Synthesis and Crystal Structure of a New Triosmium Alkylidyne Carbonyl Cluster Containing a Chiral Ferrocenylphosphine Ligand

Department of Chemistry, Hong Kong Baptist University, Waterloo Road, Kowloon Tong, Hong Kong, P.R. China
Molecules 2005, 10(6), 640-648; https://doi.org/10.3390/10060640
Submission received: 13 July 2004
/
Accepted: 3 September 2004
/
Published: 14 July 2005
(This article belongs to the Special Issue Synthesis and Chemistry of Chiral Metallocenes)
Abstract
:The synthesis, spectroscopic characterization and X-ray crystal structure of a new chiral triosmium alkylidyne carbonyl cluster, (R,S)-[Os3(μ-H)2(CO)9{μ3-CPPh2(η5-C5H4)Fe(η5-C5H3(PPh2)CH(Me)NMe2)}] (1) are described. Compound 1 crystallizes in the non-centrosymmetric space group P21 and its absolute configuration has been established. The structure consists of an Os3C metal core with one of the PPh2 moieties of the chiral ferrocenylphosphine bonded to the apical alkylidyne carbon atom to give a zwitterionic cluster complex, reminiscent of the phosphorus ylide.
Introduction
A non-centrosymmetric crystal packing of molecules is a prerequisite for second-order non-linear optical (NLO) activity and much attention has been focused on this aspect of molecular structure [1]. Introduction of a chiral centre ensures that a molecule lacks a centre of symmetry, and this is usually a favorable condition for non-centric crystal structures. In this regard, the production of chiral transition metal clusters is crucial as part of the development of useful organometallic materials, and optically active phosphines containing chiral substituents seem to be promising candidates to promote crystallization in a non-centrosymmetric fashion. The reactivity of optically active diphosphine ligands such as Ph2PCH(Me)CH2PPh2 towards metal complexes was investigated in detail [2,3,4,5,6,7]. Amongst other chiral phosphines, the chiral metalloligand (R,S)-bppfa [(–)-(R)-N,N-dimethyl-1-[(S)-1',2-bis(diphenylphosphino)ferrocenyl]ethylamine] or alike has attracted particular attention in this aspect and its tendency to act as a ligand in polynuclear metal complexes has been extensively explored [8,9,10,11,12]. These optically active ferrocenylphosphines are important in the study of modern organometallic chemistry for at least two reasons: (a) the ferrocene unit imparts excellent electron donation to the electron-withdrawing metal cluster core, thus affording a charge-separated species with a large dipole moment in the ground state; (b) enantiomerically pure ferrocenylphosphines have proved to be excellent chiral ligands for various types of transition metal catalysts and the resulting optically active ferrocene derivatives represent particularly suitable chiral reference systems for asymmetrically induced syntheses such as enantioselective alkylation, hydrogenation, hydrosilylation and cross coupling reactions [13,14,15,16,17].
