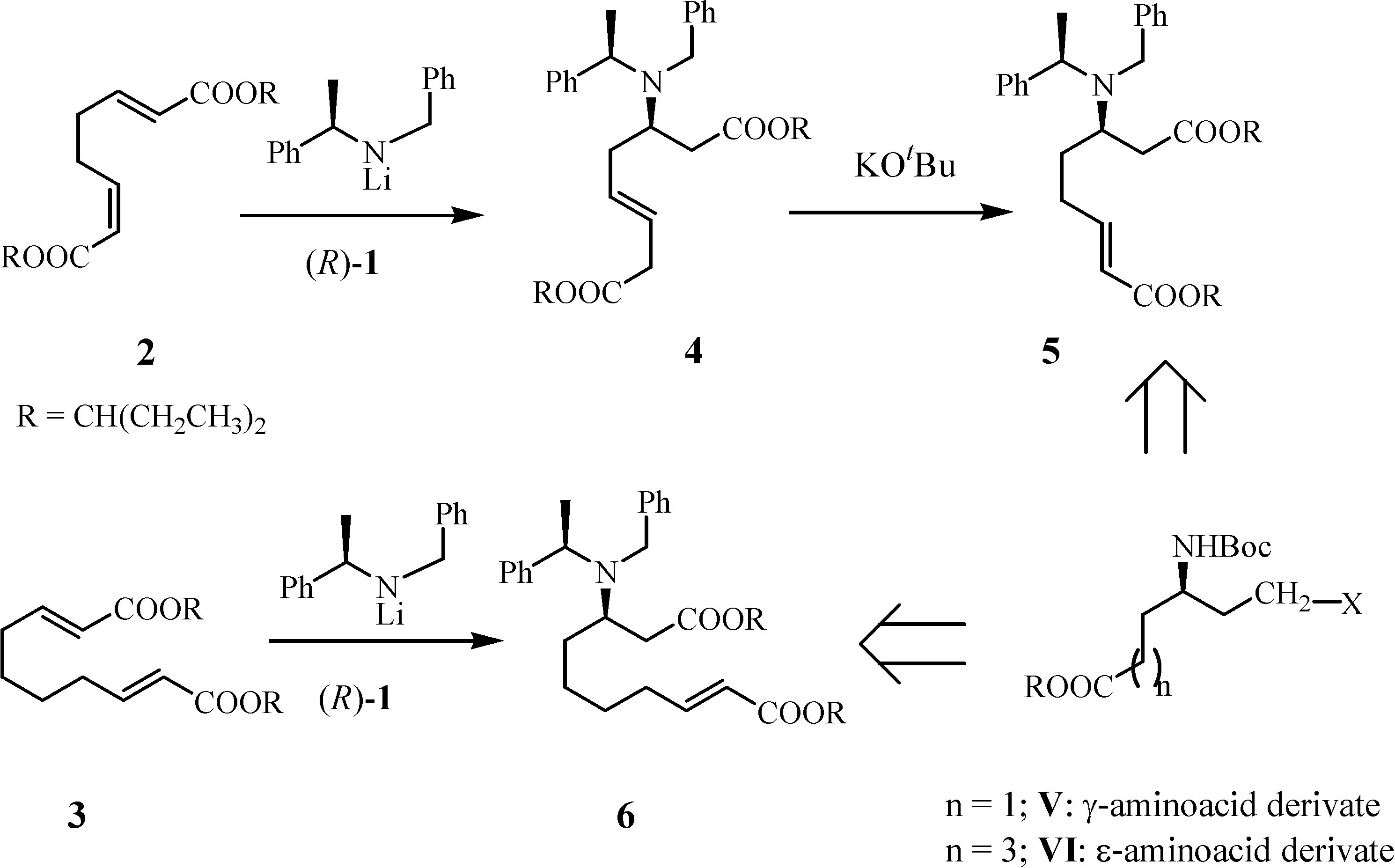
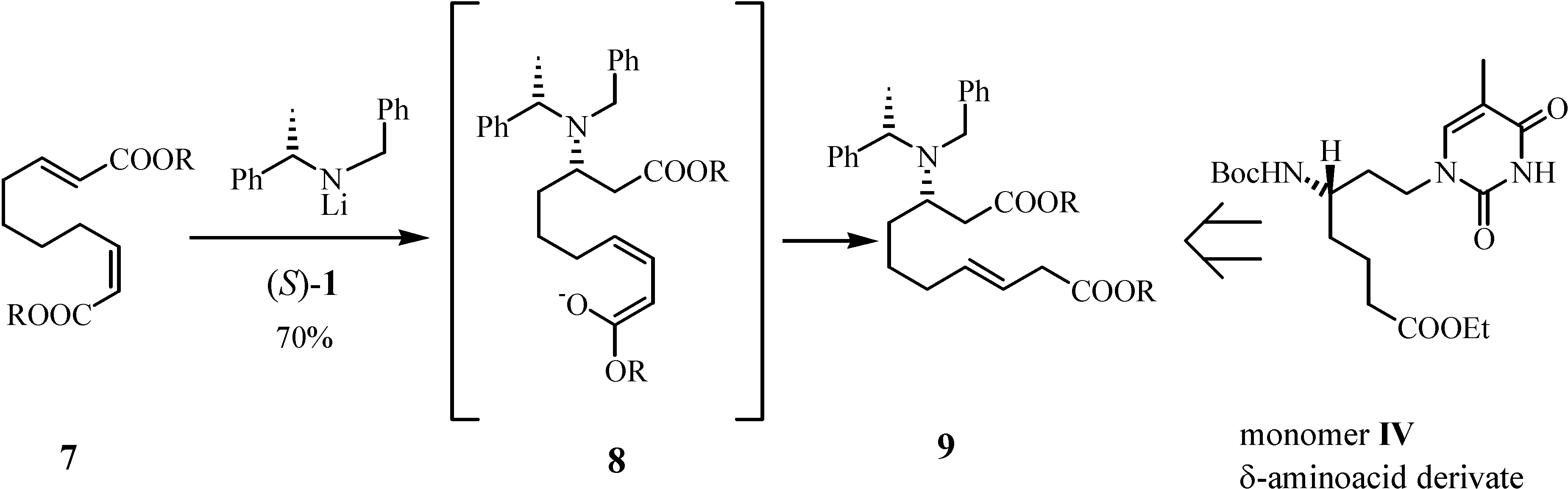
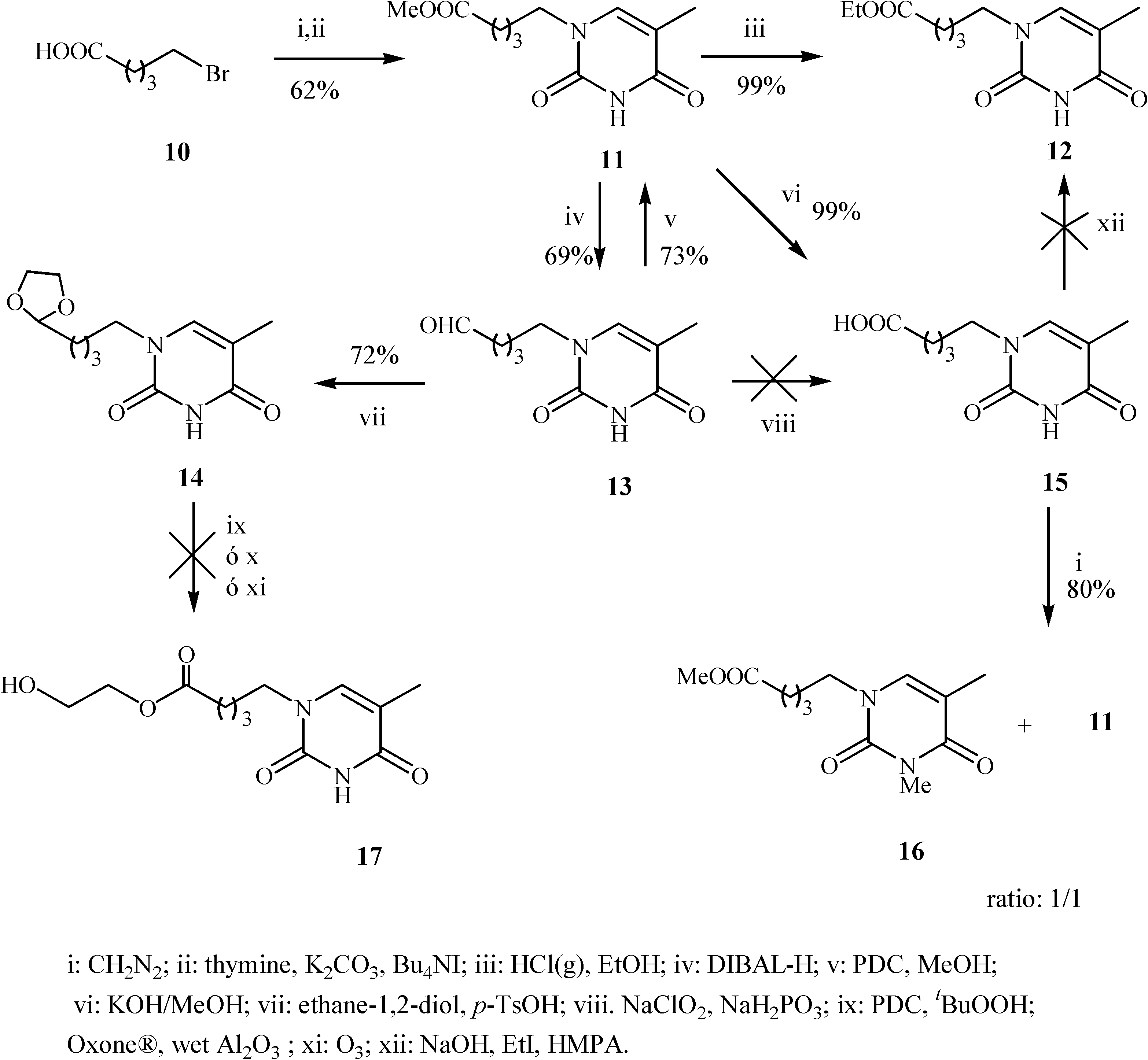
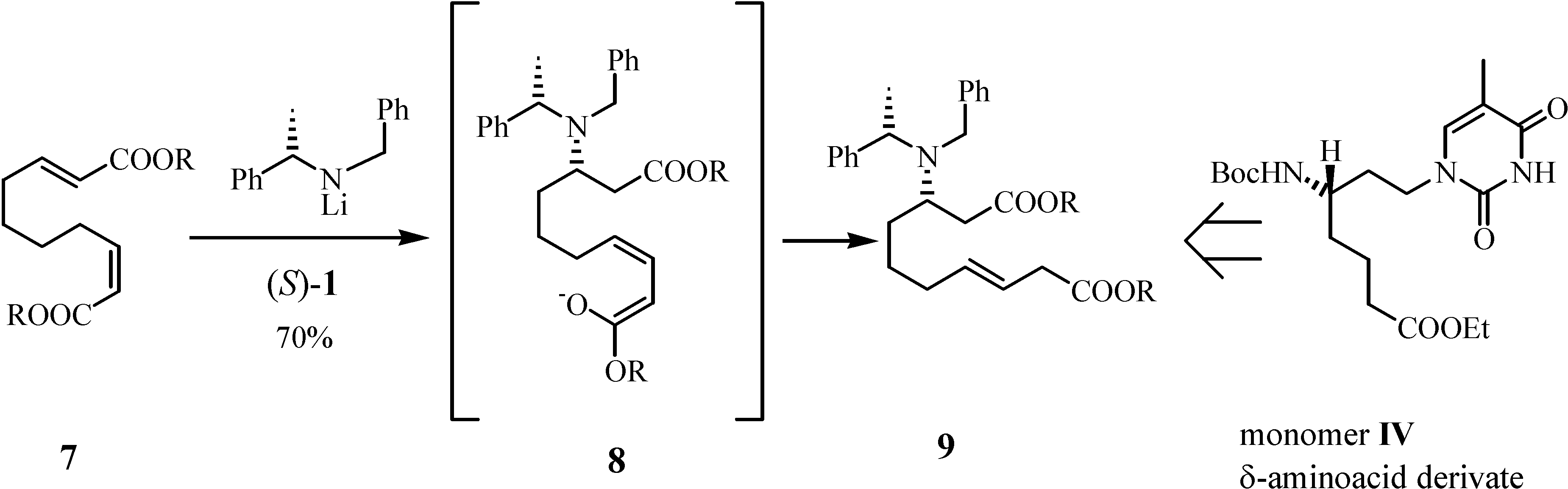
Results and Discussion
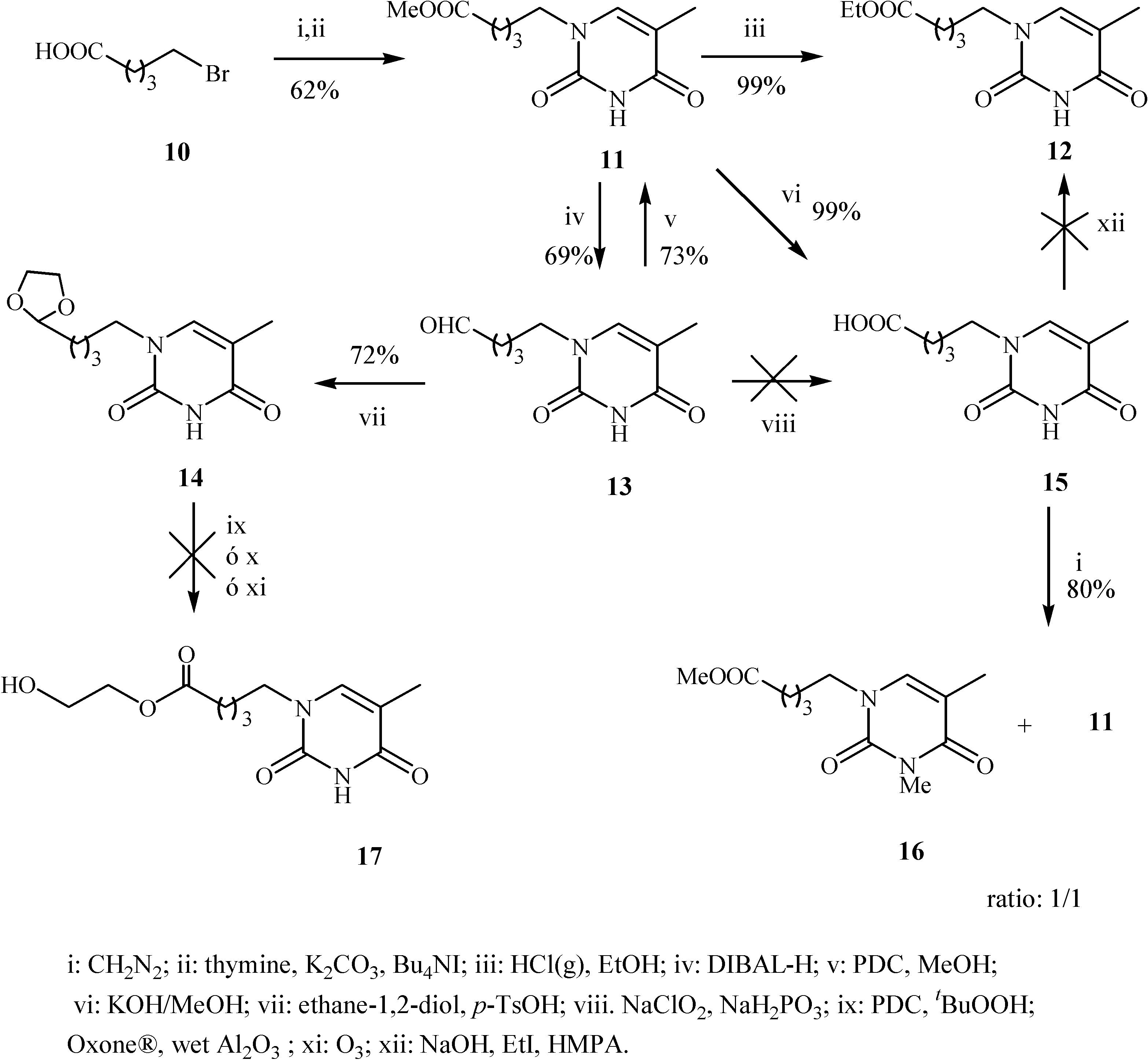
Conclusions
Experimental Section
General
Preparation of di-3-pentyl (3S,αS,7E)-2-N-benzyl-N-α-methylbenzylamino-dec-7-enedioate (9)
Preparation of 1-(4’-methoxycarbonylbutyl)-thymine (11)
Preparation of 1-(4’-ethoxycarbonylbutyl)-thymine (12)
Preparation of1-(5’-oxopentyl)-thymine (13)
Preparation of 1-(4’-[1,3]dioxolan-2-yl-butyl)-thymine (14)
Preparation of 1-(4’-methoxycarbonyl-butyl)-3-methylthymine (11)
Preparation of 1-(4’-carboxybutyl)-thymine (15)
Acknowledgements
References
- Milligan, J.F.; Matteucci, M.D.; Martin, J.C. Current concepts in antisense drug design. J. Med. Chem. 1993, 36, 1923–1937. [Google Scholar] Barrett, J.C.; Miller, P.S.; Ts’o, P.O.P. Alkyl phosphotriesters of dinucleotides and oligonucleotides. 5. Inhibitory effect of complex formation with oligodeoxyribonucleotide ethyl phosphotriesters on transfer ribonucleic acid aminoacylation. Biochemistry 1974, 13, 4897–4906. [Google Scholar]
