3.1. Validation of the Chemical Reaction Scheme
The chemical reaction scheme was applied to irreversible first-order reaction and reversible first-order reaction , such as nuclear decay and transformation of isomers. The reactions take place in a square enclosure with area 200 × 200 (lattice units). Every site was initially filled with six A-type particles and bounce-back type boundary condition was employed to all walls, where the momentum of particle is directly reversed while the substance state keeps unchanged during the collision process. Additionally, no heat transfer was taken into consideration in these two cases.
Molecules collide and react at random. Nevertheless, the time evolution of macroscopic amounts or concentrations of molecules is usually quite reproducible due to the reproducibility of experimental conditions. Such laws are called deterministic. More detailed introduction can be found in reference [
32]. For reaction
, the deterministic half-life period can be obtained by
, where
r represents the probability of one
A-type particle changes to one
B-type particle at each time, while the half-life period means the iterations needed for reducing the number of
A-type particle by half during the simulation process. The reaction probability,
r, from
A to
B was set to be 0.03, 0.04 and 0.05, and
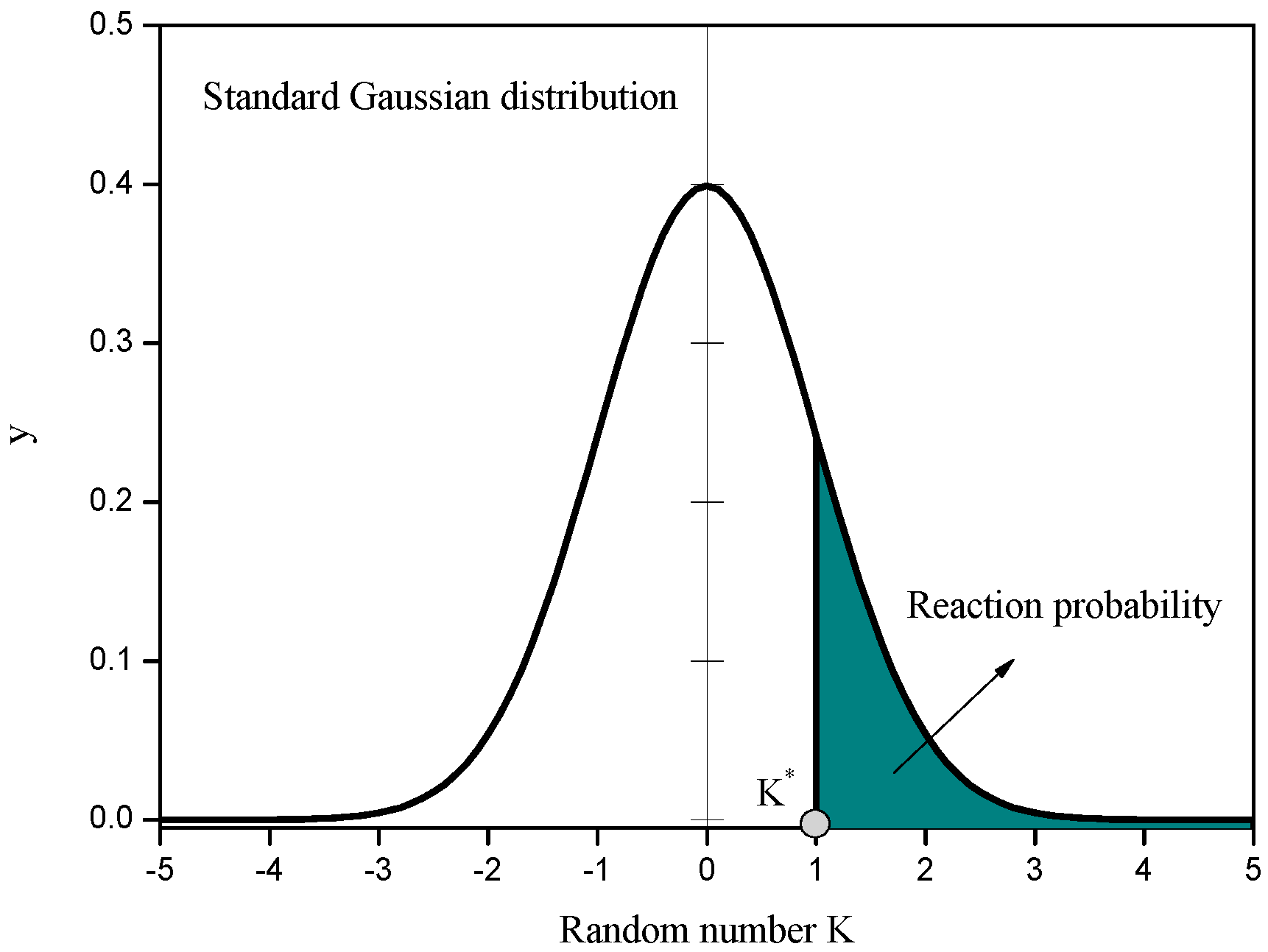
K* was determined as 1.88, 1.75 and 1.65, respectively. During the collision process, a normal distributed random number was generated to compare with
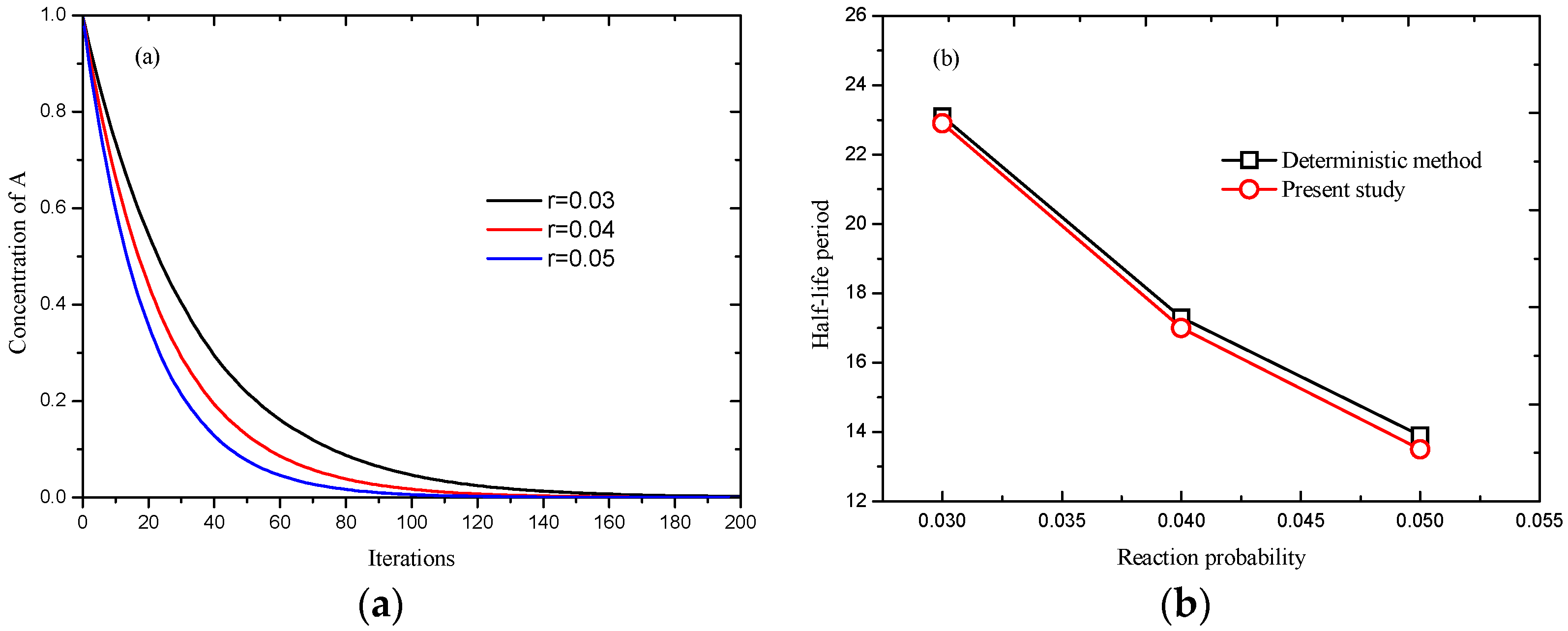
K* to decide to react or not. As shown in
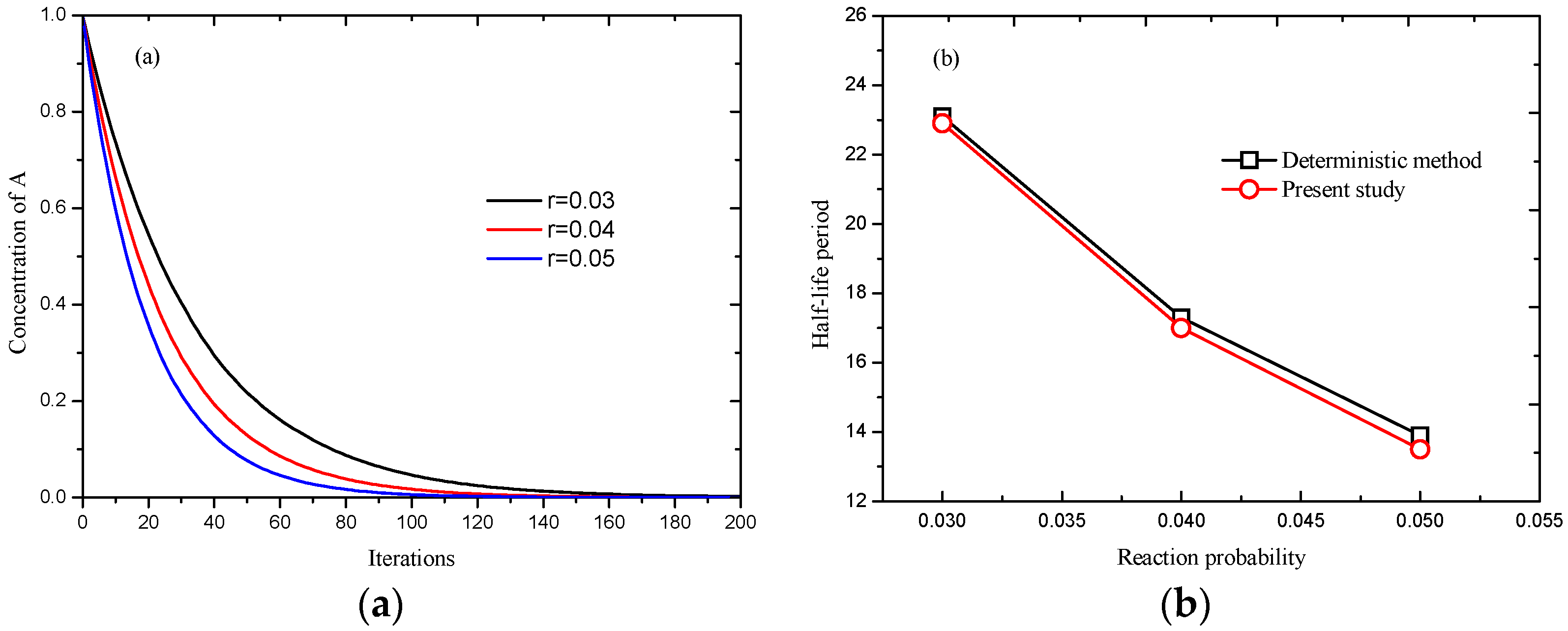
Figure 4, it can be observed that the concentration of particle
A, obtained by Equation (10), decreases with increasing reaction time, and the reaction rate,
i.e., the slope of curve in
Figure 4a, increases as reaction probability increases.
Figure 4b shows reasonable agreement has been achieved between deterministic method and present simulation. Additionally, for a system with 100 × 100, and reduction probability
r = 0.001, the deterministic half-life period is 693.1 iterations, Seybold
et al. [
17] reported 688.3 ± 9.7 iterations, and 691 iterations using present model, which also indicates the feasibility of present model in reaction systems.
