1. Introduction
Cancer is a disease characterized by the breakdown of cellular systems that exist to maintain, regulate and replicate genetic information. Progress in the field of cancer biology therefore requires a detailed understanding of these systems. Although human cells can be studied from numerous vantage points, the most fundamental genetic system is one of information flow: communication using signs, codes and signals (biological semiosis, or biosemiosis) [
1,
2]. Hence it is instructive to construct a working framework for accurately viewing the nature of biological information, its symbolic communication, and the errors which occur in its processing. DNA is the primary conduit for biological inheritance. It contains functional information in the sense that it encodes messages that execute biological functions [
3,
4]. It is recognized that the entropy of such functional information (via deleterious mutations in the form of insertions, deletions, fusion events, and aberrant epigenetic regulation) is the chief cause of cancer-related death [
5]. We propose that the theory of biosemiosis can be a very useful paradigm for guiding hypothesis-driven research, and that casting diseases such as cancer in terms of biosemiotic entropy can aid in interpreting the incoming tsunami of genomic and epigenetic data, in addition to informing the principles that shape future cancer treatment strategies.
Death of an organism (if not from violence, accidents, infection, or various “unnatural” causes) is the eventual result of biosemiotic entropy at the somatic level, which accumulates stochastically over time. It is well understood that entropy can erode usable energy gradients, and this knowledge is extremely useful in fields such as physics, chemistry and engineering; that entropy can affect something as intangible as information is intuitive, but not as easily quantified and formularized. Because entropy antagonizes order, biosemiotic entropy is the antagonist of biological order, i.e., health.
Cancer is a unique disease in that it does not typically result directly from infectious or toxic agents such as bacteria, viruses, fungi, or foreign chemicals. Instead, malignant growth is the result of the accumulation of random alterations from within, leading ultimately to a fundamental breakdown in cell cycle control in a process analogous to increasing entropy on a cellular level (though, from the cancer’s point of view, increasing fitness [
6]), often with lethal consequences at the organismal level. Hence statements like the following from the Cancer Genome Atlas Research Network [
7]:
“Cancer is a disease of genome alterations: DNA sequence changes, copy number aberrations, chromosomal rearrangements and modification in DNA methylation together drive the development and progression of human malignancies.”
The biology within our cells is a rich system of information exchange and communication, from the DNA in the nucleus to the signaling proteins excreted through the cell surface. Therefore it comes as no surprise that cancer is classified as the degradation of biological information and communication systems, at both the genetic and epigenetic levels.
2. What Is Biosemiosis?
Biosemiosis is simply sign mediated communication in living organisms resulting in biological function. The heart of all biological sign systems is the central dogma, describing how the DNA code (its signs mediated and processed as mRNA) becomes “translated” by the tRNA/ribosome interpreting complex to form polypeptides.
2.1. Why Is It Important to Recognize a Living Organism as a Semiotic System?
Recognizing living organisms as semiotic systems allows for useful analogies to be drawn from other semiotic systems. Such analogies are powerful because: (1) they give insight and understanding by relating the unfamiliar in terms of the familiar and, (2) lessons learned from other semiotic spheres (such as principles of efficient information storage and retrieval in computer science) can generate predictions and hypotheses for new frontiers in biology (such as a tree-like database structure for information storage and retrieval in the human genome). This is evidenced by the fact that biologists frequently use analogies from the familiar semiotic systems of human language and computers [
8,
9].
To optimize the usefulness of these analogies for studying genetic disease, we now underscore the legitimate similarities among these three spheres of semiotics (biology, language, and computers), while also considering some fundamental differences in order to avoid misleading inferences. While these observations are applicable to other species, our discussion will be centered on human biology, with an emphasis on cancer.
2.2. Semiosis: Signs and Meanings Connected by a Convention
Semiotic theory may cause confusion with (and is thus largely ignored by) many biologists, geneticists, and bioinformaticians because it is an unfamiliar term; yet the underlying concept is ubiquitous. Symbolic communication may be modeled as a semiotic system composed of three main parts: a set of signs and a set of meanings connected by way of an interpretive convention. Signs are arbitrary representations (e.g., words, codes, or symbols). Mapping, indexing, decoding or interpreting is accomplished by connecting these signs to an abstract icon or physical object—yielding meaning—and is fundamentally relational. Objects, entities toward which the sign points, can be anything material (e.g., parkas, pencils, proteins, or protons) or abstract (e.g., logic, ideas, or values).
The most familiar sphere of semiosis is language, whereby abstract symbols or words are connected to the world in which we live by a semantic convention or interpretive map. A relatively recently established sphere of semiosis exists in computer science, where assembly language (or some higher level language) is converted into machine code such that it can be executed for some intended output. A more recently
discovered sphere of semiosis is found in the world of genetics, where instructions for regulating cellular function and building proteins are encoded into the DNA molecule, which is then transcribed into RNA for further translation into a polypeptide molecule [
10]. This semiotic relationship is perhaps most familiar to biologists as the distinction between genotype and phenotype, in which a certain genetic makeup encodes specific phenotypic outcomes that collectively determine function and fitness. The words used to describe these biological processes (reading, copying, transcribing, and translating) reveal deep similarities between the semiotic spheres of human communication and biological life [
11], and can be extended to the sphere of computer programming [
12]. While we focus on genetics as the heart of biosemiosis, higher levels of symbolic biological communication have been noted such as cell signaling cascades, neural networks, and the immune system [
13].
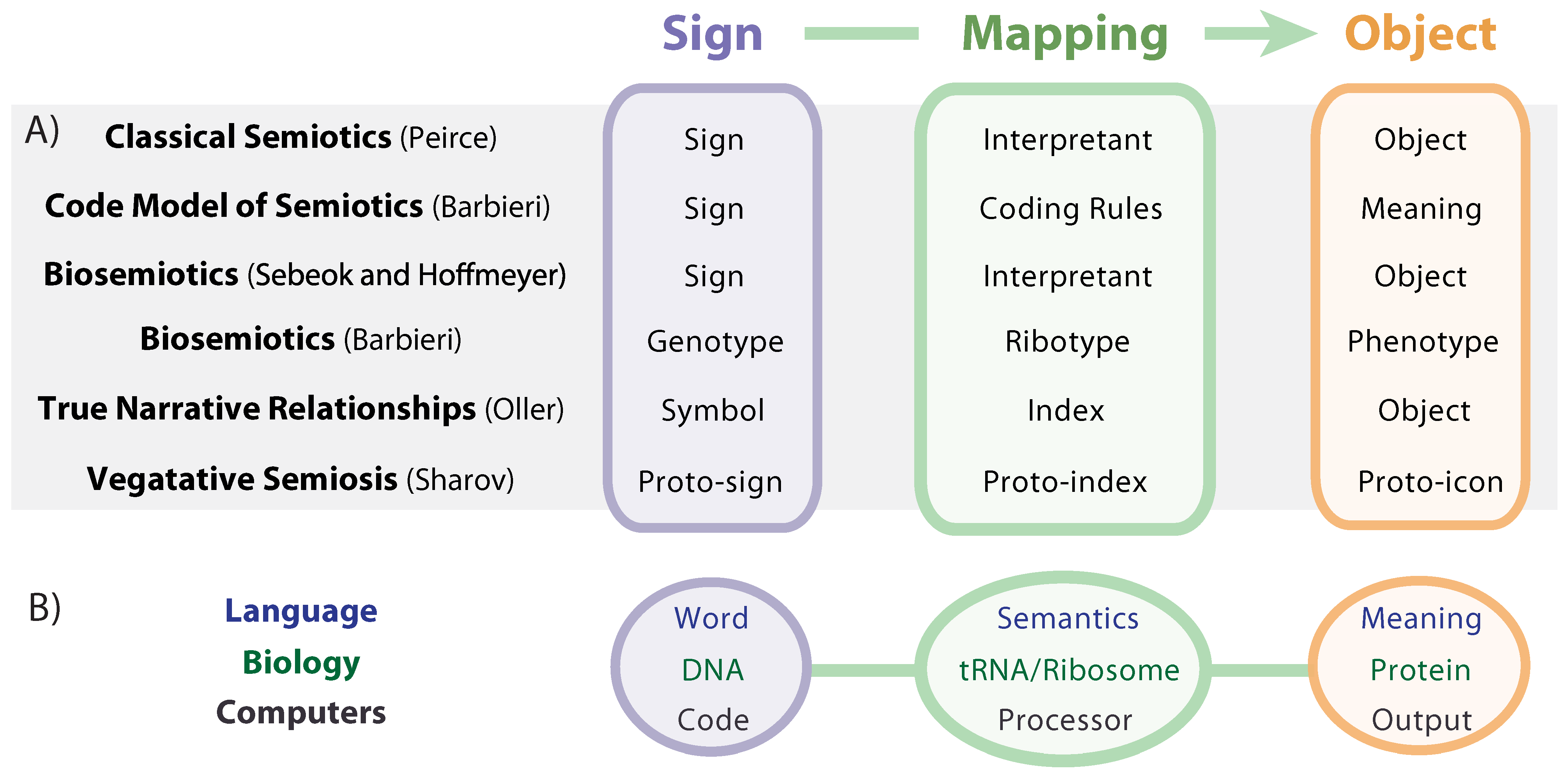
A precise delineation of biosemiosis is still being developed in the literature, but amidst the differences there are enough similarities to construct a simple but useful definition [
14]. As shown in
Figure 1A, similar sign system theories include: classical semiotics developed by Charles Sanders Peirce [
15]; biosemiosis as conceived by Sebeok [
2] and Hoffmeyer [
11], who used Peirce’s trinitarian conception of “sign-interpretant-object” to describe genetics; biosemiosis and the code model of semiosis, developed by Barbieri [
16]; and the concept of True Narrative Relationships (TNR) developed by Oller, who defines biosemiotic entropy as the breakdown of accurate (true) biological communications [
13]. A helpful distinction between human language semiosis and molecular biosemiosis, which he terms “vegetative semiosis”, is drawn by Sharov, who inserts the prefix “proto” to emphasize the “tremendous gap that separates human icons (e.g., visual shapes or sound tunes) from molecular interactions that lack any elements of perception or classification” [
4].
Figure 1.
(A) Semiotic models and (B) examples of semiotic systems.
Figure 1.
(A) Semiotic models and (B) examples of semiotic systems.
While there are many terminological options offered by the body of literature on semiotics, we use “sign”, “mapping”, and “object” for purposes of clarity, simplicity, and broad usefulness across the various spheres of semiosis. In order to flesh out a clear understanding of biosemiosis, we will make use of analogies to the semiotic spheres of language and computing (
Figure 1B), with careful attention paid to both similarities and differences.