- Braasch, D.A.; Corey, D.R. Novel antisense and peptide nucleic acid strategies for controlling gene expression. Biochemistry 2002, 41, 4503–4510. [Google Scholar] Uhlmann, E.; Peyman, A.; Breipohl, G.; Will, D.W. PNA: Synthetic polyamide nucleic acid with unusual binding properties. Angew. Chem., Int. Ed. 1998, 37, 2796–2823. [Google Scholar] De Mesmaeker, A.; Haener, R.; Martin, P.; Moser, H.E. Antisense oligonucleotides. Acc. Chem. Res. 1995, 28, 366–374. [Google Scholar]
- Nielsen, P.E. Peptide nucleic acid. A molecule with two identities. Acc. Chem. Res. 1999, 32, 624–630. [Google Scholar] Hyrup, B.; Nielsen, P.E. Peptide nucleic acid (PNA): synthesis, properties and potential applications. Bioorg. Med. Chem. 1996, 4, 5–23. [Google Scholar] Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1501. [Google Scholar]
- Govindaraju, T.; Kumar, V.A. Backbone extended pyrrolidine PNA (bepPNA): a chiral PNA for selective RNA recognition. Tetrahedron 2006, 62, 2321–2330. [Google Scholar] Balaji, B.S.; Gallazzi, F.; Jia, F.; Lewis, M.R. An efficient, convenient solid-phase synthesis of amino acid-modified peptide nucleic acid monomers and oligomers. Bioconj. Chem. 2006, 17, 551–558. [Google