Previous work has shown that the species "[Os3(µ-H)2(CO)9(µ3-C)]" behaves as a good electron sink and is readily trapped by strong nucleophiles [18,19,20,21]. So, the present strategy is to make use of an optically active phosphine complex containing the ferrocenyl group, which may afford new molecules with highly polarizable donor-acceptor properties. Herein, the preparation of a new charge-separated compound derived from the electron-accepting organometallic fragment "[Os3(µ-H)2(CO)9(µ3-C)]" with the chiral chromophore (R,S)-[(η5-C5H4)Fe(η5-C5H3(PPh2)CH(Me)NMe2)] (R,S-bppfa) as the co-ordinated ligand is described. This covalently linked donor-acceptor complex was fully characterized by spectroscopic and crystallographic methods.
Results and Discussion
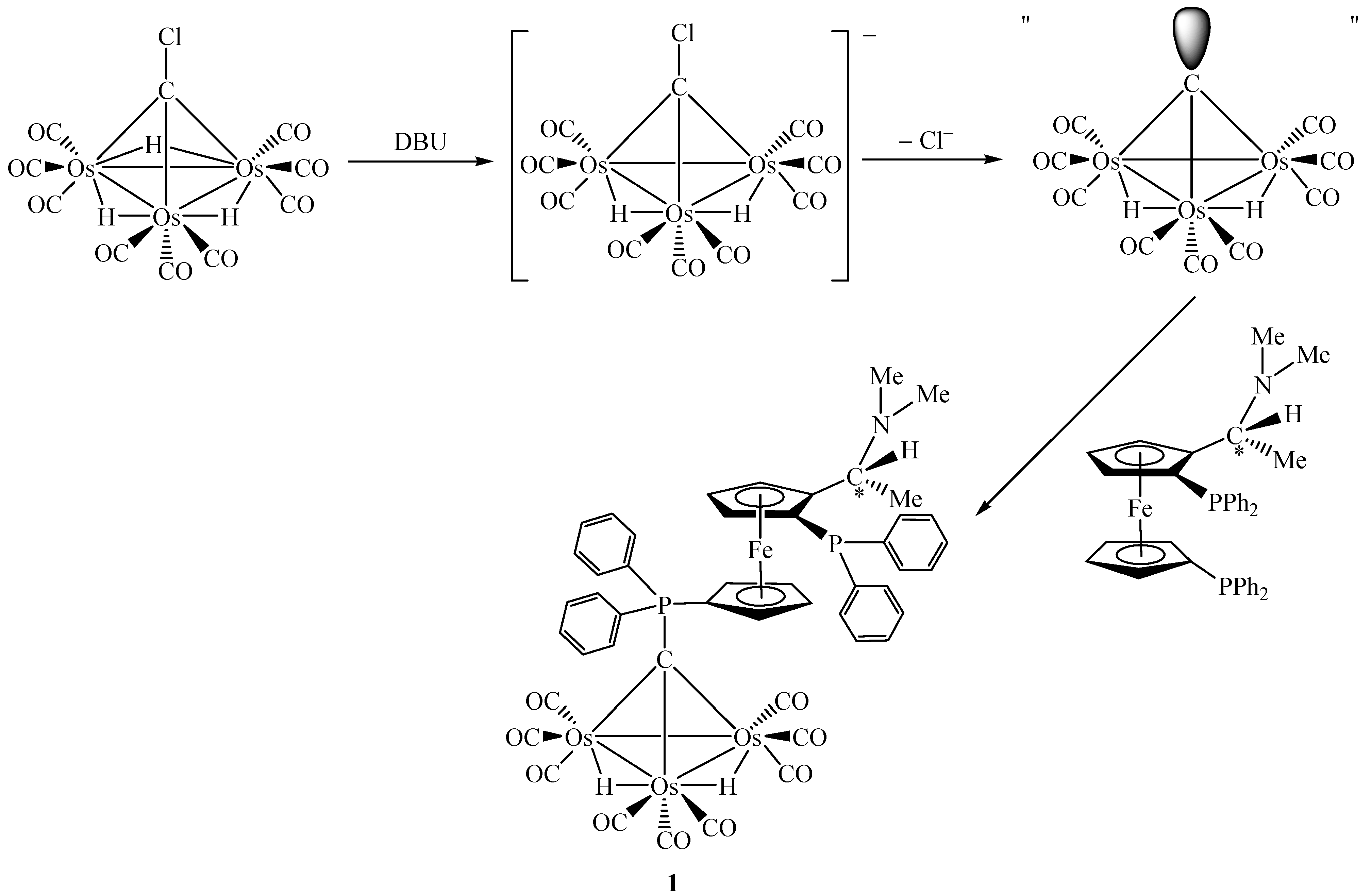
Treatment of [Os3(µ-H)3(CO)9(µ3-CCl)] with an excess of (R,S)-bppfa in the presence of a sterically hindered strong base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) at room temperature leads to the air-stable chiral complex (R,S)-[Os3(µ-H)2(CO)9{µ3-CPPh2(η5-C5H4)Fe(η5-C5H3(PPh2)-CH(Me)NMe2)}] (1) in a moderate yield. DBU was used as a deprotonating agent in this reaction. It is believed that the anionic species generated by deprotonation of [Os3(µ-H)3(CO)9(µ3-CCl)] may dissociate to give a chloride ion and a highly reactive transient species "[Os3(µ-H)2(CO)9(µ3-C)]" which is then trapped by the phosphine ligand to furnish complex 1 [21]. The key steps involved are summarized in Scheme 1.
Complex 1 displays a similar νCO absorption pattern in its IR spectrum to other clusters of the formulation [Os3(µ-H)2(CO)9(µ3-CY)] (Y = Lewis base) [18,19,20,21]. Intense molecular ion peak at m/z = 1463 is observed in the positive FAB mass spectrum of 1. The proton resonances in the 1H NMR spectrum due to the organic moieties in 1 are totally consistent with the solid-state structure. It gives a doublet hydride signal at δ −19.92 due to 31P−1H coupling with 3JP−H = 2.8 Hz. Nevertheless, no attempt was made to fully assign the signals in the range δ 3.44−4.69 due to the cyclopentadienyl rings which shows a rather complicated pattern. The 31P{1H}-NMR spectrum of 1 in CD2Cl2 exhibits two singlet signals at δ −22.44 and 41.86. The corresponding 31P resonances arising from the free ligand (R,S)-bppfa under the same experimental conditions appear at δ −21.62 and −15.74. A significant downfield shift of one of the 31P signals is observed which is caused by the electron-withdrawing nature of the alkylidyne metal core. This downfield resonance is assigned to the phosphorus atom directly coordinated to the apical carbon atom. This is consistent with the phosphorus atom in phosphonium salt.
Scheme 1.