2.3. Semiotic Comparisons between Biology and Language
2.3.1. Similarities
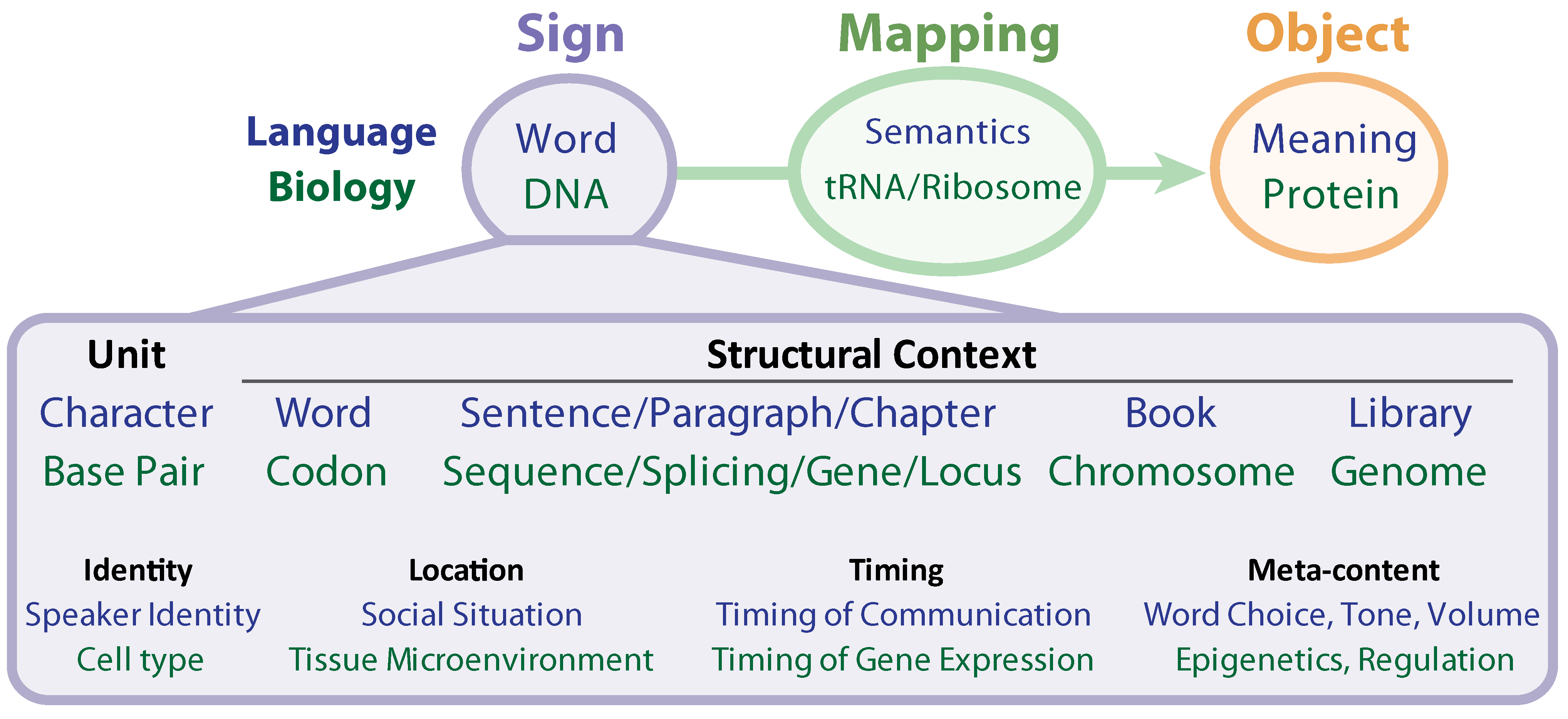
The DNA message is read, copied, edited, transcribed, and translated. It is striking that the most fitting terms used to describe the biochemical mechanics of life are rooted not in biology, chemistry or mechanics, but rather, in language. In the grammar of human language, meaning depends on immediate context (the order of words in a sentence), broader context (paragraph, chapter, and genre), temporal placement, location, and many other factors (see
Figure 2). Meaning is also highly context-dependent in biology. Differences in immediate context determine meaning; only a few amino acids may compose an active site, while a large number of neighboring residues provides the biochemical context required for function, and variation in this context provides a multiplicity of function (as seen in the splicing code). This is exemplified by such phenomena as alternative and
trans-splicing [
17]. Differences in broader context also determine meaning: the same gene may play vastly different, even opposing roles if active in a different cellular context, a different tissue, a different stage in cell differentiation, or under a different environmental stress.
Figure 2.
Similarities between Biology and Human Language.
Figure 2.
Similarities between Biology and Human Language.
The analogies of word/sentence/paragraph/chapter have not been easy to map to the genetic hierarchy in a one-to-one fashion, although DNA clearly shows a level of structure that resembles language. The difficulty may be the result of a protein-coding centric analysis. Perhaps the protein coding regions are merely the dictionary, and the higher levels of grammar (paragraphs and chapters) are to be found in their regulation via epigenetic marks and non-protein coding regions. This coincides with the observation, for instance, that the length of protein-coding genes is similar in puffer fish (30.5 Mb) and humans (31.7 Mb), whereas the non-coding region is dramatically greater in humans (2834.3 Mb) than in puffer fish (311.5 Mb), a trend of increasing non-coding elements with increased species complexity, which is consistent throughout species phylogeny [
18]. Although signs in natural language systems commonly signify objects already in existence, genetic signs usually specify arrangements of biophysical matter that have not yet been produced.
2.3.2. Differences in Event Instantiation
In spoken language, a separation exists between inward (mental processing) and outward (physical behavior) responses. While they utilize physical mediums through which they travel and subsist (light, sound, electrical impulses, chemical signals, etc.), words have abstract meanings that only exist in the mind of the reader who is interpreting them and creating imagined, virtual representations and ideas. These may or may not ever result in execution of a non-mental event. In biology, all active messages are always creating actualized physical ramifications, and do not seem to ever be mediated through a “virtual representation” in the same way human language does (although they can be represented virtually, as all information can). When we give descriptive communication through language (e.g., “the sky is blue”), the message has real meaning that creates an image in the mind of the reader rooted in their common experience, which in turn permits the interpretive function (discovery of meaning). But no action is necessitated in response. If we give command-based communication (e.g., “clean this flask”, to an undergraduate in a lab), it will (usually) result in direct physical action that resembles the desired result. Unless its function is suppressed (e.g., genes silenced in a particular cell type), biosemiotic information is always action-oriented, and without uncertainty in the language commands (given healthy and normal circumstances).
2.3.3. Differences in Plasticity
The transmittance of human language is plastic, creative, free-flowing and spontaneous in ways that the biosemiotics of the genome is not. There is a virtually infinite number of possible coherent, logical sentences that might proceed from or respond to a thought (of many types and functions, illustrative or meditative, commanding or responding, etc.), but genetic information is characterized by common, modular instructions that are processed in deterministic, temporal sequences given a particular biological context. The difference is that human language is not restricted to any particular purpose whereas genetic information is strongly proscribed by a set of required instructions. As an embryo develops, the various chapters of the DNA instruction manual unfolds in a wondrous but predictable fashion, while an author may write chapters that unfold in an unpredictable fashion. Upon fertilization, the DNA is already written, whereas new meaningful sentences may be continually written or spoken. Where the genomic content is changed (e.g., in B cell maturation), it is done so by a theoretically predictable combinatorial mechanism. Also, languages can be transferred from one medium to another (i.e., a story can be written, spoken, or typed) without losing the inherent meaning, whereas biological code cannot express its meaning (i.e., function) in any other medium.
Human language is therefore quite different from cellular biology. However, human-created instruction manuals strongly mimic, on a simpler level, genomic information. Thus, genomic information is a subset of all possible language manifestations. This allows for strong comparisons between languages and genetics and leads into a discussion of cancer as a corruption of the original, cohesive instruction set.
2.4. Semiotic Comparisons between Biology and Computers
2.4.1. Similarities
Computer-based languages operate by translating human-readable programming into increasingly simple instruction sets, which, that at their most basic level, operate transistors specially designed into logic gates: AND, OR, and NOT. Such logic gates receive high or low voltages as inputs and achieve predictable outcomes governed solely by the design of the circuit and physical law. The configuration of such gates into more complex structures allows for sophisticated functions such as arithmetic, clocks, and multiplexers. The microprocessor is in turn a complex arrangement of different units. Microprocessors are themselves arranged with other electronics into a computer, which are then operated by software programs written to run on them. For computer technology to work, logic must be translated into the fundamental level of physical law. Function is embedded within function until high-level outcomes are obtained. The data and logic used to operate computers are carefully controlled and copied at various levels of manufacture as well as use in order to ensure the stability of their expected utility.
Biological systems are similar in this regard: organisms are arranged hierarchically into organ systems with organs composed of various tissues of similar cells. Cells are differentiated according to the expression of genes, which is governed by a host of non-coding RNAs, protein pathways, and epigenetic factors. At its most fundamental level, life uses the physical laws of chemistry to execute its needed biological functions, albeit in a more stochastic manner than computer systems. Genetic information is likewise carefully copied and translated in order to achieve a predictable phenotype. Layers of nested function and precise interpretation of encoded data is highly relevant to the semiotics of both computer and biological systems.
2.4.2. Differences
Computer systems emphasize maintaining data fidelity for proper operation; they are relatively “brittle”. A single atomic change (bit, character, numeric value,
etc.) can frequently lead a program to crash (all functionality lost). In contrast, biological systems not only provide mechanisms for preserving data fidelity, but can also utilize mutation for adaption via natural selection. Self-replicating computer programs can do this only in highly contrived environments using artificial “chemistries” [
19]. Moreover, lost biological function due to deleterious changes can subsequently be restored through compensatory mutation. For computers, error-correcting codes have theoretical limits to the number of errors that can be corrected. Though a wide range of machine-learning algorithms exists to analyze data to perform complicated classification applications, such methods are constrained to work within the framework of their classification problem.
Computer systems are usually updated by making a relatively large number of simultaneous, coordinated changes. While tweaking and debugging of code does occur with only single modifications, the creation of even a small subroutine requires many concomitant additions in order to attain utility and coherence. Without such artificial intervention in biological systems, highly advantageous changes (
i.e., mutations which produce a novel complex adaptation) must as a rule be obtained gradually. Many generations may be needed in order to create significant modifications, provided viable paths to those outcomes exist in the fitness landscape. Importantly, such paths can be rare in even very simple systems [
20,
21].
Computer system signs, indices, and objects are similar to biological systems in that they are often nested hierarchically, rely on physical law at their basis, and observe careful control in the copy and use of codes. In this way, computers are more akin to biology than human language. Computer replicative cycles usually involve marked coordinated intervention at the sign level in order to achieve new object outcomes.
What can we learn from the computational world that yields useful hypotheses and predictions about cell biology? The answers to this question may fuel many new discoveries as researchers grapple with seemingly intractable amounts of information (15 trillion bytes of raw data from the ENCODE consortium alone) [
22].
3. Breaking Points in Biosemiosis
Biosemiotic entropy is the degradation of the genetic language (deleterious mutations) and/or the aberrant regulation of that language (epigenetic malfunctions). This results in reduced functionality or fitness relative to some optimum state of homeostasis, e.g., a fitness peak. Homeostasis can be defined as a healthy state of biological function, maintained by cellular systems, whose purpose is to regulate concentrations, integrity, and the status of biological materials. As more is learned about the systems under study, such homeostatic optima may be identified with increasing accuracy. Notwithstanding, it is already clear that there are three general domains of biosemiotic entropy, correlating to the three aspects of the genetic biological sign system.
3.1. Entropy in the Sign (the Gene Code): Genetic Entropy
“To be sure, there are numerous medical conditions found in children and adults that have a strong, indeed predominant, genetic basis. The continuously updated Online Mendelian Inheritance in Man (OMIM) lists many thousands of such conditions, but offers a far too narrow view of the contribution of genetics to medicine. Except for some cases of trauma, it is fair to say that virtually every human illness has a hereditary component.”