Scholar] Jia, T.; Jiang, Z.X.; Shi, C.; Li, Z.Y. Synthesis and characterization of peptide nucleic acid monomeric backbone bonded porphyrins. Chin. J. Org. Chem. 2006, 26, 223–227. [Google Scholar] Pieck, J.C.; Kuch, D.; Grolle, F.; Linne, U.; Haas, C.; Carell, T. PNA-based reagents for the direct and site-specific synthesis of thymine dimer lesions in genomic DNA. J. Am. Chem. Soc. 2006, 128, 1404–1405, and references cited therein. [Google Scholar]
- Savithri, Dh.; Leumann, Ch.; Scheffold, R. Synthesis of a Building Block for a Nucleic-Acid Analog with a Chiral, Flexible Peptide Backbone. Helv. Chim. Acta. 1996, 79, 288–294. [Google Scholar] [CrossRef]
- Urones, J.G.; Garrido, N.M.; Díez, D.; Domínguez, S.H.; Davies, S.G. Conjugate addition to (α,β)(α’,β’)-diendioate esters by lithium (α-methylbenzyl)benzylamide: tandem addition-cyclisation versus double addition. Tetrahedron: Asymmetr. 1999, 10, 1637–1641. [Google Scholar] Davies, S.G.; Díez, D.; Domínguez, S.H.; Garrido, N.M.; Kruchinin, D.; Price, P.D.; Smith, D. Cyclic β-amino acid derivates: synthesis via lithium amide promoted tandem asymmetric conjugate addition-cyclisation reactions. Org. Biomol. Chem. 2005, 3, 1284–1301. [Google Scholar]