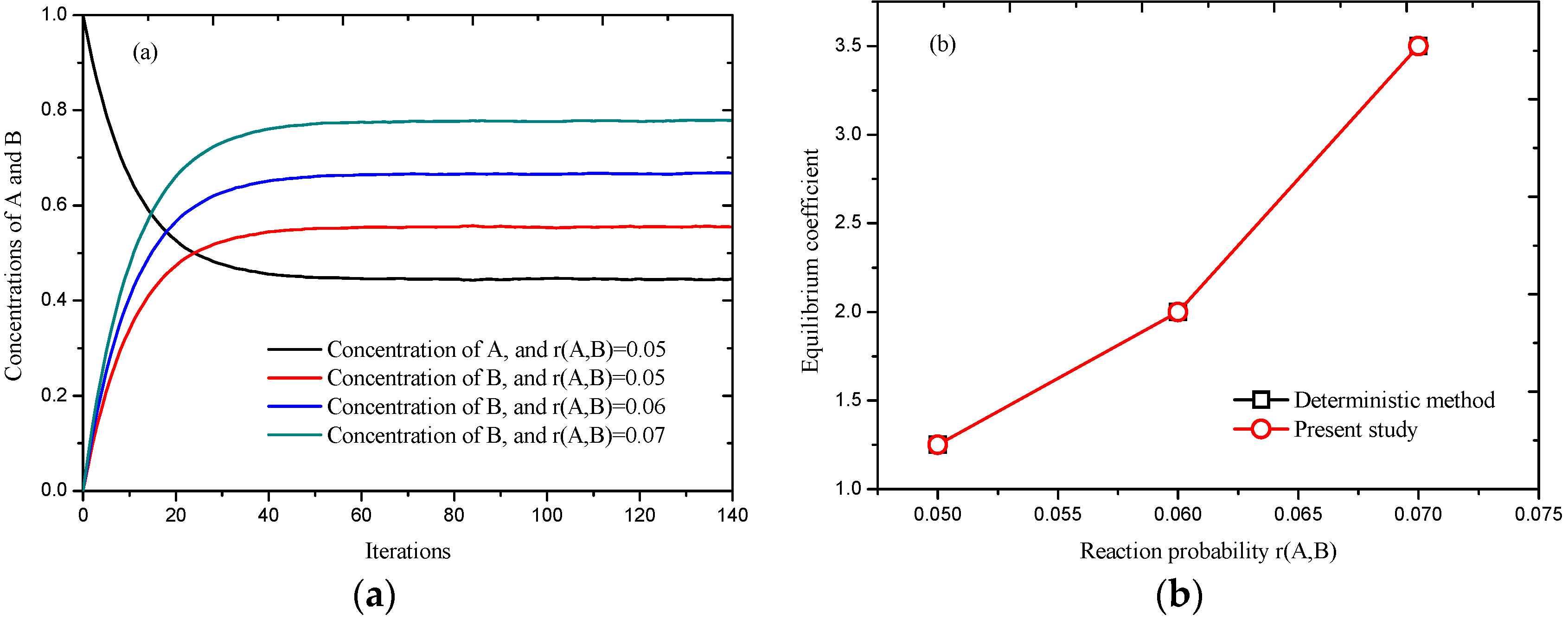
While
is an equilibrium system, from the law of mass action, the deterministic equilibrium coefficient is defined as
, and the equilibrium coefficient can be obtained by the ratio of the final concentration of
B and
A as
in a stochastic system, like simulations using lattice gas automata. The reaction probability from
A to
B was set as 0.05, 0.06, and 0.07, corresponding to the reaction probability from
B to A 0.04, 0.03, and 0.02, and for reaction probability of 0.02 and 0.07,
K* is 2.05 and 1.48, respectively. Similar results are obtained compared to
, as shown in
Figure 5, however,
Figure 5a presents a platform as time advances, meaning an equilibrium state has been reached.
Figure 5b indicates that good agreement has been achieved between the stochastic method and deterministic method. The chemical reaction scheme will be further used for the simulations of gas–solid reaction described latter.
Figure 4.
Simulation of , (a) processes at different reaction probabilities; and (b) comparison of results obtained by deterministic method and present model.
Figure 4.
Simulation of , (a) processes at different reaction probabilities; and (b) comparison of results obtained by deterministic method and present model.
Figure 5.
Simulation of , (a) processes at different reaction probabilities; and (b) comparison of results obtained by deterministic method and present model.
Figure 5.
Simulation of , (a) processes at different reaction probabilities; and (b) comparison of results obtained by deterministic method and present model.
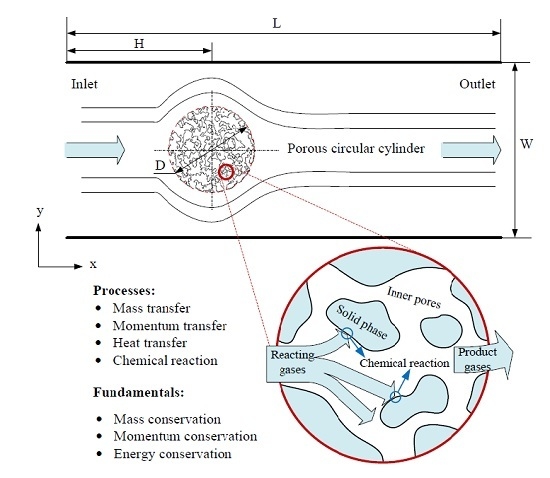
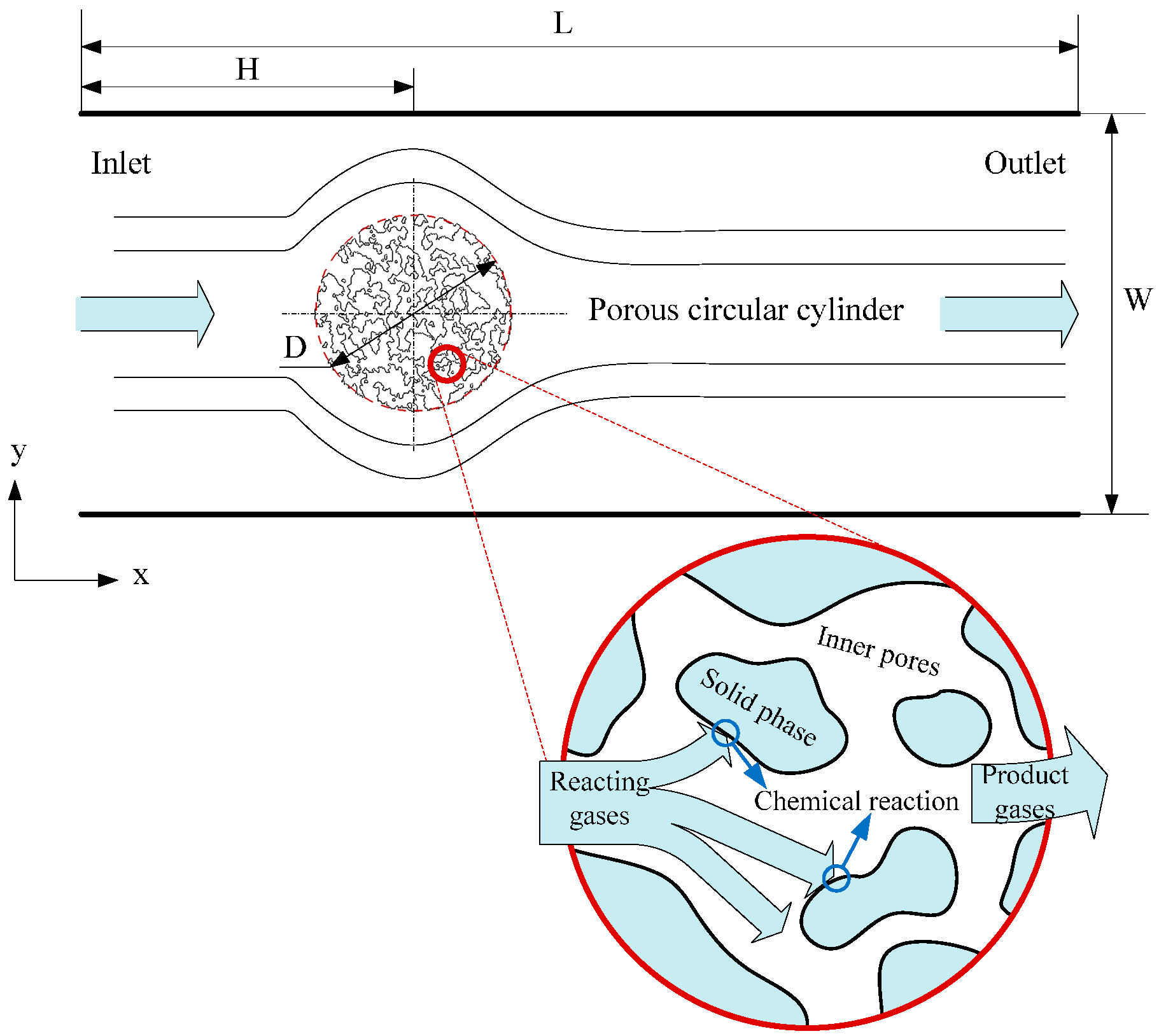
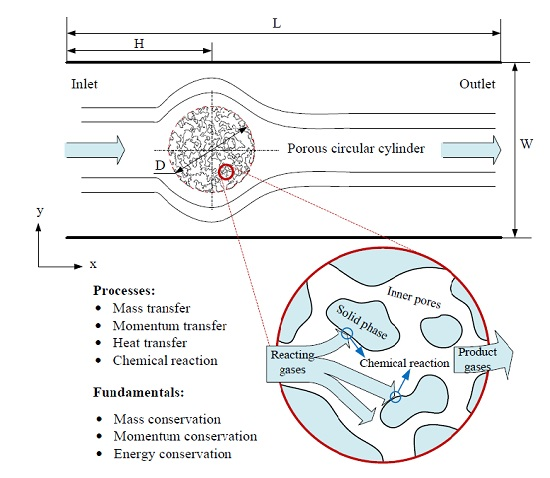
3.2. Heat Transfer and Reaction across a Porous Circular Cylinder
The characteristics of the flow and heat transfer over a solid circular cylinder in a square enclosure and a rectangular channel have been investigated by us using this model previously, showing reasonable feasibility and reliability of the application of this model to the problems of flow and heat transfer, details can be found in reference [
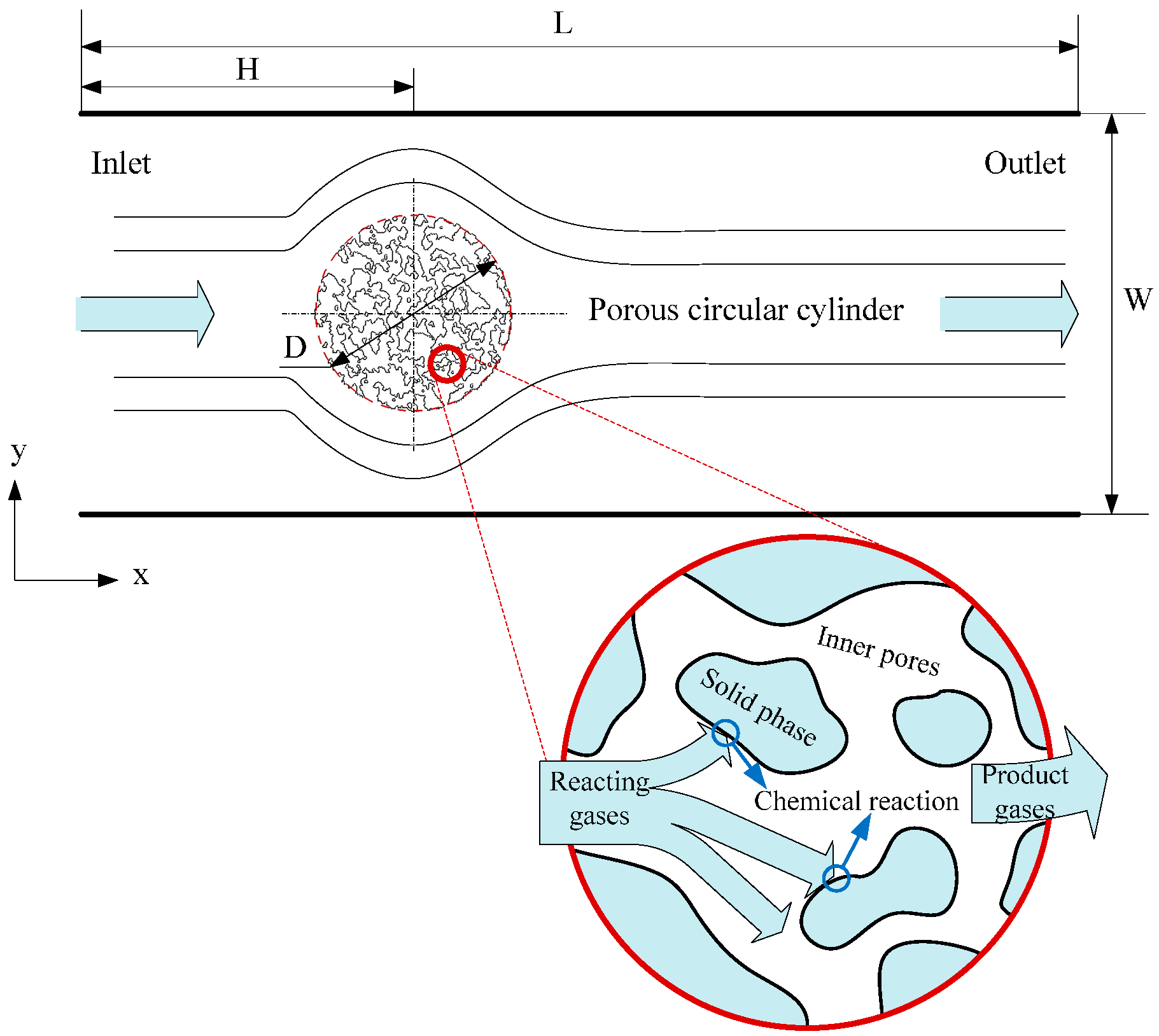
33]. Herein, simulations of flow, heat transfer and reaction around and through a porous circular cylinder in a channel were carried out. The schematic diagram of the simulated system is shown as
Figure 6, where a porous circular cylinder with diameter D = 1/4W is placed at the coordinates (x = 1/3L, y = 1/2W) of a channel with width W = 400 and length L = 1200 (lattice unit). Reactant gas entering from the inlet at a velocity
u, flows around and through and react with the porous cylinder. The effects of porous structure, reaction probability and gas velocity at inlet on the characteristics of the system will be further discussed in detail.