-Francis Collins and Victor McKusick [
23]
Entropy in the genome contributes to nearly all human disease, either through breakdown of normal physiology or increased susceptibility to infectious agents [
23]. Aging occurs at least in part as a result somatic mutation accumulation over a lifetime, increasing susceptibility to disease in a process that is progressively lethal (
Figure 3). Mutations also play a fundamental role in cellular senescence by triggering oncogene overexpression, tumor suppressor gene loss, and other forms of DNA damage [
24]. A process analogous to aging may also occur in the
germline as a result of mutation accumulation over generations, leading to the extinction of whole populations [
25,
26]. Two interrelated processes contribute to sign entropy: (1) degradation of the gene code, and (2) epigenetic abnormality and misregulation, both of which ultimately lead to corruption or aberrant expression of genetic information. Epigenetics and aberrant gene regulation will be considered separately, as higher-level errors (see
Section 5).
With the mapping of the human genome and subsequent characterization of its functional elements [
27,
28], aberrations in the genome have become increasingly tractable to characterize. They include:
Single nucleotide substitutions, where a single base pair mutates resulting in single nucleotide polymorphisms (SNPs). SNPs rarely result in a large phenotypic change, due to the nearly neutral effects of most mutations [
29] and multiple levels of redundancy and canalization built into biological systems. Single mutations within non-protein-coding regions typically have no acute effect, although there are striking exceptions [
30] and their cumulative effects can be highly negative. Also, though there are only 20 canonical amino acids utilized in most living systems (in addition to start and stop signals), a total of 64 nucleotide triplets exist, such that some nucleotide changes are synonymous (
i.e., do not change the resulting amino acid). Even mutations in protein-coding regions that do result in amino acid substitutions may not alter function significantly, although many nonsynonymous protein-coding mutations are clearly under purifying selection [
31]. When amino acid changes can reduce or destroy function, there are often duplicate (healthy) copies of the gene elsewhere in the genome, or the protein may have another isoform that can perform the same or similar function. Nevertheless, simple nucleotide substitutions are the most prominent example of genetic entropy, and cumulatively represent a significant contributor to disease [
32].
Insertions and deletions (indels), where one or more base pairs are removed or added to the sequence. This can result in the removal of a promoter region required for gene activation, can cause a frame shift in the coding region (where an entire string of signs are altered causing an entirely new, non-sense amino acid sequence), or can cause an indel in multiples of 3 to remove or add amino acids without causing a frame-shift, thus preserving the remainder of the protein’s primary sequence [
33].
Copy number variations, caused by the duplication or deletion of larger fragments of genomic DNA. Hyper-variable regions of relatively small tandem repeats are well known for their use in forensic analysis, while whole gene duplications followed by subsequent alterations in functionality are thought to lead to families of similar genes [
34]. Variations in the number of genes may alter gene expression levels through dosage effects or changes in genomic context.
Aneuploidy refers to the occurrence of aberrant numbers of single chromosomes. In humans, most are lethal; those that are not are highly deleterious. Examples of viable aneuploidies include X chromosome monosomy (Turner’s syndrome) and trisomy 21 (Down syndrome). Because of the extremely harmful effects of such large-scale changes, aneuploid individuals rarely survive to reproductive age.
Chromosomal Rearrangements, involving the breakage of DNA, include translocations and inversions. Contiguous stretches of DNA are moved intact from one location to another, possibly changing their orientation. Such changes are potentially very harmful, especially if key genomic regions are relocated.
The sign is the most intensely studied portion of the biosemiotic triad. Accumulation of entropy in the genes causes a variety of diseases at the somatic level, depending on the kind of error (as listed above) and the location of that error within the genome, which we will explore in
Section 4 and
Section 5. Concerns also exist at the level of whole populations as a result of the accumulation of germline mutations (see
Section 6.2). It is important to note that, over evolutionary time (multiple generations), the greatest threat to a species comes not from lethal or highly deleterious mutations, which are easily eliminated by purifying selection. Instead, very slightly deleterious mutations have the greatest potential to contribute to eventual species extinction [
35]. On average, such mutations escape the filter of purifying selection, accumulating slowly, while
en masse having cumulative effects that can disrupt biological functionality. A similar concept to genetic entropy has found practical application in treating HIV infection with nucleoside analogs that promote lethal mutagenesis [
36,
37]. In this case, within-host populations of viruses are driven to extinction by an elevated mutation rate alone. Mutation accumulation may also play a role in the deterioration of the human immune system during HIV infection. The elevated turnover of lymphocytes stimulated by HIV has been postulated to lead to deleterious mutation accumulation in progenitor cells, essentially causing the immune system to age at a rapid rate [
38]. It should be noted, however, that genetic entropy is distinct from (though related to) the concept of genetic load in population genetics. Importantly, genetic load may refer to the fitness decline or fertility excess required by the process of fixing a new mutation in the population. Moreover, this concept has been formulated alternatively as cost [
39] and load [
40]. Although a detailed discussion is beyond the scope of this paper, it is sufficient to conceptualize genetic entropy as a process contributing to (but not synonymous with) genetic load [
41].
3.2. Entropy in Mapping (the Ribosome Decoding Process)
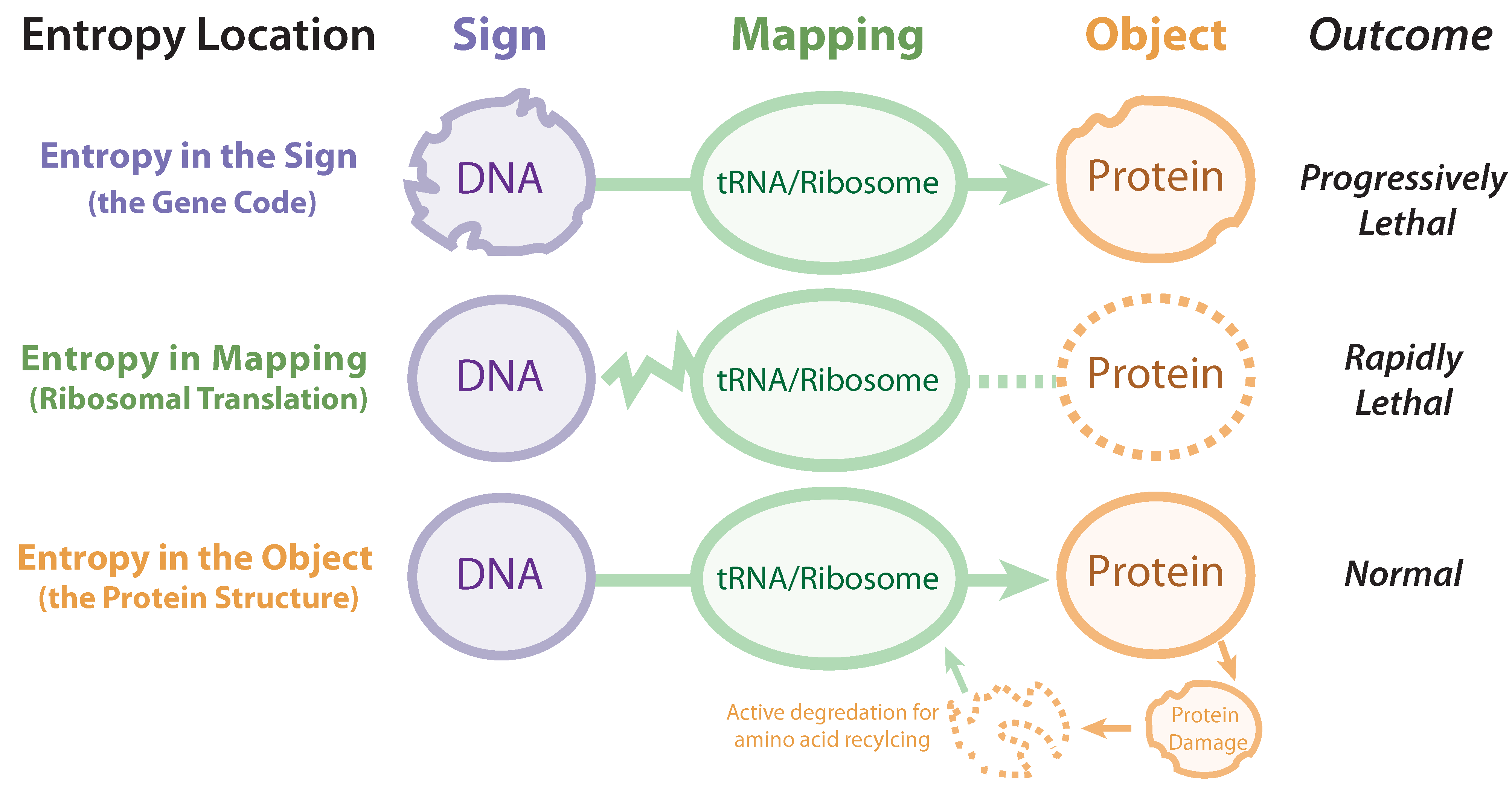
Disrupting the mapping or decoding functionality of the ribosomal interpretive index is rapidly lethal (
Figure 3), and can occur through either: (1) mutations in regions encoding ribosomal protein components, aminoacyl tRNA synthetases, and other translation factors, (2) mutations in DNA corresponding to ribosomal RNA (rRNA) or tRNA, or (3) chemical inhibition of translation. It should be noted that (1) and (2) are fundamentally caused by sign entropy, as the genome encodes the machinery that performs the indexing and interpreting. Because aberrations in the genomic regions encoding this machinery will have severe ubiquitous effects, purifying selection acts very strongly to prevent the mutational degradation of these systems.
There are 700 copies of ribosomal DNA (rDNA) in the diploid human genome, and ~60% of all RNA is rRNA, making biosemiotic mapping very robust, resistant to mutation [
42]. Therefore, the hyper-sensitivity of life to entropy in mapping is most often observed with small molecules that inhibit/perturb translation by, for instance, binding to the ribosome active site or by inhibiting tRNA synthesis [
43]. The potency of antibiotic/antibacterial agents is due to their ability to bind prokaryotic, but not eukaryotic, ribosomes [
44], while agents able to hit human ribosomes (such as ricin) are deadly in very small quantities [
45]. Therapies targeting the interpretive machinery via ricin (or similar molecules) conjugated to targeting agents (
i.e., antibodies) are being pursued as anticancer agents, but have to achieve selective delivery to cancer cells to avoid the high toxicity to healthy tissue [
46].
Figure 3.
Three areas of entropy in biosemiotics: entropy in the sign (gene code), entropy in mapping (ribosomal translation), and entropy in the object (protein structure).
Figure 3.
Three areas of entropy in biosemiotics: entropy in the sign (gene code), entropy in mapping (ribosomal translation), and entropy in the object (protein structure).
3.3. Entropy in the Object (the Protein structure)
Entropy in the primary biosemiotic output (proteins) is frequent (such as misfolding, hydrolytic degradation, or covalent modification) and even actively induced (enzymatic degradation). This is to be distinguished from protein entropy rooted in genetic/sign entropy. Since proteins do not carry the heritable information, entropy does not increase across generations (with notable epigenetic exceptions, e.g., diseases caused by prions). Proteins (not only those that are misfolded or unwanted, but also those that are fully functional) are broken down by lysosomes and proteasomes to replenish amino acids, which are then used to manufacture new proteins. This constant recycling maintains homeostasis, and thus entropy at the object level of biosemiosis can be viewed as transient and thus “healthy”. Additionally, even when undesirable entropy occurs at this level, numerous mechanisms exist to correct such errors (e.g., protein refolding by chaperones).