- Urones, J.G.; Garrido, N.M.; Díez, D.; Dominguez, S.H.; Davies, S.G. Asymmetric synthesis of (R)- and (S)- methyl (2-methoxy-carbonylcyclopent-2-enyl)acetate and (R)- and (S)-2-(2-hydroxy-methyl-cyclopent-2-enyl)ethanol. Tetrahedron: Asymmetr. 1997, 8, 2683–2685. [Google Scholar]
- Whiteshell, J.K.; Hilbling, A.M. Preparation of beta, gamma-unsatured methyl esters from allylic alcohols. J. Org. Chem. 1980, 45, 4135–4139. [Google Scholar] Uyehara, T.; Shida, N.; Yamamoto, Y. New type of cyclization of alpha, beta, chi, psi-unsaturated dioic acid esters through tandem conjugate additions by using lithium N-benzyl-N-(trimethylsilyl)amide as a nitrogen nucleophile. J. Org. Chem. 1992, 57, 3139–3145. [Google Scholar]
- Davies, S.G.; Smith, A.D.; Price, P.D. The conjugate addition of enantiomerically pure lithium amides as homochiral ammonia equivalents: scope, limitations and synthetic applications. Tetrahedron: Asymmetr. 2005, 16, 2833–2891. [Google Scholar]
- Costello, J.F.; Davies, S.G.; Ichihara, O. Origins of the high stereoselectivity in the conjugate addition of lithium (α-methylbenzyl)benzylamide to t-butyl cinnamate. Tetrahedron: Asymmetr. 1994, 5, 1999–2008. [Google Scholar]