The porous media investigated were generated by a comprehensive approach termed as quartet structure generation set (QSGS) [
34,
35], which has been demonstrated capable of generating morphological features close to many real porous media [
34]. Following the steps illustrated by Wang
et al. [
23], a porous two-dimensional cylinder can be generated with a set of three construction parameters, including (i) growing phase (fluid) distribution probability, C
d, which decides initial number of fluid seeds in the system; (ii) directional growth probability of fluid, D
i, which is considered the same for all directions in this work; and (iii) fluid volume fraction P.
Figure 6.
Schematic diagram of the processes happened in the circular cylinder with porous media structure.
Figure 6.
Schematic diagram of the processes happened in the circular cylinder with porous media structure.
Figure 7 presents the inner morphology of the porous circular cylinders of diameter D = 100 (lattice unit) resulted from different combinations of construction parameters, where shaded area is solid phase and the rest to be pores. As can be seen in
Figure 7a–d, the solid phase disappears homogenously with increasing porosity, leading to larger pore sizes; and for
Figure 7e–h, more agglomeration of solid phase,
i.e., less surface area and bigger pore size, appeared for larger directional growth probability; on the contrary, for
Figure 7i–l, more homogenous structure as well as smaller pore size of porous media can be obtained as the distribution probability increases, as a result, more react-able surface area is generated. Porous cylinders of different porosity, pore size, and surface area could be obtained by adjusting the three parameters (C
d, D
i, P), for the purpose of comparison.
Figure 7.
Porous cylinder generated with different construction parameter. (a–d) Effect of porosity on the morphology of cylinder, where Cd = 0.02, Di = 0.2 and (a) P = 0.2, (b) P = 0.4, (c) P = 0.6, and (d) P = 0.8. (e–h) Effect of directional growth probability on the morphology of cylinder, where Cd = 0.02, P = 0.2 and (e) Di = 0.05, (f) Di = 0.15, (g) Di = 0.25, and (h) Di = 0.35. (i–l) Effect of distribution probability on the morphology of cylinder, where Di = 0.2, P = 0.2 and (i) Cd = 0.005, (j) Cd = 0.015, (k) Cd = 0.025, and (l) Cd = 0.035.
Figure 7.
Porous cylinder generated with different construction parameter. (a–d) Effect of porosity on the morphology of cylinder, where Cd = 0.02, Di = 0.2 and (a) P = 0.2, (b) P = 0.4, (c) P = 0.6, and (d) P = 0.8. (e–h) Effect of directional growth probability on the morphology of cylinder, where Cd = 0.02, P = 0.2 and (e) Di = 0.05, (f) Di = 0.15, (g) Di = 0.25, and (h) Di = 0.35. (i–l) Effect of distribution probability on the morphology of cylinder, where Di = 0.2, P = 0.2 and (i) Cd = 0.005, (j) Cd = 0.015, (k) Cd = 0.025, and (l) Cd = 0.035.
Table 1 lists the simulation cases with different parameter sets, with Cases 1 to 4 selected from
Figure 7 to investigate the influence of the parameters of QSGS algorithm on the reaction process, Cases 1, 5 and 6 to study the effect of reaction probability, and Cases 1, 7 and 8 are used to investigate the inlet velocity.
Table 1.
Parameter sets for simulation cases.
Table 1.
Parameter sets for simulation cases.
| Case No. | Parameters of QSGS | r | u |
|---|
| Cd | Di | P |
|---|
| 1 | 0.02 | 0.2 | 0.2 | 0.1587 | 1.0 |
| 2 | 0.02 | 0.2 | 0.4 | 0.1587 | 1.0 |
| 3 | 0.035 | 0.2 | 0.2 | 0.1587 | 1.0 |
| 4 | 0.02 | 0.35 | 0.2 | 0.1587 | 1.0 |
| 5 | 0.02 | 0.2 | 0.2 | 0.0668 | 1.0 |
| 6 | 0.02 | 0.2 | 0.2 | 0.3085 | 1.0 |
| 7 | 0.02 | 0.2 | 0.2 | 0.1587 | 0.5 |
| 8 | 0.02 | 0.2 | 0.2 | 0.1587 | 0.8 |
A conceptual first order heterogeneous reaction between the reaction gas and the porous cylinder illustrated in Equation (10) is considered in this research, with
A being the inlet reactant gas,
B the porous solid material, and
C as the product gas.
This reaction happens at the solid surface and is simply interpreted as one reactant gas particle reacting with one solid site and generating one product gas particle at mesoscopic level, where the (microscopic level) inner structures or properties of the gas particle or solid site are ignored. The conversion of solid phase
B can be defined by the ratio of the number of reacted solid sites to the initial number of solid sites, and a formula as follows is defined
where
Nt is the number of solid sites at time t, and
N0 is the initial number of solid sites. Thus, the reaction rate can be further obtained from the time derivative of Equation (11) as
.
Instead of heat generated during the reaction process, a simplified heat transfer case is considered, where solid sites are set as hot heat source with constant temperature (i.e. 1.0). Gaseous particles will be enhanced to high energy state at impact with solid surface. The gas particles initially entering the channel are of constant low temperature state as 0.
Bounce-back and adiabatic boundary conditions are applied to the channel walls except the outlet which is a free boundary where all particles are released. Solid sites are set as bounce-back boundaries, where gaseous particles will also decide to reaction with according to the chemical reaction scheme.
The simulation carried out in such an order that reaction is not considered in the first 2000 iterations to test the coupling of flow and heat transfer only, also to ensure that the reaction takes place at a stable flow field. Reaction is introduced thereafter to investigate its effect to the flow and heat transfer.
The statistical result of temperature, component concentration, and solid phase conversion are obtained from the simulation by space average. It is noted that the statistics level has impacts on the detail of macroscopic picture. This effect is, however, not discussed in this work. Instead, to ensure consistency, the space averaged results in every 2 × 2 grids are presented for all cases.
3.2.1. Effect of Inner Porous Structure
As discussed in the previous part, the inner porous structure will be influenced by the parameters of QSGS. In this section, the influence of inner structure on the behavior of flow, heat transfer and chemical reaction around and through the cylinder will be investigated. The inlet velocity is constant as 1.0 with a site density ρ = 1.0, and based on FHP-II model, Re = 158.2 using D as the characteristics parameter.
Figure 8 shows the temperature contours around and inside the porous cylinder before chemical reaction takes place at
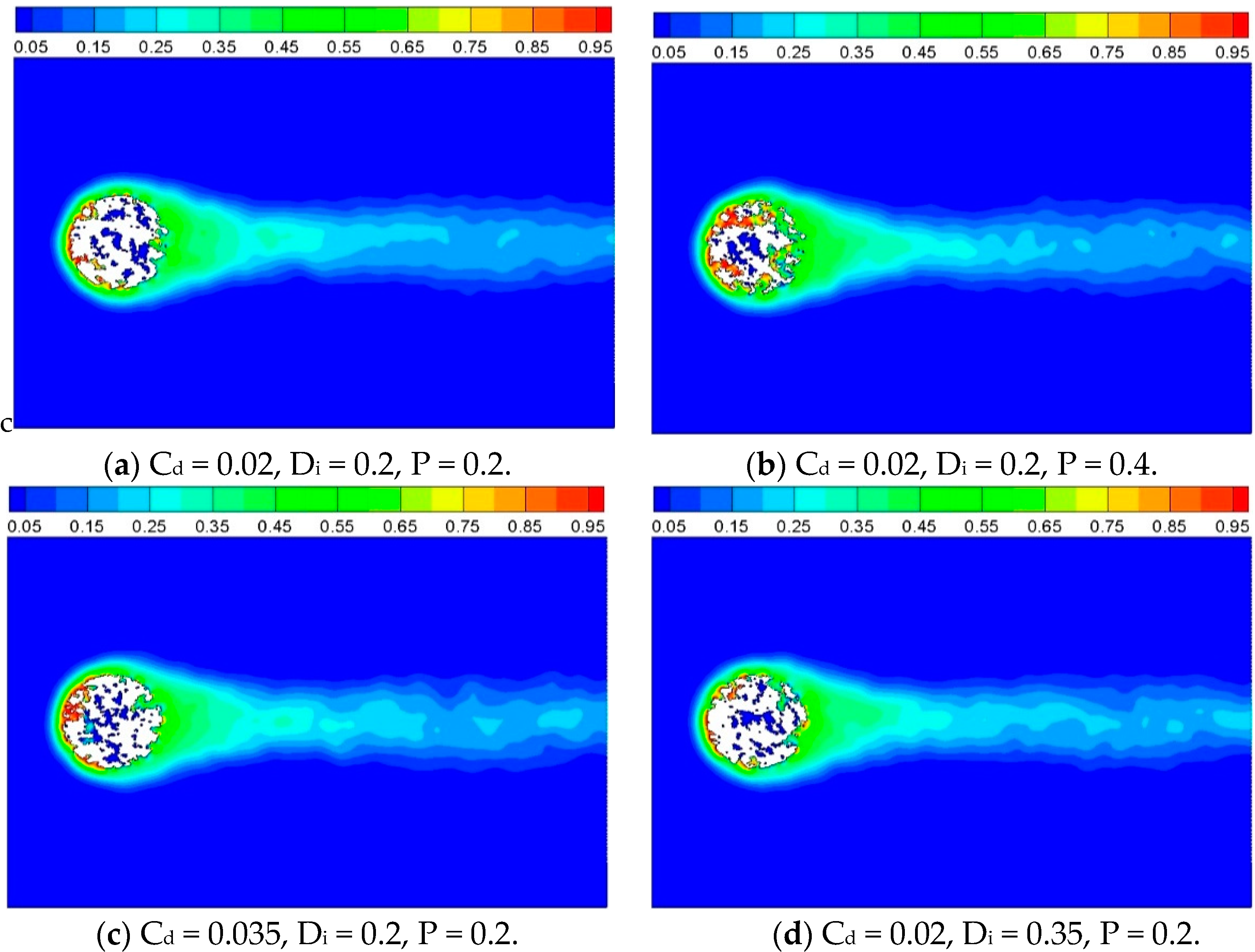
t = 2000 iterations. A large number of fluid particles are activated to high energy state when they flow over and in the porous circular cylinder, forming a high temperature zone at surrounding and inside the porous structure. As time elapses, the high temperature zone extends to the neighboring field gradually, and no clear eddies have developed behind the cylinder. The field of gas velocity also appeared to have impacts on the profile of temperature.