However, if entropy in the form of proteotoxic stress is allowed to accumulate by conditions inducing unusually high production of misfolded proteins or, by blocking protein recycling (by chemical inhibition of proteasomes, for instance), then cell death soon follows [
47]. Cancers that rapidly proliferate have a heavier entropic load at the protein level, and thus are more dependent on the proteasome, and more sensitive to proteasome inhibitors [
48]. This has enabled proteasome inhibitors to be clinically employed as a means of cancer treatment, and designing selective proteasome inhibitors is the focus of ongoing medicinal chemistry efforts for both cancer and other diseases [
49,
50].
3.4. Distinguishing Biosemiotic Entropy as Applied to Cancer
The origin or long-term trajectories of biosemiotic systems are beyond the scope of this article. Nevertheless, a brief discussion of the use of biosemiotic entropy within this study will be helpful. For present day species, biological organisms can be understood as evolving toward local fitness optima as defined by environmental and functional constraints within the context of existing biosemiotic systems. As fitness landscapes change, so can optima, and the ability of organisms to adjust and evolve toward new local optima may critically depend on their adaptability both within and possibly between biosemiotic systems.
However, we focus here on the biosemiotic entropy of cancer, where inherited biosemiotic systems—genetic or epigenetic—are fixed for a particular individual. The biosemiotic system of a person after birth is permanent, with habitat, behavior, and genetic pre-disposition now being the major players in carcinogenesis. The biosemiotic entropy of somatic cells, whose genetic programs are already set, is a more important consideration for cancer research than long-term evolution. However, evolutionary theory has direct import in understanding the development of cancer and subsequent drug resistance, and indeed, cancer can be regarded as a systematic collapse allowing for the selection of individual somatic cells at the expense of the whole organism. It is notable that positive natural selection at the organismal level does not favor such outcomes. The ability of somatic cell lines to cooperate, even sacrificially, rather than compete, promotes organismal health. Applying biosemiosis to the body’s organization and components, therefore, is a very important consideration. Biosemiotic entropy breaks down the established genetic coordination of cells, tissues, organs, and systems and replaces it with selfishly replicating tumors. Understanding the existing biosemiotic systems within individuals, the parameters affecting their entropy, and their eventual deterioration leading to cancer may aid hypothesis generation for more effective treatments. To explore this, we now turn to a more detailed characterization of entropy in these systems.
4. Entropic Pressure Points Cause Cancer
For cancer, it is not mainly the type of entropy that matters, but the location in the genome. The most carcinogenic mutations arise in genes coding for proteins that are most closely connected to the biosemiotic process, which results in a multiplicative or exponential increase in errors. This is what makes oncogenes so important in disease: they are upstream of other genes and pathways. This observation is very powerful, and deserves consideration as an expansion of the traditional tumor suppressor/oncogene paradigm.
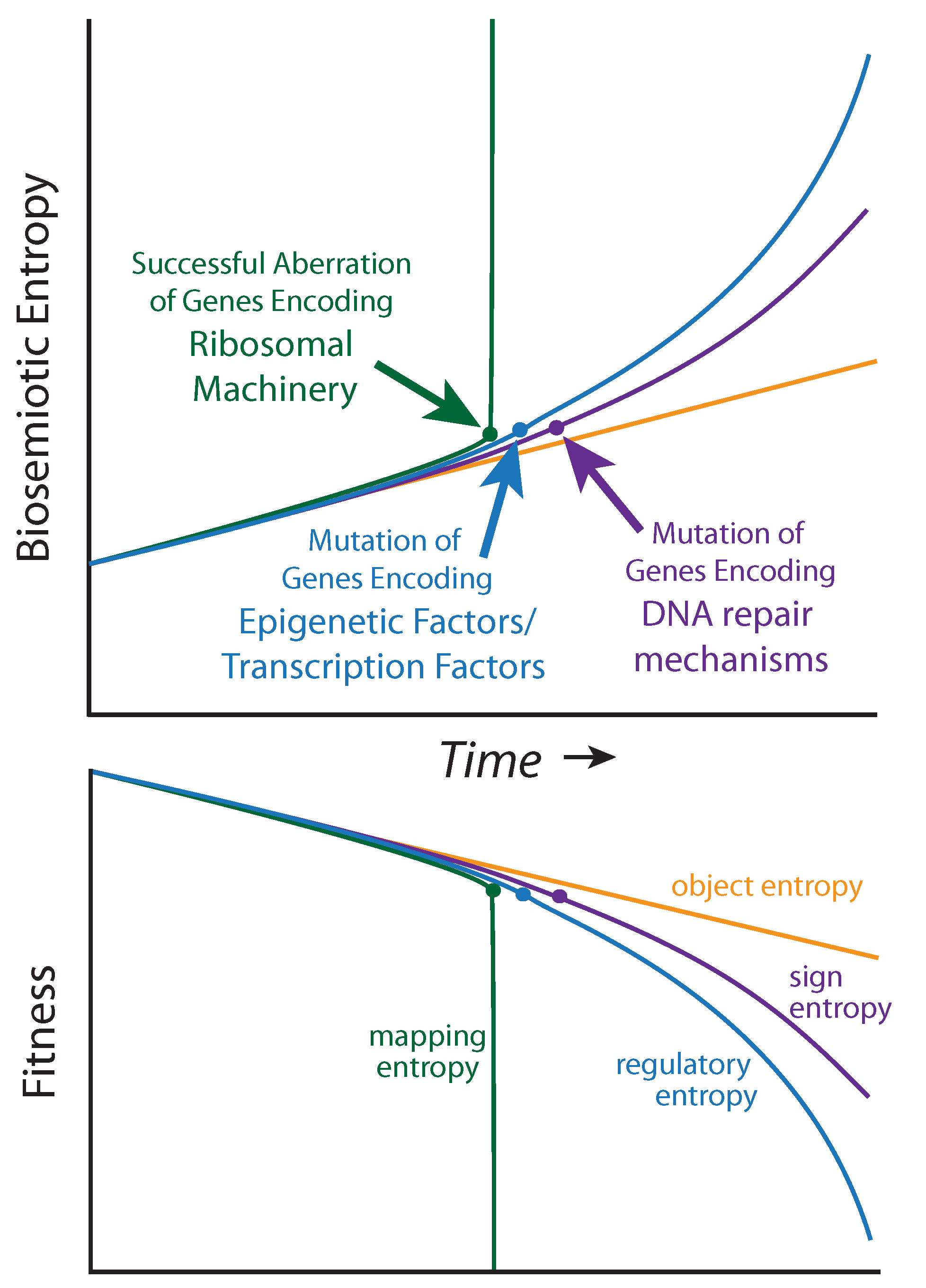
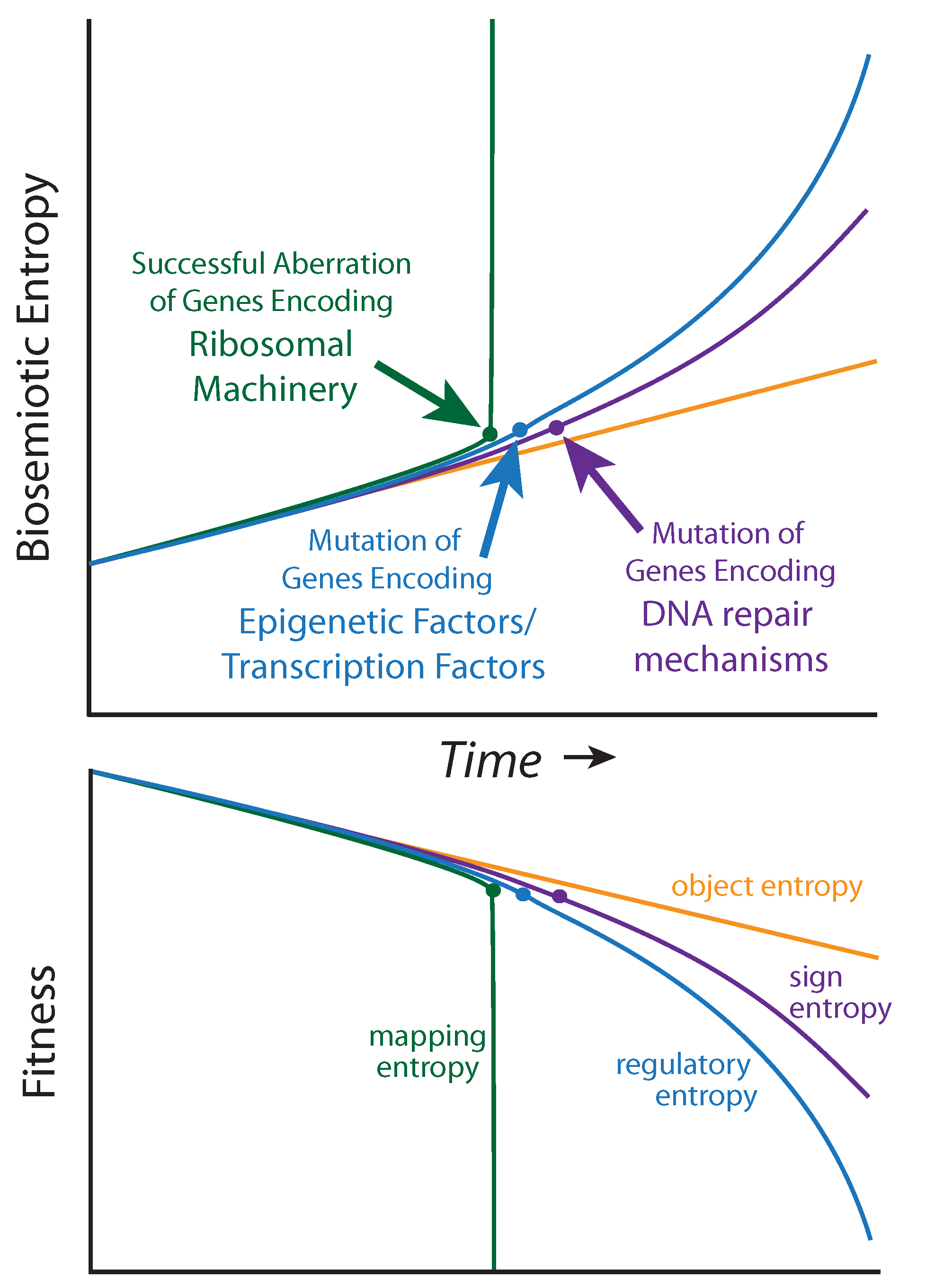
It is not yet clear how to measure biosemiotic entropy in quantitative terms, but qualitative and relative observations can be made from the mutational burden acquired by diseased tissue relative to healthy tissue within a single patient, and, to mutations that predispose individuals to cancer within a population. Biosemiotic entropy can be considered the inverse of phenotypically defined biological fitness (fitness decreases as entropy increases,
Figure 4). Aberrations of super-critical genes (such as those encoding the ribosomal machinery) are
not associated with cancers because they rapidly result in the maximal amount of biosemiotic entropy (
Figure 4), and are therefore efficiently removed by purifying selection, as fitness is reduced to zero (death). Many mutational errors are not studied because they do not promote tumorigenesis. We suggest that regulatory entropy and sign entropy are the two main types of mutations leading to cancer.
Figure 4.
Biosemiotic Pressure Points: a qualitative depiction, suggesting an inverse relationship between biosemiotic entropy and biological fitness/function.
Figure 4.
Biosemiotic Pressure Points: a qualitative depiction, suggesting an inverse relationship between biosemiotic entropy and biological fitness/function.