- Garrido, N.M.; Díez, D.; Domínguez, S.H.; Sánchez, M.R.; García, M.; Urones, J.G. in preparation.
- Kosynkina, L.; Wang, W.; Liang, T.Ch. A convenient synthesis of chiral peptide nucleic acid (PNA) monomers. Tetrahedron Lett. 1994, 35, 5173–5176. [Google Scholar] [CrossRef]
- O´Connor, B.; Just, G. A new method for the conversion of aldehydes to methyl esters using pyridinium dichromate and methanol in dimethylformamide. Tetrahedron Lett. 1987, 28, 3235–3236. [Google Scholar] [CrossRef]
- Chidambaram, N.; Bhat, S.; Chandrasekaran, S. A highly selective methodology for the direct conversion of acetals to esters. J. Org. Chem. 1992, 57, 5013–5015. [Google Scholar] [CrossRef]
- Curini, M.; Epifano, F.; Marcotullio, M.C.; Rosati, O. Oxone®: A convenient Reagent for oxidation of Acetals. Synlett 1999, 6, 777–779. [Google Scholar] Greenhaugh, R.P. Selective Oxidation of Phenyl Sulphides to Sulphoxides or Sulphones Using Oxone® and Wet Alumina. Synlett 1992, 235. [Google Scholar]