Figure 8.
Temperature contour before reaction with different inner structures, t = 2000 iterations.
Figure 8.
Temperature contour before reaction with different inner structures, t = 2000 iterations.
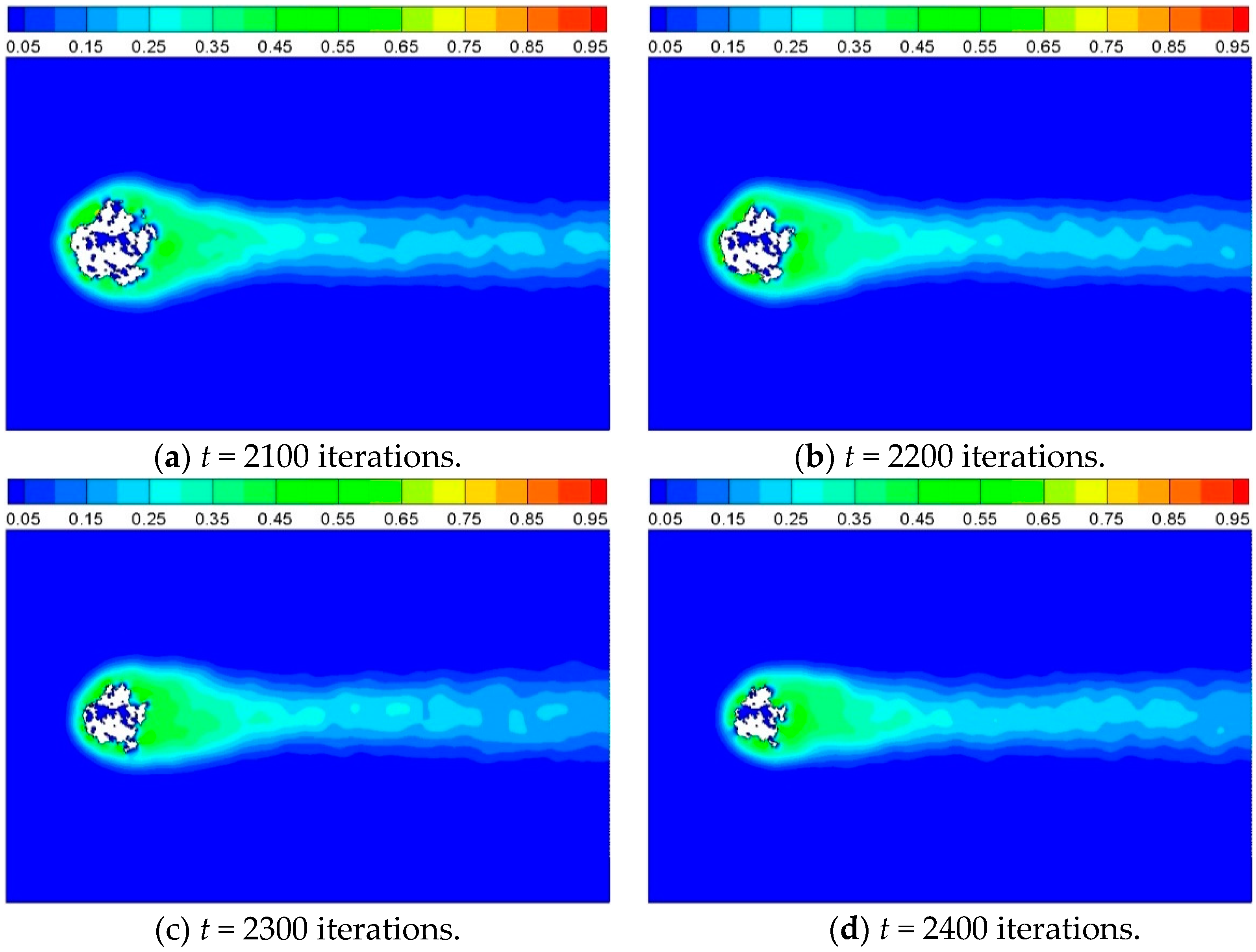
The heterogeneous reaction is started after 2000 iterations. A product zone is then formed as fluid particles flow over and through the porous cylinder and react with the solid sites according to the reaction scheme.
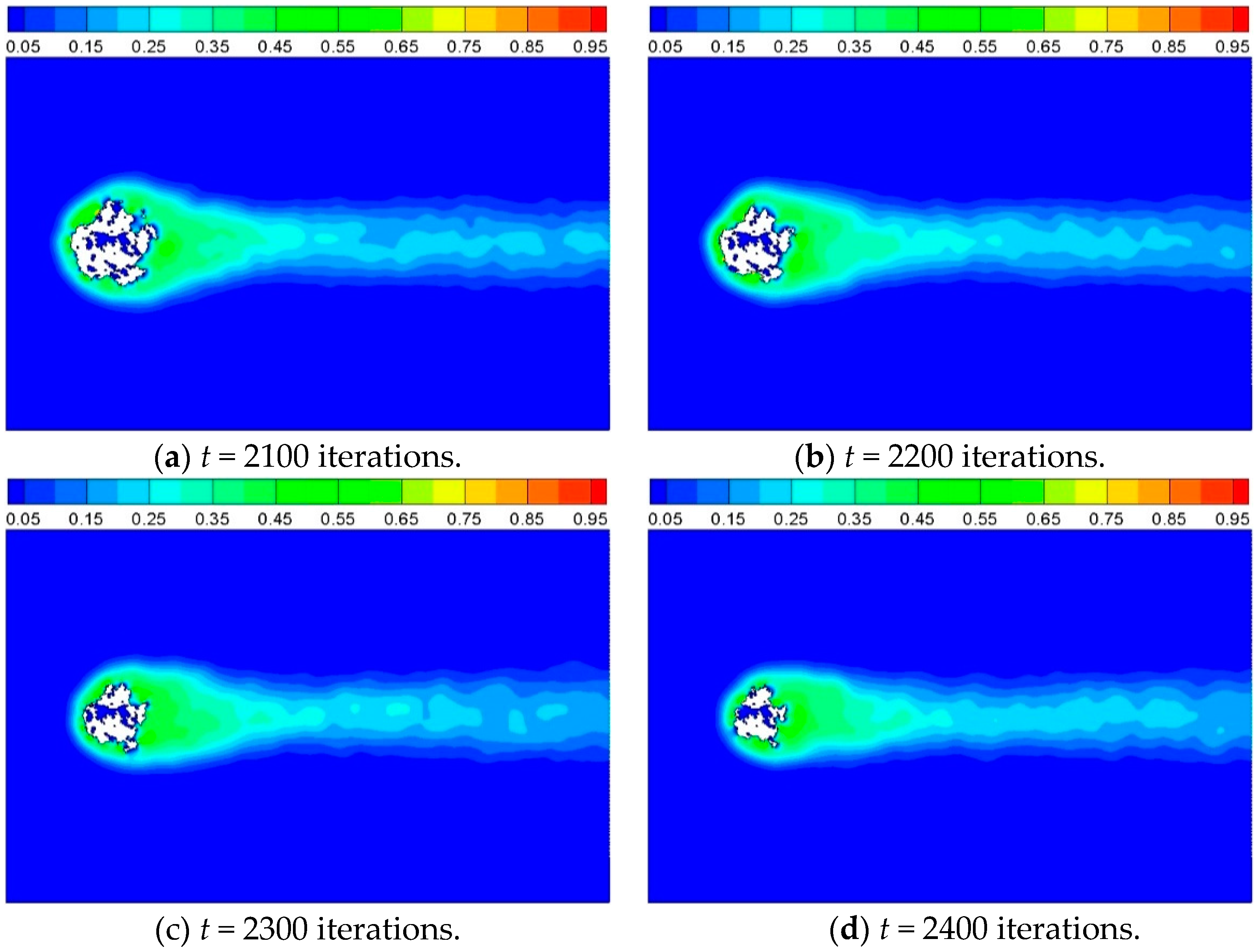
Figure 9 shows the product concentration around the solid cylinder at different times for Case 4. The solid sites disappear gradually as time goes on. It can be observed that the product emerges mainly at some hot points, and distributes homogeneously around the cylinder at the beginning, and then diffuses off from the reaction interface to the surrounding.
Figure 10 shows the corresponding temperature contour around the cylinder for
Figure 9. The structure of cylinder changes with time, thus the heat transfer characteristics changes as a result.
Figure 9.
Product concentration at different times for Case 4, where Cd = 0.02, Di = 0.35, and P = 0.2.
Figure 9.
Product concentration at different times for Case 4, where Cd = 0.02, Di = 0.35, and P = 0.2.
Figure 10.
Temperature contour at different times for Case 4, where Cd = 0.02, Di = 0.35, and P = 0.2.
Figure 10.
Temperature contour at different times for Case 4, where Cd = 0.02, Di = 0.35, and P = 0.2.