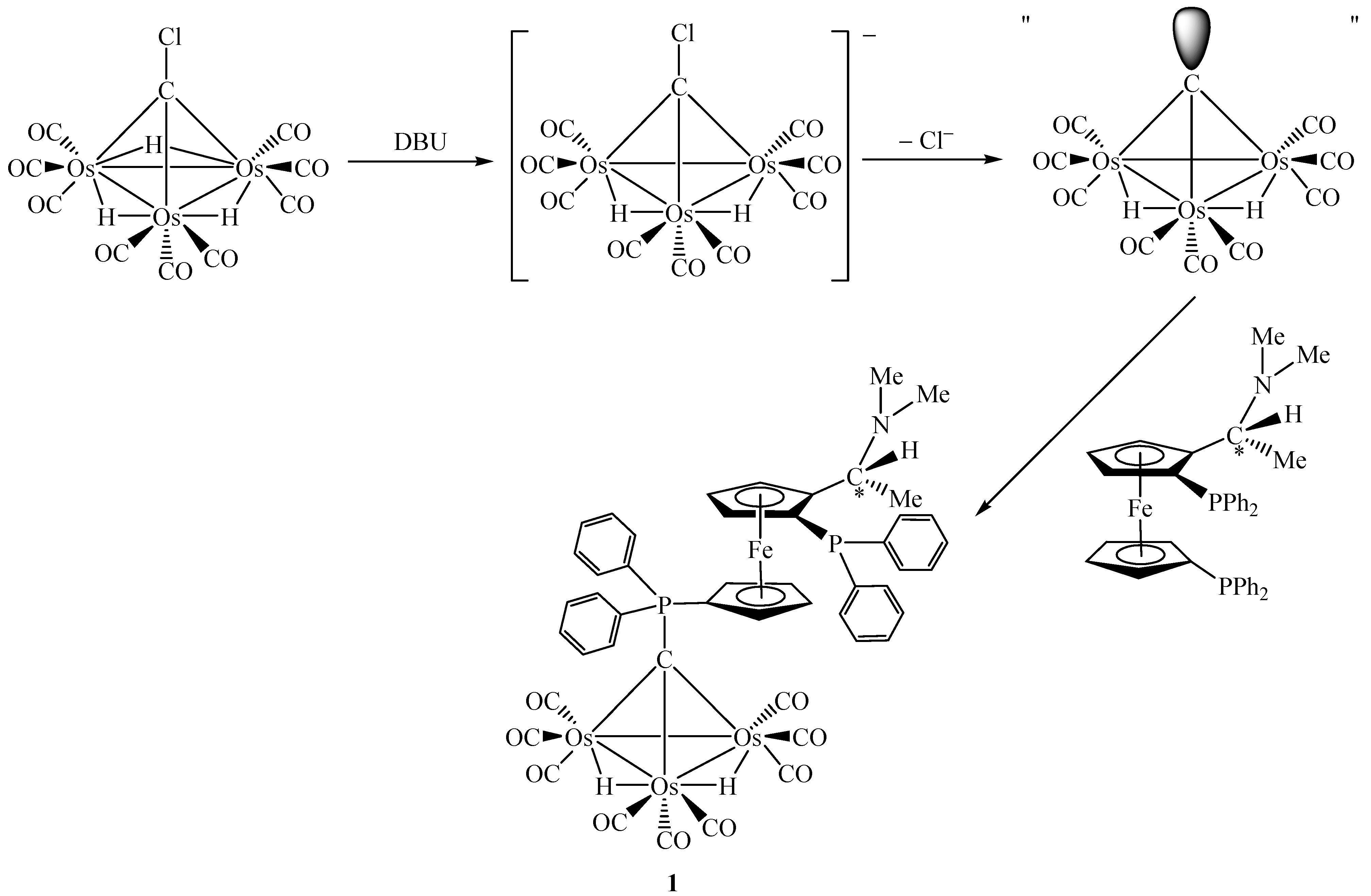
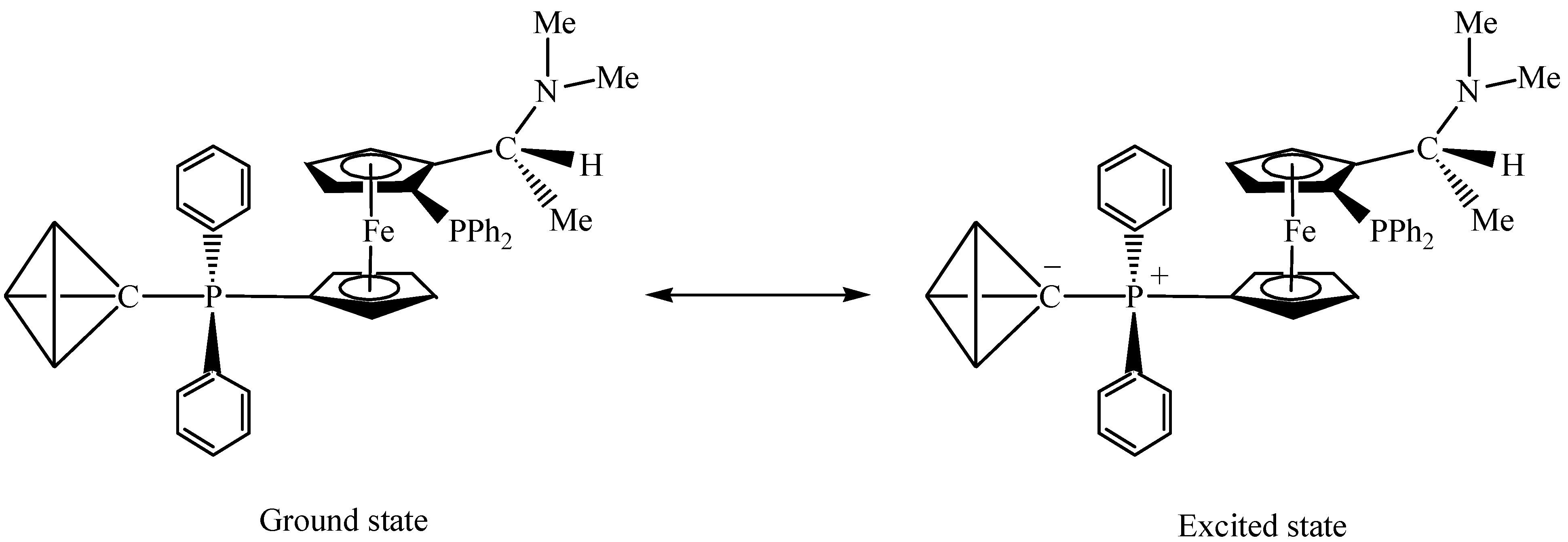
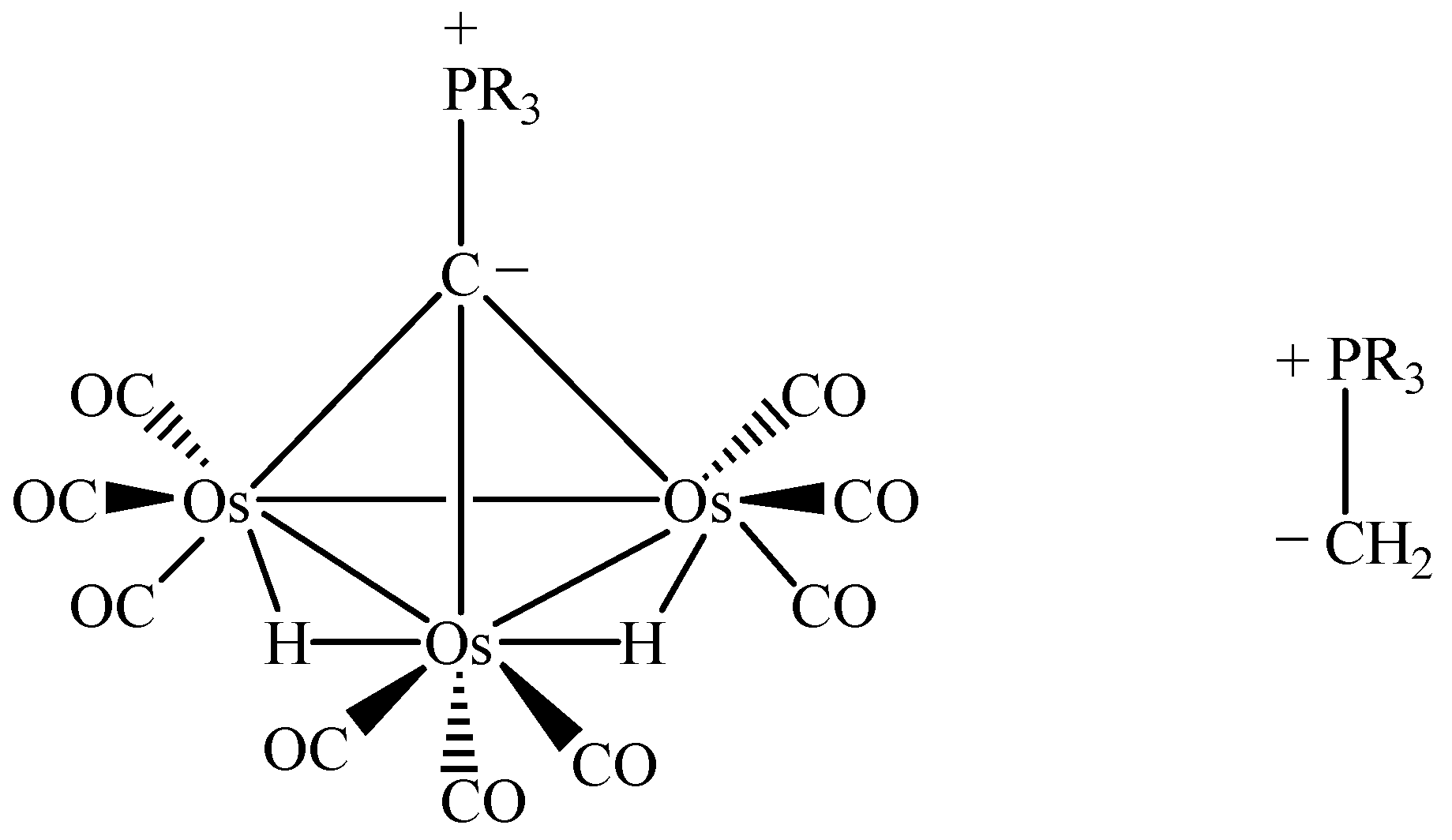
In terms of bonding description, complex 1 can be considered to be zwitterionic in nature which creates an overall asymmetric charge distribution. Both spectroscopic (IR) and structural features suggest that the tertiary phosphine molecule adds at the µ3-carbon atom to give an zwitterionic complex containing a phosphonium centre and a negative charge on the cluster framework (Scheme 2) [19,22]. To count the cluster valence electrons, complex 1 is a 47 electron system which can provide evidence for the electron-withdrawing nature of the Os3C core. Basically, such an zwitterionic formulation of the cluster may be viewed as the organometallic analogue of the phosphorus ylide R3P=CH2, which is a well-known versatile reagent in synthetic organic chemistry (see Scheme 3) [23]. It is believed that this system would provide a fertile area of metal-assisted organic reactions of phosphorus ylides and can hence gain a better understanding of some of the complicated reactions in the metal/phosphine/C1 system. As reported earlier by Vahrenkamp and co-workers, the formation of [Ru3(µ-H)2(CO)9(µ3-CPPh3)] uses up all four hydrogen atoms of methane in the sequence CH4 → CH3I → CH2=PPh2 → [Ru3(µ-H)2(CO)9(µ3-CPPh3)] [24]. The resulting cluster unit should then be a useful starting point for subsequent organic reaction. This observation has also indirectly been shown during the syntheses of the cluster compounds [Co2Fe(CO)9(µ3-CCH2PPh3)] [25] and [Co2Fe(CO)9(µ3-CCH2CH2PPh3)] [26]. The Os3C unit in 1 is isoelectronic to the one in [Ru3(µ-H)2(CO)9(µ3-CPPh3)].
Scheme 2.

Scheme 3.