- Pryor, W.A.; Giamalva, D.; Church, D.F. Kinetics of ozonation. 1. Electron-deficient alkenes. J. Am. Chem. Soc. 1983, 105, 6858–6861. [Google Scholar] Pryor, W.A.; Giamalva, D.; Church, D.F. Kinetics of ozonation. 3. Substituent effects on the rates of reaction of alkenes. J. Am. Chem. Soc. 1985, 107, 2793–2797. [Google Scholar]
- Sample Availability: Contact the authors.
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Garrido, N.M.; Díez, D.; Domínguez, S.H.; Sánchez, M.R.; García, M.; Urones, J.G. A Convenient Asymmetric Synthesis of a β-amino Ester with Additional Functionalization as a Precursor for Peptide Nucleic Acid (PNA) Monomers. Molecules 2006, 11, 435-443. https://doi.org/10.3390/11060435
Garrido NM, Díez D, Domínguez SH, Sánchez MR, García M, Urones JG. A Convenient Asymmetric Synthesis of a β-amino Ester with Additional Functionalization as a Precursor for Peptide Nucleic Acid (PNA) Monomers. Molecules. 2006; 11(6):435-443. https://doi.org/10.3390/11060435
Chicago/Turabian StyleGarrido, Narciso M., David Díez, Sara H. Domínguez, M. Rosa Sánchez, Mercedes García, and Julio G. Urones. 2006. "A Convenient Asymmetric Synthesis of a β-amino Ester with Additional Functionalization as a Precursor for Peptide Nucleic Acid (PNA) Monomers" Molecules 11, no. 6: 435-443. https://doi.org/10.3390/11060435