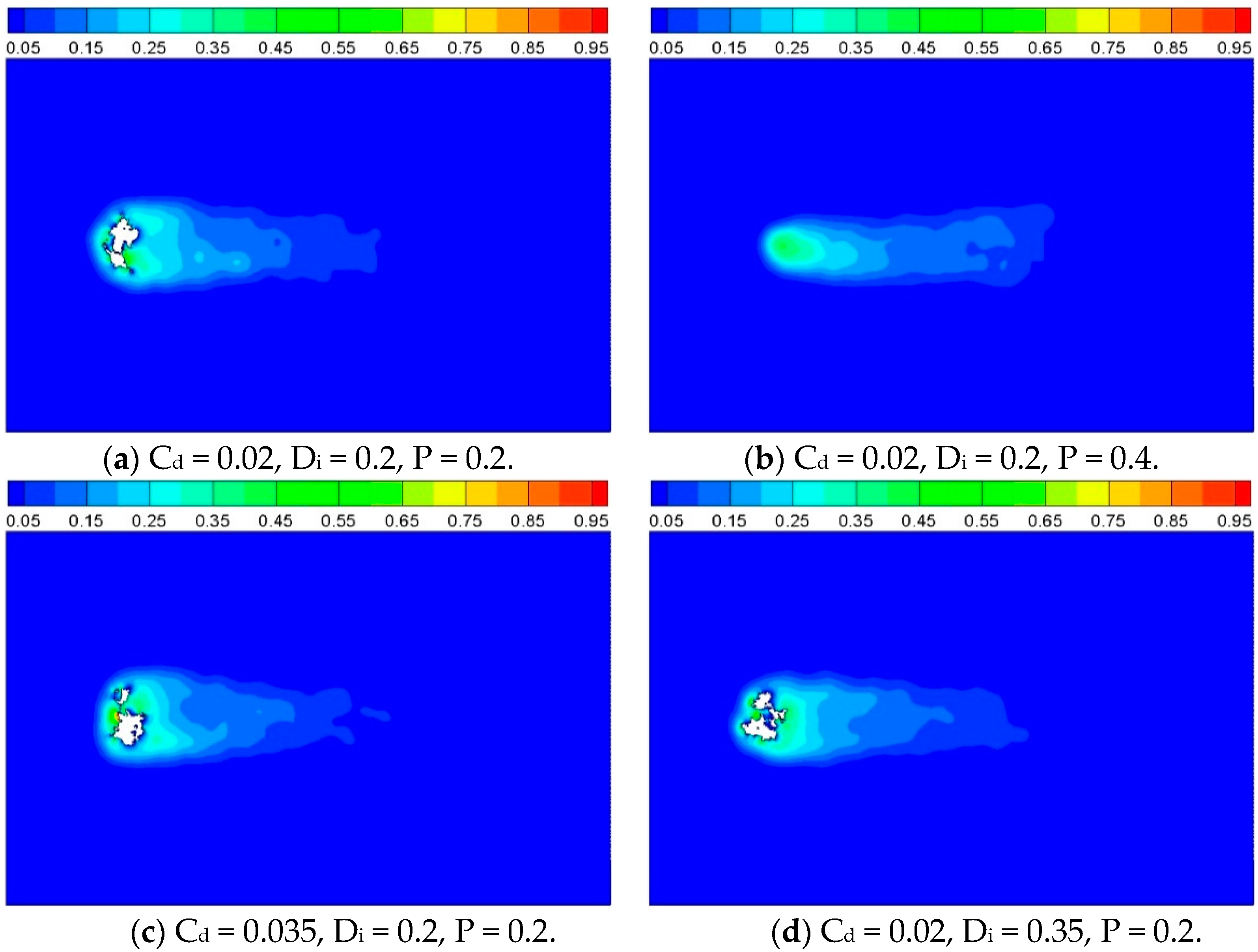
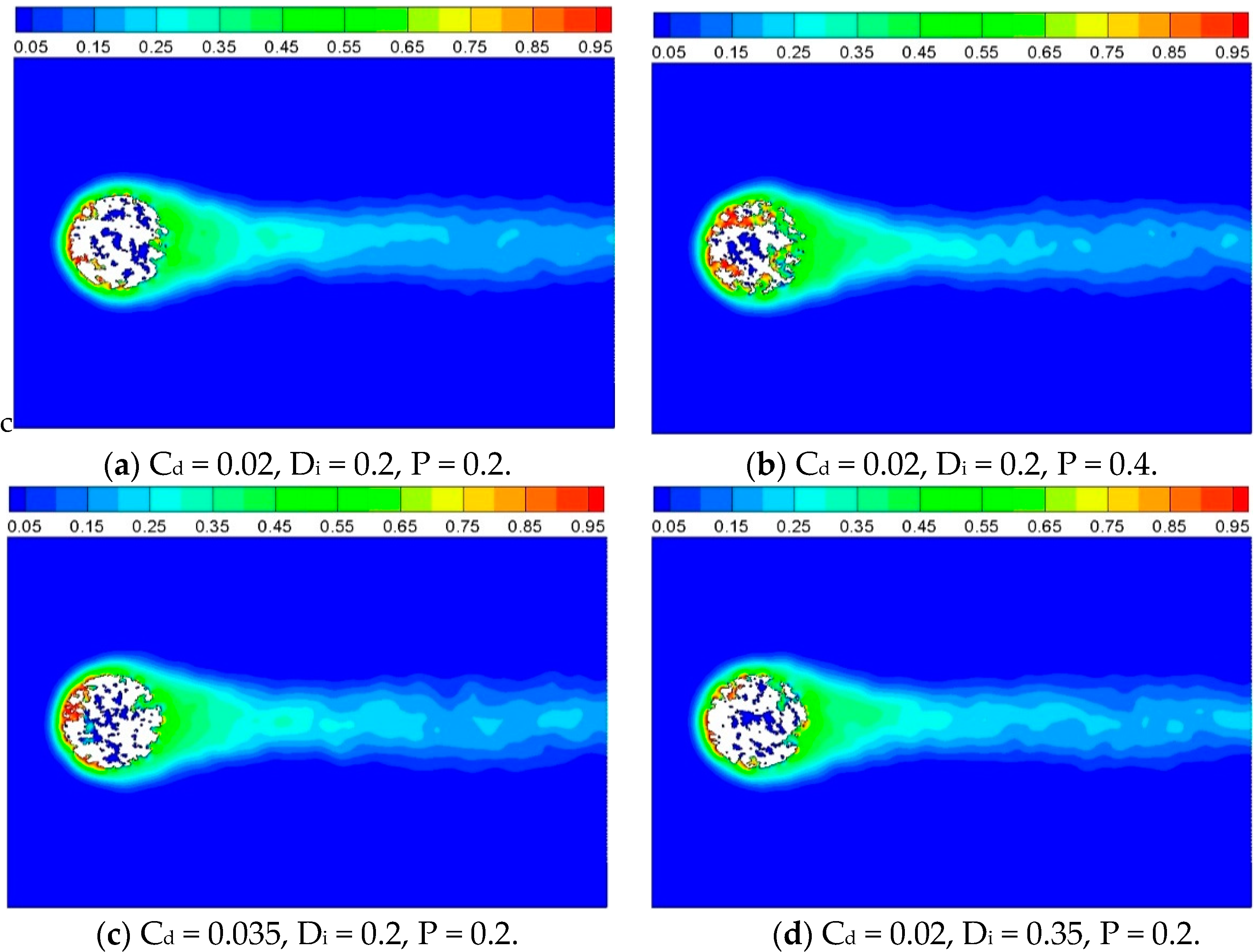
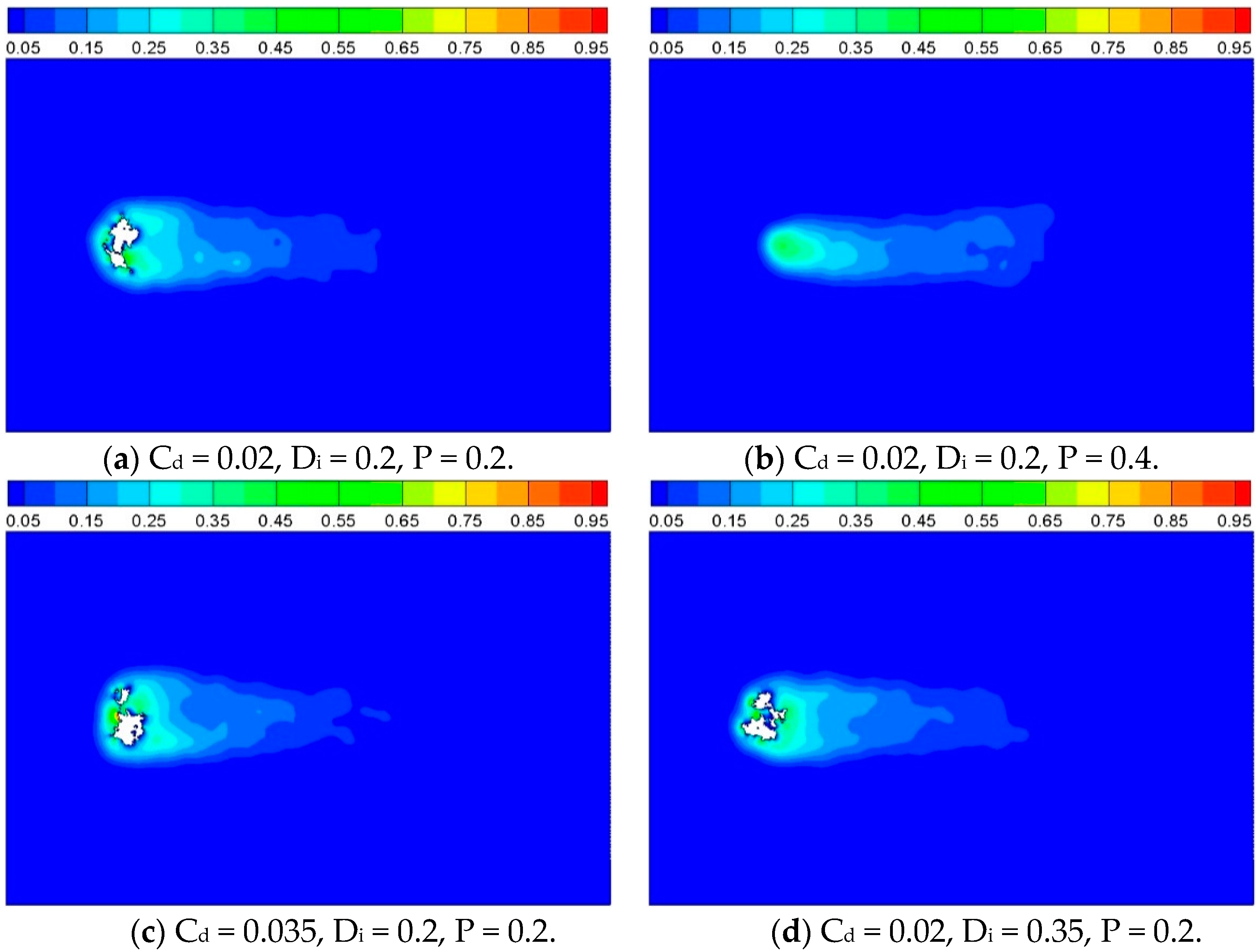
In order to compare the reaction characteristics of computational cases with porous inner structure, contours of product concentration are shown as
Figure 11, for
t = 2500. It can be observed that the solid cylinder has disappeared completely at
t = 2500 for Case 2 which has higher porosity than the other three cases.