Carcinogenic entropic pressure points broadly include degradation in genes encoding:
- (1)
DNA repair proteins (such as BRCA1/2, tumor suppressors) [
51]
- (2)
Transcription factors (such as c-Myc [
52], p53 [
53], steroid hormone receptors [
54])
- (3)
Epigenetic factors
DNA Methylation (DNMT)
Writers of histone marks (histone methyltransferase—HMT, histone acetyltransferase—HAT) [
55]
Erasers of histone marks (histone deacetylase - HDAC) [
56]
Readers of the epigenomic code (Histone acetylated lysine readers—Bromodomains, tri-methylated histone lysine (H3K4me3) readers—Tudor domains) [
57]
Genes involved in DNA repair pathways are pressure points of mutational entropy because once they are damaged, the mutation rate for all genes increases dramatically, increasing the chances that a carcinogenic combination of errors will occur. BRCA1 and BRCA2 are examples, as they protect the genome by actively responding to and repairing damaged DNA (via homologous recombination, HR), and women with a mutation in one of these genes have a 50–80% lifetime risk of developing breast cancer (and 50% lifetime risk for ovarian cancer) [
58]. Because many cancers depend on a smaller number of DNA repair pathways, they are more sensitive to drugs inhibiting remaining pathways [
59]. Nevertheless, the importance of damaged DNA repair pathways to cancer initiation has been reduced by recent large-scale studies, which show lower than expected frequency of mutations (only 14–31%) in caretaker genes involved in HR, nucleotide excision repair (NER), mismatch repair (MMR), and others [
60]. These studies had not accounted for pathways being epigenetically silenced, which needs to be considered at least as much as (if not more than) genomic entropy of repair pathways [
61]. The correlations hold strongest for mutations connected to hereditary cancers.
Genes encoding transcription factors are also most frequently associated with cancer progression, precisely because errors in them affect the transcriptome through hundreds or thousands of downstream genes, and controlling key functions such as apoptosis and cell cycle regulation [
62]. In a study performed by the Cancer Genome Atlas project, high grade ovarian cancers were characterized by a 96% frequency of mutation for
TP53 (encoding p53, the master regulatory protein and transcription factor), with mutation in BRCA1 and BRCA2 showing high correlation as well [
51]. Prostate cancer is a disease profoundly dependent upon the androgen receptor (AR), a testosterone responsive TF controlling a broad gene program [
63], which is inhibited by clinically employed antiandrogens [
64] and next generation cancer treatments such as AR-targeting nanoparticle conjugates [
65]. Mutations in the AR can cause disease onset, progression, metastasis, and resistance to therapy [
66,
67].
We predict that newly discovered genes (including non-protein-coding functional elements of DNA) most related to cancer progression will continue to be directly connected to the information flow of biosemiosis, rather than cytoplasmic and extracellular functions. Genes that are furthest downstream of this information flow will tend to have the least effect on carcinogenesis. This is certainly the case for oncogenes, but as our understanding of the functional genome extends beyond the protein-coding regions, further paradigm shifts may be needed. The third pressure point of biosemiotic entropy is in the epigenetic factors, which we consider next as in its own section.
5. Epigenetic Entropy
“Mutagenesis—permanent alteration(s) to the genetic information within previously healthy cells—has long been the main suspect in cancer progression, but the improper regulation of non-mutated DNA is turning out to be a major culprit as well. Among abnormalities that lead to cancerous phenotypes, epigenetic misregulation is reversible by definition, unlike genetic mutations or deletions.”
–Berkley Gryder, Quaovi Sodji and Adegboyega Oyelere [
68]
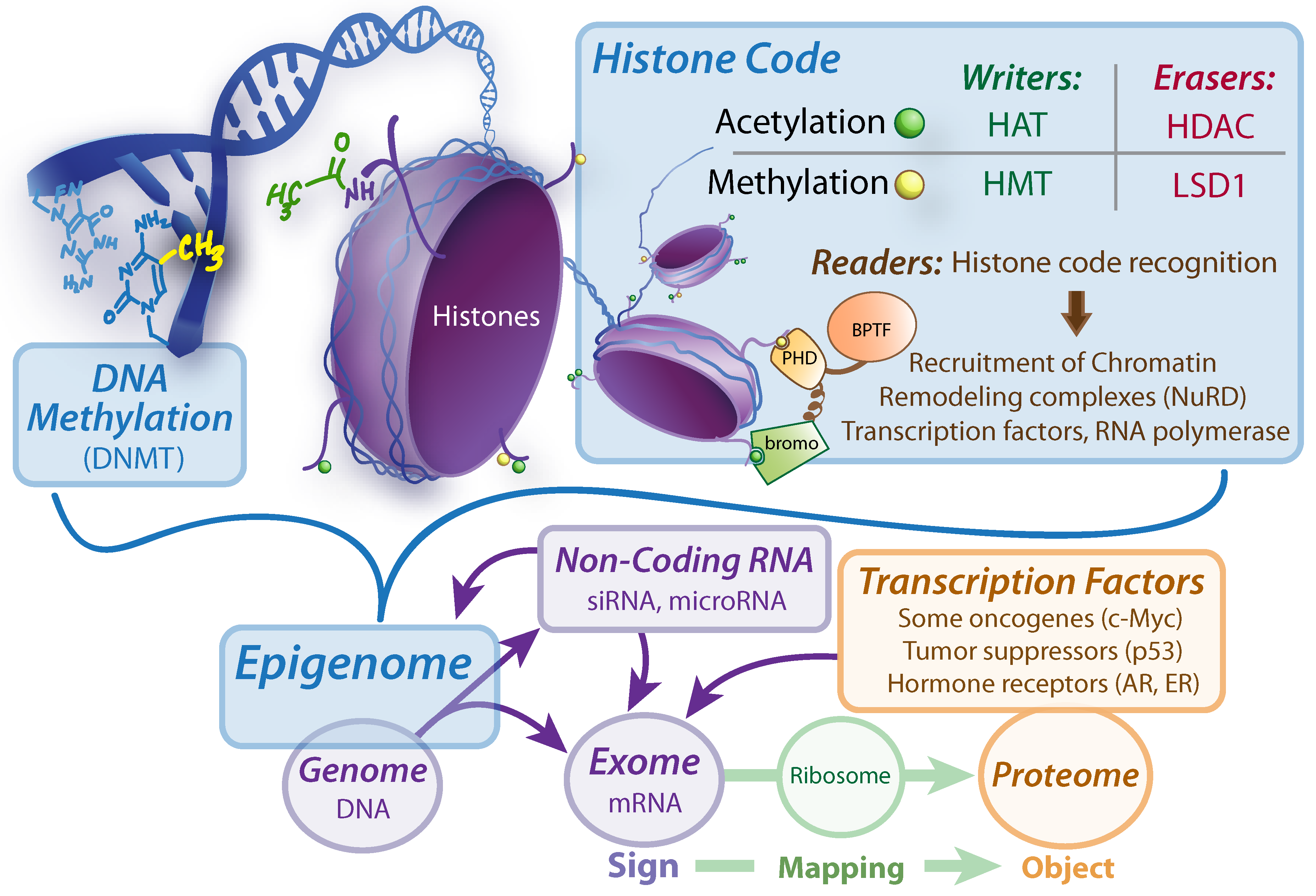
In any given language, how and when an individual says something is just as important as what is said. Genetics is no different, but it does warrant a higher-level consideration of biological biosemiosis. Cells can undergo changes in the amounts of information being generated, without breaking the sign (genes), mapping (translational machinery) or the object (protein) [
69]. The exome (
Figure 5) is a better candidate for the biosemiotic sign than the genome, because only portions of the coded information is transcribed into the directly readable mRNA, which is then translated by ribosomal mapping, and actualized into protein objects. Many factors determine the exome, including epigenomic factors, transcription factors, and non-coding RNA [
70]. In fact, the alterations required to achieve oncogenesis, while traditionally thought to be rooted primarily in a buildup of both inherited and somatic mutations to oncoproteins and tumor suppressors, cancer biology is being revolutionized by the insight that the steps toward malignancy can be caused by epigenetic malfunctions without any new somatic mutations, or a combination of both somatic mutations and aberrant epigenetic programming [
71]. Therefore, while the details of epigenetics found at the same molecular scale as DNA, understanding it requires us to zoom out from the world of genetic biosemiosis, into the world of developmental biology. The developmental pathways are defined not by changes in the DNA code, but rather in the epigenetic regulation of that code (
Figure 5). It is astounding that DNA information encoding for our brain can be found in our index finger, and vice versa. All information required to build hundreds of cell types, complex organs, and ultimately a living being, resides within the single zygote cell at conception, and virtually never changes as cells differentiate and specialize [
72]. The vast differences represented by different cell types are all due to changes in the epigenetic state that determine which sets of genes to turn on, turn off, and to what extent. In linguistic terms, this refers to what is said, when it is said, and what is left unsaid.
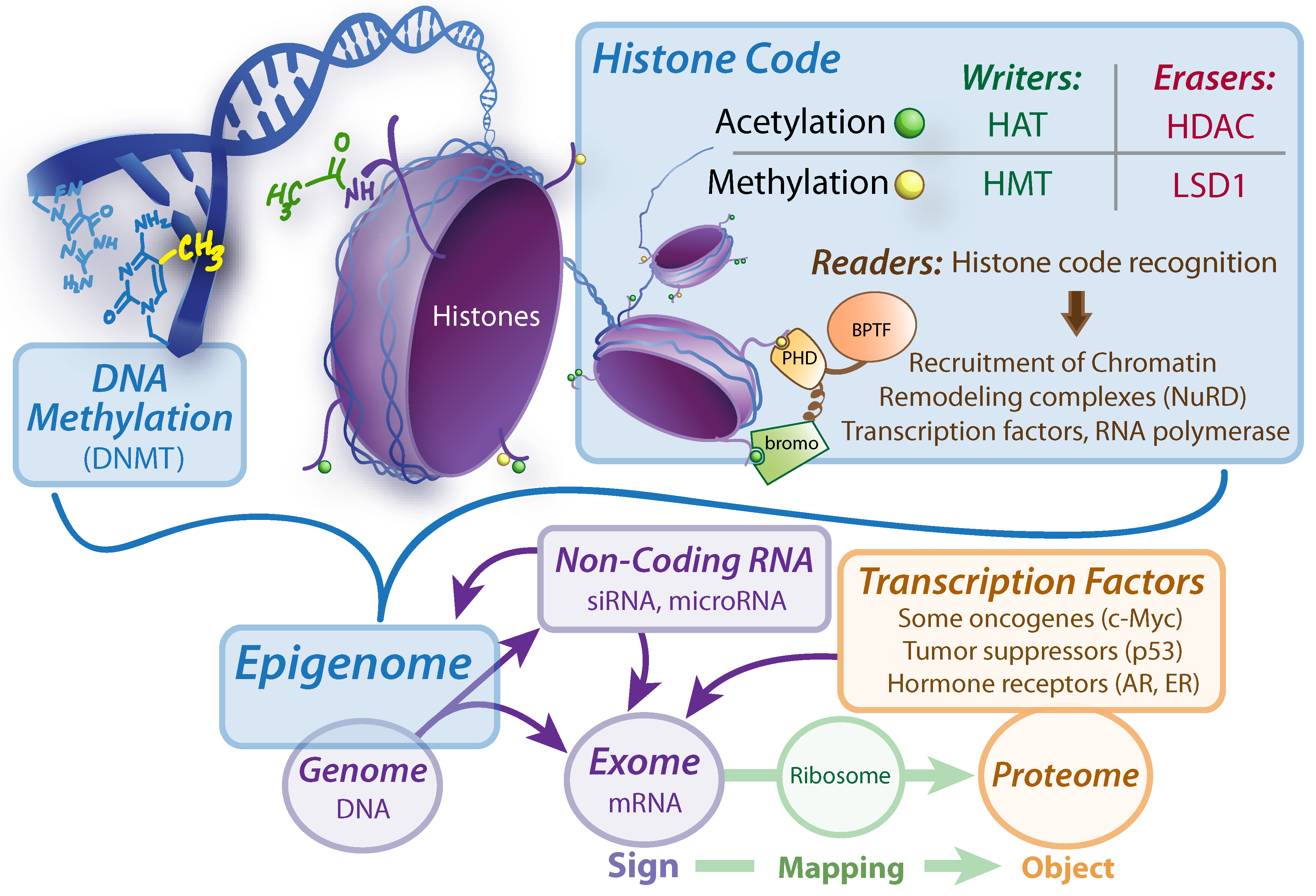
Figure 5.