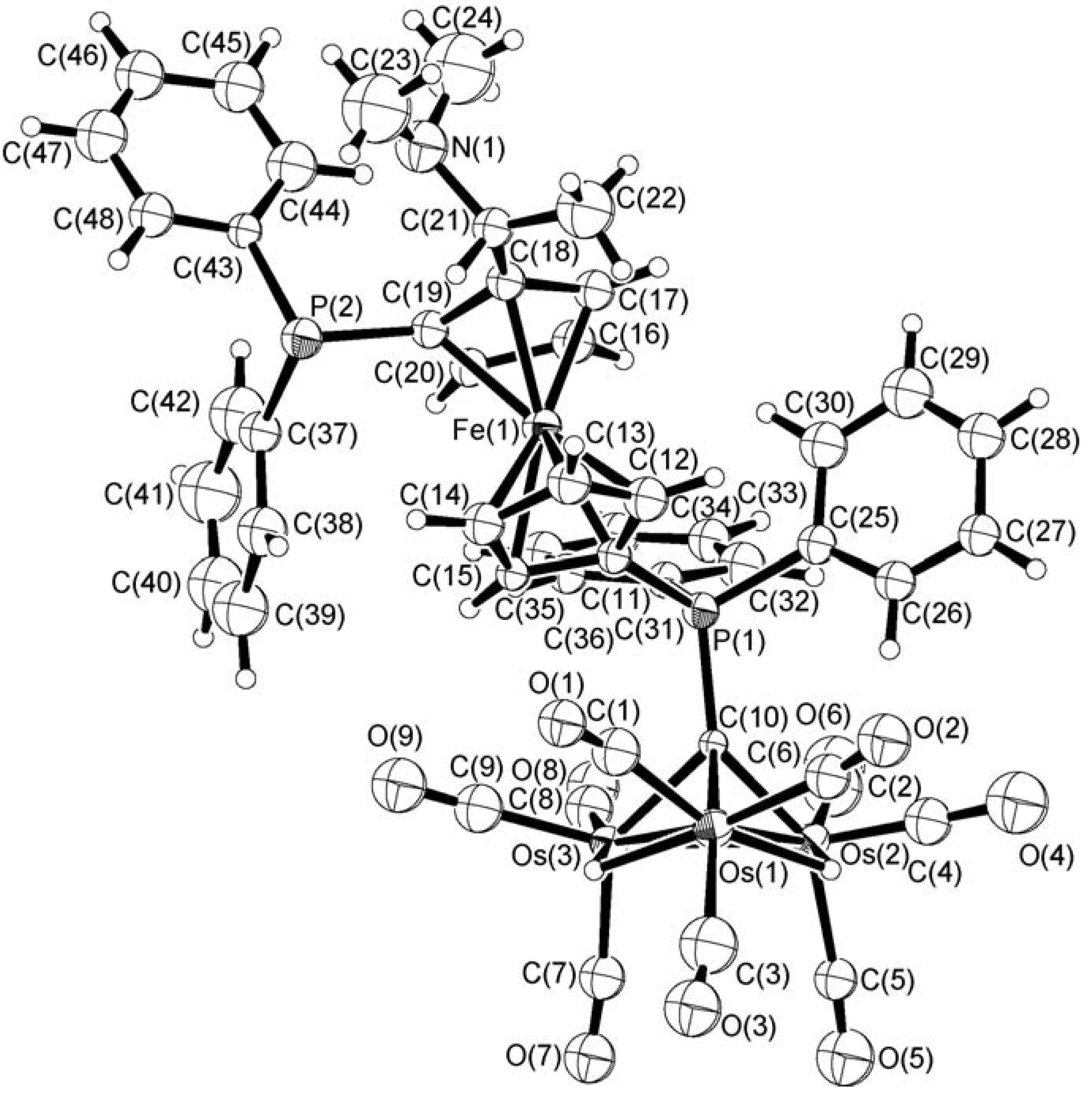
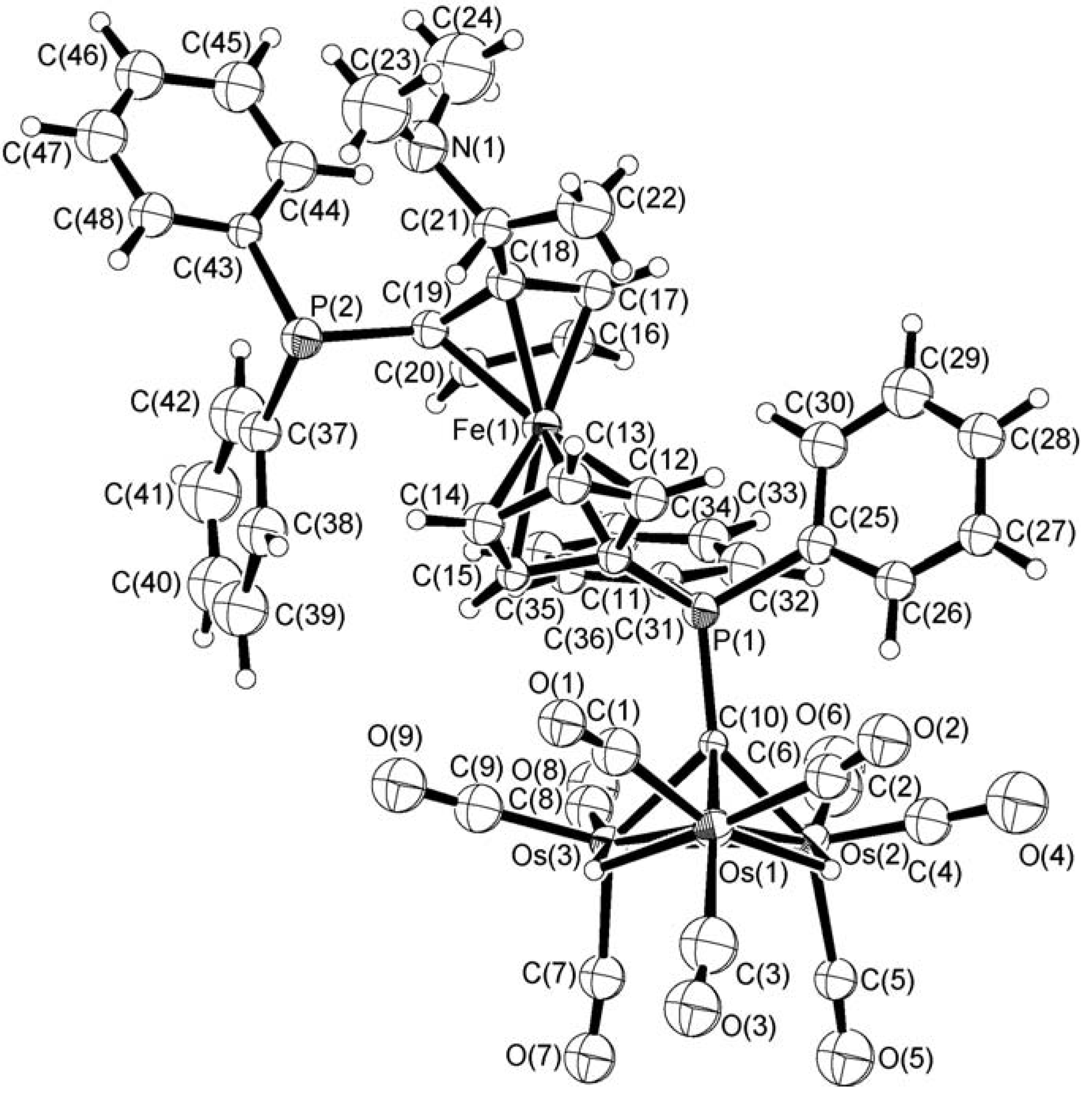
In order to establish unequivocally the exact nature of the bonding of the ferrocenylphosphine ligand to the cluster unit and the stereochemical arrangement of the chiral complex 1, an X-ray structure analysis was carried out on a single crystal obtained by slow evaporation of a n-hexane/CH2Cl2 solution of 1 at room temperature for 2 days. Figure 1 depicts the molecular structure of 1 which includes the atomic numbering scheme used. Relevant bond distances and angles are given in Table 1. Complex 1 consists of a triosmium alkylidyne metal core with the chiral ligand (R,S)-bppfa coordinated directly through the PPh2 group on the C5H4 ring to the apical carbon atom. The other PPh2 moiety on the disubstituted cyclopentadienyl ring remains pendant, probably due to steric reasons. Potential energy calculations [27] suggest that the two hydrides, which clearly show up in the 1H NMR spectrum, bridge the edges Os(1)–Os(2) [2.896(2) Å] and Os(1)–Os(3) [2.892(2) Å]. The unbridged Os−Os bond is shorter at 2.739(2). The mean Os–C(alkylidyne) bond distance is 2.08 Å. There are three terminal carbonyl ligands on each metal vertex. The dihedral angle between the η5-C5H4 and η5-C5H3 planes in the ferrocene unit is 1.5˚, whilst these two fragments deviate from the eclipsed conformation by 7.4˚. From the known absolute configuration of the starting ferrocenyl ligand which contains both central and planar elements of chirality (planar chirality due to 1,2-unsymmetrically disubstituted ferrocene structure), the configuration of 1 is determined to be (R) at C(21) and (S) for the bis(diphenylphosphino)ferrocenyl moiety by X-ray analysis.