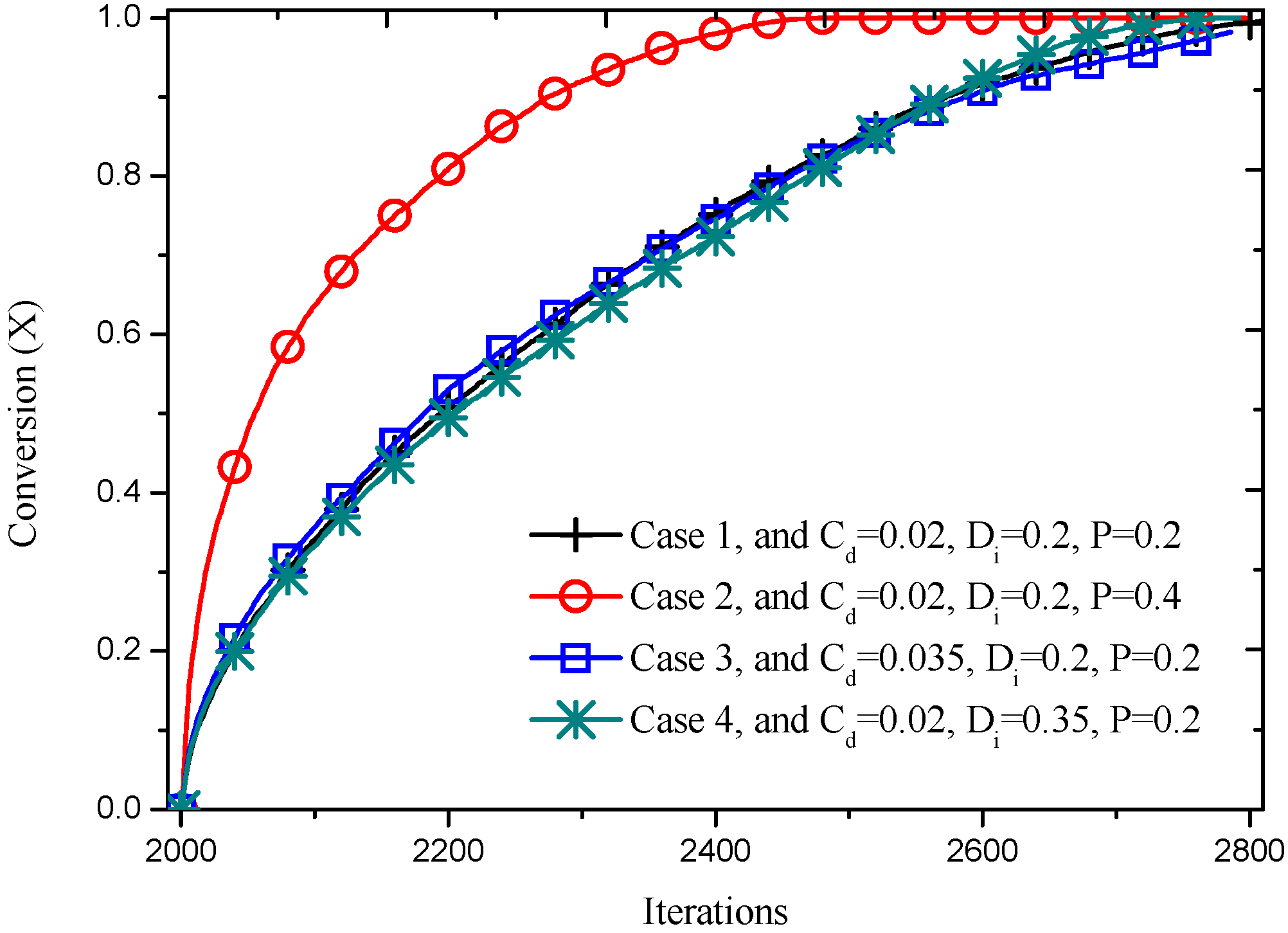
Figure 12 shows the conversion of porous cylinders with different inner structure, as a function of time. It is also notable that solid conversion of higher porosity (Case 2) is faster. This is considered due to the less amount of solid phase, as well as the larger pore size, which facilitates the diffusion of reactant and product. The other three cases with same porosity of 0.2 proceed similarly, but it can still be noted that Case 3 with larger distribution probability progress faster than the other two cases for the former part of time, about 2500 iterations. This can be attributed to the higher homogeneity resulting from larger distribution probability, leading to a larger surface area (reacting sites) to mass ratio. However, this improvement on reaction speed is limited by the relatively smaller pore size slowing down the gas diffusion. Knowing this, it is understandable that Case 4, with larger solid agglomeration and bigger pore size due to higher value of directional growth probability, has the exact opposite performance compare to Case 2.
Figure 11.
Product concentration with different inner structures, t = 2500.
Figure 11.
Product concentration with different inner structures, t = 2500.
Figure 12.
Dependence of conversion on reacting time for cylinders with different inner structure.
Figure 12.
Dependence of conversion on reacting time for cylinders with different inner structure.
3.2.2. Effect of Reaction Probability
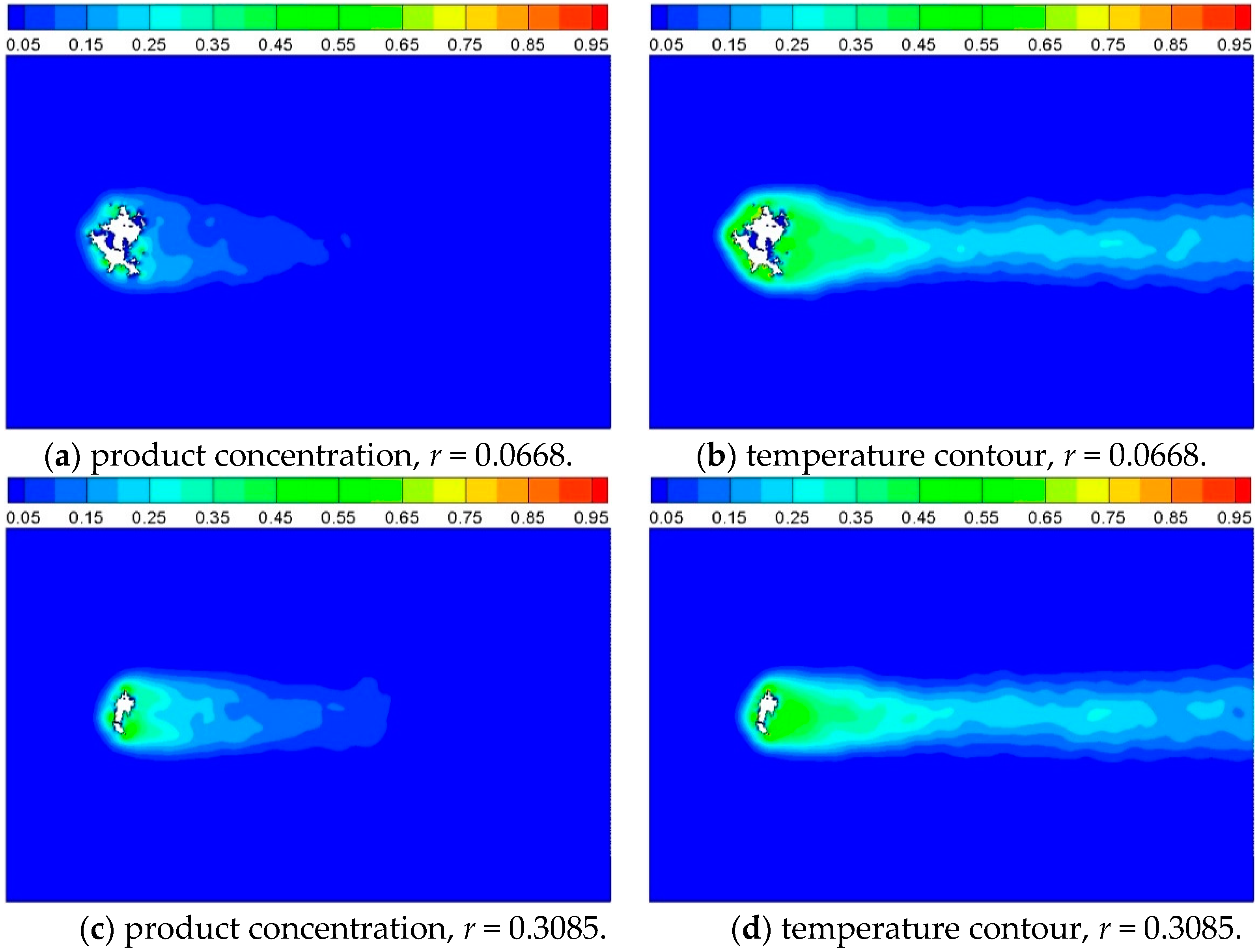
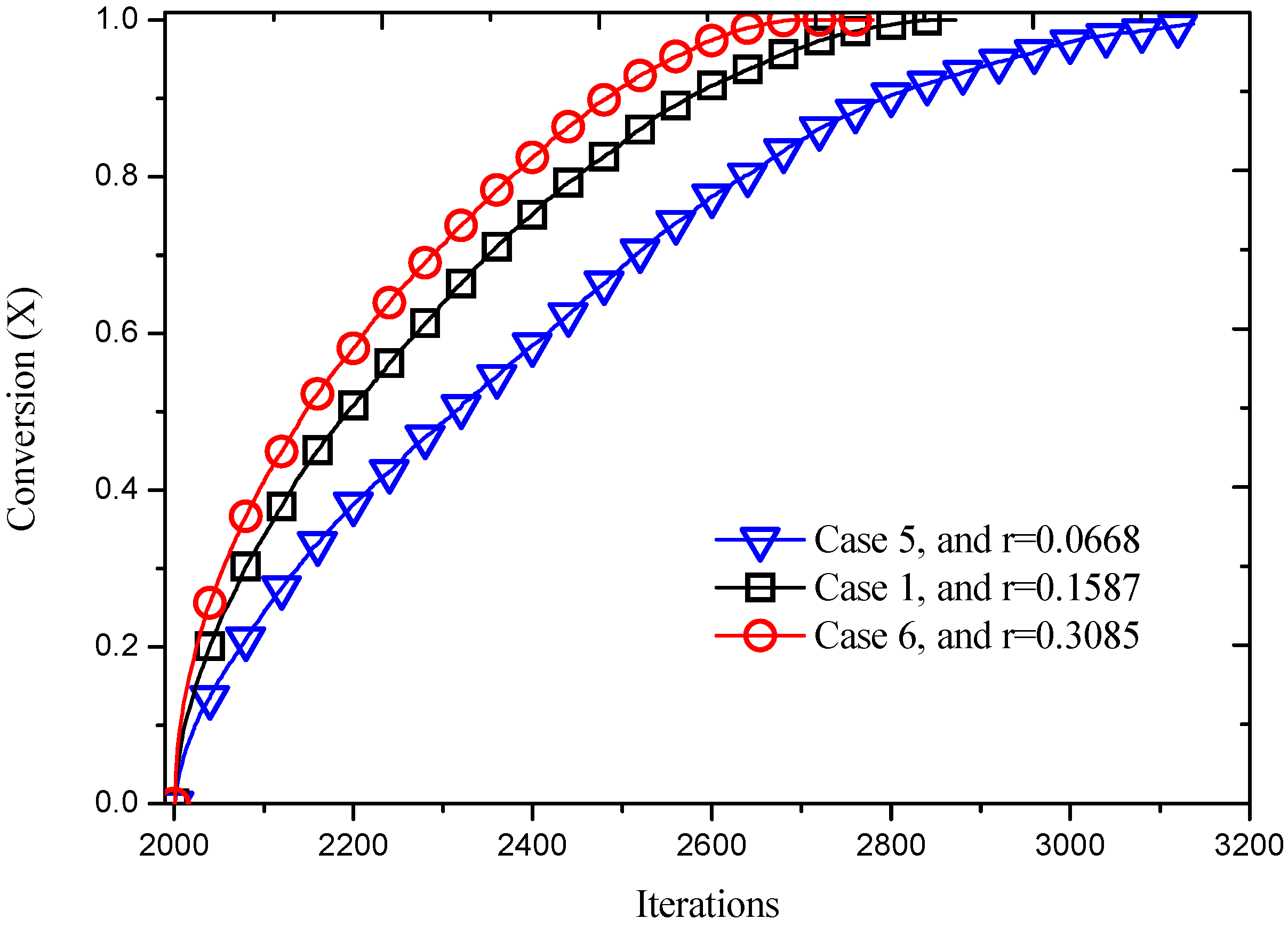
In order to obtain the influence of reaction probability on the process evolution, the value of
K* was set to be 0.5 and 1.5, and the reaction probability was determined as 0.3085 and 0.0668 according to the normal distribution function. The reaction probability decides the reaction velocity, as Equation (9). The product concentration and temperature contour around the cylinder at
t = 2500 are shown as
Figure 13. It can be obviously noted that the reaction processes faster with a higher value of probability, which also can be seen from
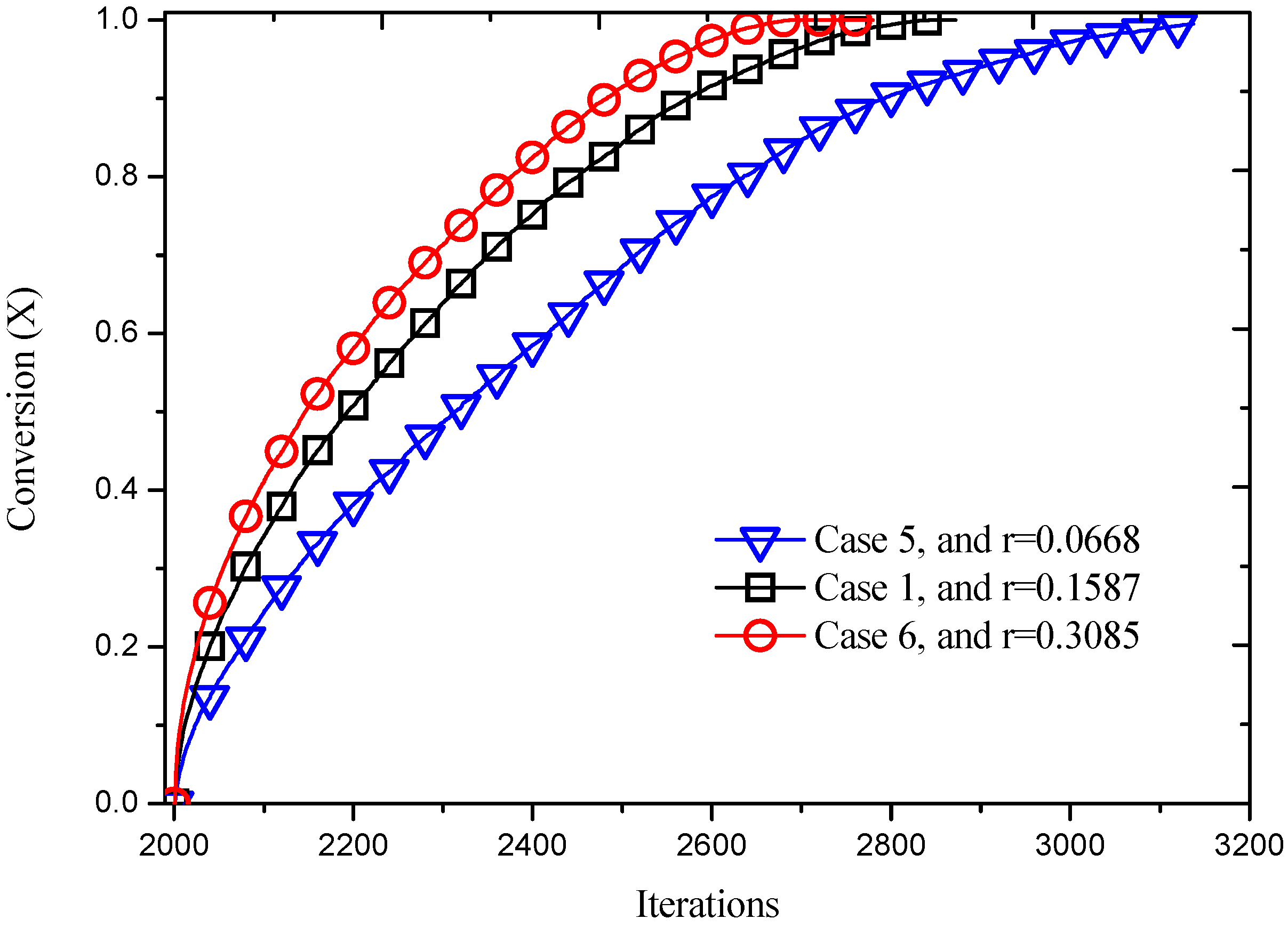
Figure 14. The conversion increases as reacting time advances and reaction probability increases. For instance, the complete reaction time increases from 696 iterations to 1140 iterations when the reaction probability decreases from 0.3085 to 0.0668.