Sign Regulators: determining the exome. DNMT, DNA Methyltransferase. HAT, histone acetyltransferase. HMT, Histone methyltransferase. HDAC, Histone deacetylase. LSD1, Lysine specific demethylase 1. BPTF, Bromodomain plant homeodomain finger transcription factor. Bromo, bromodomain. PHD, plant homeodomain. siRNA, silencing RNA. AR, Androgen Receptor. ER, Estrogen Receptor.
Figure 5.
Sign Regulators: determining the exome. DNMT, DNA Methyltransferase. HAT, histone acetyltransferase. HMT, Histone methyltransferase. HDAC, Histone deacetylase. LSD1, Lysine specific demethylase 1. BPTF, Bromodomain plant homeodomain finger transcription factor. Bromo, bromodomain. PHD, plant homeodomain. siRNA, silencing RNA. AR, Androgen Receptor. ER, Estrogen Receptor.
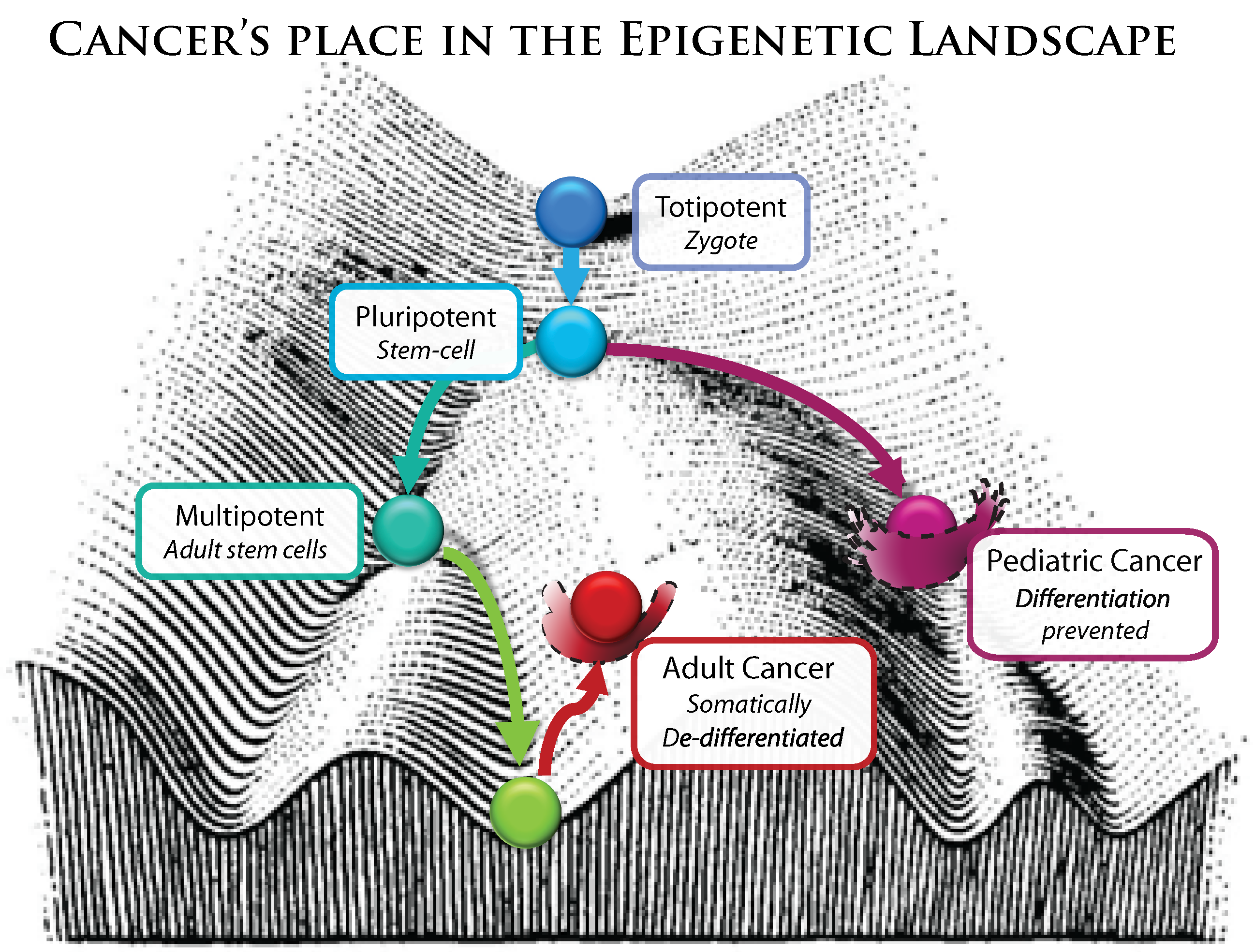
Because cancer is a multicellular phenomenon it is critical to understand it at the epigenetic level, just as much as gene regulation is critical to understand anything in developmental biology. Cancer is now recognized as a dedifferentiated phenotype, resembling pluripotent stem-cells.
There are three main sources of epigenetic malfunction:
- (1)
Direct changes in factors regulating the epigenome, without altering the underlying genetic code
- (2)
Genomic entropy in genes encoding proteins that are involved in maintaining the epigenomic homeostasis of the cell
- (3)
Genomic entropy in non-protein coding elements that regulate epigenomic homeostasis of the cell
5.1. Direct Changes in Factors Regulating the Epigenome
There are many factors which together regulate gene expression in an interconnected (but minimally understood) fashion, all of which can operate without disturbing the genetic information content. Anything directing or effecting the regulation of genes can be considered an epigenetic factor. This would include, mainly, regulatory RNA, transcription factors, and modifications to the structure of DNA and its associated protein scaffolds (known collectively as chromatin). The chromatin structure can be modified by:
Post-translational modification of histone-core octomers, primarily on side chain lysine residues, which can vary in methylation or acetylation status (most commonly), and can even be sumoylated, ubiquitinated, or phosphorylated [
57].
Chromatin remodeling complexes, such as NURF which recognizes specific histone modifications and recruits remodeling factors to uncoil nucleosomes [
73].
Methylation of the DNA base cytosine, most notably of CpG islands found in gene promoter regions [
74].
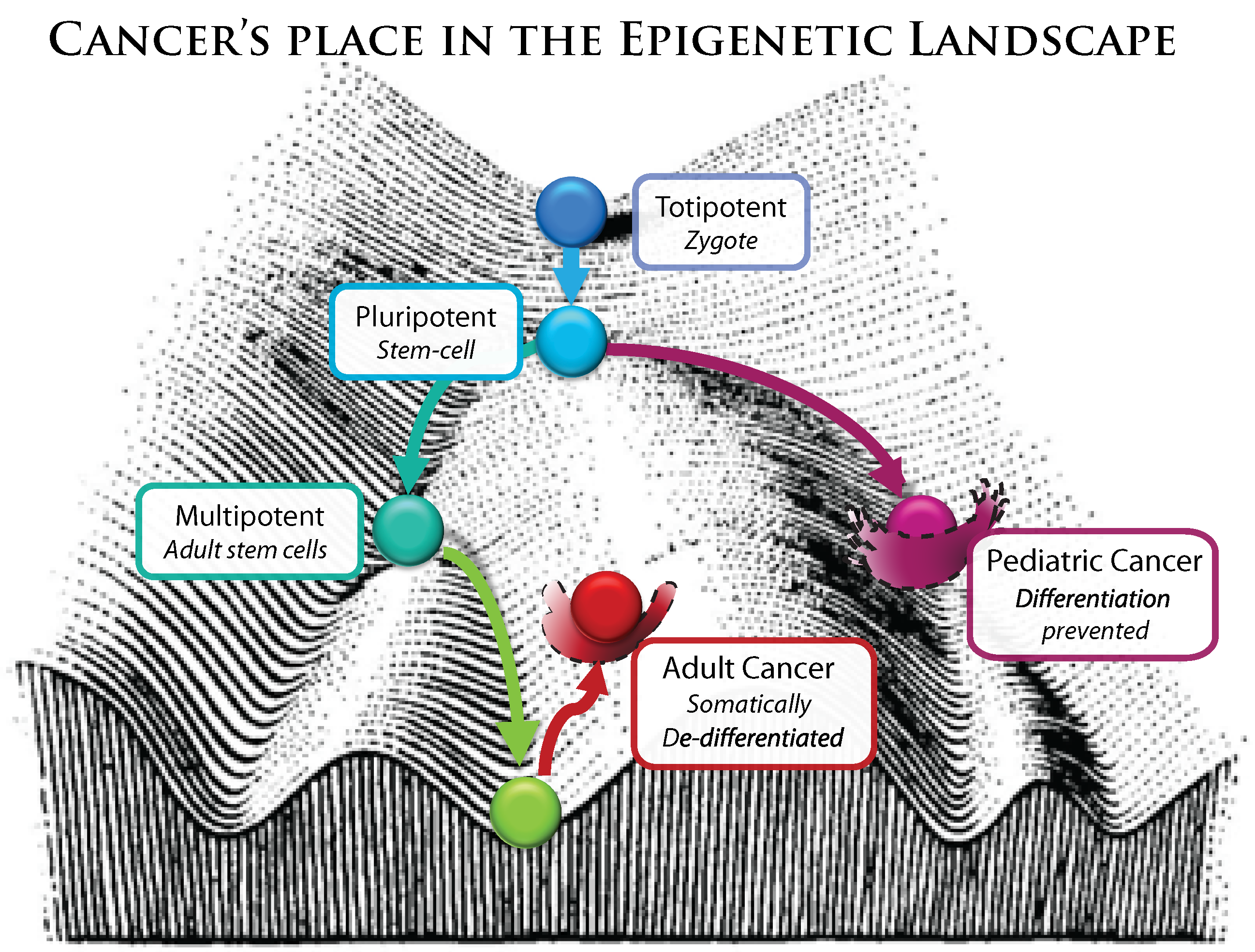
Broadly speaking, any cell stress, any cascade of signaling events or influx of small molecules (from drugs, hormones, toxins or diet) or radiation that can interact with pathways connected to the status of chromatin structure may alter the epigenetic landscape. The vast majority of epigenetic flexibility is designed to 1) maintain homeostasis by rapidly responding to environmental pressures, in addition to 2) creating a landscape of healthy, specialized cell states (thermodynamic wells) and the pathways that connect them (
Figure 6), which is the focus of stem cell and developmental biology. However, the landscape has wells and pathways outside those of normal development, where abnormal gene programs are executed, resulting in neoplastic phenotypes known as cancer. It has recently been noted that environmental stimuli altering the epigenome during fetal development can not only cause obvious birth defects, but also increases risk of carcinogenesis later in life [
75].