Figure 1.
The molecular structure of 1 with atomic labeling and thermal ellipsoids are shown at the 30% probability level.
Figure 1.
The molecular structure of 1 with atomic labeling and thermal ellipsoids are shown at the 30% probability level.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Os(1)−Os(2) | 2.896(2) | Os(1)−Os(3) | 2.892(2) | Os(2)−Os(3) | 2.739(2) | ||
| Os(1)−C(10) | 2.11(3) | Os(2)−C(10) | 2.04(3) | Os(3)−C(10) | 2.09(2) | ||
| P(1)−C(10) | 1.83(2) | P(1)−C(11) | 1.80(3) | P(1)−C(25) | 1.84(3) | ||
| P(1)−C(31) | 1.80(3) | P(2)−C(19) | 1.81(3) | P(2)−C(37) | 1.84(3) | ||
| P(2)−C(43) | 1.81(3) | Fe−ring(C5H4) | 1.631 | Fe−ring(C5H5) | 1.648 | ||
| Os(2)−Os(1)−Os(3) | 56.49(5) | Os(1)−Os(2)−Os(3) | 61.69(5) | ||||
| Os(1)−Os(3)−Os(2) | 61.82(5) | Os(1)−C(10)−P(1) | 127(1) | ||||
| Os(2)−C(10)−P(1) | 132(1) | Os(3)−C(10)−P(1) | 123(1) | ||||
| C(10)−P(1)−C(11) | 114(1) | C(11)−P(1)−C(25) | 104(1) | ||||
| C(11)−P(1)−C(31) | 109(1) | C(19)−P(2)−C(37) | 99(1) | ||||
| C(19)−P(2)−C(43) | 104(1) | N(1)−C(21)−C(18) | 106(2) | ||||
| N(1)−C(21)−C(22) | 120(2) | C(18)−C(21)−C(22) | 116(3) | ||||
Conclusions
A new triosmium alkylidyne carbonyl cluster bearing an Os3C core covalently appended to a chiral ferrocenylphosphine was prepared and structurally characterized. The isolation of complex 1 does ensure a non-centrosymmetric crystal packing due to crystallization in the non-centric space group P21 which renders the material potentially useful for second harmonic generation.