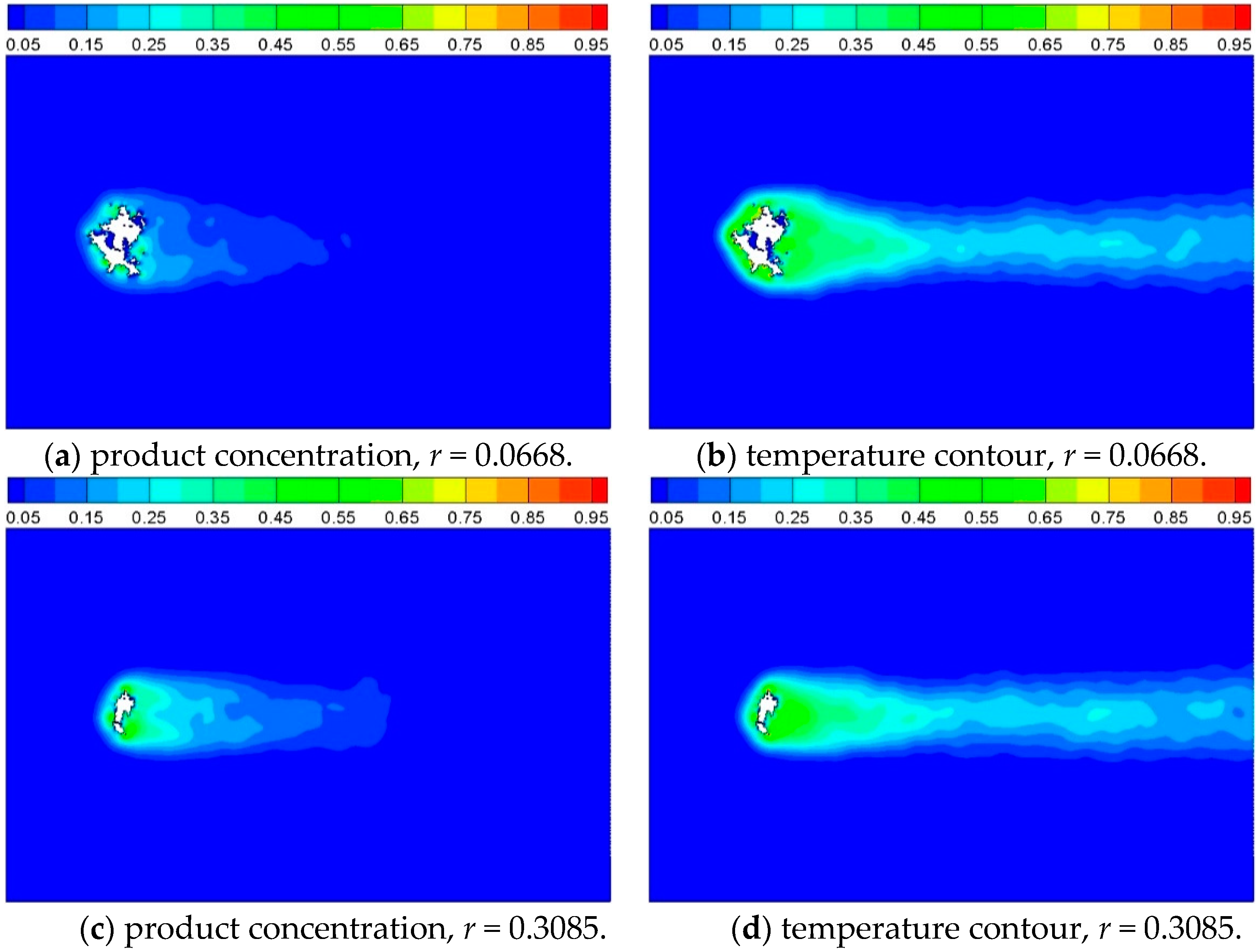
Figure 13.
Product concentration and temperature contour of Case 5 and Case 6, where reaction probability is 0.0668 and 0.3085, respectively, and t = 2500.
Figure 13.
Product concentration and temperature contour of Case 5 and Case 6, where reaction probability is 0.0668 and 0.3085, respectively, and t = 2500.
Figure 14.
Reaction processes with different reaction probabilities.
Figure 14.
Reaction processes with different reaction probabilities.
3.2.3. Effect of Inlet Gas Velocity
The equilibrium mean occupation numbers are calculated by Fermi–Dirac distribution, as follows
where
h is a real number and
is a D-dimensional vector. The two parameters are termed as Lagrange multipliers. For simplification, the Lagrange multipliers of the equilibrium distributions for lattice gas automata can be obtain by the algebra formula [
9]
where
d is equal to
for FHP-II, and
is the node velocity. Thus, the variable
at the inlet nodes can be initialized by Equation (18) with a given particle density
d and a node velocity
.
For this section, the effect of velocity at inlet on the reaction process is discussed, and the parameters are listed in
Table 1, as Cases 1, 7 and 8. Particle density ρ is fixed as 1.0 for all numerical computation cases, thus
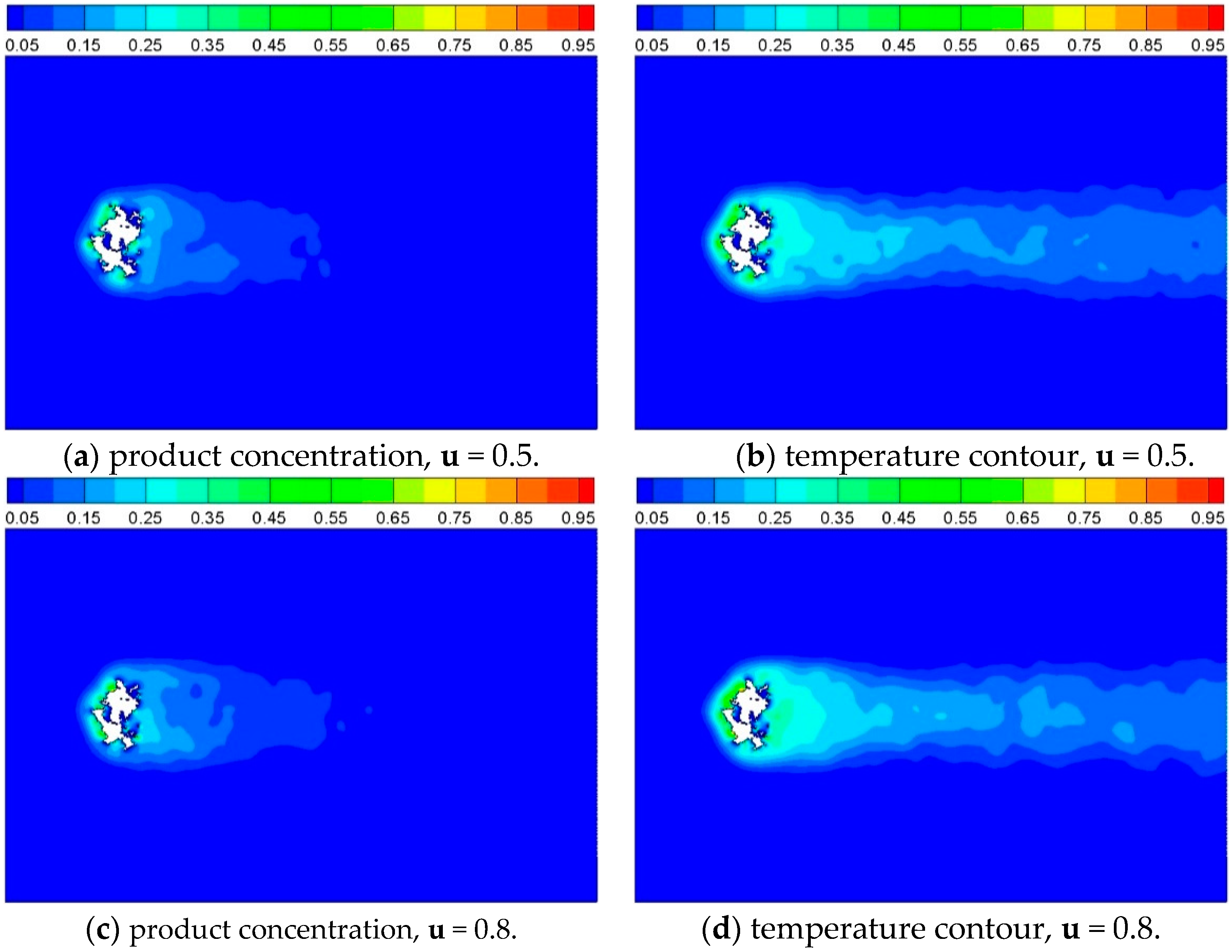
d is equal to 1/7. The product and temperature contour of cases with different inlet velocities are shown as