Changes in the epigenome (ones that that are not rooted in a genetic mutation) are an exciting target for treatment because those changes are reversible, while (as of yet) genetic errors are not [
76].
Figure 6.
Modified from C.H. Waddinton’s conception of the epigenetic landscape.
Figure 6.
Modified from C.H. Waddinton’s conception of the epigenetic landscape.
5.2. Genomic Entropy in Genes Affecting Epigenetic Factors
It is somewhat difficult to discuss epigenetic malfunctions separately from genetic malfunctions, precisely because mutations in genes encoding epigenetic and regulatory factors will have a broader biological impact. While this kind of error is rooted in an aberrant protein coding gene, the effects resulting in a malignant phenotype are mediated through many programmatic, epigenetic alterations that affect a whole host of genes. These, in effect, reshape the epigenetic landscape by disrupting its landscapers. Adult cancers are often described as “dedifferentiated” (
Figure 6), transformed cells [
77].For instance, recent studies of non-Hodgkin lymphoma revealed a very high frequency of somatic mutations in genes encoding histone methyltransferase (
MLL2, 32–89% mutated) and a gene family (
MEF2, 13–15% point mutated) that encodes calcium-dependent transcription factors which recruit HDACs and HATs to modulate histone acetylation [
78]. Child cancers such as medulloblastoma (a primary brain tumor) [
79] and rhabdomyosarcoma (cancer of connective skeletal tissue) [
80] are classified as developmental cancers, where adolescent maturation is halted for a specific progenitor cell. In the case of rhabdomyosarcoma, it is linked to the fusion oncogene PAX3-FKHR [
81], and has distinct DNA hypermethylation patterns in polycomb transcription factors [
82], which define its abnormal location in the epigenetic landscape (
Figure 6).
5.3. Genetic Entropy in Non-Protein Coding Elements
It has only recently been appreciated how incredibly narrow our view of biological functionality has been. Only 2–3% of the genome encodes proteins, the heart of the central dogma, and is thus the central sign-system studied, even when outlining cellular biosemiosis. Naturally, proteins coding regions have been the primary target for genetics-related research, from basic cell biology to population genetics and drug development initiatives worldwide. However, over the last decade especially, it has become increasingly evident that the other 97% of the genome, far from being “junk DNA”, has important functional roles to play. The myth that biosemiotic entropy was allowed to reign free and devoid of consequence in non-coding regions has been momentously dispelled by the ENCODE project, which gave the final blow with a sword of 30 papers in a single week [
83], finding that at least 80% of the genome is has a biochemical function [
28]. Some non-coding functions have been understood for decades (such as ribosomal RNA or tRNA), while many other functions are still poorly understood. So far they include (
Table 1):
Table 1.
Snapshot Overview of Non Coding Elements in Oncogenesis.
Table 1.
Snapshot Overview of Non Coding Elements in Oncogenesis.
| Non-protein coding elements | Examples of entropy resulting in cancer |
|---|
| Translation of regulatory RNA elements, such as siRNA and microRNA | Program-mediated regulatory networks: mRNAs acting as microRNA sponges can cause glioblastoma when expression levels of sponges for cancer-related genes are changed [84] |
| Translation of other functional RNA elements, such as ribosomal RNA and tRNA | Tumors can be induced by malfunctions leading to increased expression of tRNA for Methionine (which begins protein synthesis) [85] |
| Distal regulatory functions (enhancers, repressors, silencers, insulators) | Hox transcript antisense intergenic RNA (HOTAIR), a long noncoding RNA (lncRNA), is an indicator of poor prognosis for colorectal and breast cancers, via chromatin remodeling in coordination with the Polycomb complex (PRC2) and lysine specific demethylase (LSD1) [86] |
| Cis-regulatory, proximal regulatory functions (promoters, transcription factor binding sites) | Most carcinogenic events in promoter regions are due to hypermethylation, rather than nucleotide mutations. However, it has been shown that SNP (C→T) in the promoter region of MGMT (encodes for DNA a methyltransferase) predisposes this gene to hypermethylation, which is carcinogenic [87] |
| Chromosomal architecture, structural and mechanical functions (such as Telomeres and Centromeres) | Increased transcriptional activity (of the normally repressed DNA) of centromeres induces chromosomal instability (CIN) and aneuploidy, which then leads to carcinogenesis [88] |
| Pseudogenes | The PTEN pseudogene is transcribed and functions as a “sponge” for miRNAs, preventing them from binding to the PTEN gene, thus up regulating PTEN expression. As PTEN is a tumor suppressor, loss of PTEN pseudogene expression and subsequent down regulation of PTEN leads to tumor progression [89] |
Of course this requires an addition/modification to the biosemiotic framework we present here, which has a simple sign-mapping-output (protein coding centric) view of the genome, as these functions impact cellular biosemiosis at various levels. Indeed, entropy in some areas of the non-protein coding regions of the genome frequently have powerfully negative consequences leading to human disease and cancer, not because they lead to aberrations in resultant proteins, but because otherwise healthy genes are misregulated (silenced, minimally expressed, or overexpressed compared to normal cells of the same type). Beyond the obvious importance and centrality of gene-mediated biosemiotic information flow, many other layers of biosemiosis are at work in a maximally integrated fashion, including cell signaling, the immune system [
13], physiological homeostasis [
90], environmental responses, membrane signaling [
91], and water coherence domains [
92], to name a few. These must be considered both individually and collectively to obtain a full understanding of cancer etiology.
6. Fighting Biosemiotic Entropy To Maintain Homeostasis
Characterization of a healthy (or ideal) state is important for understanding the nature of possible malfunctions. Likewise, by carefully observing dysfunction biologists can also clarify normal function (
i.e., gene knockout). The onset of high-throughput genotyping and phenotyping technology has enabled dramatic progress toward characterizing healthy cell states, as well as the somatic mutations and epigenetic states featured associated with cancerous tissues [
93]. When defining the
genetic boundaries of a healthy cell type, two considerations convolute the analysis: first, there is a large amount of healthy genetic diversity within a population and second, many somatic mutations within an individual will have negligible carcinogenicity. Defining the
epigenetic boundaries of a healthy cell type is just as difficult. Measurements for methylation, histone modification, and chromosomal conformation states will be derived as an average from a heterogeneous population of cells. Moreover, clear one-to-one correlations between a single epigenetic mark and a phenotypic outcome are not universal (even within a single cell type) [
94]. The task of understanding and defining a healthy biological state is a collective, ongoing effort at many levels—the health of populations, organisms, organs, tissues, cells, regulatory networks, proteins, genes, and metabolites—all of which must be considered both individually and in connection to each other. The ideal state and mal state are defined by phenotypic output, and are rooted in genotype.
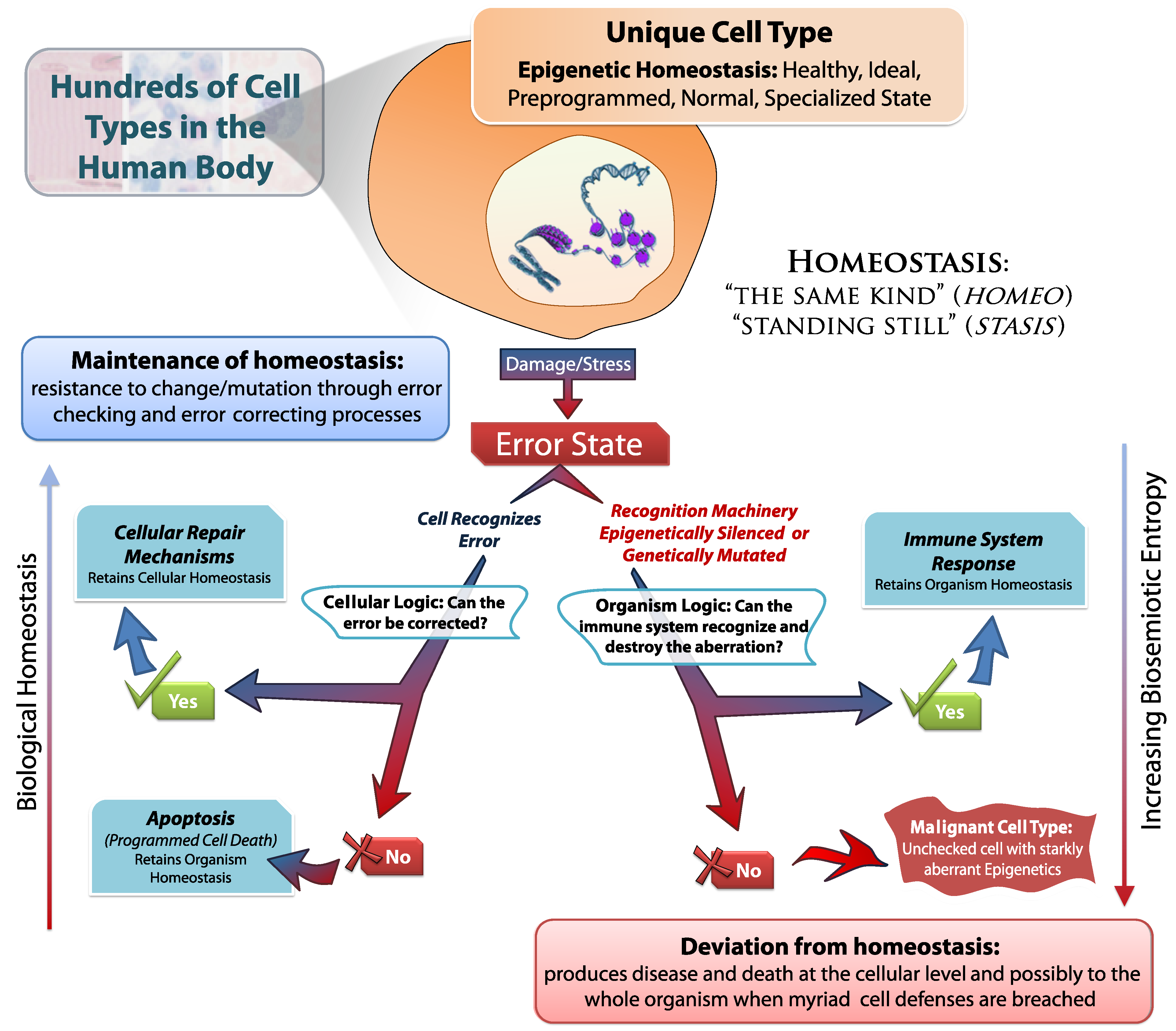
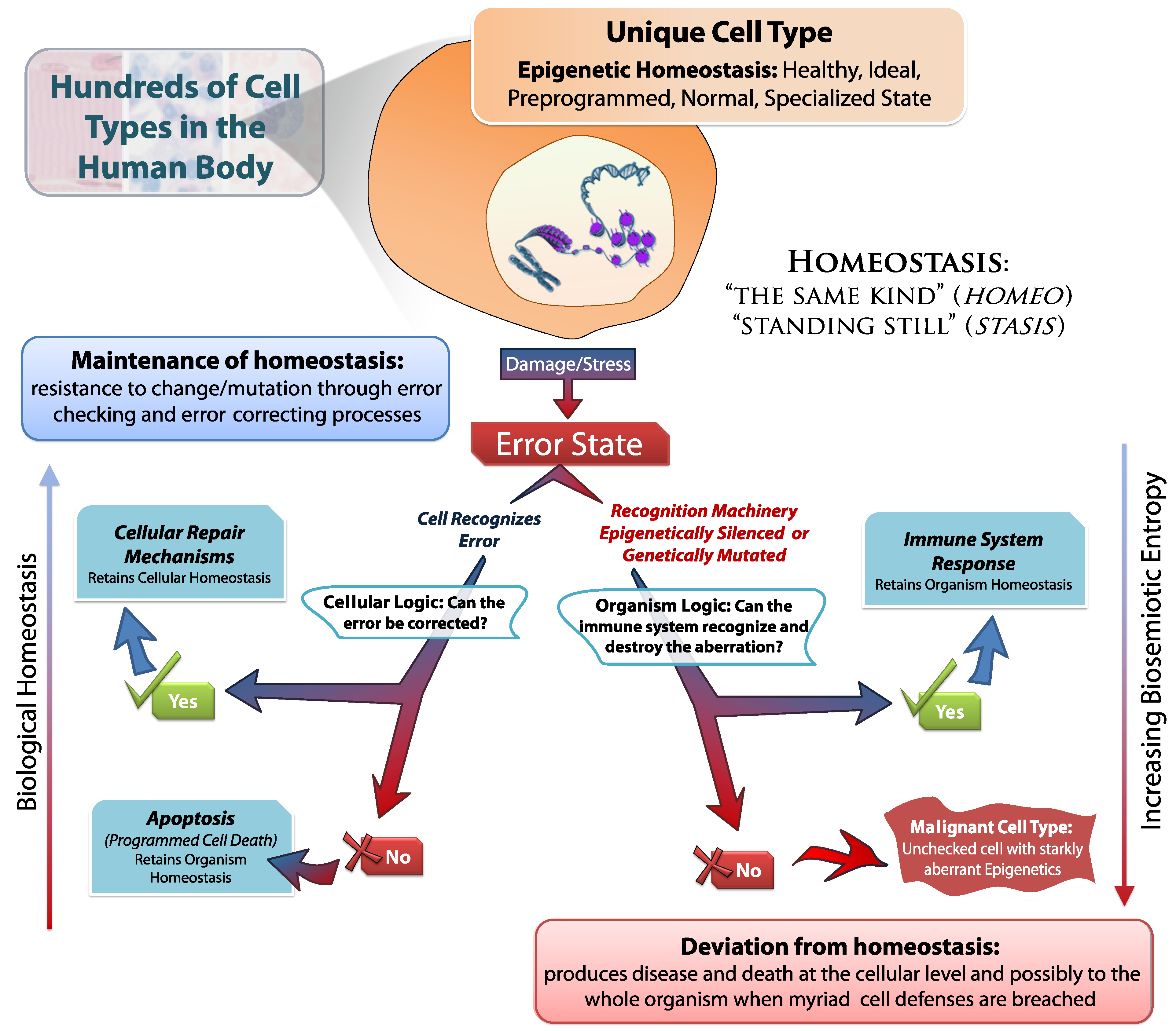
It is the task of all DNA repair pathways; cellular caretaker genes; cell cycle checkpoint machinery; and many other cellular phenomenon to maintain biological homeostasis in response to cellular stress and damage (
Figure 7). There are several levels of entropy correcting logic that must be thwarted in order for tumorigenesis to succeed; from cell cycle maintenance proteins (such as p53/p21) to immune system recognition. Each of these complex systems function to combat the inevitable increase of biosemiotic entropy.