Experimental
General
All reagents and chemicals were obtained from Aldrich Chemical Company (USA) and were used as received unless otherwise noted. The compound [Os3(µ-H)3(CO)9(µ3-CCl)] was prepared by the literature method [28]. None of the compounds reported are particularly air-sensitive, however, all reactions, unless otherwise stated, were carried out under an atmosphere of dry dinitrogen using standard Schlenk techniques and monitored by solution IR spectroscopy (CO stretching region). Solvents were dried over appropriate reagents according to literature methods and freshly distilled before use [29]. Thin layer chromatographic plates were purchased from E. Merck, consisting of 20 × 20 cm glass plate coated with Merck Kieselgel 60 GF254 silica gel of 0.25 mm thick, while thick layer chromatographic plates of the same coating were prepared in our laboratory. Column chromatography were performed in the air using silica gel of size 70−230 mesh. Infrared spectra were recorded on a Nicolet Magna 550 Series II FTIR spectrometer using CaF2 solution cells (path length 0.5 mm). The NMR spectra were recorded on a JEOL EX270 FT-NMR spectrometer using deuteriated solvents as lock and reference. Chemical shifts were reported in ppm relative to SiMe4 for 1H and 13C and 85% H3PO4 for 31P NMR spectra. Fast atom bombardment mass spectra were recorded on a Finnigan MAT 95 instrument using m-nitrobenzyl alcohol as the matrix solvent.
Synthesis of (R,S)-[Os3(μ-H)2(CO)9{μ3-CPPh2(η5-C5H4)Fe(η5-C5H3(PPh2)CH(Me)NMe2)}] (1)
To a solution of [Os3(µ-H)3(CO)9(µ3-CCl)] (30 mg, 0.03 mmol) and the chiral ferrocenylphosphine ligand (R,S)-bppfa (188 mg, 0.30 mmol) in CH2Cl2 (20 mL) was added DBU (1 equivalent) and the mixture was stirred at room temperature for 30 minutes. Following evaporation of the solvent, the residue was chromatographed by TLC using n-hexane/acetone (90:10, v/v) to afford a yellow mixture containing the title complex 1 (Rf ~ 0.2), as evidenced by IR spectroscopy. Subsequent repurification by TLC using n-hexane/acetone (85:15, v/v) led to the isolation of the pure compound 1 (Rf ~ 0.4) in 30% yield (13 mg). 1H-NMR (CD2Cl2) δ: −19.92 (d, 2H, 3JH−P = 2.8 , Os−H−Os), 1.08 (d, 3H, 3JH−H = 6.84, CHCH3), 1.67 (s, 6H, NMe2), 3.44−4.69 (m, 8H, C5H4 + C5H3 + CHCH3), 7.06−8.10 (m, 20H, C6H5); 31P-{1H} NMR (CD2Cl2) δ: −22.44 (uncoordinated PPh2), 41.86 (μ3-CPPh2); IR (CH2Cl2) cm−1: 2093s, 2055vs, 2024vs, 2015sh, 2001sh, 1979s, 1951m, 1936m; LRMS (FAB) for C48H39NFeO9Os3P2 (M+): Calcd 1463. Found 1463.
X-Ray Crystallography
A yellow single crystal of 1 was mounted on a glass fiber and intensity data were collected at room temperature on an Enraf-Nonius CAD4 diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) using the ω−2θ scan method. Intensity data were corrected for Lorentz and polarization effects and semi-empirical absorption corrections (ψ-scan method) were also applied [30]. The structure was solved by a combination of direct methods (SIR88) [31] and difference Fourier techniques and refined on F by full-matrix least-squares analysis. The hydrogen atoms of the bppfa ligand were placed in their idealized positions (C−H 0.95 Å), while all metal hydrides were estimated by potential energy calculations. The absolute configuration of 1 was established by analyzing the trends of Fo vs. Fc for all the observed reflections and its Bijvoet mate. It is consistent with the configuration of the starting phosphine ligand. The crystal data and structure refinement results are listed in Table 2 [32].
| Empirical formula | C48H39NFeO9Os3P2 |
| Molecular weight | 1462.23 |
| Crystal size (mm) | 0.38 × 0.20 × 0.16 |
| Crystal system | Monoclinic |
| Space group | P21 |
| a (Å) | 10.843(1) |
| b (Å) | 11.059(1) |
| c (Å) | 20.224(3) |
| α (°) | 90 |
| β (º) | 94.79(2) |
| γ (°) | 90 |
| U (Å3) | 2416.5(8) |
| μ (Mo−Kα) (mm−1) | 0.803 |
| Dcalc (g cm−3) | 2.009 |
| Z | 2 |
| F(000) | 1380 |
| 2θ Range (°) | 2.0−45.0 |
| Reflections collected | 3557 |
| Unique reflections | 3369 |
| Rint | 0.057 |
| Observed reflections [I > 3σ(I)] | 2645 |
| No. of parameters | 275 |
| R, Rw [I > 3σ(I)] | 0.048, 0.050 |
| Goodness-of-fit on F | 1.82 |
| Residual extrema in final diff. map (e Å−3) | 1.74 to –1.33 |
References and Notes
- Long, N. J. Angew. Chem. Int. Ed. Engl. 1995, 34, 21–38.