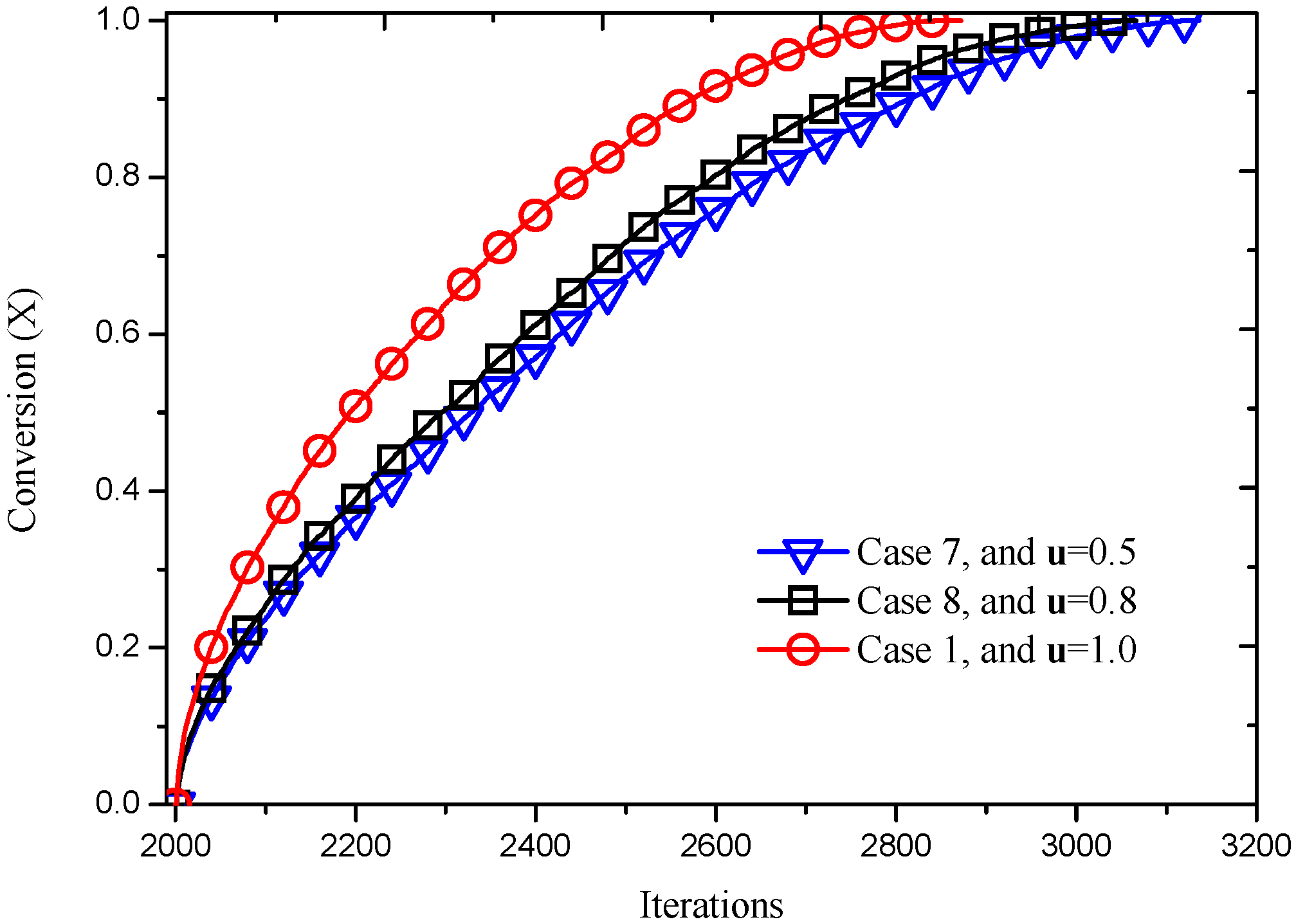
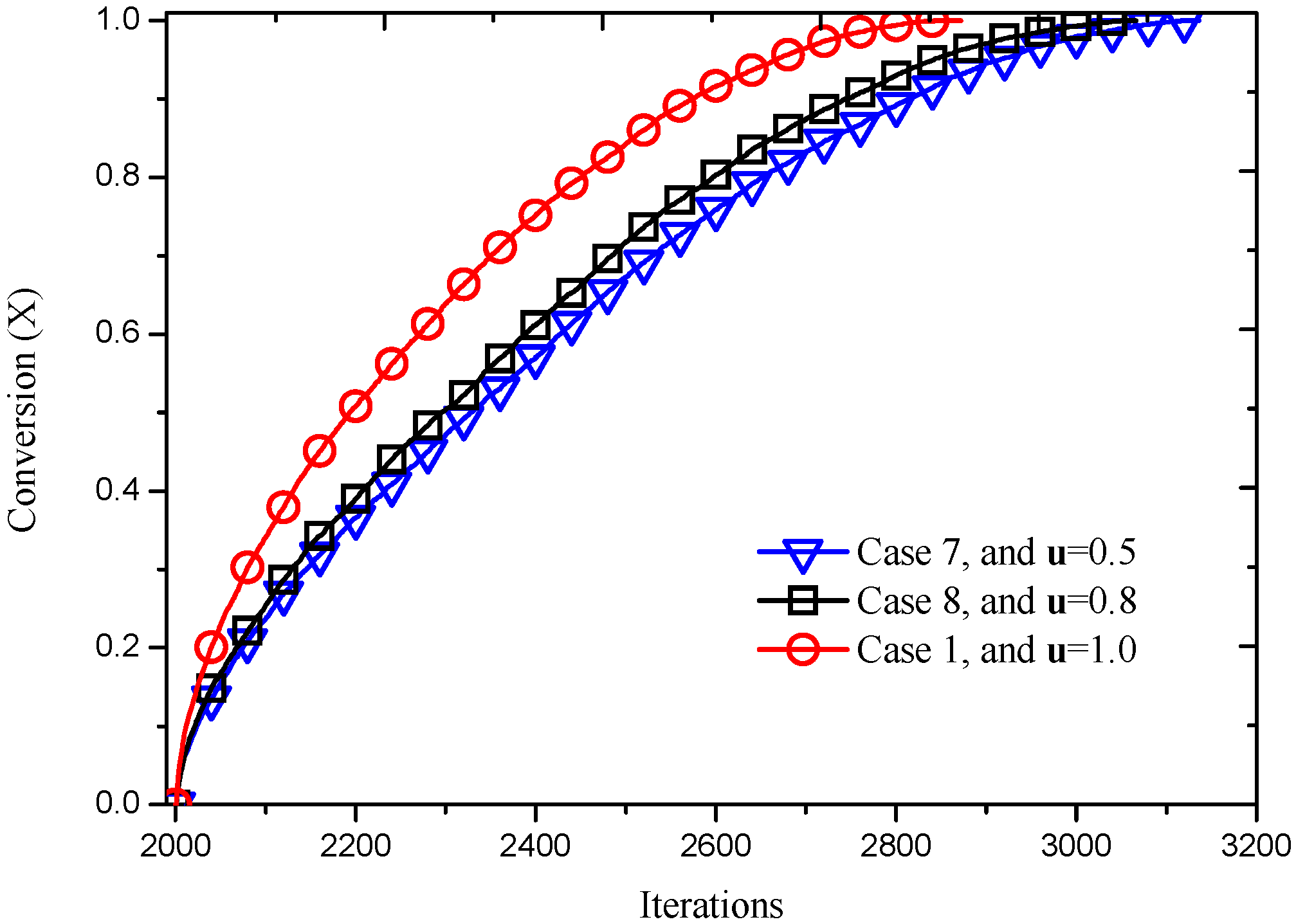
Figure 15, it can be seen that Case 8 reacts faster than Case 7 and slower than Case 1 compared with
Figure 11a, indicating that the reaction velocity increases as the velocity at inlet increases. This information can also be obtained from the dependence of conversion on the reacting time, as shown in
Figure 16, which shows that the extent of conversion increases with increasing reacting time, as well as inlet velocity. This agrees well with the theories of surface reaction in gas–solid systems, such as unreacted shrinking core model [
36] and pore model [
37], which indicate that the increase of gas velocity promotes the collision between gas particles as well as between gas particles and solid sites, facilitating the diffusion and external dispersion of reactant and product.
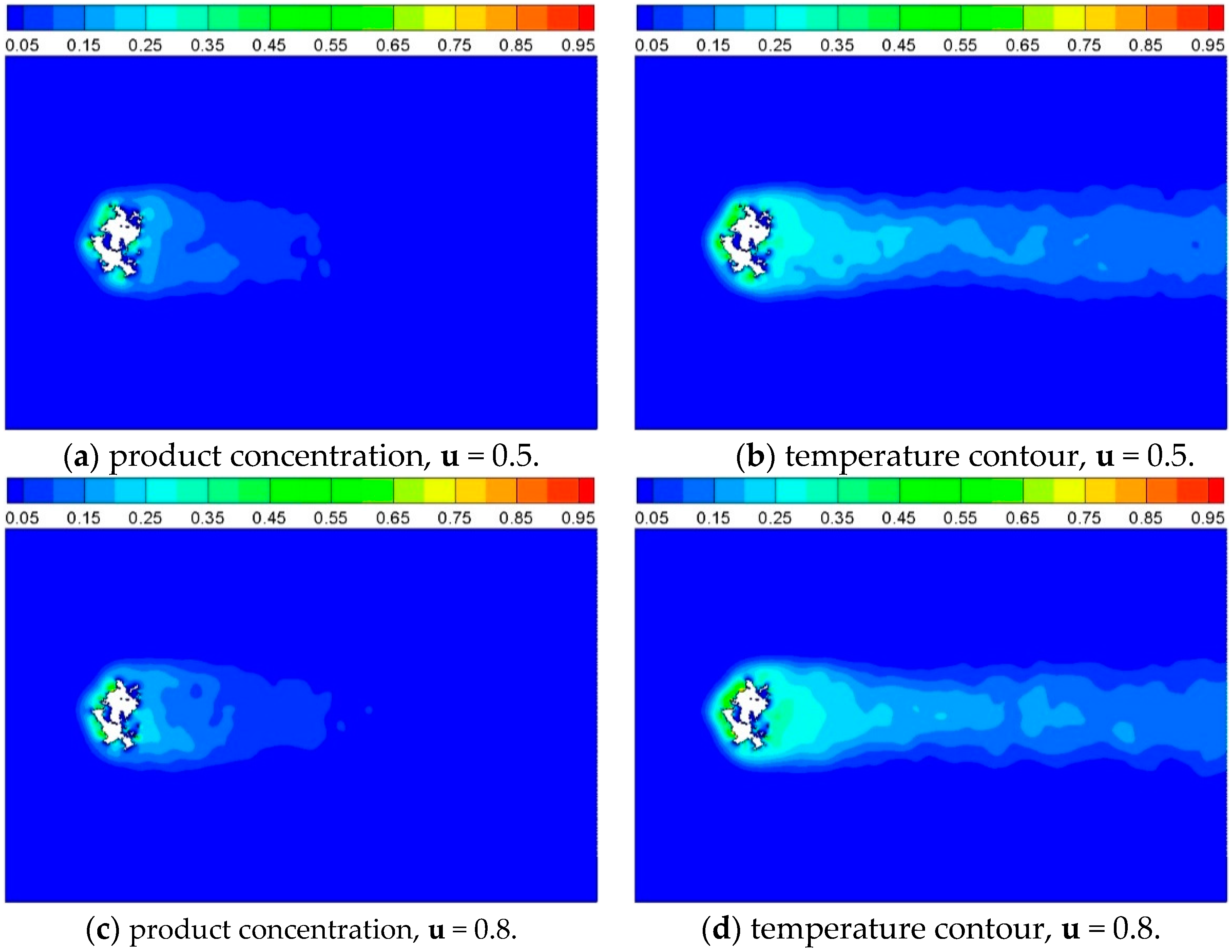
Figure 15.
Product concentration and temperature contour of Case 7 and Case 8, where inlet gas velocity is 0.5 and 0.8, respectively, and t = 2500 iterations.
Figure 15.
Product concentration and temperature contour of Case 7 and Case 8, where inlet gas velocity is 0.5 and 0.8, respectively, and t = 2500 iterations.
Figure 16.
Reaction processes at different inlet velocities.
Figure 16.
Reaction processes at different inlet velocities.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}