6.1. Various Levels of Homeostasis
The interpretation of a person, organ, tissue, cell, or protein as being normal or healthy is generally definable in a higher context than the object itself. The level of context needed is also situational. First, if one looks too broadly one may not be able to perceive the error at all. For example, genetic mutations resulting in the malfunction of L-gulono-
alpha-lactone oxidase (GLO) is understandable within the context of lost vitamin C biosynthesis; however, the strict observance of the creature at an organismal level, given a diet rich in vitamin C, will not readily expose the problem since selective pressure on GLO will have become effectively neutral [
95]. Second, one may fail to look at a sufficiently high context to ascertain the true state of health conferred by variants. It is well-known that encoding the hemoglobin S variant (protein level) leads to the dysfunctional sickle cell phenotype (cell level). Nevertheless, having both a normal hemoglobin A and hemoglobin S provides protection for humans living in areas endemic with malaria (environmental level) [
96]. Finally, cancer cell lines may be quite fit at the cellular level in terms of their reproducibility and longevity; however, at the level of the whole organism, cancer cells will eventually destroy the very system that sustains them. From these observations we suggest that the definition of a healthy or normal state should include an understanding of higher contextual levels.
Figure 7.
Maintaining Homeostasis by Fighting Biosemiotic Entropy.
Figure 7.
Maintaining Homeostasis by Fighting Biosemiotic Entropy.
Similar to the regulatory feedback of biological systems, the normal state for a computer system is designed such that deviances from it will result in pre-programmed run-time exceptions. These exceptions give the running program an opportunity to deal with problems gracefully rather than crashing and possibly bringing down the system with it. More harmful errors are unanticipated logical errors and system-catastrophic kernel panics. These can be likened to harmful genetic variants. Moreover, before computer programming can be compiled, syntax is checked and errors returned if any are found. This tight feedback ensures uniformity in the computer programming language (for those who write code) as well as its interpretations by the compiler (that which must use the code). As we have discussed, mapping (via ribosomal interpretation) is also vital to proper bio-functioning. Indeed, the standard genetic code is considered robust to translational error [
97] and is further supported by error-correcting activities in polymerases and ribosomes. Similar to biological systems, errors can also be understood in terms of higher contextual levels: an arithmetic or read error is subject to its algorithmic purpose while the algorithm itself is subject to the design of its application. The behavior of an application may in turn be understood within the context of other applications and the stability of the operating system. Just as the complexity of biological and computer systems can be understood in terms of embedded function and layered logic, the state of their health and the evaluation of their normalcy is often understood at higher contextual levels than the immediate function being performed.
6.2. Entropy at the Population Level
Sign entropy (genetic mutation) represents the most serious long-term threat to biological systems. While sign entropy may affect prokaryotes, several considerations suggest that the greatest long-term threat involves complex eukaryotes. These considerations include: (1) the relatively large effective population sizes of microorganism species, which increase the efficacy of purifying selection; (2) the lower mutation rate per effective genome in microorganisms; (3) the haploidy of microorganisms, leaving deleterious mutations relatively “exposed” as compared to diploid or polyploid organisms; (4) the relatively small size (and streamlined nature) of microorganism genomes, allowing each site to contribute a correspondingly large proportion of the total genetic information, such that deleterious mutations have larger effects on average, making purifying selection more effective; and (5) information stored and exchanged on plasmids, which can sometimes compensate for damaged genomic material in prokaryotes.
The above considerations suggest that, while the toleration of deleterious mutations is
proximally advantageous—that is, immediately advantageous to the mutant organism—it may nevertheless adversely affect long-term viability (
i.e., it is
ultimately disadvantageous). Thus, efforts to reduce human mutation rates should continue to receive high priority. Although there is evidence of widespread purifying selection in the human genome [
31], it is unclear whether this can successfully counteract genetic entropy.
It is therefore of great interest to determine the critical population sizes, mutation rates, distributions of mutational fitness effects, and effective genome sizes, at which purifying selection breaks down. Classic
in vitro serial transfer experiments make it clear that viruses can sometimes afford to lose (by deletion) the majority of their genomes excepting critical replication machinery. For example, in a classic serial transfer experiment with Qβ bacteriophage, replication rate increased by a factor of 15 and genome size decreased by 83%, with biological competency lost by the fifth transfer [
98]. Some reductions in genome size have also been observed in evolution experiments with E. coli (e.g., a 1.2% reduction [
99]). Thus, many microorganisms may have the ability to reach an effective genome size that allows persistence, as decreasing the effective genome size decreases the effective mutation rate on average. Whether complex eukaryotes could survive such a loss is less clear. Such considerations led initially to the introduction of the “junk DNA” concept for the human species, assuming a genomic mutation rate of only 1 per generation [
100]. New evidence that the majority of the human genome is biochemically active, if not biologically functional, revives the imminence of these concerns [
21] (as powerfully demonstrated by the ENCODE project [
28], discussed above). While mechanisms that maintain homeostasis and canalization certainly ensure a relatively plastic informational system, large amounts of mutational damage can still accumulate to such an extent that all functionality is lost.
6.3. Germline Predispositions towards Somatic Biosemiotic Entropy
As evolutionary biologists have long noted, genetic entropy of the germline leads to mutational change that can act slowly as it accumulates over generations, potentially leading to extinction [
25,
35,
101,
102]. On the other hand, genetic entropy of somatic cells is the cause of cancer in a single individual’s lifetime. However, germline entropy may still contribute to cancer at the population level, also exhibiting acute effects in the life of a single individual, e.g., through the accumulation of neutral mutations that predispose a genome to a cancerous state. Such mutations are themselves neutral and may become common (or even fixed) by random genetic drift. If such mutations represent a substantial fraction of the mutations that occur, the resulting entropy will go virtually undetected by purifying natural selection until enough mutations accumulate to predispose a large portion of the population to cancer. Such predisposition may represent a state that is only a handful of critical mutations removed from a cancerous state. Such considerations cannot be quantified until more is learned about what genetic changes cause cancer predisposition, and whether these changes are predominantly specific residues or more generally distributed [
25].
7. Conclusions
The biosemiotic model of the genome provides an interpretive framework that is more nuanced (as it includes epigenetic considerations) and more informative than the simple oncogene/tumor suppressor gene model, particularly for interpreting cancer. Although some of the formality is new, the biosemiotic perspective articulates many concepts that are implicit in the field [
2]. The delineation of at least three distinct but deeply layered entropic locations—sign, mapping, and object—allow increased precision in identifying various biosemiotic causes of cancer. Further, the distinction between entropy accumulation in somatic and germline cells—which have consequences over single lifetimes and many generations, respectively—allows mutation accumulation to be analyzed, prevented, and treated in its appropriate context. Sign entropy resulting from the accumulation of slightly deleterious mutations poses the greatest threat to a species’ long-term survival, while relatively drastic genomic aberrations in somatic cells can lead to cancer within an individual organism.
One useful implication of the biosemiotic framework is the existence of a healthy state. Biosemiotic entropy at the germline level [
25], which can occur despite certain levels of purifying selection, may increase genetic predisposition to entropy in somatic cells if compensating factors are not acquired. A crucial next step will be the identification of the biological parameters and mechanisms (e.g., mutation rates, population sizes, and canalization mechanisms) that are relevant to allowing this entropic accumulation, as well as their critical values and states. Such an analysis would allow the more precise identification of what point(s) in historical time (if any) biosemiotic entropy began to overwhelm error-correction mechanisms and purifying selection. This could in turn allow identification of more functional genetic states for various genes that contribute to cancer. Only by integrating the medical individual-focused view (dealing with lifetimes) and the generational population-level view (dealing with evolutionary time) can comprehensive solutions to cancer be addressed.
It is a daunting task to distill the major themes of human cancer biology into a few simple concepts. We have attempted a first construction using the framework offered by biosemiotic theory, and looked at the major ways in which the sign-mapping-object triad can be disrupted, with a special emphasis on cancer—a disease for which a wealth of genomic, epigenomic, transcriptomic, and proteomic information exists. Many of the propositions we have offered remain to be elaborated in a rigorous, mathematical fashion, leaving exciting work for future explorations.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}