- Colbert, M. C. B.; Edwards, A. J.; Lewis, J.; Long, N. J.; Page, N. A.; Parker, D. G.; Raithby, P. R. J. Soc. Dalton Trans. 1994, 2589, 2589–2590. [CrossRef]
- Consiglio, G.; Morandini, F. Chem. Rev. 1987, 87, 761–778.
- Morandini, F.; Pilloni, G.; Consiglio, G.; Sironi, A.; Moret, M. Organometallics 1993, 12, 3495–3503.
- Auburn, P. R.; Mackenzie, P. B.; Bosnich, B. J. Am. Chem. Soc. 1985, 107, 2033–2046.
- Sinigalia, R.; Michelin, R. A.; Pinna, F.; Strukui, G. Organometallics 1987, 6, 728–734.
- Wong, W.-Y.; Wong, W.-T. J. Chem. Soc. Dalton Trans. 1995, 2831–2836. [CrossRef]
- Hayashi, T.; Kumada, M.; Higuchi, T.; Hirotsu, K. J. Organomet. Chem. 1987, 334, 195–203.
- Landis, C. R.; Sawyer, R. A.; Somsook, E. Organometallics 2000, 19, 994–1002.
- Manzano, B. R.; Jalón, F. A.; Gómez-de la Torre, F.; López-Agenjo, A. M.; Rodríguez, A. M.; Mereiter, K.; Weissensteiner, W.; Sturm, T. Organometallics 2002, 21, 789–802.
- Bandoli, G.; Dolmella, A. Coord. Chem. Rev. 2000, 209, 161–196.
- Togni, A.; Hayashi, T. (Eds.) Ferrocenes; VCH: New York, 1995.
- Brunner, H.; Zettlmeier, W. Handbook of Enantioselective Catalysis with Transition Metal Compounds; VCH: Weinheim, Germany, 1993. [Google Scholar]
- Sawamura, M.; Ito, Y. Chem. Rev. 1992, 92, 857–871.
- Ojima, I. (Ed.) Catalytic Asymmetric Synthesis; VCH: Weinheim, Germany, 1993.
- Wang, L.; Kwok, W. H.; Chan, A. S. C.; Tu, T.; Hou, X.; Dai, L. Tetrahedron: Asymmetry 2003, 14, 2291–2295.
- Gotov, B.; Toma, S.; Macquarrie, D. J. New J. Chem. 2000, 24, 597–602.
- Chan, S.; Wong, W.-Y.; Wong, W.-T. J. Organomet. Chem. 1994, 474, C30–C33.
- Wong, W.-Y.; Wong, W.-T. J. Chem. Soc. Dalton Trans. 1995, 2735–2740. [CrossRef]
- Wong, W.-Y.; Wong, W.-T. J. Organomet. Chem. 1999, 584, 48–57.
- Johnson, B. F. G.; Lahoz, F. J.; Lewis, J.; Prior, N. D.; Raithby, P. R.; Wong, W.-T. J. Chem. Soc. Dalton Trans. 1992, 1701–1708. [CrossRef]
- Henrick, K.; McPartlin, M.; Deeming, A. J.; Hasso, S.; Manning, P. J. Chem. Soc. Dalton Trans. 1982, 899–906. [CrossRef]
- Albright, T. A.; Freeman, W. J.; Schweizer, E. E. J. Org. Chem. 1975, 40, 3437–3441.
- Bohle, D. S.; Heineke, D.; Tiripicchio, A.; Camellini, M. T.; Vahrenkamp, H. Angew. Chem. Int. Ed. Engl. 1990, 29, 896–906. [CrossRef]
- Albiez, T.; Vahrenkamp, H. Angew. Chem. Int. Ed. Engl. 1987, 26, 572–573. [CrossRef]
- Heineke, D.; Vahrenkamp, H. Organometallics 1990, 9, 1697–1699.
- Orpen, A. G. J. Chem. Soc. Dalton Trans. 1986, 453–455.
- Keister, J. B.; Horling, T. L. Inorg. Chem. 1980, 19, 2304–2307.
- Armarego, W. L. F.; Perrin, D. D. Purification of Laboratory Chemicals, 4th Ed. ed; Butterworth-Heinemann: Oxford, U.K., 1996. [Google Scholar]
- North, A. C. T.; Phillips, D. C.; Mathews, F. S. Acta Crystallogr. Sect. A. 1968, 24, 351–359.
- Burla, M. C.; Camalli, M.; Cascarano, G.; Giacovazzo, C.; Polidori, G.; Spagna, R.; Viterbo, D. J. Appl. Cryst. 1989, 22, 389–393. [CrossRef]
- 32. CCDC-244429 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via the URL http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223 336033; e-mail: [email protected])
- Sample Availability: Available from the authors.
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Wong, W.-Y. Synthesis and Crystal Structure of a New Triosmium Alkylidyne Carbonyl Cluster Containing a Chiral Ferrocenylphosphine Ligand. Molecules 2005, 10, 640-648. https://doi.org/10.3390/10060640
AMA Style
Wong W-Y. Synthesis and Crystal Structure of a New Triosmium Alkylidyne Carbonyl Cluster Containing a Chiral Ferrocenylphosphine Ligand. Molecules. 2005; 10(6):640-648. https://doi.org/10.3390/10060640
Chicago/Turabian StyleWong, Wai-Yeung. 2005. "Synthesis and Crystal Structure of a New Triosmium Alkylidyne Carbonyl Cluster Containing a Chiral Ferrocenylphosphine Ligand" Molecules 10, no. 6: 640-648. https://doi.org/10.3390/10060640